

Abstract
α-Synuclein (α-Syn) is a key protein that accumulates as hyperphosphorylated aggregates in pathologic hallmark features of Parkinson’s disease (PD) and other neurodegenerative disorders. Phosphorylation of this protein at serine 129 is believed to promote its aggregation and neurotoxicity suggesting that this post-translational modification could be a therapeutic target. Here, we demonstrate that protein phosphatase 2A (PP2A) dephosphorylates α-Syn at serine 129, and that this activity is greatly enhanced by carboxyl methylation of the catalytic C subunit of PP2A. α-Syn transgenic mice raised on a diet supplemented with eicosinoyl-5-hydroxytryptamide (EHT), an agent that enhances PP2A methylation, dramatically reduced both α-Syn phosphorylation at Serine 129 and α-Syn aggregation in the brain. These biochemical changes were associated with enhanced neuronal activity, increased dendritic arborizations, reduced astroglial and microglial activation, as well as improved motor performance. These findings support the notion that serine 129 phosphorylation of α-Syn is of pathogenetic significance, and that promoting PP2A activity is a viable disease modifying therapeutic strategy for α-synucleinopathies such as PD.
Introduction
α-Synuclein (α-Syn) is a key protein in Parkinson’s disease (PD) and other α-synucleinopathies due to genetic linkage and its aggregation in Lewy bodies and Lewy neurites, the pathological hallmarks of disease (reviewed in (Goedert, 2001)). The oligomerization and subsequent fibrillization of α-Syn are believed to play a major role in neuronal dysfunction and death. Postmortem human brain studies have shown that the protein is selectively and extensively phosphorylated at serine 129 in synucleinopathy lesions of PD and Dementia with Lewy Bodies (Fujiwara et al., 2002; Neumann et al., 2002; Anderson et al., 2006). Hyperphosphorylated and misfolded α-Syn also accumulates in affected neurons of transgenic mice that express human α-Syn (Neumann et al., 2002) and leads to increased α-Syn toxicity in Drosophila (Chen and Feany, 2005). In vitro, phosphorylation of α-Syn at serine 129 promotes its oligomerization and fibrillization (Fujiwara et al., 2002). Therefore, moderation of phosphorylation and aggregation of α-Syn is hypothesized to be of therapeutic value.
Levels of α-Syn phosphorylation are regulated by a balance between rates of phosphorylation by protein kinases and dephosphorylation by phosphoprotein phosphatases. Phosphorylation at Ser129 can be mediated by multiple kinases including Casein Kinases (Ishii et al., 2007), G-protein coupled receptor kinases (Pronin et al., 2000) and Polo-like kinases (Inglis et al., 2009; Mbefo et al., 2010), while the dephosphorylating enzyme has received little attention. Phosphoprotein Phosphatase 2A (PP2A) is the primary Serine/Threonine phosphatase in the brain accounting for over 50% of total brain Ser/Thr phosphatase activity (Strack et al., 1997). It exists functionally as trimeric holoenzymes whose assembly and activity are regulated by reversible carboxyl methylation of the catalytic C subunit, which in complex with a structural A subunit promotes association of the ‘AC’ dimer with regulatory B subunits that confer substrate specificity (Bryant et al., 1999; Tolstykh et al., 2000; Wu et al., 2000). The methylation status of PP2A is governed by the opposing activities of a PP2A-specific methyltransferase (PPMT) and a specific methylesterase (PME-1) (Lee and Stock, 1993; Lee et al., 1996; Ogris et al., 1999).
Here, we show that activation of PP2A stimulates the dephosphorylation of α-Syn in vitro and in vivo, and the resulting reduced level of α-Syn phosphorylation relieves the neuropathology and behavioral deficits of a transgenic mouse model of α-synucleinopathy.
Materials and Methods
Reagents
PP2A AC dimer and ABαC trimer were expressed and purified using previously published protocols (Tolstykh et al., 2000; Xing et al., 2006). PP2A specific MTase (PPMT) and MEase (PME-1) purifications were performed as described (Xing et al., 2008). PP1cα (rabbit muscle, recombinant, E. coli) was purchased from Calbiochem. EHT was synthesized at Signum Biosciences. [3H]-SAM was purchased from Perkin Elmer. Antibody sources are: anti-α-synuclein and anti-iNOS (BD Transduction Laboratories, San Jose, CA), anti-phosphoser129-α-synuclein (WAKO, Osaka, Japan), anti-demethylated PP2A, anti-PP2A-B, and anti-PP2A-C, (Millipore, Temecula, CA), anti-HDAC1, anti-MAP2 and anti-c-fos (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), anti-GFAP (DAKO, Carpinteria, CA), and anti-CD11b (AbD Serotec, Oxford, UK). The anti-demethylated PP2A antibody, 4b7, critical for these studies has well characterized specificity (Yu et al., 2001) and, in our hands, demonstrates selective recognition for demethylated over in vitro methylated PP2A.
PP2A phosphatase activity towards α-synuclein
C-terminal His6-tagged human α-synuclein was cloned into pET21b vector and expressed in E. coli BL21DE3 cells. Affinity (Ni-NTA, Sigma-Aldrich) and anion exchange chromatography (Mono-Q, GW Healthcare) was used to purify protein. Phosphorylation was performed by incubation with casein kinase 2 in 20 mM Tris, pH 7.5, 100 mM NaCl, 10 mM DTT, 5 mM MgCl2 buffer with 2 mM ATP, overnight at 37 °C. Phosphorylated α-synuclein was purified using anion exchange chromatography (Mono-Q, GW Healthcare).
To assay for phosphatase activity towards pS129 α-Syn, PP1 or PP2A (methylated or demethylated) were serially diluted to different concentrations and then incubated with 1.1 μM phosphorylated α-Syn in buffer containing 50 mM MOPS, pH 7.2, 5 mM DTT, and 100 μM Mn2+ at 30°C for 30 min. The reactions were stopped by adding 5X SDS sample buffer. Samples were resolved by SDS-PAGE, transferred onto nitrocellulose membranes and incubated with anti-pS129 α-Syn antibody overnight at 4°C. Secondary anti-rabbit antibody was applied and blots developed using ECL Plus (GE Healthcare Life Sciences). Membranes were scanned with a Storm Scanner (GE Healthcare Life Sciences).
PP2A demethylation assay
Recombinant, methylated human PP2A AC dimer ([3H]-methyl-PP2A) was freshly prepared by incubating PP2A, PPMT and [3H]-SAM at 37°C for 1hr. Unreacted SAM and PPMT were removed by ion exchange chromatography. EHT was pre-incubated with 10 nM PME-1 for 10 minutes, followed by addition of 50 nM [3H]-methyl-PP2A. Reaction was allowed to proceed for 30 minutes at room temperature and was stopped by addition of 10% TCA, and ~0.5 μg of BSA. Reactions were centrifuged for 10 minutes at 12,000 rpm to pellet the precipitated protein. An aliquot of the supernatant was added to scintillation fluid (Ecoscint, National Diagnostics) and counted on a scintillation counter (Beckman) to determine the amount of [3H]-methanol liberated by PME-1.
Cell culture and treatment
SH-SY5Y cells stably over-expressing α-Syn were cultured as described (Kanda et al., 2000). For PP2A knockdown experiments, these cells were transfected with siRNAs for 72 h at 100 nM final concentration using Lipofectamine 2000 reagent (Invitrogen). SMARTpool siRNAs of non-targeting (NT), PP2A C subunit (Cα), or Bα subunit were obtained from Thermo Scientific Dharmacon. Primary neuronal cultures were prepared from E18 rat embryos by standard procedures. Briefly, cortical neurons were dissociated by brief mechanical trituration and suspended in full neurobasal media supplemented with B27 (GIBCO), penicillin, streptomycin, and 2mM L-glutamine. After 7 days in vitro (d.i.v.), neurons were treated for 4 hours with serial dilutions of EHT in ethanol (1% v/v final concentration).
