

Abstract
Emerging evidence is implicating abnormal activation of the mechanistic target of rapamycin (mTOR) pathway in several monogenetic neuropsychiatric disorders, including Angelman syndrome (AS), which is caused by deficiency in maternally inherited UBE3A. Using an AS mouse model, we show that semi-chronic rapamycin treatment improves long-term potentiation (LTP) and actin polymerization in hippocampal slices, spine morphology, and fear-conditioning learning. Activity of mTORC1 and of its downstream substrate, S6K1, was increased in hippocampus of AS mice. However, mTORC2 activity, as reflected by PKCα levels, was decreased. Both increased mTORC1 and decreased mTORC2 activity were reversed by semi-chronic rapamycin treatment. Acute treatment of hippocampal slices from AS mice with rapamycin or an S6K1 inhibitor, PF4708671, improved LTP, restored actin polymerization, and normalized mTORC1 and mTORC2 activity. These treatments also reduced Arc levels in AS mice. Treatment with Torin 1, an inhibitor of both mTORC1 and mTORC2, partially rescued LTP and actin polymerization in hippocampal slices from AS mice, while partially impairing them in wild-type (WT) mice. Torin 1 decreased mTORC1 and increased mTORC2 activity in slices from AS mice but inhibited mTORC1 and decreased mTORC2 in WT mice. Finally, an mTORC2 activator, A-443654, increased hippocampal LTP in AS mice and actin polymerization in both WT and AS mice. Collectively, these results indicate that events set in motion by increased mTORC1 and decreased mTORC2 activities, including increased Arc translation and impaired actin remodeling, are crucial in AS pathogenesis. Therefore, selectively targeting these two master kinase complexes may provide new therapeutic approaches for AS treatment.
Keywords: S6K1 inhibitor, mTORC2 activator, GluA1, cognitive deficits, dendritic spine, phosphorylation, translation
Introduction
Angelman syndrome (AS) is a genetic neurodevelopmental disorder characterized by severe developmental delay, language and cognition deficits, motor impairment [1,2], and, in many patients, seizure activity and autistic behavior [1,3,4]. AS is caused by the deficient expression of the maternally inherited UBE3A gene in neurons [5–7]. Mice with maternal Ube3A deficiency (AS mice) exhibit several features of the human disease, including reduced brain size, abnormal electroencephalogram, learning and memory deficits and impairment in long-term potentiation (LTP) [8–11], and motor dysfunction [12,9,13]. However, the molecular bases of these alterations are not completely clear. Increased expression of the immediate-early gene Arc (activity-regulated cytoskeleton-associated protein) was proposed to be involved in synaptic plasticity impairment, after it was identified as a substrate of UBE3A and postulated to be degraded by UBE3A-mediated ubiquitination [14]. However, a recent study argued that UBE3A regulates Arc at the transcriptional level rather than through direct ubiquitination [15]. Nevertheless, increased Arc levels could account for LTP impairment, since Arc promotes AMPAR (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor) internalization, thereby reducing AMPAR-mediated synaptic responses [16–18]. Increased Arc levels have also been shown to disrupt LTP induction and maintenance in AS mice by interrupting brain-derived neurotrophic factor (BDNF) signaling [19].
Abnormal signaling of the mechanistic target of rapamycin (mTOR) pathway has been implicated in multiple monogenetic neurodevelopmental disorders linked to autism spectrum disorders [20]. mTOR consists of two distinct complexes, mTORC1 and mTORC2, each of which critically contributes to synaptic plasticity and memory [21], although much less is known regarding the roles of mTORC2. Both mTORC1 downstream targets, 4E-BPs and S6K1/2, and upstream effectors of mTOR such as TSC1/2 have been implicated in synaptic plasticity and memory [22]. Several groups have shown that mTORC2 activity is crucial for actin polymerization and memory [23–26]. We recently reported that imbalanced mTORC1 and mTORC2 activities play important roles in motor dysfunction and abnormal dendritic spine morphology of Purkinje neurons in AS mice [27]. These findings prompted us to investigate whether Ube3A deficiency affected mTOR signaling in hippocampus of AS mice. Here we report that Ube3A deficiency results in increased mTORC1 activity as evidenced by increased S6K1 and S6 phosphorylation, and decreased mTORC2 activity with reduced PKCα levels. These alterations are causally associated with impairments in hippocampal LTP, actin polymerization, and fear-conditioning learning in AS mice, since either inhibition of mTORC1-S6K1 or enhancement of mTORC2 activity significantly ameliorates these impairments.
Materials and methods
Animals
Ube3Atm1Alb/J mice (AS mice) were purchased from The Jackson Laboratory (Bar Harbor, MN). Mice were maintained on the hybrid B6;129 background. Wild-type (WT) and AS mice were obtained through in-house breeding of heterozygous females with WT males (both on the hybrid background). Genotype was determined as previously described [8]. Animal handling and experimental use followed protocols approved by the local Institutional Animal Care and Use Committee (IACUC). In all experiments, male AS mice and WT littermates aged between 2–4 months were used. Mice were kept on a 12-h light/dark cycle, with food and water available ad libitum and were housed in groups of 2–3 per cage.
Rapamycin treatment
Rapamycin (LC Laboratories, Woburn, MA) was prepared as previously described [27]. For details, see Supplementary materials and methods. AS and WT mice were randomly assigned to either drug or vehicle groups and injected intraperitoneally (i.p.) with either rapamycin (5 mg/kg) or vehicle once a day for a total of five days before the following experiments.
Fear conditioning
Rapamycin- or vehicle-treated AS and WT mice were tested blindly by an observer. Fear conditioning was performed using a standard technique, as described previously [28]. For details concerning the fear conditioning training and testing, see Supplementary materials and methods.
Tissue collection, P2/S2 fractionation and Western blot analysis
Twenty-four hours after the last rapamycin treatment, mice were deeply anesthetized using gaseous isoflurane and decapitated. The whole brain was removed and various brain regions including hippocampus were dissected. Brain tissues were immediately frozen on dry ice and stored at −80 °C until further processed.
P2/S2 fractionation and Western blots were performed according to published protocols [27]. For details concerning homogenization buffers, P2/S2 fractionation and Western blot procedures, and sources and concentrations of antibodies, see Supplementary materials and methods.
Dendritic spine analysis
Twenty-four hours after the last rapamycin treatment, mice were deeply anesthetized using gaseous isoflurane and then decapitated. The brain was rapidly removed and Golgi impregnation was performed according to our published protocol [27] and outlined in the FD Rapid GolgiStain Kit (FD Neurotechnologies, Ellicott, MD). The number of spines located on randomly selected dendritic branches was counted manually by an investigator blind to genotype and treatment. Spine density was calculated by dividing the number of spines on a segment by the length of the segment and was expressed as the number of spines per μm of dendrite. Five to seven dendritic branches between 10 and 20 μm in length were analyzed and averaged to provide a section mean.
Immunohistochemistry
After behavioral testing, mice were sacrificed under deep anesthesia using pentobarbital (i.p.) and were perfused through the left cardiac ventricle with phosphate buffered saline (PBS) followed by 4% paraformaldehyde (PFA). Free-floating sections (20 μm) were processed for immunohistochemical staining as previously described [27]. For details, see Supplementary materials and methods.
