

Abstract
Sleep disorders are prevalent in Alzheimer’s disease (AD) and a major cause of institutionalization. Like AD pathology, sleep abnormalities can appear years before cognitive decline and may be predictive of dementia. A bidirectional relationship between sleep and amyloid β (Aβ) has been well established with disturbed sleep and increased wakefulness leading to increased Aβ production and decreased Aβ clearance; whereas Aβ deposition is associated with increased wakefulness and sleep disturbances. Aβ fluctuates with the sleep wake cycle and is higher during wakefulness and lower during sleep. This fluctuation is lost with Aβ deposition, likely due to its sequestration into amyloid plaques. As such, Aβ is believed to play a significant role in the development of sleep disturbances in the preclinical and clinical phase of AD. In addition to Aβ, the influence of tau AD pathology is likely important to the sleep disturbances observed in AD. Abnormal tau is the earliest observable AD-like pathology in the brain with abnormal tau phosphorylation in many sleep regulating regions such as the locus coeruleus, dorsal raphe, tuberomammillary nucleus, parabrachial nucleus, and basal forebrain prior to the appearance of amyloid or cortical tau pathology. Furthermore, human tau mouse models exhibit AD-like sleep disturbances and sleep changes are very common in other tauopathies including frontotemporal dementia and progressive supranuclear palsy. Together these observations suggest that tau pathology can induce sleep disturbances and may play a large role in the sleep disruption seen in AD. To elucidate the relationship between sleep and AD it will be necessary to not only understand the role of amyloid but also tau and how these two pathologies, together with comorbid pathology such as alpha-synuclein, interact and affect sleep regulation in the brain.
Keywords: Alzheimer’s Disease; sleep, amyloid; tau; alpha-synuclein
1. Introduction
For over 25 years, a high prevalence of sleep disorders has been identified in patients with Alzheimer’s disease (AD). Many studies have estimated that 25–66% of AD patients exhibit sleep disorders of some kind and sleep disturbances are one of the leading causes of AD patient institutionalization (Bianchetti et al. 1995, Guarnieri et al. 2012, Moran et al. 2005). Sleep is an important biological function and although sleep disturbances were once considered to be a bi-product of neurodegeneration, they are not limited to advanced disease states but occur even before cognitive decline and may be predictive of neurodegeneration (Hahn et al. 2013, Lim et al. 2013). AD pathology has been shown to begin long before the onset of the clinical cognitive impairment characteristic of AD. The preclinical stage of AD is characterized by the aggregation of amyloid-β (Aβ) peptide into amyloid plaques in the brain and a decrease of Aβ42 in the cerebrospinal fluid (CSF) as well as tau phosphorylation and aggregation into neurofibrillary tangles (NFT) and neuropil threads in neocortical regions (Holtzman, Morris and Goate 2011). This molecular pathology, especially tau pathology, is then linked with neuronal death, synaptic loss, cognitive impairment, and AD diagnosis (Jack and Holtzman 2013, Holtzman et al. 2011). The presence of sleep disturbances throughout preclinical and clinical disease progression underscores the important role sleep may play in disease pathology and progression.
Studies in humans and mouse models have begun to unravel the relationship between sleep, AD, and cognitive impairment, but much work remains. Due to the abundance of mouse models that develop amyloid deposition and biomarker tests available, much is known about the bidirectional interaction between sleep, Aβ, and its aggregation in the brain. However, to fully understand the interaction between AD and sleep, the contribution of tau and other pathologies cannot be ignored. Here, we will review the clinical relationship between AD and sleep and how amyloid pathology contributes to this interaction. We will further highlight how tau pathology in AD may interact with sleep disturbances including evidence from tauopathies such as frontotemporal dementia (FTD). Lastly, up to 60% of AD patients also have the presence of alpha-synuclein aggregation in the brain and we will highlight ways in which this pathology may contribute to sleep disturbances seen in AD as well as similarities between AD and dementia with Lewy bodies (DLB)(Hamilton 2000).
2. Sleep and circadian disturbances in AD patients
AD patients exhibit a wide array of sleep, electroencephalography (EEG), and circadian dysfunction that is well reviewed in the literature (Musiek, Xiong and Holtzman 2015, Peter-Derex et al. 2015, Lim, Gerstner and Holtzman 2014b). Excessive daytime sleepiness (EDS), sundowning, and insomnia are among the most common reported disturbances in AD and stem from changes in sleep architecture and circadian rhythm. AD patients have been shown to have decreased sleep efficiency, non-rapid eye movement (NREM) sleep and slow wave sleep (SWS) as well as decreased rapid eye movement (REM) sleep and increased latency to REM sleep (Vitiello et al. 1990, Bliwise et al. 1989, Bonanni et al. 2005). Consequently, AD patients display an increase in wake that is characterized by an increased number of awakenings at night (Moe et al. 1995, Vitiello et al. 1990, Bonanni et al. 2005). Taken together these changes result in an observed increase in sleep fragmentation in AD patients (Vitiello et al. 1990). Sleep disturbances are found to occur throughout clinical disease with most changes visible in mild disease stages and these disruptions worsen with disease severity (Vitiello et al. 1990, Bonanni et al. 2005, Liguori et al. 2014b). Increased REM sleep latency and wakefulness as well as decreased REM sleep and sleep efficiency are also found to correlate with impaired cognitive function (Moe et al. 1995, Liguori et al. 2014b). The abundance of sleep disturbances in Alzheimer’s disease patients even in early stages of cognitive impairment may be due to neuronal and synaptic loss that is already occurring at these stages of disease.
Prior to clinical diagnosis, sleep disturbances are also a risk factor for developing cognitive dysfunction and AD. A recent study in a preclinical population has shown using actigraphy that a decrease in sleep efficiency is visible even in cognitively normal people that are amyloid positive compared to those that are amyloid negative, suggesting a role for Aβ aggregation in preclinical sleep changes (Ju et al. 2013). Furthermore, clinical follow up studies have shown that cognitively normal older individuals with high sleep fragmentation had a 1.5-fold increased risk of developing AD and self-reported reduced sleep was associated with a 2-fold increased risk of AD development (Hahn et al. 2013, Lim et al. 2013). These early, preclinical changes in sleep suggest that poor sleep may be a risk factor and biomarker for AD and cognitive decline and demonstrates that pathological AD-type changes may be leading to sleep abnormalities prior to obvious cognitive symptoms. This supports the idea that sleep disruption may be prognostic for future cognitive decline.
Changes in sleep architecture are also accompanied by changes in EEG power. Quantitative EEG studies show that AD patients exhibit EEG slowing in both wake and REM sleep that increases with disease severity, and studies suggest that REM sleep EEG slowing is a robust biomarker for AD severity (see (**Petit et al. 2004) for in depth review). Markers of circadian dysfunction have also been identified in AD patients and include increased nocturnal activity, decreased diurnal activity, core body temperature phase delay and amplitude decrease, clock gene phase changes across brain regions, and changes in sleep regulating hormones such as decreased nocturnal melatonin (sleep promoting) and increased hypocretin/orexin (wake promoting) (Volicer et al. 2001, Harper et al. 2005, Liguori et al. 2014b, Wu et al. 2003, Cermakian et al. 2011). The relationship between AD progression and sleep modulating hormones is not straightforward however. Studies have shown that CSF orexin levels in moderate to severe AD are significantly increased compared to controls and high orexin levels were appropriately associated with increased nocturnal disturbance (Liguori et al. 2014b). However, post mortem analysis shows decreased hypocretin in the CSF and a 40% reduction in hypocretin-1 immunoreactive neurons in the hypothalamus demonstrating a potential, intricate relationship between sleep, AD pathology, and neurodegeneration (Fronczek et al. 2012).
The exact cause of sleep and circadian changes in AD is unknown but likely stems from pathology induced breakdown in crucial sleep regulating areas and pathways in the brain. Sleep centers are located throughout the basal forebrain, hypothalamus, thalamus, midbrain, pons, and brainstem, areas that can be affected by AD pathology. Changes may also result from loss of specific neuronal populations important to sleep regulation. A recent study shows a loss of galanin positive neurons in the ventrolateral preoptic (VLPO)/intermediate nucleus in AD patients and less galanin neurons correlated with increased sleep fragmentation (Lim et al. 2014a). VLPO galanin neurons are sleep active, inhibit wake promoting neurons, and loss of these neurons is a possible mechanisms for decreased NREM sleep and increased awakenings in AD (Saper, Scammell and Lu 2005). Loss of cholinergic neurons in the basal forebrain or noradrenergic neurons in the locus coeruleus in AD could also not only affect cognition but sleep/wake regulation as well (Iraizoz et al. 1999, Rogers, Brogan and Mirra 1985, Brunnström et al. 2011, Wilson et al. 2013). Scattered AD pathology in the suprachiasmatic nucleus (SCN) has also been established and this circadian rhythm ‘pacemaker’ of the brain degenerates in AD (Stopa et al. 1999). The loss of vasopressin and neurotensin positive neurons in the SCN is also correlated with increased activity rhythm fragmentation and increased nocturnal activity, respectively (Harper et al. 2008, Stopa et al. 1999). These observations demonstrate a direct relationship between AD pathology and sleep degeneration.