Animals and EHT administration
All animal procedures were approved by the UMDNJ – Robert Wood Johnson Medical School Institutional Animal Care and Use Committee and were performed according to the NIH Guide for the Care and Use of Laboratory Animals. Male mice transgenic for human wild-type α-Syn under the control of the Thy-1 promoter (Rockenstein et al., 2002) and wild-type littermates were housed 2 per cage under a 12-hour dark-light cycle with ad libitum access to food and water. The transgenic line was maintained on the mixed C57BL/6 – DBA/2 background by breeding mutant females with wild-type males. Upon weaning, animals were placed on a diet containing 0.01% EHT (n = 13 α-SynTg; n = 10 wild-type), 0.1% EHT (n = 12 α-SynTg; n = 9 wild-type) or control chow (n = 12 α-SynTg; n = 9 wild-type). Mice were weighed and their food intake quantified weekly. Behavioral tests were performed every 3 months. At 9 months of age, animals were anesthetized, perfused with PBS (pH 7.4), and the brains quickly dissected. One hemi-brain was immediately frozen for Western blot analyses and biochemical assays, and the other hemi-brain post-fixed in 4% paraformaldehyde in PBS (pH 7.4) at 4°C overnight for immunohistochemical stains.
Behavioral tests
Sensorimotor behavioral assessments were performed at 3, 6 and 9 months of age. The rotarod test was performed using the automated TSE system. Mice were placed on the rod at a constant speed of 4 rpm for 30 seconds, and then tested with an accelerating speed of 0.2 rpm/sec for a total of three trials. Latency to fall was measured for each trial and the average of three trials was recorded. Nesting behavior was performed as described (Sager et al., 2010) by placing the animals in a new cage with a 5 cm tightly packed cotton square Nestlet. Eighteen hours later, nest formation was rated by a blinded observer on a scale of 0 = non shredded to 5 = maximally shredded (Deacon, 2006).
Western blotting and Immunohistochemistry
For Western blot analysis, brain tissue was homogenized in 4°C lysis RIPA buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 1% NP-40, 0.1% SDS, 0.5% sodium deoxycholate) containing phosphatase inhibitor cocktail set II (Calbiochem, La Jolla, CA) and protease inhibitor cocktail set V (Calbiochem, La Jolla, CA), and then centrifuged at 14,000 rpm at 4°C for 30 min. The supernatants (soluble fraction) were saved, and the pellets (insoluble fraction) were lysed with buffer (1% SDS in PBS) containing protease and phosphatase inhibitor cocktail. Each lane was loaded with 20 μg protein. Separated proteins were transferred onto a PVDF membrane (Bio-Rad, Hercules, CA, USA), and the membranes were blocked with 5% non-fat dry milk in Tris-buffered saline and 0.1% Tween 20. Immunoblots were developed using ECL plus (PerkinElmer, Boston, MA). Isolation of nuclear and cytoplasmic fractions was performed by the Nuclear Extract Kit (Active Motif, Carlsbad, CA) according to the manufacturer’s protocol. Specificity of the nuclear fraction was assessed by SDS-PAGE followed by Western blot analysis with anti-HDAC1 antibody.
For immunohistochemistry, 40 μm thick coronal free-floating sections were blocked by 5% BSA. Biotinylated HRP complex (Vector Laboratories, Burlingame, CA) and 3.3′-diaminobenzidine were used for color development. For image analysis, computer-assisted image analysis program Bioquant Nova Prime Version 6.90 (Carl Zeiss, Germany) was used. Analysis of the in vivo data was confirmed by blinded members of the research team.
Statistical analysis
Data are presented as means ± S.E.M. and analyzed by one-way analysis of variance (ANOVA) followed by the Newman-Keuls multiple range test. Significance was determined at p < 0.05.
Results
PP2A dephosphorylates pS129 α-Syn in a methylation dependent manner
To determine the dephosphorylating enzyme, purified human α-Syn was incubated in vitro with casein kinase 2, a major enzyme in brain that phosphorylates serine 129 of α-Syn (Ishii et al., 2007). The resulting phosphorylated α-Syn was incubated with different concentrations of purified and reconstituted AC dimers of Phosphoprotein Phosphatase 2A (PP2A) or purified Phosphoprotein Phosphatase 1 (PP1). Western blotting of reaction products using p-S129 α-Syn antibody indicated marked dephosphorylation by PP2A but not PP1 (Fig. 1A, B). The role of PP2A was also confirmed in human dopaminergic neuroblastoma SH-SY5Y cells stably over-expressing α-Syn. Incubating these cells with the broad specificity protein phosphatase inhibitor okadaic acid resulted in increased p-S129 α-Syn whereas the PP1 specific inhibitor tautomycetin showed little or no effect (Fig. 1C). In this cellular system, tautomycetin was able to inhibit PP1 significantly while having no effect on PP2A (Fig. 1D). Okadaic acid has a broad spectrum and can inhibit additional phosphatases at nM concentrations (Swingle et al., 2007). To confirm the role of PP2A in dephosphorylating α-Syn, siRNA to the C or Bα subunits were transfected into α-Syn over-expressing SH-SY5Y cells, and a significant increase in p-S129 α-Syn was detected by Western blotting (Fig. 1E, F). These in vitro and cell-based findings coupled with the high expression of PP2A in the brain (Strack et al., 1997) indicate that PP2A is a major Ser/Thr phosphatase of α-Syn.
Figure 1.
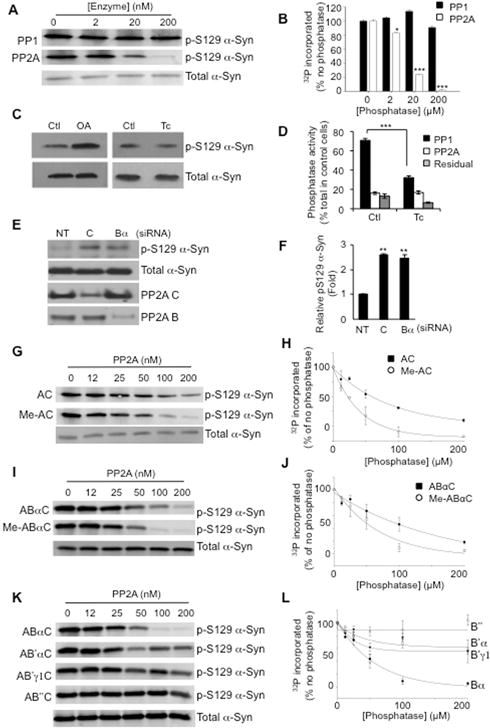
PP2A catalyzes the dephosphorylation of pS129 α-Syn in a methylation dependent manner. A, In vitro dephosphorylation reactions performed by incubating phosphorylated α-Syn with PP1 (upper panel) or PP2A (lower panel) demonstrate that PP2A, but not PP1 dephosphoylates α-Syn in a concentration dependent manner. B, Quantification of the effects of PP1 and PP2A on α-Syn dephosphorylation. * p < 0.05; *** p < 0.005. C, Western blotting of lysates from SH-SY5Y cells stably expressing α-Syn treated for 1 hour with 250 nM Okadaic acid (OA) or 1 μM Tautomycetin (Tc) with p-S129 α-Syn antibody shows that inhibition of PP2A but not PP1 modulates α-Syn phosphorylation (upper panels). Total levels of α-Syn are unaffected by OA or Tc treatment (lower panels). D, Tautomycetin is active on PP1 in SH-SY5Y cells. Cells incubated with ethanol (control) or 1 μM Tc for 20 min were lysed, and in vitro dephosphorylation reactions performed using 32P-labelled phosphorylase a. Activities defined as: PP1 the fraction sensitive to 2 μM OA but insensitive to 5 nM OA; PP2A the fraction sensitive to 5 nM OA, and residual fraction insensitive to 2 μM OA. *** p < 0.005. E. Knockdown of PP2A subunits results in increased p-S129 α-Syn. SH-SY5Y cells expressing α-Syn were transfected with siRNA of non-targeting (NT), catalytic C subunit, or Bα subunit. Cell lysates were subjected to Western blotting for p-S129 α-Syn, total α-Syn, PP2A C subunit, PP2A B subunit, and β-actin (loading control). Representative data from 3 separate experiments are shown. F, Quantification of p-S129 α-Syn compared to total α-Syn with knockdown of PP2A subunits. ** p < 0.01. G, Western blots with pS129 α-Syn antibody of in vitro dephosphorylation reactions performed by incubating phosphorylated α-Syn with PP2A AC dimer (AC, upper panel) or methylated AC dimer (Me-AC, lower panel) indicate that methylation increases the ability of PP2A AC to dephosphorylate α-Syn at Ser129. H, Quantification of the effects of methylation of AC dimers on PP2A activity toward phosphorylated α-Syn. I, Heterotrimeric assemblies of PP2A ABαC subunits (ABαC, upper panel) increase activity to dephosphorylate α-Syn subsequent to methylation (Me- ABαC, lower panel). J, Quantification of the effects of methylation of ABαC heterotrimers on PP2A activity toward phosphorylated α-Syn. K, Different regulatory B subunits strongly influence the ability of PP2A to dephosphorylate α-Syn at Ser129. L, Quantification of the effects of B subunit composition on PP2A activity toward phosphorylated α-Syn.