Acute hippocampal slice preparation, electrophysiology, and in situ phalloidin labeling
Acute hippocampal transversal slices (350 μm-thick) were prepared from adult male mice as previously described [8], and recording was done according to published protocols [28]. For details, see Supplementary materials and methods. Rapamycin (50 nM), PF-4708671 (5 μM), Torin 1 (250 nM), or A-443654 (500 nM) were applied to slices for 30 minutes before theta-burst stimulation (TBS). Some of the slices were processed for either P2/S2 fractionation and Western blots or actin polymerization assay. Phalloidin staining of filamentous actin (F-actin) was performed as previously described [8]. All images were taken in CA1 stratum radiatum between the stimulating and recording electrodes.
Actin polymerization assay
Actin polymerization was quantified by measurement of “rhodamine-phalloidin fluorescent enhancement”, as previously described with minor modifications [29]. For details, see Supplementary materials and methods.
Statistical analysis
Error bars indicate standard errors of the mean. To compute p values, two-way ANOVA with Newman-Keuls post-test was used.
Results
1. Semi-chronic rapamycin treatment promotes LTP, improves dendritic spine morphology and learning and memory performance in AS mice
We first determined the effects of semi-chronic rapamycin treatment on LTP in hippocampal slices from AS mice and WT littermates. As previously reported [8,11,28], TBS elicited LTP in field CA1 of hippocampal slices in vehicle-treated WT mice, whereas it only elicited transient facilitation in vehicle-treated AS mice (Fig. 1a,b). Systemic treatment with rapamycin (5 mg/kg) for 5 days improved TBS-elicited LTP in hippocampal slices from AS mice (Fig. 1a,b), while it did not affect TBS-induced LTP in slices from WT mice (Fig. 1a,b). We also determined the effect of rapamycin treatment on TBS-induced actin polymerization using Alexa 568-conjugated phalloidin, which selectively binds to F-actin. TBS elicited a clear increase in the number of F-actin-positive puncta in slices from WT but not AS mice. Semi-chronic rapamycin treatment markedly enhanced TBS-induced actin polymerization in slices from AS mice, but had no effect in WT mice (Fig. 1c,d), nor did it affect F-actin basal levels (Figure S1).
Fig. 1.
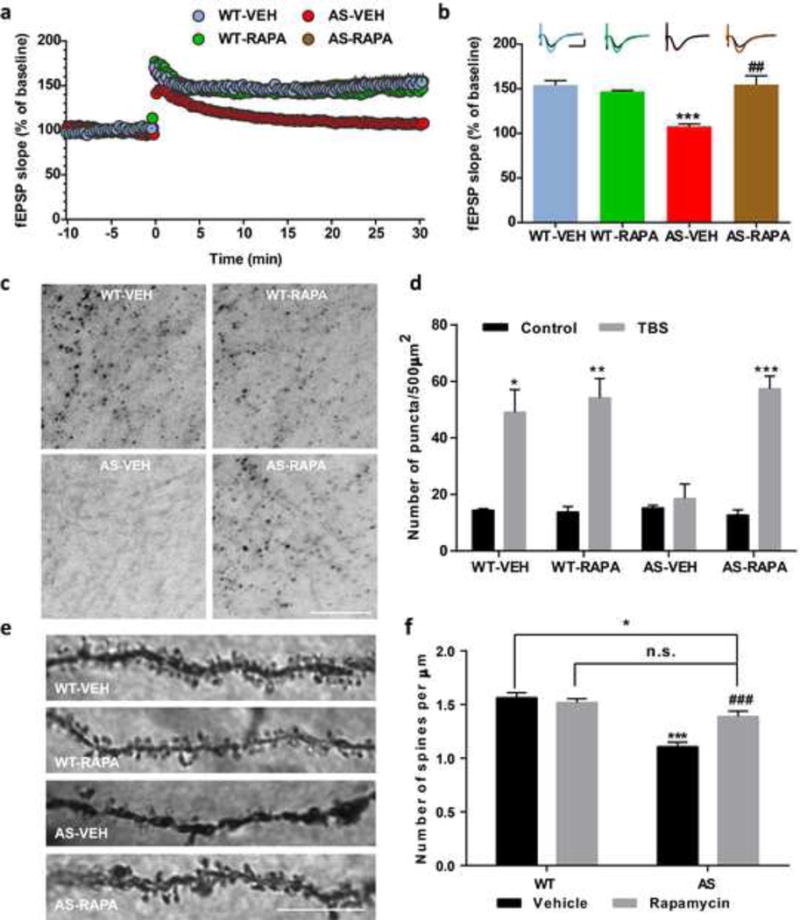
Effects of semi-chronic rapamycin treatment on LTP and dendritic spine morphology in hippocampus of WT and AS mice. (a) Reversal of LTP impairment in AS mice by semi-chronic rapamycin treatment. Slopes of fEPSPs were normalized to the average values recorded during the 10 min baseline. (b) Means ± S.E.M. of fEPSPs measured 30 min after TBS in different groups. N = 3–5 slices from 3–5 mice. Insert shows representative traces of evoked fEPSPs before and 30 min after TBS. Scale bar: 0.5 mV/10 ms. (c–d) Rapamycin treatment promotes TBS-induced actin polymerization in hippocampal slices from AS mice. (c) Representative images of in situ phalloidin staining after TBS in CA1 region of hippocampus from vehicle- or rapamycin-treated WT or AS mice. Scale bar = 20 μm. (d) Quantitative analysis of F-actin staining. Results are means ± S.E.M. *p < 0.05, **p < 0.01, ***p < 0.001 (n=3 for each experimental group; two-way ANOVA followed by Newman-Keuls post-test). (e-f) Effects of rapamycin treatment on dendrites and spines of CA1 pyramidal neurons in WT and AS mice. (e) Representative light micrograph images from Golgi-impregnated CA1 pyramidal neurons. Scale bar = 10 μm. (f) Quantitative analysis of dendritic spine density shown in e (means ± SEM from 10 slices). *p<0.05, ***p<0.001, as compared to vehicle-treated wild-type mice, and ##p<0.01, ###p<0.001, as compared to vehicle-treated AS mice, two-way ANOVA with Newman-Keuls post-test. n.s., not significant
We also performed Golgi staining in hippocampal CA1 region of WT and AS mice treated with rapamycin or vehicle. As previously reported [30], spine density was lower in AS mice, as compared with WT mice (Fig. 1e,f). Rapamycin treatment partially normalized number and morphology of dendritic spines of hippocampal pyramidal neurons in AS mice, but did not affect these parameters in WT mice (Fig. 1e,f).
We used the fear-conditioning paradigm to determine whether the 5-day rapamycin treatment could also reverse learning impairment in AS mice. As previously reported [28], AS mice were impaired in context- and tone-dependent fear learning, and rapamycin treatment significantly enhanced their performance in both types of learning (Fig. 2). Under our experimental conditions (3 conditioned stimuli (CS) paired with 3 unconditioned stimuli (US)), rapamycin treatment did not affect learning in WT mice (Fig. 2). Additionally, there was no difference in freezing time in the pre-conditioning period, or before tone application in the testing period between all experimental groups (Fig. 2).
Fig. 2.