3. Amyloid and sleep
Aβ aggregation begins early during preclinical, or pre-symptomatic Alzheimer’s disease (Jack and Holtzman 2013, Holtzman et al. 2011). Similarly, sleep changes can begin early in AD and be predictive of neurodegeneration or increase risk of developing cognitive decline in AD. As such, Aβ has so far been a focus in AD sleep research which is aided by the availability of many animal models that overexpress and develop Aβ pathology.
3.1. Amyloid-precursor protein (APP)/Aβ overproduction induces sleep changes in animal models
APP/Aβ overproduction models recapitulate many of the sleep changes observed in AD patients and provide a unique opportunity to explore the direct effect of APP/Aβ on sleep without the presence of other pathological characteristics of AD such as tau aggregation. Increases in wake and decreases in both NREM and REM sleep have been reported in most mouse models studied including APP/PS1, 5× FAD, PDAPP, TgCRND8, and Tg2576, similar to changes seen in AD patients (Roh et al. 2012, Schneider et al. 2014, Huitrón-Reséndiz et al. 2002, Colby-Milley et al. 2015, Zhang et al. 2005). The normalization of these deficits by Aβ immunization in APP/PS1 and Tg2576 mice demonstrates the ability of Aβ pathology itself (distinct from APP) to induce changes in sleep architecture (Wisor et al. 2005, Roh et al. 2012). Furthermore, APP transgenic mice have increased sleep fragmentation, increased latency to REM sleep, and decreased number of REM sleep bouts, changes that are reminiscent of those seen in clinical AD (Colby-Milley et al. 2015, Sethi et al. 2015, Zhang et al. 2005). Sleep changes in multiple models have also been shown to occur early in disease progression when plaque pathology is low and worsen with age, exemplifying the importance of Aβ aggregation on sleep disruption in early and advanced stages of AD (Colby-Milley et al. 2015, Huitrón-Reséndiz et al. 2002, Zhang et al. 2005). The effects of Aβ on sleep are not limited to mice and humans, but extend to Drosophila where expressing Aβ results in decreased and fragmented sleep (Tabuchi et al. 2015). These studies across organisms demonstrate the importance and robust effect of amyloid pathology on sleep. EEG power also appears to be affected in most mouse models. Despite differences between models, there is a prevailing observation of decreased power with disease progression in the delta and/or theta frequencies (Colby-Milley et al. 2015, Zhang et al. 2005, Schneider et al. 2014). Furthermore, unlike humans with AD who display EEG showing, these mouse models show an increase in higher EEG frequencies, demonstrating that Aβ mouse models do not recapitulate all aspects of sleep changes seen in clinical AD (Colby-Milley et al. 2015, Zhang et al. 2005).
The ability of amyloid pathology to induce many changes similar to that seen in AD suggests that amyloid may be a driving force behind sleep changes in early AD and throughout disease progression. How Aβ induces sleep changes is still to be determined and is likely the result of many mechanisms. Changes could be due to the effect of Aβ and amyloid pathology on neuronal populations or brain regions crucial to sleep/wake systems, although amyloid pathology in the subcortical and brainstem nuclei are shown to occur later in disease and cannot explain early changes in sleep (Serrano-Pozo et al. 2011). Worsening of sleep deficits however may be due to progression of pathology spreading from cortical to subcortical and later brainstem structures, as suggested in the CRND8 mouse model (Colby-Milley et al. 2015). In AD brain, diffuse plaques are found throughout the hypothalamus that contains many sleep regulating regions, including the VLPO, the main sleep promoting region of the brain where NFTs are infrequent, and amyloid pathology in this region is one possible mechanism for the sleep loss observed in AD (Standaert et al. 1991). Amyloid plaques are also the dominant clinical AD pathology observed in the periaqueductal grey matter (PAG), with 81% of AD cases having amyloid pathology in this dopaminergic wake active area, although tau pathology is observed early in disease progression as well (Parvizi, Van Hoesen and Damasio 2000, Stratmann et al. 2016). Sleep changes could also be the result of neuronal circuit dysfunction, such as Aβ induced breakdown of slow oscillations and coherence in the neocortex, thalamus and hippocampus and deterioration of slow-wave activity (Busche et al. 2015). These are but a few of the possible mechanisms by which Aβ may induce changes in sleep.
3.2. The bidirectional relationship between Aβ and sleep disturbance
In addition to AD patients and APP/Aβ overexpressing animal models exhibiting sleep disturbances it has been well documented that sleep disruption can alter soluble Aβ levels acutely and Aβ deposition chronically, thus demonstrating a bidirectional relationship between Aβ and sleep (see (Ju, Lucey and Holtzman 2014) for in depth review). In imaging studies in adults without cognitive decline, sleep disruptions such as shorter sleep time, decreased sleep efficiency, and increased latency to sleep are shown to be associated with Aβ deposition in the brain (Ju et al. 2013, Brown et al. 2016, Branger et al. 2016, Spira et al. 2013). Furthermore, SWS arousals are correlated with Aβ42 levels in humans with mild cognitive impairment but not controls (Sanchez-Espinosa, Atienza and Cantero 2014). The importance of sleep in amyloid AD pathogenesis has been further elucidated in sleep deprivation studies in humans and animal models. A single night of sleep deprivation in healthy young adults significantly increased morning Aβ42 levels (Ooms et al. 2014). Sleep deprivation in Aβ overexpressing mice also induces an increase of monomeric Aβ acutely in the brain (Kang et al. 2009). Over time, sleep deprivation induced monomeric Aβ increase can have a dramatic disease promoting effect. This was demonstrated in the Tg2576 and APP/PS1 amyloid mouse models which both had a significant increase in amyloid plaque pathology following 6 weeks of sleep deprivation (Kang et al. 2009). Conversely, 8 weeks of increased sleep by treatment with an orexin receptor antagonist significantly decreased amyloid pathology in APP/PS1 mice, suggesting that sleep is a potent modulator of amyloid both acutely, of monomeric Aβ, and over time, of amyloid pathology progression (Kang et al. 2009). Drosophila that overexpress Aβ demonstrate the same increase in Aβ accumulation with sleep deprivation and decrease in accumulation with sleep increase, further supporting the strong effect of sleep on amyloid pathology (Tabuchi et al. 2015).
The cause of Aβ increases with sleep disturbance could be the results of increased Aβ secretion, decreased clearance, or both mechanisms. In Drosophila, sleep deprivation induced Aβ increases are lost when neuronal hyperexcitability is inhibited, suggesting that Aβ increases with sleep deprivation are due to increased neuronal activity that accompanies wakefulness (Tabuchi et al. 2015). This observation is supported in mice where both sleep deprivation and neuronal activity increases Aβ secretion into the interstitial fluid (ISF) (Cirrito et al. 2005, Kang et al. 2009). Sleep has also been shown to facilitate clearance of Aβ from the ISF by means of the glymphatic system, and thus the decrease of clearance in this fashion may also contribute to the increases in Aβ when sleep is lost (Xie et al. 2013).
3.3. Amyloid-β and circadian rhythm
Circadian dysfunction is well documented in AD patients and studies have demonstrated a delicate interaction between AD, circadian function, and amyloid (see (Musiek et al. 2015) for in depth review). Aβ, like many proteins in the brain, exhibits a circadian fluctuation and this observation is seen in both humans and mice. Humans have increased CSF Aβ throughout the daytime that then decreases at night and these Aβ levels are inversely correlated with sleep (Huang et al. 2012). Mouse ISF Aβ exhibits the same circadian fluctuation in both wild type and APP/Aβ overexpression models (Roh et al. 2012, Kang et al. 2009). The ability of sleep deprivation to raise Aβ levels in the brain could be a direct result of this fluctuation. Lack of sleep may inhibit the reduction of Aβ that would normally occur during the restful period, which could affect amyloid pathology development in AD over a longer period of time. Furthermore, AD pathology affects this Aβ fluctuation. In humans, the amplitude of Aβ fluctuation decreases both with age as well as the presence of amyloid aggregation and amyloid positive patients with familial dominant AD have a loss of Aβ fluctuation (Roh et al. 2012, Huang et al. 2012). Lower CSF Aβ42 levels in humans is also associated with lower hypocretin levels and increased amyloid pathology, further suggesting the important relationship between abnormal circadian function and AD amyloid pathology (Slats et al. 2012). Aβ overexpressing APP/PS1 mice display a similar loss of circadian Aβ fluctuation in the ISF, likely due to sequestration of Aβ42 into amyloid plaques, a phenomenon also likely occurring in humans (Roh et al. 2012). The loss of fluctuation occurs with the onset of amyloid pathology and is rescued by Aβ42 immunization, demonstrating the causative role of amyloid pathology in the loss of Aβ fluctuation (Roh et al. 2012). In addition to Aβ fluctuation, amyloid overexpression mouse models also show changes in circadian processes such as decreased thermoregulation and increased period, suggesting that some form of Aβ is sufficient to induce circadian dysregulation similar to that seen in ageing and AD (Wisor et al. 2005, Huitrón-Reséndiz et al. 2002, Song et al. 2015). Mechanistically, APP/Aβ overexpression may affect circadian features through SCN degeneration, which is observed in 3xTg mice and humans with AD (Sterniczuk et al. 2010, Harper et al. 2008). Although rare, diffuse amyloid plaques are seen in the SCN of AD patients (Stopa et al. 1999). Aβ in culture and mouse models is also shown to induce changes in clock genes, such as decreases in PER2 and BMAL1, which could induce circadian dysregulation outside the SCN (Song et al. 2015). Taken together, these studies demonstrate an important interaction between Aβ and circadian rhythms that could contribute to and possibly accelerate disease progression.