The impact of PP2A methylation and subunit composition on p-S129 α-Syn was investigated next. Phosphorylated human α-Syn was incubated with different concentrations of unmethylated or in vitro methylated PP2A AC dimers. Western blotting for p-S129 α-Syn showed significantly greater dephosphorylation at a lower concentration of methylated AC dimer compared to the unmethylated form (Fig. 1G, H). In addition to this direct effect on PP2A phosphatase activity, the importance of methylating the C subunit of PP2A in this process was also confirmed and extended with a trimeric form of PP2A. Demethylated-AC or methylated-AC dimers were reconstituted with Bα subunit to yield ABαC and Meth-ABαC holoenzymes, respectively, and incubated with p-S129 α-Syn. The more efficient dephosphorylation of p-S129 α-Syn occurred with Meth-ABαC (Fig. 1I, J). To further assess the specificity of PP2A’s activity on p-S129 α-Syn, we examined the efficiency of dephosphorylation by alternative heteromeric PP2A assemblies. The trimeric holoenzyme containing Bα subunit (ABαC) dephosphorylated p-S129 α-Syn more efficiently than holoenzymes containing the B′α (AB′αC), B′γ1 (AB′ γ1) or B″ (AB″C) isoforms (Fig. 1K, L). These results suggest that methylated ABαC PP2A isoform is the major phosphatase acting on p-S129 α-Syn.
Based on reported aberrations in PP2A in the brains of Alzheimer’s disease (AD) patients (Sontag et al., 2004a; Sontag et al., 2004b), we sought to build on our findings with α-Syn and identify agents that inhibit the demethylation of PP2A as potential therapeutics for PD. In an in vitro screen of natural products, inhibitory activity to demethylation was identified in coffee extracts. Since a negative correlation has been observed between coffee consumption and the incidence of both PD and AD (Barranco Quintana et al., 2007; Saaksjarvi et al., 2008), this activity was intriguing for further study. The active agent was purified to homogeneity and identified as eicosinoyl-5-hydroxytryptamide (EHT). Synthetic EHT exhibited the same ability to inhibit PP2A carboxyl demethylation as EHT derived from coffee. EHT inhibits PME-mediated demethylation of methylated PP2A under defined conditions with the purified enzymes (IC50 = 3.9 μM, Fig. 2A). Importantly, EHT was efficacious in cells, inhibiting the demethylation of PP2A in cultured hippocampal neurons (Fig. 2B).
Figure 2.
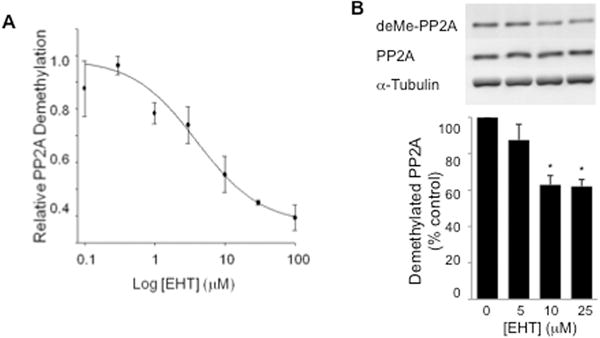
EHT inhibits PP2A demethylation. A, In vitro PP2A demethylation is inhibited by EHT. In vitro assay performed with PP2A AC dimers, methylated and labeled using 3H-S-adenosylmethionine and PP2A methyl transferase, incubated with varying concentrations of EHT and 400 nM PP2A methyl esterase. Amount of 3H remaining in PP2A was assessed using liquid scintillation counting. B, EHT inhibits demethylation of PP2A in cultured neurons. Primary hippocampal neurons were incubated with varying concentrations of EHT for 4 hours and harvested for Western blotting. Demethylation of PP2A was inhibited in a dose dependent manner by EHT (upper panel and graph, * p < 0.05), while total levels of PP2A and α-tubulin (lower panels) were unchanged (n = 3 experiments).
Enhancing PP2A methylation prevents α-Syn phosphorylation and aggregation in transgenic mice
As enhancing PP2A methylation increases its activity towards α-Syn in vitro (Fig. 1G–J), we postulated that inhibiting the demethylation of PP2A could increase its activity and reduce levels of phosphorylated α-Syn in vivo, leading to decreased protein aggregation and neuroprotection.
Pharmacokinetic studies delivering [3H]-EHT intraperitoneally revealed that the compound is able to cross the blood-brain barrier achieving μM levels of 3H-label, sufficient to inhibit PP2A demethylation, indicating its feasibility for systemic administration. Male mouse pups transgenic for human α-Syn (α-SynTg) under the control of the Thy-1 promoter (Rockenstein et al., 2002) and wild-type littermates were placed upon weaning on a diet containing two different concentrations of EHT (0.01% and 0.1%) or control feed, and chow consumption was measured weekly. Given ad libitum access to feed, these EHT concentrations translate to 12 mg/kg/day and 120 mg/kg/day, respectively. Motor behavior was tested every 3 months and animals were sacrificed at 9 months for biochemical and immunohistochemical studies.
Consistent with the pharmacological action of EHT in cultured hippocampal neurons, Western blotting of cortical tissue lysates showed that the percentage of demethylated PP2A relative to total PP2A expression decreased by 76% in EHT treated wild-type mice and by 58% in α-SynTg mice compared with their respective control chow fed littermates (Fig. 3A–D). As total PP2A expression was not affected by this treatment, the decline in the levels of demethylated enzyme indicates a dramatic decrease in the rate of PME’s demethylation activity towards PP2A. This was associated with a significant 45% decline in the amount of p-Ser129-α-Syn in the insoluble fraction of brain lysates from animals fed with 0.1% of the compound (Fig. 3B, E). A dose-dependent decrease in high-molecular weight insoluble α-Syn aggregation by Western blotting was also seen with EHT treatment compared to controls (Fig. 3B, F), while, levels of monomeric α-Syn exhibited no significant alterations (Fig 3B). Subcellular fractionation of brain lysates performed to isolate nuclear and cytoplasmic compartments indicated that p-Ser129-α-Syn is localized primarily in the cytoplasm (Fig. 3G).
Figure 3.