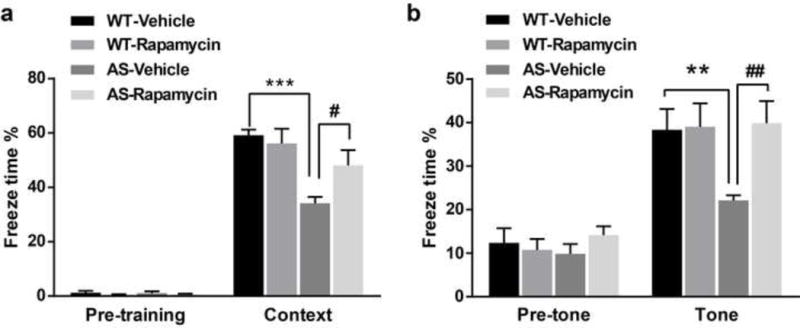
Effects of semi-chronic rapamycin treatment on fear-conditioning learning in WT and AS mice. (a) Percent freezing for different experimental groups in context memory (means ± S.E.M. of 6–9 mice). (b) Percent freezing for different experimental groups in tone memory (means ± S.E.M. of 6–9 mice). **p<0.01, ***p<0.001, as compared to WT controls; #p<0.05, ##p<0.01, as compared to vehicle-treated AS mice; two-way ANOVA with Newman-Keuls post-hoc analysis
2. mTORC1 signaling is increased while mTORC2 activity is reduced in hippocampus of AS mice; both are normalized by rapamycin treatment
We next analyzed the phosphorylation/activation status of mTOR in hippocampal crude membrane (P2) fractions from rapamycin- and vehicle-treated mice. Levels of mTOR phosphorylated at Ser2448 and Ser2481 were increased in AS mice (Table 1, Figure S2A), and rapamycin treatment significantly reduced them to control values, while it had no effect in WT mice (Table 1, Figure S2A). Levels of TSC2, an inhibitory regulator of mTOR, were significantly higher in AS mice, as compared to WT mice. However, inhibitory phosphorylation of TSC2 at Thr1462 (p-TSC2) was also significantly increased, although the ratio of p-TSC2/TSC2 was only slightly increased in P2 fractions of AS mice (Table 1, Figure S2B). Rapamycin treatment significantly reduced p-TSC2 levels in AS and WT mice, but did not alter total TSC2 levels (Table 1, Figure S2B). These results suggest that increased TSC2 inhibitory phosphorylation may reduce its inhibition of mTOR and that phosphorylation of TSC2 at Thr1462 is mediated by a rapamycin-sensitive kinase, which is consistent with what we reported in cerebellum of AS mice [27].
Table 1.
Effects of rapamycin treatment on mTOR signaling pathway in hippocampus of WT and AS mice
| WT/VEH | WT/RAPA | AS/VEH | AS/RAPA | |
|---|---|---|---|---|
| p-mTOR 2448/mTOR |
100.0 ± 6.9 | 95.8 ± 2.3 | 185.1 ± 9.0*** | 109.3 ± 9.8### |
| p-mTOR 2481/mTOR |
100.0 ± 6.5 | 76.1 ± 5.9 | 143.4 ± 9.8* | 69.2 ± 7.9### |
| p-TSC2/TSC2 | 100.0 ± 6.7 | 74.7 ± 6.1* | 127.4 ± 7.4* | 83.5 ± 11.3# |
| p-S6K1/S6K1 | 100.0 ± 10.2 | 61.4 ± 11.5 | 158.6 ± 17.8* | 43.7 ± 10.8## |
| p-S6 235/236/S6 | 100.0 ± 11.5 | 44.2 ± 3.9** | 130.0 ± 4.0* | 36.4 ± 6.4### |
| p-S6 240/244/S6 | 100.0 ± 6.7 | 49.5 ± 9.1* | 183.2 ± 9.0*** | 42.1 ± 9.7### |
| PKCα/β-actin | 100.0 ± 5.0 | 117.7 ± 4.3* | 77.3 ± 3.8** | 100.4 ± 3.2## |
| p-Rictor/Rictor | 100.0 ± 7.1 | 13.2 ± 0.1*** | 129.0 ± 6.3* | 13.5 ± 4.0### |
WT and AS mice were treated with vehicle or rapamycin as described in Materials and methods. Animals were sacrificed at the end of the 5-day treatment, and hippocampus were dissected and P2 fraction was prepared.
All quantitative data of Western blot analyses are presented as means ± SEM from 3–6 mice;
p<0.05,
p<0.01,
p<0.001, as compared to vehicle-treated wild-type mice, and
p<0.05,
p<0.01,
p<0.001, as compared to vehicle-treated AS mice, two-way ANOVA with Newman-Keuls post-test.
We next evaluated mTORC1 and mTORC2 activity by analyzing the phosphorylation status of some of their respective downstream substrates. Levels of mTORC1 downstream proteins, p-S6K1-Thr389 and p-S6-Ser235/236/p-S6-Ser240/244, were significantly increased in AS mice, as compared to WT mice (Table 1, Figure S2C) and the increases in AS mice were significantly reduced by rapamycin treatment (Table 1, Figure S2C). Rapamycin treatment also significantly reduced S6 phosphorylation in WT mice (Table 1, Figure S2C). Immunofluorescence examination of p-S6K1-Thr389 confirmed the western blot results (Figure S3).
We then determined levels of PKCα, since previous reports indicated that mTORC2-mediated PKCα phosphorylation was critical for its stability [31,32]. PKCα levels were significantly reduced in AS mice, as compared to WT mice, an effect that was reversed by rapamycin treatment (Table 1, Figure S4). Rapamycin treatment also increased levels of PKCα in P2 fractions of WT mice (Table 1, Figure S4). Levels of inhibitory phosphorylation (at Thr1135) of Rictor, an mTORC2 component [33], were significantly increased in AS mice, as compared to WT mice (Table 1, Figure S4), confirming that mTORC2 activity was reduced. Rapamycin treatment markedly decreased p-Rictor levels in both AS and WT mice (Table 1, Figure S4).
3. Acute application of rapamycin or the S6K inhibitor PF-4708671 normalizes hippocampal LTP and promotes actin polymerization in AS mice
We next asked whether acute inhibition of mTORC1 or S6K could improve synaptic plasticity in hippocampal slices of AS mice. Pre-incubation of hippocampal slices from AS mice with rapamycin (50 nM) or PF-4708671 (5 μM), a novel and specific S6K1 inhibitor, for 30 min reestablished TBS-elicited LTP to levels identical to those in WT mice (Fig. 3a–d). Rapamycin or PF-4708671 at the concentrations used did not affect TBS-induced LTP in slices from WT mice (Fig. 3a–d).
Fig. 3.