4. Tau and sleep
The hyperphosphorylation and intercellular aggregation of tau protein is the second hallmark of AD and unlike Aβ, total and phosphorylated tau in the CSF has been shown to predict and correlate with cognitive decline in preclinical and clinical AD (Fagan et al. 2007, Mattsson et al. 2009). Furthermore, tau pathology has recently been shown to be the earliest observable AD-like change in human brain, with abnormal tau phosphorylation and aggregation in the locus coeruleus (LC) beginning as early as young adulthood and extending to other connected regions even before amyloid is detected (Braak et al. 2011, Stratmann et al. 2016). Despite these observations, very little is known about the effect of tau pathology on sleep in preclinical or clinical AD. Currently, our understanding of the possible role of tau in AD related sleep disturbance is based off the presence of NFTs in sleep centers of the brain, limited studies in AD mouse models containing both APP and tau transgenes or a human tau transgene alone, and what is known about sleep in other tauopathies such as FTD. Together, these areas highlight the important role tau may play in sleep dysregulation in AD.
4.1. Neurofibrillary tangles in sleep, wake, and circadian centers of the brain during AD pathogenesis
Sleep/wake cycles are tightly regulated in the brain by many brain regions, neurotransmitters, and flip-flop switches. Sleep regulating areas are found throughout brain, including the brainstem, midbrain, thalamus, hypothalamus, and basal forebrain (Figure 1) (Saper et al. 2005). These many regions regulate arousal (ascending arousal pathway), promote and inhibit NREM and REM sleep, as well as regulate circadian rhythms and the biological clock (Figure 1). Many of these regions, especially the wake promoting arousal system, are shown to be affected by abnormal tau pathology in the human brain with many of the changes occurring during pretangle stages or stage 0 of tau pathology by Braak and Braak staging, prior to any cortical tau or amyloid pathology development (Figure 1) (Braak et al. 2011, Stratmann et al. 2016, Stern and Naidoo 2015, Braak and Del Tredici 2011). Here we will discuss sleep, wake, and circadian regulating areas of the brain known to be affected by tau pathology to underscore the potential importance of tau in sleep dysregulation.
Fig 1.
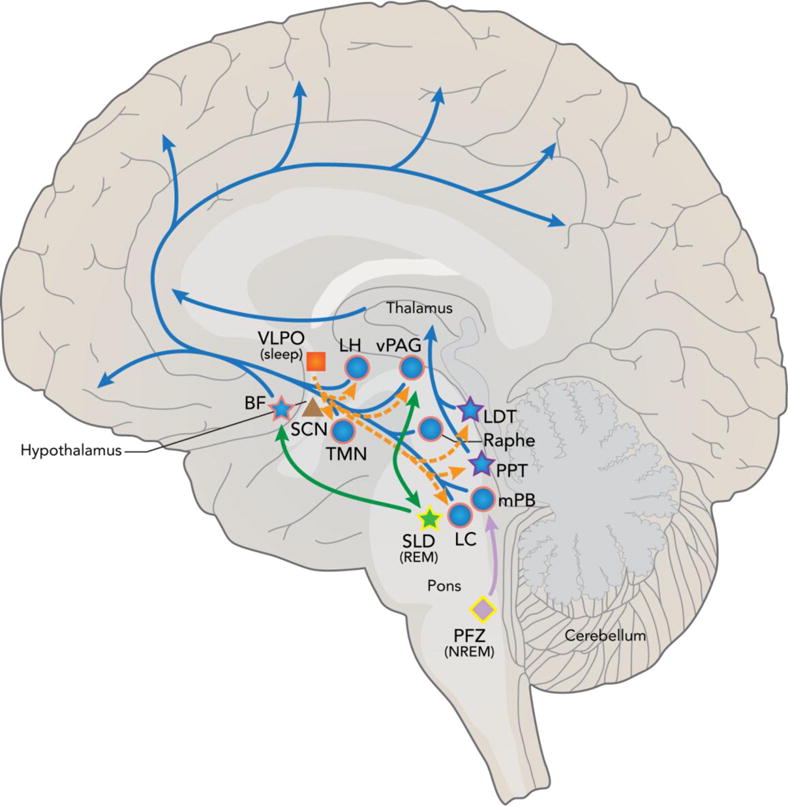
Schematic of tau pathology in sleep, wake, and circadian brain regions and pathways. The ascending arousal system (blue) leading to cortical arousal is composed of brain regions active in wake (blue circles) or both wake and REM sleep (blue stars). Many of these regions are shown to be affected by abnormal tau pathology in pretangle stages, prior to cortical tau or Aβ pathology (pink outline) or in clinical AD (purple outline), suggesting a role for tau in wake/sleep dysregulation in aging and AD. The hypothalamic VLPO (orange square) sleep generating region inhibits wake active areas of the ascending arousal system (orange dash). However, the VLPO as well as the circadian master clock SCN (brown triangle) have not been shown to have significant tau pathology (no outline) in AD. The REM sleep producing SLD (green), which interacts with the REM-off ventrolateral PAG, and NREM/SWS generating PFZ (light purple) have not yet been studied for the presence of tau pathology (yellow outline) but could play a role in sleep loss seen in AD. Adapted by permission from Macmillan Publishers Ltd: Neuron (Saper et al. 2005).
4.1.1. Ascending arousal system
The ascending arousal system is composed of wake active neuronal groups and includes many brain areas known to contain abnormal tau phosphorylation pathology early in life and with AD disease progression (Stern and Naidoo 2015, Braak et al. 2011, Braak and Del Tredici 2011). Tau pathology first appears in the brainstem LC and extends to the medial parabrachial nucleus (mPB), the dorsal raphe (DR), and the midbrain PAG (Braak et al. 2011, Stratmann et al. 2016, Braak and Del Tredici 2011). Pretangle tau pathology is also found in the hypothalamic tuberomammilary nucleus (TMN), lateral hypothalamus (LH), and the basal forebrain (BF) (Braak et al. 2011, Stratmann et al. 2016, Braak and Del Tredici 2011) (Figure 1). All of these areas have abnormal tau phosphorylation prior to cortical tau or amyloid pathology and may play a role in tau induced sleep dysregulation during normal aging (Braak et al. 2011, Stratmann et al. 2016, Braak and Del Tredici 2011). In AD, many regions of the ascending arousal system, including the pedunculopontine tegmental nucleus (PPT) and laterodorsal tegmental nucleus (LDT), have been shown to have robust NFT pathology, further supporting a role of tau in AD sleep disturbances (Figure 1) (Parvizi, Van Hoesen and Damasio 2001, Stern and Naidoo 2015).
Noradrenergic wake active neurons in the LC play an excitatory role in the ascending arousal system of the cortex and inhibit the sleep producing VLPO neurons (Figure 1) (Saper et al. 2005, Stern and Naidoo 2015). Braak et al. demonstrates that abnormal tau species are found in this area in a high percentage of young adults (70% at 20 years of age) prior to cortical tau or amyloid burden (Braak et al. 2011). Furthermore, NFTs are reproducibly found in AD patients as well as cognitively normal and MCI adults without full blown dementia (Grudzien et al. 2007, Dugger et al. 2012). In AD, LC noradrenergic neurons are lost and LC neuron number is found to be correlated to cognitive decline in a non-demented cohort (Wilson et al. 2013, Brunnström et al. 2011). Increased NFTs in the LC is also related to more rapid cognitive decline (Wilson et al. 2013). NFTs in and loss of LC neurons may therefore be playing a role in sleep disturbances and EDS found in preclinical and early AD (Stern and Naidoo 2015). In addition to AD pathology driving loss of LC neurons, these neurons are also susceptible to increased wakefulness (Zhang et al. 2014). Since increased wake is characteristic of AD, it follows that AD may be able to induce degeneration of this area, further contributing to disease and sleep problems. Lastly, LC degeneration has also been shown to promote Aβ pathology in APP23 mice and result in increased neuronal loss and memory deficits (Heneka et al. 2006). Tau pathology in the LC or wake induced neuronal loss could also affect amyloid pathology development and thus disease progression in AD. Taken together, tau pathology in the LC may have a profound effect on both sleep and cognition in AD pathogenesis and begin years before other pathologies are present, making it an important area for study of the interaction between sleep, tau, and AD. Furthermore, early pathology in the LC alone is not recapitulated, to our knowledge, in any AD or human tau (htau) mouse models. Such models would be invaluable in studying this interaction.