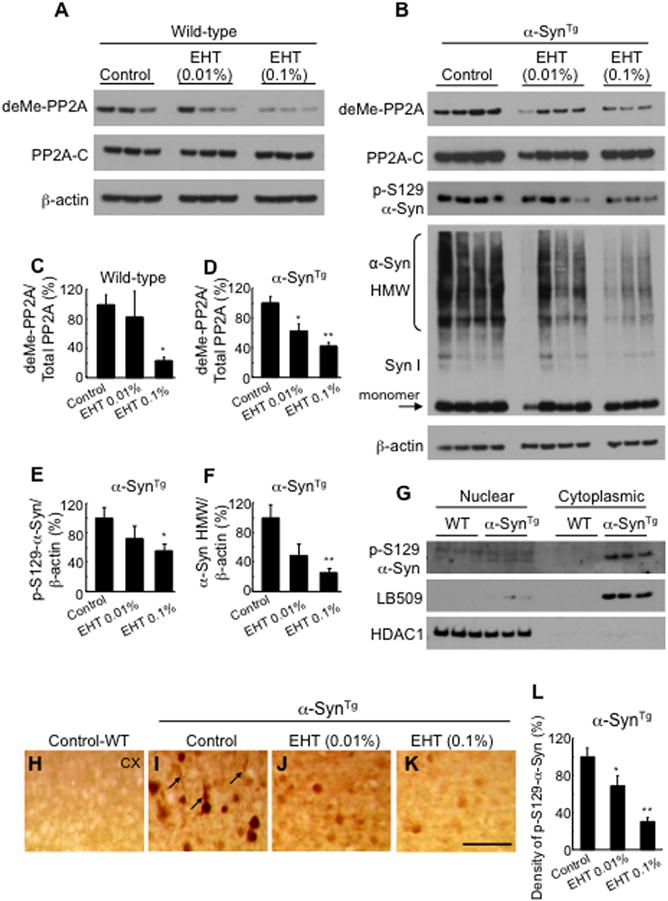
EHT administration modulates PP2A methylation and prevents α-Syn phosphorylation and aggregation in transgenic mice. A, Western blots of cortical tissue lysates from wild-type mice. RIPA insoluble fraction was solubilized with 1% SDS and probed with antibodies to demethylated PP2A and total PP2A-C subunit. B, Western blots for PP2A as in Fig. 2A with lysates from α-SynTg mice. These samples were also probed with phospho-S129-α-Syn antibody and total α-Syn antibody (Syn 1) showing reduction of high molecular weight aggregation. C, Quantification of band intensities in Fig. 2A showing significant reduction with 0.1% EHT administration (n = 3 for each bar). D–F, Quantification of band intensities in Fig. 2B showing significant decrease in demethylated PP2A, phospho-S129-α-Syn and aggregated α-Syn (n for control feed = 4; low dose = 4; high dose = 3). G, Western blots of fractionated brain lysates with phospho-S129-α-Syn antibody and LB509 (human monoclonal α-Syn antibody) show the phosphorylated transgene expressed primarily in the cytoplasmic fraction, while the nuclear marker HDAC1 is in the nuclear fraction. H–K, Immunohistochemistry with phospho-S129-α-Syn antibody of cortical tissue from α-SynTg mice treated with control, 0.01% and 0.1% EHT, respectively, demonstrating significant decrease in the number of immunoreactive neurons and neurites (arrows). Scale bar = 100 μm. L, Quantification of the number of cortical phospho-S129-α-Syn immunoreactive neurons (n for control feed=6; low dose = 5; high dose = 4). * p < 0.05, ** p < 0.001.
Immunohistochemical analysis confirmed the effects of EHT administration on p-α-Syn accumulation (Fig. 3H–L). The number of p-Ser129-α-Syn immunoreactive neurons in the cortex of EHT treated α-SynTg mice decreased dramatically in a dose dependent manner, with a 31% decrease in low dose treated animals and 70% in the high dose group (Fig. 3L). Similarly, a substantial decrease in p-Ser129-α-Syn immunoreactive neurites was also noted in the cortex of EHT treated mice (Fig. 3I–K).
Improved neuronal activity and dendritic arborization by EHT treatment
The neuropathological impact of α-Syn over-expression and the effect of chronic EHT treatment were addressed next. α-SynTg mice have substantial depletion of the cytoskeletal microtubule associated protein 2 (MAP2) in the cortex (Fig. 4A, B, I) indicative of reduced dendritic complexity (Harada et al., 2002). However, animals treated with EHT demonstrated a noticeable, dose-dependent, increase in MAP2 immunoreactive neurites (Fig. 4C, D, I). In addition, a marked depletion of the number of neurons immunoreactive to the immediate early gene product c-Fos (Palop et al., 2003), which is a surrogate marker for neuronal activity, was noted in the dentate gyrus of the hippocampi of α-SynTg mice compared to wild-type animals (Fig. 4E, F, J). This was prevented in EHT treated animals (Fig. 4G, H, J). EHT did not impact the number of c-fos immunoreactive neurons in wild-type mice (Fig. 4K–N). The α-SynTg mice used in these studies do not demonstrate degeneration of nigral dopaminergic neurons by 9 months of age. They also do not have appreciable deficits in striatal nerve terminal integrity assessed by synaptophysin immunohistochemistry nor do they have significant depletion of striatal catecholamines or TH content.
Figure 4.
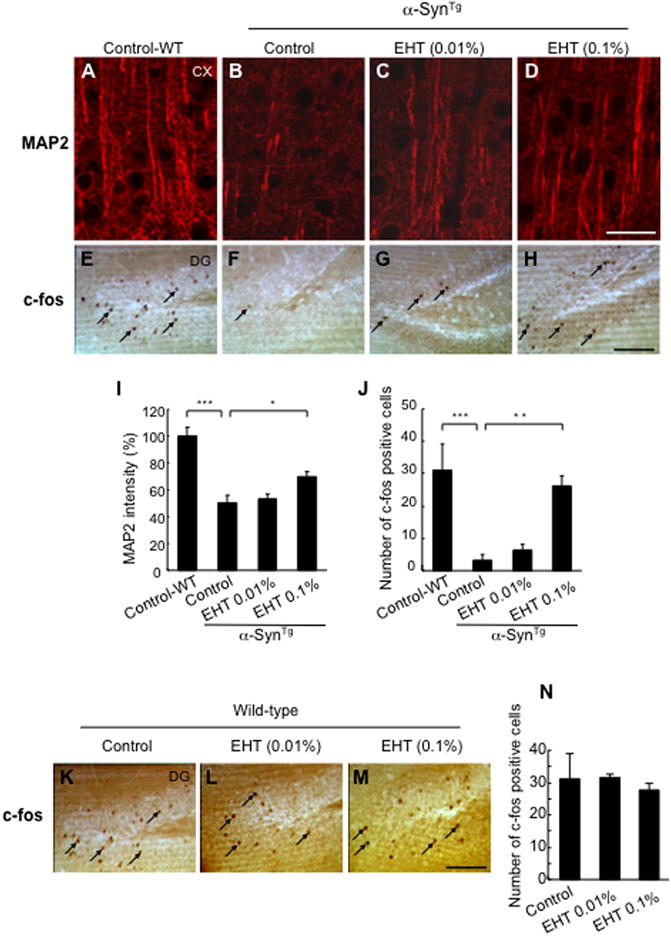
Improved dendritic arborization and neuronal activity by EHT. A–D, Confocal micrographs of cortical sections stained with MAP2. Scale bar = 25 μm. E–H, c-fos immunoreactive neurons in the hippocampus. Arrows point to some of the c-fos positive neurons. Scale bar = 100 μm. I, Quantification of MAP2 immunoreactive dendrites by ImageJ (n for each of the four groups = 5.) * p < 0.05, *** p < 0.001. J, Quantification of c-fos positive neurons in the dentate gyrus. (n for control fed wild-type mice = 4, control fed α-SynTg = 6, low dose α-SynTg = 4, high dose α-SynTg = 4).** p < 0.01, *** p < 0.001. K–N. c-fos immunoreactive neurons in the dentate gyrus of the hippocampus in wild-type mice. Arrows point to some of the c-fos positive neurons. No difference was found with EHT administration (n for control fed = 4; low dose = 3; high dose = 3). Scale bar = 100 μm.
Repression of astrocytic and microglial activation by EHT treatment
Histopathological studies of non-neuronal cells also revealed significant changes with α-Syn overexpression and EHT treatment. α-SynTg mice given control feed demonstrated marked astroglial activation as detected by increased numbers of GFAP immunoreactive cells in cerebral cortex and hippocampus (Fig. 5A, B, E, F). EHT treatment resulted in the presence of fewer GFAP positive cells (Fig. 5C, D, G, H). These changes in astrocytic cells with transgenic α-Syn expression and EHT treatment paralleled the profile of GFAP expression observed by Western blotting (Fig. 5M, N). A similar trend of microglial proliferation was noted in the cortex of untreated transgenic mice and diminished by EHT administration (Fig. 5I–L). Consistent with the latter finding, Western blot analysis showed that the expression of inducible nitric oxide synthase (iNOS) in the cortex of untreated transgenic mice was significantly greater than in wild-type littermates but decreased with EHT treatment (Fig. 5M, O). Notably, this treatment had no effect on GFAP or iNOS expression in wild-type animals (Fig. 5M–O). These findings collectively point to a significant astroglial and microglial activation by transgenic over-expression of human α-Syn, and the diminution of this reactive process by EHT.