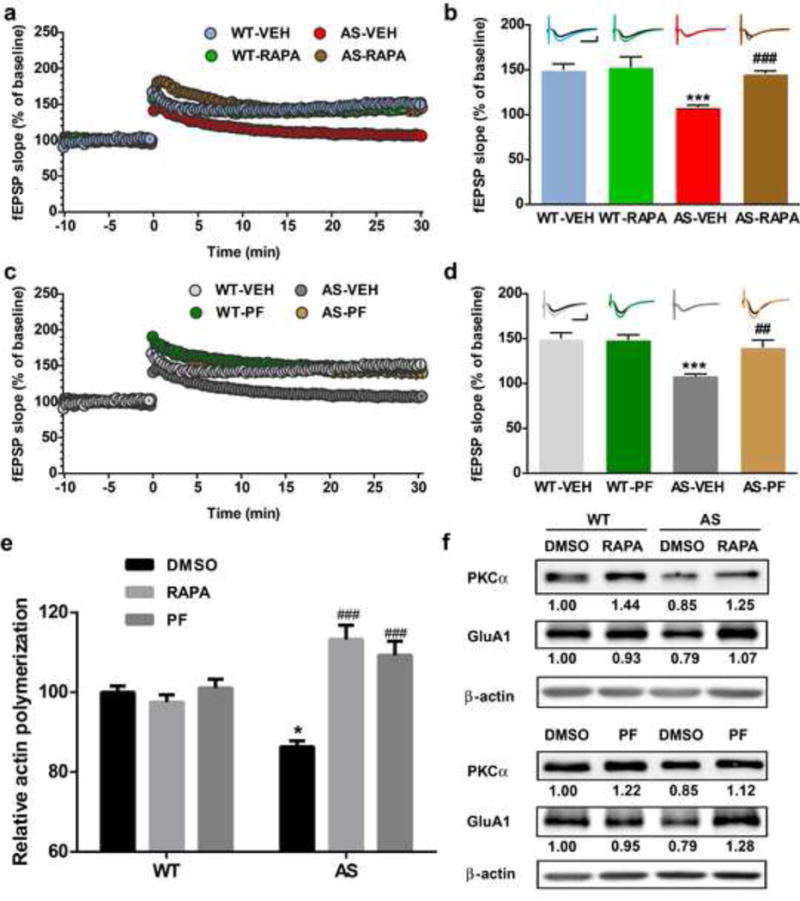
Effects of acute rapamycin or PF-4708671 treatment on LTP, actin polymerization and PKCα and GluA1 levels in hippocampus slices of WT and AS mice. (a) Reversal of LTP impairment in AS mice by acute rapamycin (50 nM) treatment. Slopes of fEPSPs were normalized to the average values recorded during the 10 min baseline. (b) Means ± S.E.M. of fEPSPs measured 30 min after TBS in different groups. N = 3–5 slices from 3–5 mice. Insert shows representative traces of evoked fEPSPs before and 30 min after TBS. Scale bar: 0.5 mV/10 ms. (c) Reversal of LTP impairment in AS mice by acute PF-4708671 (5 μM) treatment. Slopes of fEPSPs were normalized to the average values recorded during the 10 min baseline. (d) Means ± S.E.M. of fEPSPs measured 30 min after TBS in different groups. N = 3–5 slices from 3–5 mice. Insert shows representative traces of evoked fEPSPs before and 30 min after TBS. Scale bar: 0.5 mV/10 ms. (e) Effects of rapamycin (50 nM) and PF-4708671 (5 μM) treatment on actin polymerization in hippocampal slices from WT and AS mice. Data represent means ± SEM from 3 mice. (f) Representative images and quantitative analysis of Western blots labeled with PKCα, GluA1 and β-actin antibodies in P2 fractions of WT and AS hippocampal slices treated with vehicle, rapamycin or PF-4708671. *p<0.05, ***p<0.001, as compared to vehicle-treated WT slices, and ##p<0.01, ###p<0.001, as compared to vehicle-treated AS slices, two-way ANOVA with Newman-Keuls post-test
We then determined the effects of rapamycin or PF-4708671 treatment of hippocampal slices on basal levels of actin polymerization. Actin polymerization in hippocampal slices from AS mice was decreased as compared to WT mice. Inhibition of mTORC1 by rapamycin or PF-4708671 significantly increased actin polymerization in AS hippocampal slices, while it had no effect in WT slices (Fig. 3e). Western blot results showed that treatment with rapamycin or PF-4708671 increased mTORC2 activity, as assessed with increased PKCα levels in hippocampal slices from both WT and AS mice (Fig. 3f). It has been previously reported that actin dynamics regulate AMPA receptor trafficking during synaptic plasticity [34]. We therefore analyzed GluA1 levels in P2 fractions from acute hippocampal slices from WT and AS mice treated with vehicle, rapamycin, or PF-4708671. In vehicle-treated animals, levels of GluA1 were slightly lower in slices from AS mice as compared to those from WT mice; these alterations were reversed by treatment with either rapamycin or PF4708671 (Fig. 3f).
4. mTORC1/S6K1 inhibition in AS mice is associated with reduction in Arc levels
We analyzed Arc levels in P2 fractions from acute hippocampal slices treated with vehicle, rapamycin or PF-4708671. In vehicle-treated slices, levels of Arc were significantly higher in AS mice as compared to WT mice; these alterations were reversed by treatment with either rapamycin or PF4708671 in hippocampal slices (Fig. 4a,b). Of note, changes in Arc levels were paralleled with changes in S6 phosphorylation (Fig. 4a,b). Together, these results suggest that mTORC1/S6K1/S6 over-activation contributes to increased Arc levels in AS mice. Acute rapamycin or PF4708671 treatment also decreased Arc levels in WT hippocampal slices (Fig. 4a,b).
Fig. 4.
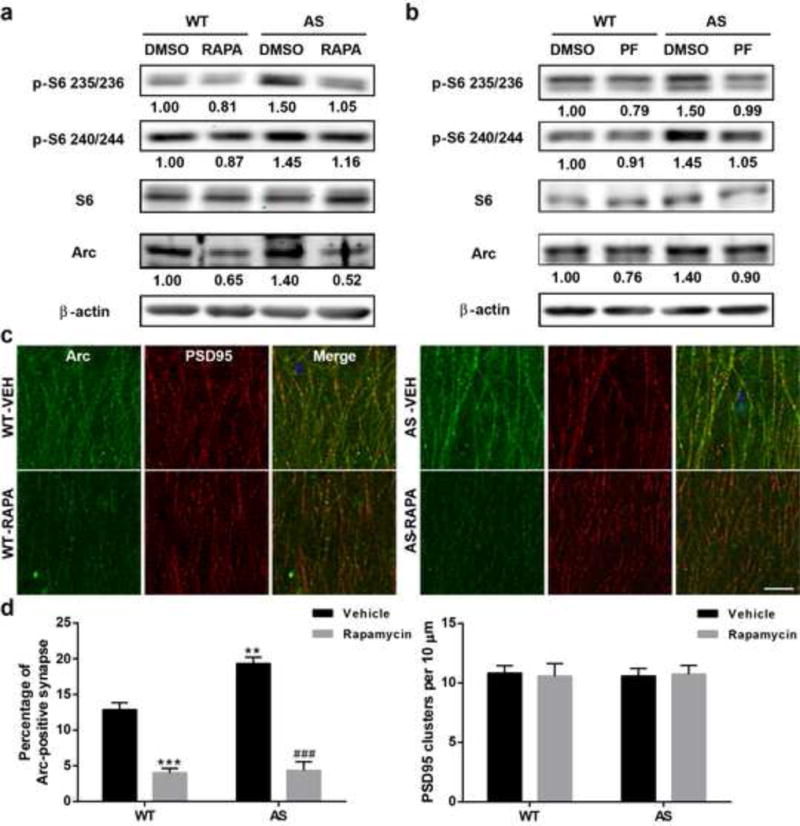
Effects of rapamycin or PF-4708671 treatment on mTORC1 signaling and Arc levels in hippocampus of WT and AS mice. (a–b) Representative images and quantitative analysis of Western blots labeled with p-S6 (235/236 and 240/244), S6, Arc, and β-actin antibodies in P2 fractions of WT and AS hippocampal slices treated with vehicle, rapamycin or PF-4708671. (c) Representative immunostaining images of Arc (green) and PSD95 (red) in dendritic CA1 region of hippocampus from vehicle- or rapamycin- treated mice after fear conditioning test. Scale bar = 20 μm. (d) Quantitative analysis of percentage of Arc and PSD95 dually-stained puncta/synapses and the number of PSD95-immunoreactive puncta. Shown are means ± SEM from 3 mice; **p<0.01, ***p<0.001, as compared to vehicle-treated WT slices, and ###p<0.001, as compared to vehicle-treated AS slices, two-way ANOVA with Newman-Keuls post-test
Immunofluorescence examination of Arc in CA1 dendritic field showed that Arc-immunoreactive puncta were distributed along apical dendrites and partially co-localized with PSD95-immunopositive profiles (Fig. 4c). Quantitative analysis showed that the percentage of the puncta dually-stained with Arc and PSD95 was significantly increased in AS mice, an effect which was reversed by semi-chronic rapamycin treatment (Fig. 4d). There was no significant difference in the overall number of PSD95-immunopositive puncta between all experimental groups (Fig. 4d).