The serotonergic neurons of the DR are also wake active and in addition to pretangle stage detection of tau pathology in this region, NFT pathology in the DR was also identified in preclinical AD patients (stage 0, 1 and 2) as well as those with a clinical AD diagnosis (Saper et al. 2005, Stern and Naidoo 2015, Chen et al. 2000, Stratmann et al. 2016, Rüb et al. 2000, Grinberg et al. 2009). Beyond the presence of NFTs in the DR, neuron density is also decreased in AD patients (Chen et al. 2000). Although this loss of neurons or NFT number in the DR of AD patients was not correlated with cognitive decline, it is possible that loss of serotinergic neurons and NFT induced changes in this region contributes to sleep dysregulation in AD patients (Chen et al. 2000). The brainstem mPB is also wake active and inhibited by the parafacial zone (PFZ) during NREM sleep (Anaclet et al. 2014). Abnormal tau pathology in the mPB was identified at stage 0 and NFTs are found in the mPB of up to 91% of postmortem clinical AD brains in the absence of amyloid pathology in this region, supporting a link between clinical symptoms and tau pathology in this region (German, White and Sparkman 1987, Parvizi et al. 2001, Stratmann et al. 2016).
Cholinergic neurons in the PPT and LTD are active during wake and REM sleep (Figure 1) (Stern and Naidoo 2015, Saper et al. 2005). Although studies of stage 0 AD tau pathology have not reported early tau pathology in these areas, NFTs are common in the LDT and PPT in clinical AD (Dugger et al. 2011, Dugger et al. 2012, Parvizi et al. 2001). One study showed that 69% of AD postmortem brains contained NFTs in the PPT without amyloid pathology present there, even in patients with short disease duration (Parvizi et al. 2001). Furthermore, 66% of patients had NFT pathology in the LDT and the presence of NFTs in this area correlated with disease progression (Parvizi et al. 2001). The wake active, REM inhibiting PAG of the midbrain has Stage 0 development of tau pathology (Stratmann et al. 2016). However, in the PAG one study reported that only 9% of clinical AD brains autopsied had NFT pathology, while 81% contained amyloid plaques (Parvizi et al. 2000). Therefore the effect of NFTs in this area is unclear.
In the hypothalamus, abnormal tau pathology is present in the pretangle, precortical tau stage in the TMN and LH (Figure 1) (Stratmann et al. 2016). Furthermore, in patients who died with dementia due to AD, hypothalamic NFTs are numerous and concentrated in the TMN and accompanied by wake active histaminergic neuronal lost (Nakamura et al. 1993, Airaksinen et al. 1991a, Stern and Naidoo 2015, Standaert et al. 1991). This focus of pathology suggests a role of tau in neuronal loss in the TMN and possible contribution to sleep disruption, but the influence of amyloid cannot be ignored since diffuse amyloid plaques are also present throughout the hypothalamus in the AD brain (Standaert et al. 1991). Although most prominent in the TMN, NFTs can also be seen in the LH in clinical AD (Standaert et al. 1991). LH neurons are wake active and produce orexin, which acts to stabilize the sleep/wake switch allowing for sustained arousal and sleep (Saper et al. 2005). Pretangle tau pathology has also been reported specifically in the prefornical area which contains the orexinergic neurons (Stratmann et al. 2016). Although it is not known if tau pathology affects orexinergic neurons, orexin is an important marker of sleep/wake regulation that is shown to increase in the CSF of moderate to severe AD patients and is positively correlated to total tau and related to sleep impairment in AD (Liguori et al. 2014b). Furthermore, orexin levels in cognitively normal elderly correlate with phosphorylated tau, suggesting that tau could be a driving factor in orexin and wakefulness changes in the brain (Osorio et al. 2016).
Tau pathology also reaches the magnocellular nuclei of the basal forebrain in pre-tangle stages of AD development, MCI, and early AD, as well as in cognitively normal age matched controls (Braak et al. 2011, Mesulam et al. 2004, Stratmann et al. 2016, Iraizoz et al. 1999). Cholinergic neurons in the basal forebrain (nucleus basalis of Meynert (NBM)) are active during wake and REM sleep and also play an important role in cognition with excitatory projection to the cortex (Figure 1) (Saper et al. 2005, Stern and Naidoo 2015). NBM neurons are lost in AD and cell number decreases with disease progression (Rogers et al. 1985, Iraizoz et al. 1999). NFTs may be to blame for this cell loss since remaining neurons in this area contain NFTs but very few, if any, amyloid plaques are present (Iraizoz et al. 1999, Rogers et al. 1985). Furthermore, NFTs in the NBM correlate with higher mental deterioration and decreased BF volume in AD correlates with increased cortical amyloid plaque burden, suggesting that tau pathology and loss of neurons could play a role in disease progression as well as cognitive decline and sleep disruption (Kerbler et al. 2015, Iraizoz et al. 1999).
Many regions of the ascending arousal system are affected by tau pathology early in life and prior to preclinical amyloid pathology development, supporting that tau may play a crucial role in sleep disturbance and sleep/wake system dysregulation not only in clinical AD but also in preclinical AD and normal aging. More work is needed to determine if tau pathology in these areas induces changes in sleep/wake regulation. It is known that lesions in some of the wake regulating areas discussed here do not induce changes in sleep/wake architecture (Fuller et al. 2011, Gompf et al. 2010). However, tau pathology may induce dysregulation by affecting multiple areas, specific cell types, or in conjunction with pathology in sleep promoting regions.
4.1.2. Sleep promoting regions
AD pathology in and degeneration of sleep promoting regions of the brain could explain the loss of sleep seen in clinical AD. One of the main sleep promoting regions of the brain is the VLPO. The VLPO, located in the hypothalamus, projects to the ascending arousal system and inhibits this system during sleep (Figure 1) (Saper et al. 2005). Surprisingly, the VLPO has not been shown to contain prominent NFT pathology (Standaert et al. 1991). However, there are other regions in the brain that function to generate NREM and REM sleep, such as the PFZ and the sublaterodorsal area (SLD), which may be affected by tau pathology (Figure 1). The PFZ, a group of GABAergic neurons in the brainstem, is a SWS generating region recently identified in mice (Anaclet et al. 2014). Disruption of the PFZ could result in decreased SWS as seen in AD, but to our knowledge pathology in this region remains unstudied (Anaclet et al. 2014). The SLD and pre coeruleus is a REM generating region in the brain also elucidated in mice and rats (Krenzer et al. 2011, Lu et al. 2006). The presence of tau pathology in the SLD has not been specifically studied. However, the SLD REM producing region is located near the LC, the earliest location of tau pathology, so it is very possible that tau pathology could accumulate in this region throughout aging and the progression of AD. Furthermore, the SLD receives input from the ventrolateral PAG which serves as a REM off region, and the PAG has also been shown to have early presence of tau pathology, strengthening the possible effect of tau in the SLD (Krenzer et al. 2011, Lu et al. 2006). Disruption to or cell loss in the SLD or disruption of the REM on/off balance could be a possible mechanism for loss of REM sleep and increased REM latency in AD. Other brain areas that are active in wake are also active during REM sleep and affected by tau pathology in pre-tangle stages, the BF, and in clinical AD, the LDT and PPT (figure 1). Degeneration in these regions could further decrease REM sleep pathway activation and sustenance.
4.1.3. Circadian regions
The SCN is the master pacemaker of the body and regulates circadian rhythm and inputs (Figure 1). There is not significant tau pathology in this region with only scattered NFTs as well as rare, diffuse amyloid plaques observed in the SCN of AD brains (Stopa et al. 1999). Although the impact of this pathology on circadian rhythm is unknown it may contribute to the loss of vasopressin and neurotensin neurons in the SCN and circadian disruption observed in AD patients (Musiek et al. 2015, Stopa et al. 1999). The TMN has also been shown to project to the SCN and contain tau pathology which could possibly affect input and regulation of the SCN circadian clock (Stern and Naidoo 2015, Airaksinen et al. 1991b, Braak et al. 2011, Standaert et al. 1991). More work is needed to identify if tau induces circadian changes or if tau itself is regulated in a circadian fashion to determine if there is a tau/circadian interaction.
4.2. Sleep changes in htau expressing mice
Mice expressing human tau transgenes with disease promoting mutations that cause FTD have been studied in recent years, but only a small number of sleep studies in these mice have been conducted to assess the ability of tau pathology to induce sleep disruption. In an effort to model AD, mice containing both amyloid pathology and human forms of tau have been studied. Circadian changes reminiscent of AD have been noted in the 3xTg mouse model, which has decreased vasopressin neurons in the SCN, increased daytime activity, and shorter circadian period (Sterniczuk et al. 2010). This demonstrates that tau and Aβ pathology, either alone or together, can induce circadian changes. A second amyloid and tau model, PLB1, has shown changes in sleep architecture similar to AD patients and Aβ-depositing mice including increased wake, decreased NREM and REM sleep, less frequent and shorter REM bouts, increased number of short NREM bouts, and overall sleep fragmentation that worsens with age (Jyoti et al. 2015, Platt et al. 2011). This model was further analyzed for the effect of the htau P301L transgene alone by extraction of the amyloid pathology producing portion of the transgene (Koss et al. 2016). Analysis of PLB1Tau mice demonstrated for the first time that forebrain mutant htau expression alone alters sleep. Specifically, PLB1Tau mice have increased wake and decreased NREM sleep, as well as more robust changes in EEG power than observed in PLB1 mice with both tau and amyloid (Koss et al. 2016). This work is the first evidence that tau pathology alone can induce sleep changes reminiscent of neurodegenerative disease. However, more work is needed in this model to determine what brain regions tau pathology accumulates in as well as other tau models to understand how tau may be inducing changes in sleep. Lastly, loss of tau in tau knock out mice induces changes in sleep including increased wake, decreased NREM sleep, and sleep fragmentation, demonstrating that endogenous tau function is important to sleep regulation and loss of tau function in tauopathies like AD, not just tau pathology, could contribute to the sleep changes observed (Cantero et al. 2010).