Figure 5.
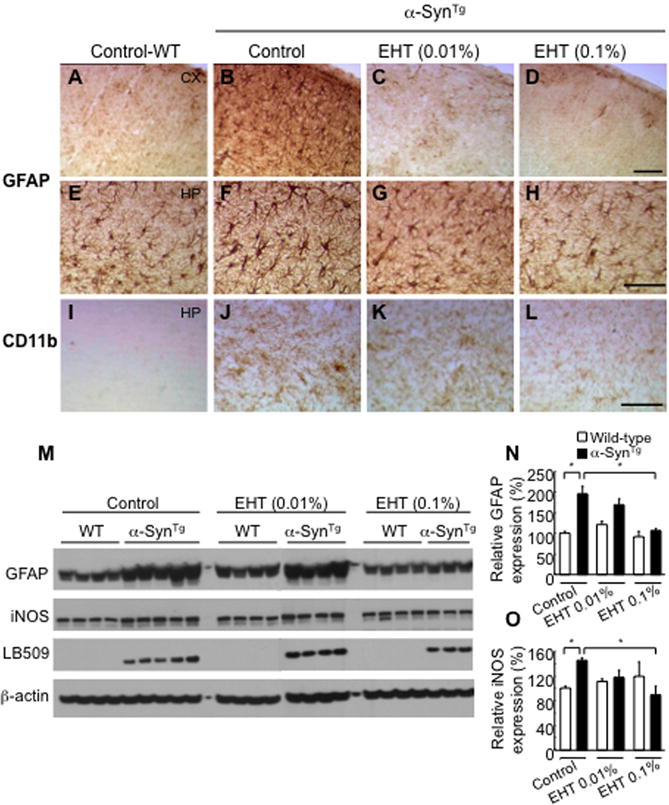
Repression of glial activation by EHT administration. A–H, Immunohistochemistry for GFAP in cortex (top row) and hippocampus (second row) demonstrating significantly more intense staining in α-SynTg mice compared to wild-type littermates and reduction with EHT treatment. Scale bar = 100 μm. I–L, Staining with the microglial marker CD11b showing the same trend. M, Western blotting of cortical tissue lysates (soluble fraction) with GFAP, iNOS and LB509 (human monoclonal α-Syn antibody). N, Quantification of GFAP band intensities relative to β-actin shown in panel M. O, Quantification of iNOS band intensities shown in panel M. * p < 0.05.
Improved behavioral outcome of α-SynTg mice with EHT treatment
Sensorimotor behavior assessments showed significant improvement in transgenic mice raised on EHT containing diet. Rotarod performance, which is markedly impaired in transgenic mice compared to wild-type littermates on control feed, was significantly improved with 0.1% EHT at 3 months (Fig. 6A) and 6 months of age (Fig. 6B). Nesting behavior, which requires shredding cotton from a tightly packed square, was also markedly impaired in α-SynTg mice at 9 months but was significantly improved in EHT treated animals (Fig. 6C). Although nesting behavior has been used to assess nigrostriatal sensorimotor function in rodents (Szczypka et al., 2001; Deacon, 2006), the α-SynTg mice used in the present study are significantly impaired with respect to this complex behavior, as has been described previously (Fleming et al., 2004), despite lack of appreciable dopaminergic deficits by 9 months of age. This behavioral impairment has been attributed to a decline in fine motor skills or reduction in the shredding urge (Fleming et al., 2004), perhaps due to cortical dysfunction related to high levels of α-Syn expression (Kolb and Whishaw, 1985; Rockenstein et al., 2002).
Figure 6.
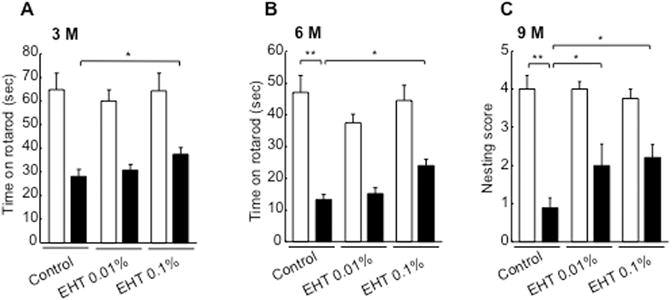
Amelioration of α-Syn mediated behavioral deficits by EHT administration. A, Performance on the rotarod at 3 months of age. (n for control fed wild-type mice = 9; control fed α-SynTg = 12; low dose wild-type = 10, low dose α-SynTg = 13, high dose wild-type = 9; high dose α-SynTg = 12). B, Performance on the rotarod at 6 months. (n for control fed wild-type mice = 9; control fed α-SynTg = 12; low dose wild-type = 10, low dose α-SynTg = 11, high dose wild-type = 9; high dose α-SynTg = 10). C, Nesting behavior scored at 9 months (n for control fed wild-type mice = 4; control fed α-SynTg = 4; low dose wild-type = 4, low dose α-SynTg = 6, high dose wild-type = 6; high dose α-SynTg = 5). * p < 0.05; ** p < 0.01.
Discussion
The findings presented here demonstrate that PP2A catalyzes the dephosphorylation of phospho-Ser129 α-Syn and that the methylation state of the PP2A C subunit regulates this activity. Furthermore, enhancement of PP2A methylation is beneficial in reducing α-Syn phosphorylation and aggregation, preventing neuropathological features associated with α-Syn, and improving behavioral outcome in α-Syn transgenic mice. Interplay between α-Syn and PP2A in regulating tyrosine hydroxylase activity in dopaminergic neurons has previously been described (Saraf et al., 2007; Lou et al., 2010). The results reported here suggest that PP2A acts as an upstream regulator of α-Syn phosphorylation so that agents that enhance PP2A phosphatase activity toward phospho-Ser129 α-Syn may provide therapeutic benefits.
Our results also support a significant role of α-Syn phosphorylation at serine 129 in neuronal dysfunction in vivo. Prior studies concerning the role of this phosphorylation using genetic or viral vector mediated delivery of Ser129Asp or Ser129Ala α-Syn mutants in rodent and Drosophila models have yielded inconsistent results (Chen and Feany, 2005; Gorbatyuk et al., 2008; Azeredo da Silveira et al., 2009; McFarland et al., 2009). Although designed as a phospho-mimetic, in vitro biophysical studies indicate that S129D does not faithfully reproduce the effects of native phosphorylation (Paleologou et al., 2008). On the other hand, our present observations are based on pharmacological manipulation of the native phosphorylation state of α-Syn in the brain.
The mechanism(s) of the protective effects of EHT downstream of α-Syn in this mouse model of α-synucleinopathy deserve additional consideration. The marked depletion of c-fos immunoreactive neurons in the hippocampus of α-SynTg mice indicates severely impaired neuronal activity as a result of α-Syn overexpression. The substantial improvement of this marker with EHT treatment suggests protection either directly through dephosphorylation of p-Ser129 α-Syn or indirectly through some off-target effect of EHT that may or may not be related to increased PP2A activity. The fact that EHT did not impact c-fos immunoreactive neurons in wild-type mice supports the notion that this protective effect is directly associated with the modulation of α-Syn phosphorylation. Preservation of MAP2 positive dendritic arborizations implies improved neuronal integrity. This could also be related to maintenance of microtubule networks through decreased phosphorylation of tau, another target of the ABαC neuronal variant of PP2A (Liu et al., 2005). Molecular, genetic and neuropathological observations point to an association between α-Syn and tau, and phosphorylated tau inclusions have been detected in another α-Syn transgenic mouse line (Frasier et al., 2005). Whether the effects of EHT on α-Syn or tau predominate in explaining the results seen in the present study has not been definitively proven, although tau phosphorylation appears to be downstream of α-Syn (Frasier et al., 2005; Khandelwal et al., 2010). Since the mice used in this study do not have demonstrable synaptic terminal deficits by 9 months of age as determined by synaptophysin immunoreactivity, protecting these terminals anatomically is an unlikely mechanism. However, it is still possible that synaptic transmitter release is impaired in these mice, as has been suggested in other murine models (Abeliovich et al., 2000; Yavich et al., 2004), and that EHT treatment could have prevented this impairment. It is unlikely that the effects we have observed are through modulating phosphorylation of multiple PP2A substrates as methylation only strongly affects the assembly of Bα containing PP2A holoenzymes (Longin et al., 2007). Therefore, the effects of EHT, through modulating PP2A methylation, could only influence the demonstrated substrates of this isoform, namely α-Syn (the present study), tau (Liu et al., 2005), β-catenin (Zhang et al., 2009) and Raf (Kao et al., 2004). The strong correlation between our dose-dependent effects on PP2A methylation and α-Syn dephosphorylation make this the most obvious pathway, although others cannot be completely discounted.