5. Effects of Torin 1, an inhibitor of both mTORC1 and mTORC2, and of A-443654, an mTORC2 activator, on hippocampal LTP in WT and AS mice
To further verify the role of mTORC2 in LTP, we pretreated hippocampal slices for 30 min with Torin 1, a potent inhibitor of both mTORC1 and mTORC2. Torin 1 treatment significantly reduced LTP in WT hippocampal slices (Fig. 5a,b). We also determined the effect of Torin 1 treatment on TBS-induced actin polymerization. TBS elicited a clear increase in the number of F-actin-positive puncta in WT slices, but not in Torin 1-treated WT slices (Fig. 5c). In contrast, Torin 1 treatment partially rescued LTP and actin polymerization in AS hippocampal slices (Fig. 5a–c). It has been shown that Torin 1 inhibits mTORC1 and mTORC2 in cell-free kinase assays with IC50 values of 2 nM and 10 nM, respectively [35,36]. It is possible that in AS mice, Torin 1-mediated direct inhibition of mTORC2 was overcome by mTORC1 inhibition-induced disinhibition of mTORC2. We therefore analyzed levels of PKCα and p-S6 in P2 fractions from Torin 1- or vehicle-treated acute hippocampal slices. In Torin 1-treated WT slices, levels of PKCα were slightly lower as compared to vehicle-treated WT slices, while in Torin 1-treated AS slices, levels of PKCα were higher as compared to vehicle-treated AS slices (Fig. 5d). In contrast, Torin 1 treatment reduced levels of p-S6 in both WT and AS slices (Fig. 5d).
Fig. 5.
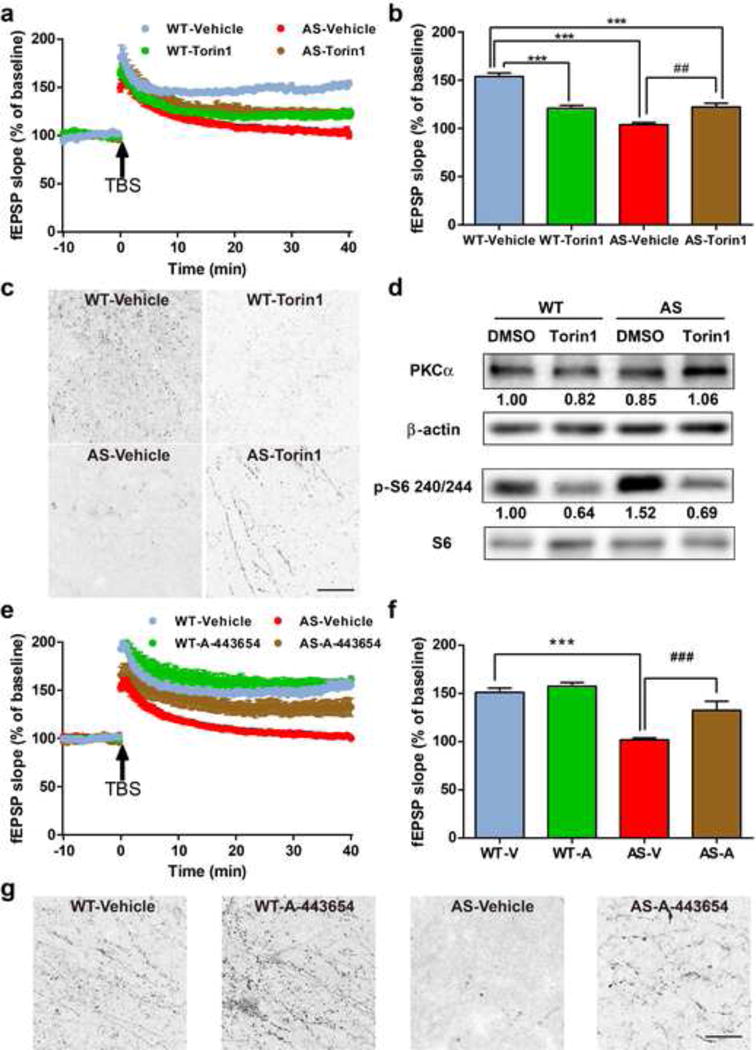
Effects of acute Torin 1 or A-443654 treatment on LTP, actin polymerization and mTOR signaling in hippocampus slices of WT and AS mice. (a) Effect of acute Torin 1 (250 nM) treatment on LTP. Slopes of fEPSPs were normalized to the average values recorded during the 10 min baseline. (b) Means ± S.E.M. of fEPSPs measured 40 min after TBS in different groups. N = 3–5 slices from 3–5 mice. (c) Effects of Torin 1 (250 nM) treatment on TBS-induced actin polymerization in hippocampal slices from WT and AS mice. Scale bar = 20 μm. (d) Representative images and quantitative analysis of Western blots labeled with PKCα, β-actin, p-S6 240/244, and S6 antibodies in P2 fractions of WT and AS hippocampal slices treated with vehicle or Torin 1. (e) Effect of acute A-443654 (500 nM) treatment on LTP. Slopes of fEPSPs were normalized to the average values recorded during the 10 min baseline. (f) Means ± S.E.M. of fEPSPs measured 40 min after TBS in different groups. N = 3–5 slices from 3–5 mice. (g) Effects of A-443654 (500 nM) treatment on TBS-induced actin polymerization in hippocampal slices from WT and AS mice. Scale bar = 20 μm. ***p<0.001, as compared to vehicle-treated WT slices, and ##p<0.01, ###p<0.001, as compared to vehicle-treated AS slices, two-way ANOVA with Newman-Keuls post-test
We also pretreated hippocampal slices with A-443654, which increases mTORC2-mediated phosphorylation of Akt at Ser473 (independently of mTORC1) [37]. It has been reported that A-443654 promoted mTORC2 activity and actin polymerization in WT slices, but not in mTORC2-deficient slices [24]. Pre-incubation of A-443654 for 30 min improved TBS-elicited LTP in hippocampal slices from AS mice (Fig. 5e,f), while it did not affect TBS-induced LTP in slices from WT mice (Fig. 5e,f). Of note, acute A-443654 treatment markedly enhanced TBS-induced actin polymerization in both WT and AS slices (Fig. 5g). P-Akt levels in P2 fractions from acute hippocampal slices of WT and AS mice were also increased after A-443654 treatment (Figure S5).