Tau may not only affect sleep and wake parameters but it is possible that sleep disruption could contribute to tau pathology progression, as seen with Aβ. One study in 3xTg mice showed an increase in insoluble tau and MC1 staining for conformational tau changes following 2 months of sleep deprivation by light cycle manipulation (Di Meco, Joshi and Praticò 2014). This suggests a possible bi-directional relationship between tau pathology and sleep, similar to that seen with Aβ, which could accelerate disease progression. However, this model also includes APP/Aβ overexpression so the effect of amyloid in the observed changes cannot be ignored (Di Meco et al. 2014). Neuronal activity has also been shown to increase tau in the ISF, raising the possibility for extended wakefulness to impact tau pathology progression (Yamada et al. 2014). Although much work remains, these studies demonstrate that tau may be an important component of sleep/wake dysregulation in the development of AD and needs to be taken into consideration when dissecting and understanding the cause and effect of sleep changes in AD.
4.3. Sleep disturbances in tauopathies
The FTD spectrum includes multiple disorders, many of which contain tau pathology. Tau aggregation occurs in approximately half of the patients with behavioral variant FTD (bvFTD), which affects the frontal and temporal lobe, and 70% of nonfluent/agrammatic variant FTD cases, which affects the posterior frontal lobe and insula (Pressman and Miller 2014, Pan and Chen 2013). There are also 2 other pure tauopathies: progressive supranuclear palsy (PSP) and corticobasal degeneration (CBD) (Pan and Chen 2013). PSP is characterized by globose NFTs that affect largely the brainstem and basal ganglia while CBD is characterized by ballooned neurites, globose corticobasal bodies, and fine filamentous inclusions in the cortex as well as the basal ganglia, subcortical nuclei, and brainstem (Dickson 1999, Pan and Chen 2013). In both of these disorders, there are also astrocytic tau inclusions (Dickson 1999). Despite the variation in FTD, studies have shown that patients suffer from sleep disturbances as frequently as or possibly more than AD. One large clinical study showed that 65.7% of AD patients and 76% of FTD patients had sleep disturbances of some kind including insomnia, restless leg syndrome, sleep disordered breathing, and EDS (Guarnieri et al. 2012). This and other studies suggest that sleep is possibly better preserved in AD than FTD (Bonakis et al. 2014, Guarnieri et al. 2012, Pistacchi et al. 2014).
FTD patients experience many similar sleep disturbances as AD. Studies in bvFTD patients have shown decreased total sleep time and percent sleep, decreased sleep efficiency that is worse in MRI abnormal FTD, decreased REM sleep, and changes in NREM sleep (Anderson et al. 2009, Bonakis et al. 2014). When compared to AD patients, there are no striking differences in bvFTD patient sleep parameters. One study showed no differences in any sleep parameters between AD and bvFTD and while a second showed AD and bv-FTD to both display EDS and altered NREM sleep compared to controls, only the NREM/REM ratio was significantly decreased in FTD compared to AD, although REM sleep and total sleep were decreased in FTD only compared to controls (Bonakis et al. 2014, Kundermann et al. 2011). Finally, when analyzing orexin, no difference has been observed between FTD and controls but FTD orexin levels are decreased compared to AD (Çoban et al. 2013, Liguori et al. 2014a).
Studies in pure tauopathies have also shown these patients to exhibit sleep disturbances similar to those in AD. Patients with PSP have decreased total sleep time and sleep efficiency, decreased sleep spindles, increased sleep latency, and decreased REM sleep characterized by decreased number and duration of REM episodes (Gama et al. 2010, Montplaisir et al. 1997, Petit et al. 2004). The observed changes in REM sleep in PSP are suggestive of PPT degeneration (Montplaisir et al. 1997). As previously discussed, the PPT contains robust tau pathology in AD, and thus tau pathology in this brain region may contribute to the loss of REM in AD. Furthermore, PSP exhibits slowing of wake EEG as well as EDS that correlates with disease progression, similar to changes observed in AD (Montplaisir et al. 1997, Gama et al. 2010, Petit et al. 2004). A patient with a TAU G389R mutation causing tau pathology and hereditary Pick’s disease also showed sleep fragmentation and decreased SWS (Gemignani et al. 2005). These observations demonstrate the ability of tau pathology to induce profound changes in sleep not unlike those seen in AD and supports a possible role for tau pathology in AD sleep disruption.
5. Alpha-synuclein aggregation, DLB, and sleep
Alpha-synuclein is a 140 amino acid protein predominantly expressed at pre-synaptic terminals of neurons and is thought to play a role in vesicle trafficking and release (Bendor, Logan and Edwards 2013). Intracytoplasmic aggregates of alpha-synuclein, present in Lewy bodies and Lewy neurites, are pathological hallmarks of a group of neurodegenerative diseases such as Parkinson disease, multiple system atrophy, and DLB. Up to 60% of patients with sporadic AD also have Lewy body pathology in the brain (Hamilton 2000). AD cases with Lewy bodies show pathology consistently in the amygdala but this can also be observed to spread through the neocortex with limited brainstem and substantia nigra involvement (Hamilton 2000). Dominantly inherited AD patients also have a high prevalence of Lewy bodies in the brain that are concentrated in the amygdala but also found in limbic, neocortical, and brainstem regions (Lippa et al. 1998, Leverenz et al. 2006). Dominantly inherited AD patients with Lewy bodies outside of the amygdala also had a longer course of disease (Leverenz et al. 2006). Furthermore, there is some overlap in symptoms and pathology of AD and DLB which makes it worthwhile to examine the relationship between DLB and sleep here (McKeith 2000, Lippa and Knopman 2007, McKeith et al. 2005).
DLB is characterized by the onset of dementia like symptoms, cognitive dysfunction and visual hallucinations coincident with Parkinsonism. In addition to the brain stem and striatum, DLB features synuclein deposits in the insula, cingulate cortex, amygdala and hippocampus (Dickson et al. 1994, Tsuboi and Dickson 2005, Harding and Halliday 2001, Bertrand et al. 2004, Aarsland, Ballard and Halliday 2004). DLB cases can further present with extensive Aβ deposits in the striatum(Serrano-Pozo et al. 2011). Parkinson’s disease with dementia is similar to DLB except that Parkinsonism precedes cognitive impairment by at least one year and usually longer. A number of sleep disorders present along with DLB and some reports estimate that ~50% of DLB patients have sleep disorders and this can increase to over 70% in moderate to severe disease (Borroni, Agosti and Padovani 2008, Farina et al. 2009). When compared to AD, it has also been suggested that DLB has an overall greater sleep disturbance with DLB patients exhibiting twice as many nocturnal sleep disturbances as AD (Grace, Walker and McKeith 2000, Bliwise et al. 2011). There is also some evidence to suggest measurable changes in sleep characteristics in DLB patients. Although research is limited, one study reported decreased stage 1 NREM sleep and increased stage 2 NREM sleep in DLB patients when compared with Parkinson disease patients (Terzaghi et al. 2013). Quantitative EEG studies in DLB have further reported EEG slowing as well as high variation and fluctuation of mean EEG power in wake (Petit et al. 2004, Walker et al. 2000). There is evidence of accumulation of synuclein pathology in the form of Lewy bodies in wake active neurons of various brain regions (LH, LC, TMN, NBM, DR) which could contribute to the sleep disorders and disturbances observed in DLB (Stern and Naidoo 2015). Furthermore, like in AD and other neurodegenerative disorders, DLB patients show underlying circadian dysregulation. One study suggests that DLB patients have greater disruption in locomotor activity than AD patients and non-demented controls with significantly decreased diurnal activity (Harper et al. 2004). DLB also trended towards increased nocturnal activity compared to controls but remained less than AD (Harper et al. 2004). Temperature circadian phase occurred significantly later in both AD and DLB, although DLB patients showed lower core-body temperature amplitude than AD and was not significantly different from non-demented controls (Harper et al. 2004).