Our data point to an additional mechanism for the protective activity of EHT through inhibition of microglial/astroglial activation and iNOS expression. Neuroinflammation is a key component of PD (Tansey and Goldberg, 2010), and can influence α-Syn aggregation (Gao et al., 2008). Although α-Syn mediated microglial activation has been reported in mouse models of α-Syn overexpression (Su et al., 2008; Theodore et al., 2008), it is unknown whether S129 phosphorylated α-Syn is a stronger trigger of this reactive process than unphosphorylated α-Syn. Additionally, EHT could have a direct anti-inflammatory effect through activation of PP2A (Shanley et al., 2001). This would be responsible for reduced glial activation independent of its actions on α-Syn phosphorylation.
Modulation of PP2A provides a novel approach for treating PD and other α-synucleinopathies. Interestingly, the active compound in these studies, eicosanoyl-5-hydroxytryptamide (EHT) was identified as the major PP2A demethylation inhibitor in coffee. A number of epidemiological studies have suggested that consumption of this beverage is associated with a reduced risk of developing PD (Hellenbrand et al., 1996; Fall et al., 1999; Benedetti et al., 2000). Some of these studies have suggested caffeine to be the active ingredient in this inverse association (Ross et al., 2000; Ascherio et al., 2001), and the inhibitory effect of caffeine on adenosine A2a receptors is implicated in preclinical studies to protect dopaminegic neurons against toxins such as MPTP (Chen et al., 2001). The present observations provide an additional active agent in coffee that appears to exert beneficial pharmacological effects in the brain through α-Syn. Appreciable levels of EHT and closely related N-alkanoyl-5-hydoxytryptamides are present in coffee, averaging 0.5 mg/g (Lang, 2005), suggesting that human exposure to EHT through coffee consumption may be a contributing factor to the putative beneficial effect of this beverage in reducing PD risk.
Taken together, the above in vitro and in vivo findings suggest that targeting PP2A is a promising therapeutic strategy to mitigate the neuropathology of α-synucleinopathies. This approach appears particularly effective considering the fact that multiple kinases phosphorylate α-Syn at serine 129 (Pronin et al., 2000; Anderson et al., 2006; Inglis et al., 2009; Mbefo et al., 2010) and, therefore, inhibiting a single kinase is unlikely to impact significantly the extent of this post-translational modification. Recent studies with sodium selenate, a PP2A enhancer, have demonstrated that such an approach may also be relevant in reducing tau hyperphosphorylation in aged mice and in transgenic tauopathy models of AD (Corcoran et al., 2010; van Eersel et al., 2010). PP2A modulation may, therefore, provide a common strategy for treating neurodegenerative proteinopathies associated with hyperphosphorylated pathogenic proteins (Hanger et al., 2009).
Acknowledgments
The authors thank Teresa Chan for technical assistance and Yulia Frenkel for manuscript preparation. This work was supported by a grant from Signum Biosciences to M.M.M. who is the William Dow Lovett Professor of Neurology. J.B.S. is supported in part by a grant from the American Parkinson Disease Association.
Footnotes
Disclosure: J.B.S. is a founder of Signum Biosciences, Inc., which employs the indicated co-authors.
References
- Abeliovich A, Schmitz Y, Farinas I, Choi-Lundberg D, Ho WH, Castillo PE, Shinsky N, Verdugo JM, Armanini M, Ryan A, Hynes M, Phillips H, Sulzer D, Rosenthal A. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron. 2000;25:239–252. doi: 10.1016/s0896-6273(00)80886-7. [DOI] [PubMed] [Google Scholar]
- Anderson JP, Walker DE, Goldstein JM, de Laat R, Banducci K, Caccavello RJ, Barbour R, Huang J, Kling K, Lee M, Diep L, Keim PS, Shen X, Chataway T, Schlossmacher MG, Seubert P, Schenk D, Sinha S, Gai WP, Chilcote TJ. Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J Biol Chem. 2006;281:29739–29752. doi: 10.1074/jbc.M600933200. [DOI] [PubMed] [Google Scholar]
- Ascherio A, Zhang SM, Hernan MA, Kawachi I, Colditz GA, Speizer FE, Willett WC. Prospective study of caffeine consumption and risk of Parkinson’s disease in men and women. Ann Neurol. 2001;50:56–63. doi: 10.1002/ana.1052. [DOI] [PubMed] [Google Scholar]
- Azeredo da Silveira S, Schneider BL, Cifuentes-Diaz C, Sage D, Abbas-Terki T, Iwatsubo T, Unser M, Aebischer P. Phosphorylation does not prompt, nor prevent, the formation of alpha-synuclein toxic species in a rat model of Parkinson’s disease. Hum Mol Genet. 2009;18:872–887. doi: 10.1093/hmg/ddn417. [DOI] [PubMed] [Google Scholar]
- Barranco Quintana JL, Allam MF, Serrano Del Castillo A, Fernandez-Crehuet Navajas R. Alzheimer’s disease and coffee: a quantitative review. Neurol Res. 2007;29:91–95. doi: 10.1179/174313206X152546. [DOI] [PubMed] [Google Scholar]
- Benedetti MD, Bower JH, Maraganore DM, McDonnell SK, Peterson BJ, Ahlskog JE, Schaid DJ, Rocca WA. Smoking, alcohol, and coffee consumption preceding Parkinson’s disease: a case-control study. Neurology. 2000;55:1350–1358. doi: 10.1212/wnl.55.9.1350. [DOI] [PubMed] [Google Scholar]
- Bryant JC, Westphal RS, Wadzinski BE. Methylated C-terminal leucine residue of PP2A catalytic subunit is important for binding of regulatory Balpha subunit. Biochem J. 1999;339(Pt 2):241–246. [PMC free article] [PubMed] [Google Scholar]
- Chen JF, Xu K, Petzer JP, Staal R, Xu YH, Beilstein M, Sonsalla PK, Castagnoli K, Castagnoli N, Jr, Schwarzschild MA. Neuroprotection by caffeine and A(2A) adenosine receptor inactivation in a model of Parkinson’s disease. J Neurosci. 2001;21:RC143. doi: 10.1523/JNEUROSCI.21-10-j0001.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Feany MB. Alpha-synuclein phosphorylation controls neurotoxicity and inclusion formation in a Drosophila model of Parkinson disease. Nat Neurosci. 2005;8:657–663. doi: 10.1038/nn1443. [DOI] [PubMed] [Google Scholar]
- Corcoran NM, Martin D, Hutter-Paier B, Windisch M, Nguyen T, Nheu L, Sundstrom LE, Costello AJ, Hovens CM. Sodium selenate specifically activates PP2A phosphatase, dephosphorylates tau and reverses memory deficits in an Alzheimer’s disease model. J Clin Neurosci. 2010;17:1025–1033. doi: 10.1016/j.jocn.2010.04.020. [DOI] [PubMed] [Google Scholar]
- Deacon RM. Assessing nest building in mice. Nat Protoc. 2006;1:1117–1119. doi: 10.1038/nprot.2006.170. [DOI] [PubMed] [Google Scholar]
- Fall PA, Fredrikson M, Axelson O, Granerus AK. Nutritional and occupational factors influencing the risk of Parkinson’s disease: a case-control study in southeastern Sweden. Mov Disord. 1999;14:28–37. doi: 10.1002/1531-8257(199901)14:1<28::aid-mds1007>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- Fleming SM, Salcedo J, Fernagut PO, Rockenstein E, Masliah E, Levine MS, Chesselet MF. Early and progressive sensorimotor anomalies in mice overexpressing wild-type human alpha-synuclein. J Neurosci. 2004;24:9434–9440. doi: 10.1523/JNEUROSCI.3080-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frasier M, Walzer M, McCarthy L, Magnuson D, Lee JM, Haas C, Kahle P, Wolozin B. Tau phosphorylation increases in symptomatic mice overexpressing A30P alpha-synuclein. Exp Neurol. 2005;192:274–287. doi: 10.1016/j.expneurol.2004.07.016. [DOI] [PubMed] [Google Scholar]
- Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, Shen J, Takio K, Iwatsubo T. alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol. 2002;4:160–164. doi: 10.1038/ncb748. [DOI] [PubMed] [Google Scholar]
- Gao HM, Kotzbauer PT, Uryu K, Leight S, Trojanowski JQ, Lee VM. Neuroinflammation and oxidation/nitration of alpha-synuclein linked to dopaminergic neurodegeneration. J Neurosci. 2008;28:7687–7698. doi: 10.1523/JNEUROSCI.0143-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M. Alpha-synuclein and neurodegenerative diseases. Nat Rev Neurosci. 2001;2:492–501. doi: 10.1038/35081564. [DOI] [PubMed] [Google Scholar]
- Gorbatyuk OS, Li S, Sullivan LF, Chen W, Kondrikova G, Manfredsson FP, Mandel RJ, Muzyczka N. The phosphorylation state of Ser-129 in human alpha-synuclein determines neurodegeneration in a rat model of Parkinson disease. Proc Natl Acad Sci U S A. 2008;105:763–768. doi: 10.1073/pnas.0711053105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanger DP, Anderton BH, Noble W. Tau phosphorylation: the therapeutic challenge for neurodegenerative disease. Trends Mol Med. 2009;15:112–119. doi: 10.1016/j.molmed.2009.01.003. [DOI] [PubMed] [Google Scholar]
- Harada A, Teng J, Takei Y, Oguchi K, Hirokawa N. MAP2 is required for dendrite elongation, PKA anchoring in dendrites, and proper PKA signal transduction. J Cell Biol. 2002;158:541–549. doi: 10.1083/jcb.200110134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellenbrand W, Boeing H, Robra BP, Seidler A, Vieregge P, Nischan P, Joerg J, Oertel WH, Schneider E, Ulm G. Diet and Parkinson’s disease. II: A possible role for the past intake of specific nutrients. Results from a self-administered food-frequency questionnaire in a case-control study. Neurology. 1996;47:644–650. doi: 10.1212/wnl.47.3.644. [DOI] [PubMed] [Google Scholar]
- Inglis KJ, Chereau D, Brigham EF, Chiou SS, Schobel S, Frigon NL, Yu M, Caccavello RJ, Nelson S, Motter R, Wright S, Chian D, Santiago P, Soriano F, Ramos C, Powell K, Goldstein JM, Babcock M, Yednock T, Bard F, Basi GS, Sham H, Chilcote TJ, McConlogue L, Griswold-Prenner I, Anderson JP. Polo-like kinase 2 (PLK2) phosphorylates alpha-synuclein at serine 129 in central nervous system. J Biol Chem. 2009;284:2598–2602. doi: 10.1074/jbc.C800206200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii A, Nonaka T, Taniguchi S, Saito T, Arai T, Mann D, Iwatsubo T, Hisanaga S, Goedert M, Hasegawa M. Casein kinase 2 is the major enzyme in brain that phosphorylates Ser129 of human alpha-synuclein: Implication for alpha-synucleinopathies. FEBS Lett. 2007;581:4711–4717. doi: 10.1016/j.febslet.2007.08.067. [DOI] [PubMed] [Google Scholar]
- Kanda S, Bishop JF, Eglitis MA, Yang Y, Mouradian MM. Enhanced vulnerability to oxidative stress by alpha-synuclein mutations and C-terminal truncation. Neuroscience. 2000;97:279–284. doi: 10.1016/s0306-4522(00)00077-4. [DOI] [PubMed] [Google Scholar]
- Kao G, Tuck S, Baillie D, Sundaram MV. C. elegans SUR-6/PR55 cooperates with LET-92/protein phosphatase 2A and promotes Raf activity independently of inhibitory Akt phosphorylation sites. Development. 2004;131:755–765. doi: 10.1242/dev.00987. [DOI] [PubMed] [Google Scholar]
- Khandelwal PJ, Dumanis SB, Feng LR, Maguire-Zeiss K, Rebeck G, Lashuel HA, Moussa CE. Parkinson-related parkin reduces alpha-Synuclein phosphorylation in a gene transfer model. Mol Neurodegener. 2010;5:47. doi: 10.1186/1750-1326-5-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolb B, Whishaw IQ. Neonatal frontal lesions in hamsters impair species-typical behaviors and reduce brain weight and neocortical thickness. Behav Neurosci. 1985;99:691–706. doi: 10.1037//0735-7044.99.4.691. [DOI] [PubMed] [Google Scholar]
- Lang RaH T. A versatile method for the quantitative determination of beta-N-alkanoyl-5-hydroxytryptamides in roasted coffee. Eur Food Res Technol. 2005;220:638–643. [Google Scholar]
- Lee J, Chen Y, Tolstykh T, Stock J. A specific protein carboxyl methylesterase that demethylates phosphoprotein phosphatase 2A in bovine brain. Proc Natl Acad Sci U S A. 1996;93:6043–6047. doi: 10.1073/pnas.93.12.6043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Stock J. Protein phosphatase 2A catalytic subunit is methyl-esterified at its carboxyl terminus by a novel methyltransferase. J Biol Chem. 1993;268:19192–19195. [PubMed] [Google Scholar]
- Liu F, Grundke-Iqbal I, Iqbal K, Gong CX. Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. Eur J Neurosci. 2005;22:1942–1950. doi: 10.1111/j.1460-9568.2005.04391.x. [DOI] [PubMed] [Google Scholar]
- Longin S, Zwaenepoel K, Louis JV, Dilworth S, Goris J, Janssens V. Selection of protein phosphatase 2A regulatory subunits is mediated by the C terminus of the catalytic Subunit. J Biol Chem. 2007;282:26971–26980. doi: 10.1074/jbc.M704059200. [DOI] [PubMed] [Google Scholar]
- Lou H, Montoya SE, Alerte TN, Wang J, Wu J, Peng XM, Hong CS, Friedrich EE, Mader SA, Pedersen CJ, Marcus BS, McCormack AL, Di Monte DA, Daubner SC, Perez RG. Serine 129 phosphorylation reduces {alpha}-synuclein’s ability to regulate tyrosine hydroxylase and protein phosphatase 2A in vitro and in vivo. J Biol Chem. 2010;285:17648–17661. doi: 10.1074/jbc.M110.100867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mbefo MK, Paleologou KE, Boucharaba A, Oueslati A, Schell H, Fournier M, Olschewski D, Yin G, Zweckstetter M, Masliah E, Kahle PJ, Hirling H, Lashuel HA. Phosphorylation of synucleins by members of the Polo-like kinase family. J Biol Chem. 2010;285:2807–2822. doi: 10.1074/jbc.M109.081950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarland NR, Fan Z, Xu K, Schwarzschild MA, Feany MB, Hyman BT, McLean PJ. Alpha-synuclein S129 phosphorylation mutants do not alter nigrostriatal toxicity in a rat model of Parkinson disease. J Neuropathol Exp Neurol. 2009;68:515–524. doi: 10.1097/NEN.0b013e3181a24b53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, Kahle PJ, Giasson BI, Ozmen L, Borroni E, Spooren W, Muller V, Odoy S, Fujiwara H, Hasegawa M, Iwatsubo T, Trojanowski JQ, Kretzschmar HA, Haass C. Misfolded proteinase K-resistant hyperphosphorylated alpha-synuclein in aged transgenic mice with locomotor deterioration and in human alpha-synucleinopathies. J Clin Invest. 2002;110:1429–1439. doi: 10.1172/JCI15777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogris E, Du X, Nelson KC, Mak EK, Yu XX, Lane WS, Pallas DC. A protein phosphatase methylesterase (PME-1) is one of several novel proteins stably associating with two inactive mutants of protein phosphatase 2A. J Biol Chem. 1999;274:14382–14391. doi: 10.1074/jbc.274.20.14382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paleologou KE, Schmid AW, Rospigliosi CC, Kim HY, Lamberto GR, Fredenburg RA, Lansbury PT, Jr, Fernandez CO, Eliezer D, Zweckstetter M, Lashuel HA. Phosphorylation at Ser-129 but Not the Phosphomimics S129E/D Inhibits the Fibrillation of {alpha}-Synuclein. J Biol Chem. 2008;283:16895–16905. doi: 10.1074/jbc.M800747200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Jones B, Kekonius L, Chin J, Yu GQ, Raber J, Masliah E, Mucke L. Neuronal depletion of calcium-dependent proteins in the dentate gyrus is tightly linked to Alzheimer’s disease-related cognitive deficits. Proc Natl Acad Sci U S A. 2003;100:9572–9577. doi: 10.1073/pnas.1133381100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pronin AN, Morris AJ, Surguchov A, Benovic JL. Synucleins are a novel class of substrates for G protein-coupled receptor kinases. J Biol Chem. 2000;275:26515–26522. doi: 10.1074/jbc.M003542200. [DOI] [PubMed] [Google Scholar]