Discussion
Abnormal mTOR signaling has been implicated in a number of monogenetic and sporadic brain disorders, including tuberous sclerosis, Fragile X syndrome, AS, Alzheimer’s disease, schizophrenia, and epilepsy [38–40]. Thus, manipulations of the mTOR signaling pathway represent attractive drug development approaches, and as a result, a few compounds have entered clinical trials for several neurological disorders [39]. However, signaling along the mTOR kinase complexes and their downstream cascades is nothing but complicated. Although mTORC1 and mTORC2 share common subunit proteins, they also have distinctive components, which are activated by different upstream factors, and once activated, mTORC1 and mTORC2 initiate different signaling cascades. We previously reported that mTORC1 activity was increased while mTORC2 activity was decreased in cerebellar Purkinje neurons in AS mice. Here we report similar imbalanced mTORC1 and mTORC2 activities in hippocampus of AS mice. We showed that increased mTORC1 activity contributes to cognitive deficits in AS mice, since both acute and semi-chronic rapamycin applications rescued hippocampal LTP and actin polymerization and semi-chronic application also significantly improved fear-conditioning learning. Furthermore, acute application of an S6K1 inhibitor, PF-4708671, also rescued hippocampal LTP and actin polymerization. Of note, Bhattacharya et al [41] recently showed that PF-4708671 reversed several phenotypes in Fragile X syndrome mice. On the other hand, decreased mTORC2 activity was equally important, as Torin 1 partially rescued LTP and actin polymerization in AS mice, while partially impairing them in WT mice. In addition, directly activating mTORC2 with A-443654 also rescued LTP and actin polymerization in AS mice. Finally, since both rapamycin and PF-4708671 treatments also increased mTORC2 activity in AS mice, mTORC1-S6K1 over-activation is responsible for reduced mTORC2 activation, most likely through S6K1-mediated inhibitory phosphorylation of Rictor [42].
What could be the downstream effectors of mTORC1 and mTORC2 that contribute to synaptic plasticity and learning deficits in AS mice? The best characterized function of mTORC1 is to stimulate protein translation by phosphorylating/activating 4E-BP and S6K1 [39,43]. The immediate-early gene product, Arc, is locally synthesized in an activity-dependent manner [44] and its levels have been shown to be elevated in AS mice [14,15]. We found that increased Arc expression in AS mice was normalized by treatment of hippocampal slices with rapamycin or PF-4708671. It is therefore conceivable that over-activation of the mTORC1-S6K1 pathway results in synaptic plasticity impairment in AS mice by increasing Arc translation, since Arc induces AMPA receptor endocytosis. In support of this argument, both rapamycin and PF-4708671 treatment increased GluA1 synaptic levels in AS mice. Additionally, Cao et al [19] reported that increased Arc levels in AS mice inhibited LTP induction and maintenance by reducing BDNF signaling through blocking the interaction between BDNF receptor TrkB and PSD95. We previously reported that treatment with a positive AMPA receptor regulator also restored LTP in AS mice, possibly through increased BDNF release [8]. Since BDNF signaling is impaired in AS mice, whether rapamycin treatment improves BDNF signaling remains an interesting question to be addressed. It is noteworthy that a recent study showed that rapamycin treatment also restored exogenous BDNF-induced LTP in a mouse model of Down syndrome [45].
In contrast to mTORC1, much less is known regarding the roles of mTORC2 in hippocampal synaptic plasticity and learning and memory. Huang and colleagues [24] recently showed that selective reduction of mTORC2 activity in postnatal murine forebrain by deletion of Rictor greatly impaired both long-term memory and the late phase of hippocampal LTP. They further demonstrated that mTORC2 facilitated LTP and memory by promoting actin polymerization. Subsequently, they showed that brain mTORC2 activity is reduced with age in both fruit flies and rodents in parallel with reduced actin polymerization [46]. These findings are not surprising, as mTORC2 has been shown to participate in actin polymerization [23–26]. Knockout of Rictor, either in whole brain or specifically in Purkinje neurons, resulted in changes in neuronal morphology, especially in Purkinje neurons in a PKC-dependent manner [26]. Additionally, it has been suggested that many of the phenotypes observed in Rictor-deficient mice may be due to abnormal PKC signaling [47]. Thus, decreased mTORC2 activity and the resulting decrease in PKCα levels could be, at least partially, responsible for the decreased spine density of hippocampal pyramidal neurons in AS mice reported here as well as in previous studies [30]. Reduced actin polymerization in hippocampus of AS mice, which is likely due to reduced mTORC2 activity, could account for alterations in dendritic spine morphology. We previously reported that correcting mTORC2 activity was associated with normalizing spine morphology in cerebellar Purkinje neurons in AS mice [27]. These results indicate that mTORC2 plays important roles in maintaining neuronal morphology through regulation of the cytoskeletal network, either directly or indirectly via PKCα and other downstream effectors. Along this line, emerging evidence indicates that manipulation of the evolutionary conserved cell motility machinery may provide promising treatment for cognitive disorders (see [48] for a recent review).
In summary, using rapamycin, Torin 1, PF-4708671, and A-443654, we provide evidence that Ube3A deficiency in hippocampus of AS mice resulted in increased activities of mTORC1 and of its downstream target S6K1, which reduced the activities of mTORC2 through Rictor inhibitory phosphorylation. Our results also showed that enhanced mTORC1-S6K1 activities may contribute to LTP and learning impairment through increased Arc protein synthesis, while reduced mTORC2 activities may be responsible for abnormal actin cytoskeleton dynamics in AS mice. These results are of considerable importance because they demonstrate that, although abnormal mTOR signaling has been implicated in a number of neurological and neuropsychological disorders, the two mTOR complexes may exhibit different, and sometime opposite, alterations. They also suggest that for AS patients, manipulations of mTOR pathways should be directed at reestablishing a normal balanced activation of mTORC1 and mTORC2, by using an mTORC1 inhibitor, PF-4708671 or an mTORC2 activator, A-443654.
Supplementary Material
Acknowledgments
This work was supported by grants P01NS045260 (PI: Dr. C.M. Gall) and R01NS057128 from NINDS to MB and R15MH101703 from NIMH to XB. XB is also supported by funds from the Daljit and Elaine Sarkaria Chair.
Abbreviations
- AMPAR
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor
- Arc
Activity-regulated cytoskeleton-associated protein
- AS
Angelman syndrome
- BDNF
Brain-derived neurotrophic factor
- CS
Conditioned stimuli
- F-actin
Filamentous actin
- i.p.
Intraperitoneally
- LTP
Long-term potentiation
- mTOR
Mechanistic target of rapamycin
- PBS
Phosphate buffered saline
- PFA
Paraformaldehyde
- TBS
Theta-burst stimulation
- US
Unconditioned stimuli
- WT
Wild-type
Footnotes
Conflict of interest
The authors declare no conflict of interest.