Outside of these generalized sleep and circadian disturbances, DLB is characterized by several specific sleep conditions that also aid in differentiating it from AD and other dementias including REM sleep behavior disorder (RBD), EDS, and periodic limb movement disorder (PLMD) (Borroni et al. 2008, Farina et al. 2009). RBD, a condition where patients physically ‘act out’ their vivid dreams during REM sleep, is now considered a part of the diagnostic criteria for DLB (Boeve et al. 1998, Iranzo et al. 2014, Ferman et al. 2011, McKeith et al. 2005). In fact, RBD can precede any dementia-like symptoms and can be considered to be the prodromal phase of DLB (Boeve et al. 2013, Donaghy and McKeith 2014). Multiple longitudinal studies tracking frequency and emergence of neurodegenerative symptoms in patients diagnosed with RBD have found that a large portion of these patients go on to develop Parkinsonism or dementia. About 30–40% of patients were found to have Parkinsonism or dementia within 5 years of an RBD diagnosis, and about 80% of RBD patients develop Parkinsonism or dementia within 12 years (Boeve et al. 2013, Schenck, Bundlie and Mahowald 1996, Schenck, Boeve and Mahowald 2013, Postuma et al. 2009, Iranzo et al. 2006, Iranzo et al. 2013). Both LBD and RBD have been associated with loss of cholinergic neurons in the brain stem including the LC, PPT/LDT and substantia nigra (Dugger et al. 2012, Uchiyama et al. 1995). One study in dementia patients found that patients with DLB are much more likely to have RBD than patients with AD (Boeve et al. 2001). Although RBD is a not a common feature of AD, it is interesting to note that patients with AD do show loss of neurons in LC and tau pathology is present in the PPT and LDT (Dugger et al. 2011, Brunnström et al. 2011, Parvizi et al. 2001). PLMD is more commonly associated with restless leg syndrome in PD, but is also found separately in patients with DLB (Fantini et al. 2002). A study comparing patients with AD and DLB found a higher frequency of PLMD in DLB patients (Hibi et al. 2012). A lot remains unknown about PLMD, such as its etiology, true prevalence, and mechanism.
EDS is another sleep condition that although prevalent in AD, is shown to be more frequent in DLB (Boddy et al. 2007). As the name suggests patients with EDS have difficulty maintaining wakefulness during the day. Like RBD, EDS is more prevalent in patients with DLB than AD as measured and confirmed by polysomnography and multiple sleep latency test (Boddy et al. 2007, Ferman et al. 2011). Although the mechanism(s) underlying EDS is not completely clear, there are some indications that orexinergic neurons in the lateral hypothalamus are lost in both DLB and AD, which might contribute to EDS in both diseases (Fronczek et al. 2007, Fronczek et al. 2012, Lessig et al. 2010). Furthermore, orexinergic neurons modulate the sleep-wake switch by virtue of their reciprocal actions on ascending arousal wakefulness neurons, many of which are known to be affected by AD and DLB pathology and could contribute to sleep disturbances in both diseases (Saper et al. 2010).
6. Conclusion
Sleep disturbances are prevalent in AD and may play a significant role in the advancement of pathology and disease progression. Although Aβ and amyloid pathology has been established to interact with sleep and circadian rhythms, tau pathology and also the co-morbidity of alpha-synuclein aggregation may be important contributors to sleep system dysfunction in AD. Tau pathology in the brainstem is the earliest AD-like pathological change that occurs during normal aging, and abnormal tau pathology is present in many sleep centers of the brain even before the onset of amyloid pathology. Early studies in an htau expressing mouse model shows sleep disturbances with tau pathology alone. These observations combined with the high prevalence of sleep disorders in other tauopathies exemplifies the need to understand the interaction between tau and sleep in addition to and in conjunction with Aβ pathology.
Acknowledgments
This work was supported by National Institute of Health (grants P01NS074969 (DMH), F32NS089381 (JH)), and a grant from the Tau consortium (DMH).
Abbreviations
- Aβ
amyloid β
- AD
Alzheimer’s disease
- APP
amyloid precursor protein
- BF
basal forebrain
- bvFTD
behavioral variant FTD
- CBD
corticobasal degeneration
- CSF
cerebrospinal fluid
- DLB
dementia with Lewy bodies
- DR
dorsal raphe
- EDS
excessive daytime sleepiness
- EEG
electroencephalography
- FTD
Frontotemporal Dementia
- htau
human tau
- ISF
interstitial fluid
- LC
locus coeruleus
- LDT
laterodorsal tegmental nucleus
- LH
lateral hypothalamus
- mPB
medial parabrachial nucleus
- NBM
nucleus basalis of Meynert
- NFT
neurofibrillary tangle
- NREM
non-rapid eye movement
- PAG
periaqueductal gray matter
- PLMD
periodic limb movement disorder
- PPT
pedunculopontine tegmental nucleus
- PSP
progressive supranuclear palsy
- PFZ
parafacial zone
- RBD
REM sleep behavior disorder
- REM
rapid eye movement
- SCN
suprachiasmatic nucleus
- SLD
sublaterodorsal area
- SWS
slow wave sleep
- TMN
tuberomammillary nucleus
- VLPO
ventrolateral preoptic nucleus
- vPAG
ventral periaqueductal gray
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aarsland D, GBallard C, Halliday G. Are Parkinson’s disease with dementia and dementia with Lewy bodies the same entity? J Geriatr Psychiatry Neurol. 2004;17:137–45. doi: 10.1177/0891988704267470. [DOI] [PubMed] [Google Scholar]
- Airaksinen MS, Paetau A, Paljärvi L, Reinikainen K, Riekkinen P, Suomalainen R, Panula P. Histamine neurons in human hypothalamus: anatomy in normal and Alzheimer diseased brains. Neuroscience. 1991a;44:465–81. doi: 10.1016/0306-4522(91)90070-5. [DOI] [PubMed] [Google Scholar]
- Airaksinen MS, Reinikainen K, Riekkinen P, Panula P. Neurofibrillary tangles and histamine-containing neurons in Alzheimer hypothalamus. Agents Actions. 1991b;33:104–7. doi: 10.1007/BF01993139. [DOI] [PubMed] [Google Scholar]
- Anaclet C, Ferrari L, Arrigoni E, Bass CE, Saper CB, Lu J, Fuller PM. The GABAergic parafacial zone is a medullary slow wave sleep-promoting center. Nat Neurosci. 2014;17:1217–24. doi: 10.1038/nn.3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson KN, Hatfield C, Kipps C, Hastings M, Hodges JR. Disrupted sleep and circadian patterns in frontotemporal dementia. Eur J Neurol. 2009;16:317–23. doi: 10.1111/j.1468-1331.2008.02414.x. [DOI] [PubMed] [Google Scholar]
- Bendor JT, Logan TP, Edwards RH. The function of alpha-synuclein. Neuron. 2013;79:1044–66. doi: 10.1016/j.neuron.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand E, Lechowicz W, Szpak GM, Lewandowska E, Dymecki J, Wierzba-Bobrowicz T. Limbic neuropathology in idiopathic Parkinson’s disease with concomitant dementia. Folia Neuropathol. 2004;42:141–50. [PubMed] [Google Scholar]
- Bianchetti A, Scuratti A, Zanetti O, Binetti G, Frisoni GB, Magni E, Trabucchi M. Predictors of mortality and institutionalization in Alzheimer disease patients 1 year after discharge from an Alzheimer dementia unit. Dementia. 1995;6:108–12. doi: 10.1159/000106930. [DOI] [PubMed] [Google Scholar]
- Bliwise DL, Mercaldo ND, Avidan AY, Boeve BF, Greer SA, Kukull WA. Sleep disturbance in dementia with Lewy bodies and Alzheimer’s disease: a multicenter analysis. Dement Geriatr Cogn Disord. 2011;31:239–46. doi: 10.1159/000326238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliwise DL, Tinklenberg J, Yesavage JA, Davies H, Pursley AM, Petta DE, Widrow L, Guilleminault C, Zarcone VP, Dement WC. REM latency in Alzheimer’s disease. Biol Psychiatry. 1989;25:320–8. doi: 10.1016/0006-3223(89)90179-0. [DOI] [PubMed] [Google Scholar]