- Rockenstein E, Mallory M, Hashimoto M, Song D, Shults CW, Lang I, Masliah E. Differential neuropathological alterations in transgenic mice expressing alpha-synuclein from the platelet-derived growth factor and Thy-1 promoters. J Neurosci Res. 2002;68:568–578. doi: 10.1002/jnr.10231. [DOI] [PubMed] [Google Scholar]
- Ross GW, Abbott RD, Petrovitch H, Morens DM, Grandinetti A, Tung KH, Tanner CM, Masaki KH, Blanchette PL, Curb JD, Popper JS, White LR. Association of coffee and caffeine intake with the risk of Parkinson disease. JAMA. 2000;283:2674–2679. doi: 10.1001/jama.283.20.2674. [DOI] [PubMed] [Google Scholar]
- Saaksjarvi K, Knekt P, Rissanen H, Laaksonen MA, Reunanen A, Mannisto S. Prospective study of coffee consumption and risk of Parkinson’s disease. Eur J Clin Nutr. 2008;62:908–915. doi: 10.1038/sj.ejcn.1602788. [DOI] [PubMed] [Google Scholar]
- Sager TN, Kirchhoff J, Mork A, Van Beek J, Thirstrup K, Didriksen M, Lauridsen JB. Nest building performance following MPTP toxicity in mice. Behav Brain Res. 2010;208:444–449. doi: 10.1016/j.bbr.2009.12.014. [DOI] [PubMed] [Google Scholar]
- Saraf A, Virshup DM, Strack S. Differential expression of the B’beta regulatory subunit of protein phosphatase 2A modulates tyrosine hydroxylase phosphorylation and catecholamine synthesis. J Biol Chem. 2007;282:573–580. doi: 10.1074/jbc.M607407200. [DOI] [PubMed] [Google Scholar]
- Shanley TP, Vasi N, Denenberg A, Wong HR. The serine/threonine phosphatase, PP2A: endogenous regulator of inflammatory cell signaling. J Immunol. 2001;166:966–972. doi: 10.4049/jimmunol.166.2.966. [DOI] [PubMed] [Google Scholar]
- Sontag E, Hladik C, Montgomery L, Luangpirom A, Mudrak I, Ogris E, White CL., 3rd Downregulation of protein phosphatase 2A carboxyl methylation and methyltransferase may contribute to Alzheimer disease pathogenesis. J Neuropathol Exp Neurol. 2004a;63:1080–1091. doi: 10.1093/jnen/63.10.1080. [DOI] [PubMed] [Google Scholar]
- Sontag E, Luangpirom A, Hladik C, Mudrak I, Ogris E, Speciale S, White CL., 3rd Altered expression levels of the protein phosphatase 2A ABalphaC enzyme are associated with Alzheimer disease pathology. J Neuropathol Exp Neurol. 2004b;63:287–301. doi: 10.1093/jnen/63.4.287. [DOI] [PubMed] [Google Scholar]
- Strack S, Westphal RS, Colbran RJ, Ebner FF, Wadzinski BE. Protein serine/threonine phosphatase 1 and 2A associate with and dephosphorylate neurofilaments. Brain Res Mol Brain Res. 1997;49:15–28. doi: 10.1016/s0169-328x(97)00117-4. [DOI] [PubMed] [Google Scholar]
- Su X, Maguire-Zeiss KA, Giuliano R, Prifti L, Venkatesh K, Federoff HJ. Synuclein activates microglia in a model of Parkinson’s disease. Neurobiol Aging. 2008;29:1690–1701. doi: 10.1016/j.neurobiolaging.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swingle M, Ni L, Honkanen RE. Small-molecule inhibitors of ser/thr protein phosphatases: specificity, use and common forms of abuse. Methods Mol Biol. 2007;365:23–38. doi: 10.1385/1-59745-267-X:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczypka MS, Kwok K, Brot MD, Marck BT, Matsumoto AM, Donahue BA, Palmiter RD. Dopamine production in the caudate putamen restores feeding in dopamine-deficient mice. Neuron. 2001;30:819–828. doi: 10.1016/s0896-6273(01)00319-1. [DOI] [PubMed] [Google Scholar]
- Tansey MG, Goldberg MS. Neuroinflammation in Parkinson’s disease: its role in neuronal death and implications for therapeutic intervention. Neurobiol Dis. 2010;37:510–518. doi: 10.1016/j.nbd.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theodore S, Cao S, McLean PJ, Standaert DG. Targeted overexpression of human alpha-synuclein triggers microglial activation and an adaptive immune response in a mouse model of Parkinson disease. J Neuropathol Exp Neurol. 2008;67:1149–1158. doi: 10.1097/NEN.0b013e31818e5e99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolstykh T, Lee J, Vafai S, Stock JB. Carboxyl methylation regulates phosphoprotein phosphatase 2A by controlling the association of regulatory B subunits. Embo J. 2000;19:5682–5691. doi: 10.1093/emboj/19.21.5682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Eersel J, Ke YD, Liu X, Delerue F, Kril JJ, Gotz J, Ittner LM. Sodium selenate mitigates tau pathology, neurodegeneration, and functional deficits in Alzheimer’s disease models. Proc Natl Acad Sci U S A. 2010;107:13888–13893. doi: 10.1073/pnas.1009038107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Tolstykh T, Lee J, Boyd K, Stock JB, Broach JR. Carboxyl methylation of the phosphoprotein phosphatase 2A catalytic subunit promotes its functional association with regulatory subunits in vivo. EMBO J. 2000;19:5672–5681. doi: 10.1093/emboj/19.21.5672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing Y, Li Z, Chen Y, Stock JB, Jeffrey PD, Shi Y. Structural mechanism of demethylation and inactivation of protein phosphatase 2A. Cell. 2008;133:154–163. doi: 10.1016/j.cell.2008.02.041. [DOI] [PubMed] [Google Scholar]
- Xing Y, Xu Y, Chen Y, Jeffrey PD, Chao Y, Lin Z, Li Z, Strack S, Stock JB, Shi Y. Structure of protein phosphatase 2A core enzyme bound to tumor-inducing toxins. Cell. 2006;127:341–353. doi: 10.1016/j.cell.2006.09.025. [DOI] [PubMed] [Google Scholar]
- Yavich L, Tanila H, Vepsalainen S, Jakala P. Role of alpha-synuclein in presynaptic dopamine recruitment. J Neurosci. 2004;24:11165–11170. doi: 10.1523/JNEUROSCI.2559-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu XX, Du X, Moreno CS, Green RE, Ogris E, Feng Q, Chou L, McQuoid MJ, Pallas DC. Methylation of the protein phosphatase 2A catalytic subunit is essential for association of Balpha regulatory subunit but not SG2NA, striatin, or polyomavirus middle tumor antigen. Mol Biol Cell. 2001;12:185–199. doi: 10.1091/mbc.12.1.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Yang J, Liu Y, Chen X, Yu T, Jia J, Liu C. PR55 alpha, a regulatory subunit of PP2A, specifically regulates PP2A-mediated beta-catenin dephosphorylation. J Biol Chem. 2009;284:22649–22656. doi: 10.1074/jbc.M109.013698. [DOI] [PMC free article] [PubMed] [Google Scholar]