References
- 1.Barry RJ, Leitner RP, Clarke AR, Einfeld SL. Behavioral aspects of Angelman syndrome: a case control study. Am J Med Genet A. 2005;132A(1):8–12. doi: 10.1002/ajmg.a.30154. [DOI] [PubMed] [Google Scholar]
- 2.Chamberlain SJ, Chen PF, Ng KY, Bourgois-Rocha F, Lemtiri-Chlieh F, Levine ES, Lalande M. Induced pluripotent stem cell models of the genomic imprinting disorders Angelman and Prader-Willi syndromes. Proc Natl Acad Sci U S A. 2010;107(41):17668–17673. doi: 10.1073/pnas.1004487107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dan B. Angelman syndrome: current understanding and research prospects. Epilepsia. 2009;50(11):2331–2339. doi: 10.1111/j.1528-1167.2009.02311.x. [DOI] [PubMed] [Google Scholar]
- 4.Peters SU, Beaudet AL, Madduri N, Bacino CA. Autism in Angelman syndrome: implications for autism research. Clin Genet. 2004;66(6):530–536. doi: 10.1111/j.1399-0004.2004.00362.x. [DOI] [PubMed] [Google Scholar]
- 5.Lalande M, Calciano MA. Molecular epigenetics of Angelman syndrome. Cell Mol Life Sci. 2007;64(7–8):947–960. doi: 10.1007/s00018-007-6460-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Albrecht U, Sutcliffe JS, Cattanach BM, Beechey CV, Armstrong D, Eichele G, Beaudet AL. Imprinted expression of the murine Angelman syndrome gene, Ube3a, in hippocampal and Purkinje neurons. Nat Genet. 1997;17(1):75–78. doi: 10.1038/ng0997-75. [DOI] [PubMed] [Google Scholar]
- 7.Gustin RM, Bichell TJ, Bubser M, Daily J, Filonova I, Mrelashvili D, Deutch AY, Colbran RJ, Weeber EJ, Haas KF. Tissue-specific variation of Ube3a protein expression in rodents and in a mouse model of Angelman syndrome. Neurobiol Dis. 2010;39(3):283–291. doi: 10.1016/j.nbd.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baudry M, Kramar E, Xu X, Zadran H, Moreno S, Lynch G, Gall C, Bi X. Ampakines promote spine actin polymerization, long-term potentiation, and learning in a mouse model of Angelman syndrome. Neurobiol Dis. 2012;47(2):210–215. doi: 10.1016/j.nbd.2012.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jiang YH, Armstrong D, Albrecht U, Atkins CM, Noebels JL, Eichele G, Sweatt JD, Beaudet AL. Mutation of the Angelman ubiquitin ligase in mice causes increased cytoplasmic p53 and deficits of contextual learning and long-term potentiation. Neuron. 1998;21(4):799–811. doi: 10.1016/s0896-6273(00)80596-6. [DOI] [PubMed] [Google Scholar]
- 10.van Woerden GM, Harris KD, Hojjati MR, Gustin RM, Qiu S, de Avila Freire R, Jiang YH, Elgersma Y, Weeber EJ. Rescue of neurological deficits in a mouse model for Angelman syndrome by reduction of alphaCaMKII inhibitory phosphorylation. Nat Neurosci. 2007;10(3):280–282. doi: 10.1038/nn1845. [DOI] [PubMed] [Google Scholar]
- 11.Kaphzan H, Hernandez P, Jung JI, Cowansage KK, Deinhardt K, Chao MV, Abel T, Klann E. Reversal of impaired hippocampal long-term potentiation and contextual fear memory deficits in Angelman syndrome model mice by ErbB inhibitors. Biol Psychiatry. 2012;72(3):182–190. doi: 10.1016/j.biopsych.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heck DH, Zhao Y, Roy S, LeDoux MS, Reiter LT. Analysis of cerebellar function in Ube3a-deficient mice reveals novel genotype-specific behaviors. Hum Mol Genet. 2008;17(14):2181–2189. doi: 10.1093/hmg/ddn117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang HS, Burns AJ, Nonneman RJ, Baker LK, Riddick NV, Nikolova VD, Riday TT, Yashiro K, Philpot BD, Moy SS. Behavioral deficits in an Angelman syndrome model: effects of genetic background and age. Behav Brain Res. 2013;243:79–90. doi: 10.1016/j.bbr.2012.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Greer PL, Hanayama R, Bloodgood BL, Mardinly AR, Lipton DM, Flavell SW, Kim TK, Griffith EC, Waldon Z, Maehr R, Ploegh HL, Chowdhury S, Worley PF, Steen J, Greenberg ME. The Angelman Syndrome protein Ube3A regulates synapse development by ubiquitinating arc. Cell. 2010;140(5):704–716. doi: 10.1016/j.cell.2010.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kuhnle S, Mothes B, Matentzoglu K, Scheffner M. Role of the ubiquitin ligase E6AP/UBE3A in controlling levels of the synaptic protein Arc. Proc Natl Acad Sci U S A. 2013;110(22):8888–8893. doi: 10.1073/pnas.1302792110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chowdhury S, Shepherd JD, Okuno H, Lyford G, Petralia RS, Plath N, Kuhl D, Huganir RL, Worley PF. Arc/Arg3.1 interacts with the endocytic machinery to regulate AMPA receptor trafficking. Neuron. 2006;52(3):445–459. doi: 10.1016/j.neuron.2006.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rial Verde EM, Lee-Osbourne J, Worley PF, Malinow R, Cline HT. Increased expression of the immediate-early gene arc/arg3.1 reduces AMPA receptor-mediated synaptic transmission. Neuron. 2006;52(3):461–474. doi: 10.1016/j.neuron.2006.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shepherd JD, Rumbaugh G, Wu J, Chowdhury S, Plath N, Kuhl D, Huganir RL, Worley PF. Arc/Arg3.1 mediates homeostatic synaptic scaling of AMPA receptors. Neuron. 2006;52(3):475–484. doi: 10.1016/j.neuron.2006.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cao C, Rioult-Pedotti MS, Migani P, Yu CJ, Tiwari R, Parang K, Spaller MR, Goebel DJ, Marshall J. Impairment of TrkB-PSD-95 signaling in Angelman syndrome. PLoS Biol. 2013;11(2):e1001478. doi: 10.1371/journal.pbio.1001478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ehninger D, Silva AJ. Rapamycin for treating Tuberous sclerosis and Autism spectrum disorders. Trends Mol Med. 2011;17(2):78–87. doi: 10.1016/j.molmed.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Costa-Mattioli M, Monteggia LM. mTOR complexes in neurodevelopmental and neuropsychiatric disorders. Nat Neurosci. 2013;16(11):1537–1543. doi: 10.1038/nn.3546. [DOI] [PubMed] [Google Scholar]
- 22.Costa-Mattioli M, Sossin WS, Klann E, Sonenberg N. Translational control of long-lasting synaptic plasticity and memory. Neuron. 2009;61(1):10–26. doi: 10.1016/j.neuron.2008.10.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.He Y, Li D, Cook SL, Yoon MS, Kapoor A, Rao CV, Kenis PJ, Chen J, Wang F. Mammalian target of rapamycin and Rictor control neutrophil chemotaxis by regulating Rac/Cdc42 activity and the actin cytoskeleton. Mol Biol Cell. 2013;24(21):3369–3380. doi: 10.1091/mbc.E13-07-0405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang W, Zhu PJ, Zhang S, Zhou H, Stoica L, Galiano M, Krnjevic K, Roman G, Costa-Mattioli M. mTORC2 controls actin polymerization required for consolidation of long-term memory. Nat Neurosci. 2013;16(4):441–448. doi: 10.1038/nn.3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jacinto E, Loewith R, Schmidt A, Lin S, Ruegg MA, Hall A, Hall MN. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol. 2004;6(11):1122–1128. doi: 10.1038/ncb1183. [DOI] [PubMed] [Google Scholar]