- Boddy F, Rowan EN, Lett D, O’Brien JT, McKeith IG, Burn DJ. Subjectively reported sleep quality and excessive daytime somnolence in Parkinson’s disease with and without dementia, dementia with Lewy bodies and Alzheimer’s disease. Int J Geriatr Psychiatry. 2007;22:529–35. doi: 10.1002/gps.1709. [DOI] [PubMed] [Google Scholar]
- Boeve BF, Silber MH, Ferman TJ, Kokmen E, Smith GE, Ivnik RJ, Parisi JE, Olson EJ, Petersen RC. REM sleep behavior disorder and degenerative dementia: an association likely reflecting Lewy body disease. Neurology. 1998;51:363–70. doi: 10.1212/wnl.51.2.363. [DOI] [PubMed] [Google Scholar]
- Boeve BF, Silber MH, Ferman TJ, Lin SC, Benarroch EE, Schmeichel AM, Ahlskog JE, Caselli RJ, Jacobson S, Sabbagh M, Adler C, Woodruff B, Beach TG, Iranzo A, Gelpi E, Santamaria J, Tolosa E, Singer C, Mash DC, Luca C, Arnulf I, Duyckaerts C, Schenck CH, Mahowald MW, Dauvilliers Y, Graff-Radford NR, Wszolek ZK, Parisi JE, Dugger B, Murray ME, Dickson DW. Clinicopathologic correlations in 172 cases of rapid eye movement sleep behavior disorder with or without a coexisting neurologic disorder. Sleep Med. 2013;14:754–62. doi: 10.1016/j.sleep.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeve BF, Silber MH, Ferman TJ, Lucas JA, Parisi JE. Association of REM sleep behavior disorder and neurodegenerative disease may reflect an underlying synucleinopathy. Mov Disord. 2001;16:622–30. doi: 10.1002/mds.1120. [DOI] [PubMed] [Google Scholar]
- Bonakis A, Economou NT, Paparrigopoulos T, Bonanni E, Maestri M, Carnicelli L, Di Coscio E, Ktonas P, Vagiakis E, Theodoropoulos P, Papageorgiou SG. Sleep in frontotemporal dementia is equally or possibly more disrupted, and at an earlier stage, when compared to sleep in Alzheimer’s disease. J Alzheimers Dis. 2014;38:85–91. doi: 10.3233/JAD-122014. [DOI] [PubMed] [Google Scholar]
- Bonanni E, Maestri M, Tognoni G, Fabbrini M, Nucciarone B, Manca ML, Gori S, Iudice A, Murri L. Daytime sleepiness in mild and moderate Alzheimer’s disease and its relationship with cognitive impairment. J Sleep Res. 2005;14:311–7. doi: 10.1111/j.1365-2869.2005.00462.x. [DOI] [PubMed] [Google Scholar]
- Borroni B, Agosti C, Padovani A. Behavioral and psychological symptoms in dementia with Lewy-bodies (DLB): frequency and relationship with disease severity and motor impairment. Arch Gerontol Geriatr. 2008;46:101–6. doi: 10.1016/j.archger.2007.03.003. [DOI] [PubMed] [Google Scholar]
- Braak H, Del Tredici K. The pathological process underlying Alzheimer’s disease in individuals under thirty. Acta Neuropathol. 2011;121:171–81. doi: 10.1007/s00401-010-0789-4. [DOI] [PubMed] [Google Scholar]
- Braak H, Thal DR, Ghebremedhin E, Del Tredici K. Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol. 2011;70:960–9. doi: 10.1097/NEN.0b013e318232a379. [DOI] [PubMed] [Google Scholar]
- Branger P, Arenaza-Urquijo EM, Tomadesso C, Mézenge F, André C, de Flores R, Mutlu J, de La Sayette V, Eustache F, Chételat G, Rauchs G. Relationships between sleep quality and brain volume, metabolism, and amyloid deposition in late adulthood. Neurobiol Aging. 2016;41:107–14. doi: 10.1016/j.neurobiolaging.2016.02.009. [DOI] [PubMed] [Google Scholar]
- Brown BM, Rainey-Smith SR, Villemagne VL, Weinborn M, Bucks RS, Sohrabi HR, Laws SM, Taddei K, Macaulay SL, Ames D, Fowler C, Maruff P, Masters CL, Rowe CC, Martins RN, A. R. Group The Relationship between Sleep Quality and Brain Amyloid Burden. Sleep. 2016;39:1063–8. doi: 10.5665/sleep.5756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunnström H, Friberg N, Lindberg E, Englund E. Differential degeneration of the locus coeruleus in dementia subtypes. Clin Neuropathol. 2011;30:104–10. doi: 10.5414/npp30104. [DOI] [PubMed] [Google Scholar]
- Busche MA, Kekuš M, Adelsberger H, Noda T, Förstl H, Nelken I, Konnerth A. Rescue of long-range circuit dysfunction in Alzheimer’s disease models. Nat Neurosci. 2015;18:1623–30. doi: 10.1038/nn.4137. [DOI] [PubMed] [Google Scholar]
- Cantero JL, Hita-Yañez E, Moreno-Lopez B, Portillo F, Rubio A, Avila J. Tau protein role in sleep-wake cycle. J Alzheimers Dis. 2010;21:411–21. doi: 10.3233/JAD-2010-100285. [DOI] [PubMed] [Google Scholar]
- Cermakian N, Lamont EW, Boudreau P, Boivin DB. Circadian clock gene expression in brain regions of Alzheimer ‘s disease patients and control subjects. J Biol Rhythms. 2011;26:160–70. doi: 10.1177/0748730410395732. [DOI] [PubMed] [Google Scholar]
- Chen CP, Eastwood SL, Hope T, McDonald B, Francis PT, Esiri MM. Immunocytochemical study of the dorsal and median raphe nuclei in patients with Alzheimer’s disease prospectively assessed for behavioural changes. Neuropathol Appl Neurobiol. 2000;26:347–55. doi: 10.1046/j.1365-2990.2000.00254.x. [DOI] [PubMed] [Google Scholar]
- Cirrito JR, Yamada KA, Finn MB, Sloviter RS, Bales KR, May PC, Schoepp DD, Paul SM, Mennerick S, Holtzman DM. Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron. 2005;48:913–22. doi: 10.1016/j.neuron.2005.10.028. [DOI] [PubMed] [Google Scholar]
- Çoban A, Bilgiç B, Lohmann E, Küçükali C, Benbir G, Karadeniz D, Hanagasi HA, Tüzün E, Gürvit H. Reduced orexin-A levels in frontotemporal dementia: possible association with sleep disturbance. Am J Alzheimers Dis Other Demen. 2013;28:606–11. doi: 10.1177/1533317513494453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colby-Milley J, Cavanagh C, Jego S, Breitner JC, Quirion R, Adamantidis A. Sleep-Wake Cycle Dysfunction in the TgCRND8 Mouse Model of Alzheimer’s Disease: From Early to Advanced Pathological Stages. PLoS One. 2015;10:e0130177. doi: 10.1371/journal.pone.0130177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Meco A, Joshi YB, Praticò D. Sleep deprivation impairs memory, tau metabolism, and synaptic integrity of a mouse model of Alzheimer’s disease with plaques and tangles. Neurobiol Aging. 2014;35:1813–20. doi: 10.1016/j.neurobiolaging.2014.02.011. [DOI] [PubMed] [Google Scholar]
- Dickson DW. Neuropathologic differentiation of progressive supranuclear palsy and corticobasal degeneration. J Neurol. 1999;246(Suppl 2):II6–15. doi: 10.1007/BF03161076. [DOI] [PubMed] [Google Scholar]
- Dickson DW, Schmidt ML, Lee VM, Zhao ML, Yen SH, Trojanowski JQ. Immunoreactivity profile of hippocampal CA2/3 neurites in diffuse Lewy body disease. Acta Neuropathol. 1994;87:269–76. doi: 10.1007/BF00296742. [DOI] [PubMed] [Google Scholar]
- Donaghy PC, McKeith IG. The clinical characteristics of dementia with Lewy bodies and a consideration of prodromal diagnosis. Alzheimers Res Ther. 2014;6:46. doi: 10.1186/alzrt274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugger BN, Murray ME, Boeve BF, Parisi JE, Benarroch EE, Ferman TJ, Dickson DW. Neuropathological analysis of brainstem cholinergic and catecholaminergic nuclei in relation to rapid eye movement (REM) sleep behaviour disorder. Neuropathol Appl Neurobiol. 2012;38:142–52. doi: 10.1111/j.1365-2990.2011.01203.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugger BN, Tu M, Murray ME, Dickson DW. Disease specificity and pathologic progression of tau pathology in brainstem nuclei of Alzheimer’s disease and progressive supranuclear palsy. Neurosci Lett. 2011;491:122–6. doi: 10.1016/j.neulet.2011.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagan AM, Roe CM, Xiong C, Mintun MA, Morris JC, Holtzman DM. Cerebrospinal fluid tau/beta-amyloid(42) ratio as a prediction of cognitive decline in nondemented older adults. Arch Neurol. 2007;64:343–9. doi: 10.1001/archneur.64.3.noc60123. [DOI] [PubMed] [Google Scholar]
- Fantini ML, Michaud M, Gosselin N, Lavigne G, Montplaisir J. Periodic leg movements in REM sleep behavior disorder and related autonomic and EEG activation. Neurology. 2002;59:1889–94. doi: 10.1212/01.wnl.0000038348.94399.f6. [DOI] [PubMed] [Google Scholar]