- 26.Thomanetz V, Angliker N, Cloetta D, Lustenberger RM, Schweighauser M, Oliveri F, Suzuki N, Ruegg MA. Ablation of the mTORC2 component rictor in brain or Purkinje cells affects size and neuron morphology. J Cell Biol. 2013;201(2):293–308. doi: 10.1083/jcb.201205030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sun J, Liu Y, Moreno S, Baudry M, Bi X. Imbalanced mechanistic target of rapamycin C1 and C2 activity in the cerebellum of Angelman syndrome mice impairs motor function. J Neurosci. 2015;35(11):4706–4718. doi: 10.1523/JNEUROSCI.4276-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sun J, Zhu G, Liu Y, Standley S, Ji A, Tunuguntla R, Wang Y, Claus C, Luo Y, Baudry M, Bi X. UBE3A Regulates Synaptic Plasticity and Learning and Memory by Controlling SK2 Channel Endocytosis. Cell Rep. 2015;12(3):449–461. doi: 10.1016/j.celrep.2015.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Briz V, Zhu G, Wang Y, Liu Y, Avetisyan M, Bi X, Baudry M. Activity-dependent rapid local RhoA synthesis is required for hippocampal synaptic plasticity. J Neurosci. 2015;35(5):2269–2282. doi: 10.1523/JNEUROSCI.2302-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dindot SV, Antalffy BA, Bhattacharjee MB, Beaudet AL. The Angelman syndrome ubiquitin ligase localizes to the synapse and nucleus, and maternal deficiency results in abnormal dendritic spine morphology. Hum Mol Genet. 2008;17(1):111–118. doi: 10.1093/hmg/ddm288. [DOI] [PubMed] [Google Scholar]
- 31.Bornancin F, Parker PJ. Phosphorylation of protein kinase C-alpha on serine 657 controls the accumulation of active enzyme and contributes to its phosphatase-resistant state. J Biol Chem. 1997;272(6):3544–3549. doi: 10.1074/jbc.272.6.3544. [DOI] [PubMed] [Google Scholar]
- 32.Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol. 2004;14(14):1296–1302. doi: 10.1016/j.cub.2004.06.054. [DOI] [PubMed] [Google Scholar]
- 33.Oh WJ, Jacinto E. mTOR complex 2 signaling and functions. Cell Cycle. 2011;10(14):2305–2316. doi: 10.4161/cc.10.14.16586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gu J, Lee CW, Fan Y, Komlos D, Tang X, Sun C, Yu K, Hartzell HC, Chen G, Bamburg JR, Zheng JQ. ADF/cofilin-mediated actin dynamics regulate AMPA receptor trafficking during synaptic plasticity. Nat Neurosci. 2010;13(10):1208–1215. doi: 10.1038/nn.2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu Q, Chang JW, Wang J, Kang SA, Thoreen CC, Markhard A, Hur W, Zhang J, Sim T, Sabatini DM, Gray NS. Discovery of 1-(4-(4-propionylpiperazin-1-yl)-3-(trifluoromethyl)phenyl)-9-(quinolin-3-yl)benz o[h][1,6]naphthyridin-2(1H)-one as a highly potent, selective mammalian target of rapamycin (mTOR) inhibitor for the treatment of cancer. J Med Chem. 2010;53(19):7146–7155. doi: 10.1021/jm101144f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, Reichling LJ, Sim T, Sabatini DM, Gray NS. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem. 2009;284(12):8023–8032. doi: 10.1074/jbc.M900301200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Han EK, Leverson JD, McGonigal T, Shah OJ, Woods KW, Hunter T, Giranda VL, Luo Y. Akt inhibitor A-443654 induces rapid Akt Ser-473 phosphorylation independent of mTORC1 inhibition. Oncogene. 2007;26(38):5655–5661. doi: 10.1038/sj.onc.1210343. [DOI] [PubMed] [Google Scholar]
- 38.Bi X, Sun J, Ji AX, Baudry M. Potential therapeutic approaches for Angelman syndrome. Expert Opin Ther Targets. 2015:1–13. doi: 10.1517/14728222.2016.1115837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lipton JO, Sahin M. The neurology of mTOR. Neuron. 2014;84(2):275–291. doi: 10.1016/j.neuron.2014.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sawicka K, Zukin RS. Dysregulation of mTOR signaling in neuropsychiatric disorders: therapeutic implications. Neuropsychopharmacology. 2012;37(1):305–306. doi: 10.1038/npp.2011.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bhattacharya A, Mamcarz M, Mullins C, Choudhury A, Boyle RG, Smith DG, Walker DW, Klann E. Targeting Translation Control with p70 S6 Kinase 1 Inhibitors to Reverse Phenotypes in Fragile X Syndrome Mice. Neuropsychopharmacology. 2015 doi: 10.1038/npp.2015.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Julien LA, Carriere A, Moreau J, Roux PP. mTORC1-activated S6K1 phosphorylates Rictor on threonine 1135 and regulates mTORC2 signaling. Mol Cell Biol. 2010;30(4):908–921. doi: 10.1128/MCB.00601-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hoeffer CA, Klann E. mTOR signaling: at the crossroads of plasticity, memory and disease. Trends Neurosci. 2010;33(2):67–75. doi: 10.1016/j.tins.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Steward O, Wallace CS, Lyford GL, Worley PF. Synaptic activation causes the mRNA for the IEG Arc to localize selectively near activated postsynaptic sites on dendrites. Neuron. 1998;21(4):741–751. doi: 10.1016/s0896-6273(00)80591-7. [DOI] [PubMed] [Google Scholar]
- 45.Andrade-Talavera Y, Benito I, Casanas JJ, Rodriguez-Moreno A, Montesinos ML. Rapamycin restores BDNF-LTP and the persistence of long-term memory in a model of Down’s syndrome. Neurobiol Dis. 2015;82:516–525. doi: 10.1016/j.nbd.2015.09.005. [DOI] [PubMed] [Google Scholar]
- 46.Johnson JL, Huang W, Roman G, Costa-Mattioli M. TORC2: a novel target for treating age-associated memory impairment. Sci Rep. 2015;5:15193. doi: 10.1038/srep15193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Angliker N, Ruegg MA. In vivo evidence for mTORC2-mediated actin cytoskeleton rearrangement in neurons. Bioarchitecture. 2013;3(4):113–118. doi: 10.4161/bioa.26497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Baudry M, Bi X. Learning and memory: an emergent property of cell motility. Neurobiol Learn Mem. 2013;104:64–72. doi: 10.1016/j.nlm.2013.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.