- Farina E, Baglio F, Caffarra P, Magnani G, Scarpini E, Appollonio I, Bascelli C, Cheldi A, Nemni R, Franceschi M, Messa G, Mantovani F, Bellotti M, Olivotto F, Alberoni M, Isella V, Regazzoni R, Schiatti E, Vismara C, Falautano M, Barbieri A, Restelli I, Fetoni V, Donato M, Zuffi M, Castiglioni S, I. G. f. L. B. D. a. D. A. t. Parkinsonism Frequency and clinical features of Lewy body dementia in Italian memory clinics. Acta Biomed. 2009;80:57–64. [PubMed] [Google Scholar]
- Ferman TJ, Boeve BF, Smith GE, Lin SC, Silber MH, Pedraza O, Wszolek Z, Graff-Radford NR, Uitti R, Van Gerpen J, Pao W, Knopman D, Pankratz VS, Kantarci K, Boot B, Parisi JE, Dugger BN, Fujishiro H, Petersen RC, Dickson DW. Inclusion of RBD improves the diagnostic classification of dementia with Lewy bodies. Neurology. 2011;77:875–82. doi: 10.1212/WNL.0b013e31822c9148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fronczek R, Overeem S, Lee SY, Hegeman IM, van Pelt J, van Duinen SG, Lammers GJ, Swaab DF. Hypocretin (orexin) loss in Parkinson’s disease. Brain. 2007;130:1577–85. doi: 10.1093/brain/awm090. [DOI] [PubMed] [Google Scholar]
- Fronczek R, van Geest S, Frölich M, Overeem S, Roelandse FW, Lammers GJ, Swaab DF. Hypocretin (orexin) loss in Alzheimer’s disease. Neurobiol Aging. 2012;33:1642–50. doi: 10.1016/j.neurobiolaging.2011.03.014. [DOI] [PubMed] [Google Scholar]
- Fuller PM, Fuller P, Sherman D, Pedersen NP, Saper CB, Lu J. Reassessment of the structural basis of the ascending arousal system. J Comp Neurol. 2011;519:933–56. doi: 10.1002/cne.22559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gama RL, Távora DG, Bomfim RC, Silva CE, de Bruin VM, de Bruin PF. Sleep disturbances and brain MRI morphometry in Parkinson’s disease, multiple system atrophy and progressive supranuclear palsy – a comparative study. Parkinsonism Relat Disord. 2010;16:275–9. doi: 10.1016/j.parkreldis.2010.01.002. [DOI] [PubMed] [Google Scholar]
- Gemignani A, Pietrini P, Murrell JR, Glazier BS, Zolo P, Guazzelli M, Ghetti B. Slow wave and rem sleep mechanisms are differently altered in hereditary pick disease associated with the TAU G389R mutation. Arch Ital Biol. 2005;143:65–79. [PubMed] [Google Scholar]
- German DC, White CL, Sparkman DR. Alzheimer’s disease: neurofibrillary tangles in nuclei that project to the cerebral cortex. Neuroscience. 1987;21:305–12. doi: 10.1016/0306-4522(87)90123-0. [DOI] [PubMed] [Google Scholar]
- Gompf HS, Mathai C, Fuller PM, Wood DA, Pedersen NP, Saper CB, Lu J. Locus ceruleus and anterior cingulate cortex sustain wakefulness in a novel environment. J Neurosci. 2010;30:14543–51. doi: 10.1523/JNEUROSCI.3037-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace JB, Walker MP, McKeith IG. A comparison of sleep profiles in patients with dementia with lewy bodies and Alzheimer’s disease. Int J Geriatr Psychiatry. 2000;15:1028–33. doi: 10.1002/1099-1166(200011)15:11<1028::aid-gps227>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- Grinberg LT, Rüb U, Ferretti RE, Nitrini R, Farfel JM, Polichiso L, Gierga K, Jacob-Filho W, Heinsen H, B. B. B. S. Group The dorsal raphe nucleus shows phospho-tau neurofibrillary changes before the transentorhinal region in Alzheimer’s disease. A precocious onset? Neuropathol Appl Neurobiol. 2009;35:406–16. doi: 10.1111/j.1365-2990.2009.00997.x. [DOI] [PubMed] [Google Scholar]
- Grudzien A, Shaw P, Weintraub S, Bigio E, Mash DC, Mesulam MM. Locus coeruleus neurofibrillary degeneration in aging, mild cognitive impairment and early Alzheimer’s disease. Neurobiol Aging. 2007;28:327–35. doi: 10.1016/j.neurobiolaging.2006.02.007. [DOI] [PubMed] [Google Scholar]
- Guarnieri B, Adorni F, Musicco M, Appollonio I, Bonanni E, Caffarra P, Caltagirone C, Cerroni G, Concari L, Cosentino FI, Ferrara S, Fermi S, Ferri R, Gelosa G, Lombardi G, Mazzei D, Mearelli S, Morrone E, Murri L, Nobili FM, Passero S, Perri R, Rocchi R, Sucapane P, Tognoni G, Zabberoni S, Sorbi S. Prevalence of sleep disturbances in mild cognitive impairment and dementing disorders: a multicenter Italian clinical cross-sectional study on 431 patients. Dement Geriatr Cogn Disord. 2012;33:50–8. doi: 10.1159/000335363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn EA, Wang HX, Andel R, Fratiglioni L. A Change in Sleep Pattern May Predict Alzheimer Disease. Am J Geriatr Psychiatry. 2013 doi: 10.1016/j.jagp.2013.04.015. [DOI] [PubMed] [Google Scholar]
- Hamilton RL. Lewy bodies in Alzheimer’s disease: a neuropathological review of 145 cases using alpha-synuclein immunohistochemistry. Brain Pathol. 2000;10:378–84. doi: 10.1111/j.1750-3639.2000.tb00269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding AJ, Halliday GM. Cortical Lewy body pathology in the diagnosis of dementia. Acta Neuropathol. 2001;102:355–63. doi: 10.1007/s004010100390. [DOI] [PubMed] [Google Scholar]
- Harper DG, Stopa EG, Kuo-Leblanc V, McKee AC, Asayama K, Volicer L, Kowall N, Satlin A. Dorsomedial SCN neuronal subpopulations subserve different functions in human dementia. Brain. 2008;131:1609–17. doi: 10.1093/brain/awn049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper DG, Stopa EG, McKee AC, Satlin A, Fish D, Volicer L. Dementia severity and Lewy bodies affect circadian rhythms in Alzheimer disease. Neurobiol Aging. 2004;25:771–81. doi: 10.1016/j.neurobiolaging.2003.04.009. [DOI] [PubMed] [Google Scholar]
- Harper DG, Volicer L, Stopa EG, McKee AC, Nitta M, Satlin A. Disturbance of endogenous circadian rhythm in aging and Alzheimer disease. Am J Geriatr Psychiatry. 2005;13:359–68. doi: 10.1176/appi.ajgp.13.5.359. [DOI] [PubMed] [Google Scholar]
- Heneka MT, Ramanathan M, Jacobs AH, Dumitrescu-Ozimek L, Bilkei-Gorzo A, Debeir T, Sastre M, Galldiks N, Zimmer A, Hoehn M, Heiss WD, Klockgether T, Staufenbiel M. Locus ceruleus degeneration promotes Alzheimer pathogenesis in amyloid precursor protein 23 transgenic mice. J Neurosci. 2006;26:1343–54. doi: 10.1523/JNEUROSCI.4236-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibi S, Yamaguchi Y, Umeda-Kameyama Y, Yamamoto H, Iijima K, Momose T, Akishita M, Ouchi Y. The high frequency of periodic limb movements in patients with Lewy body dementia. J Psychiatr Res. 2012;46:1590–4. doi: 10.1016/j.jpsychires.2012.07.007. [DOI] [PubMed] [Google Scholar]
- Holtzman DM, Morris JC, Goate AM. Alzheimer’s disease: the challenge of the second century. Sci Transl Med. 2011;3:77sr1. doi: 10.1126/scitranslmed.3002369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Potter R, Sigurdson W, Santacruz A, Shih S, Ju YE, Kasten T, Morris JC, Mintun M, Duntley S, Bateman RJ. Effects of age and amyloid deposition on Aβ dynamics in the human central nervous system. Arch Neurol. 2012;69:51–8. doi: 10.1001/archneurol.2011.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huitrón-Reséndiz S, Sánchez-Alavez M, Gallegos R, Berg G, Crawford E, Giacchino JL, Games D, Henriksen SJ, Criado JR. Age-independent and age-related deficits in visuospatial learning, sleep-wake states, thermoregulation and motor activity in PDAPP mice. Brain Res. 2002;928:126–37. doi: 10.1016/s0006-8993(01)03373-x. [DOI] [PubMed] [Google Scholar]
- Iraizoz I, Guijarro JL, Gonzalo LM, de Lacalle S. Neuropathological changes in the nucleus basalis correlate with clinical measures of dementia. Acta Neuropathol. 1999;98:186–96. doi: 10.1007/s004010051068. [DOI] [PubMed] [Google Scholar]
- Iranzo A, Gelpi E, Tolosa E, Molinuevo JL, Serradell M, Gaig C, Santamaria J. Neuropathology of prodromal Lewy body disease. Mov Disord. 2014;29:410–5. doi: 10.1002/mds.25825. [DOI] [PubMed] [Google Scholar]
- Iranzo A, Molinuevo JL, Santamaría J, Serradell M, Martí MJ, Valldeoriola F, Tolosa E. Rapid-eye-movement sleep behaviour disorder as an early marker for a neurodegenerative disorder: a descriptive study. Lancet Neurol. 2006;5:572–7. doi: 10.1016/S1474-4422(06)70476-8. [DOI] [PubMed] [Google Scholar]
- Iranzo A, Tolosa E, Gelpi E, Molinuevo JL, Valldeoriola F, Serradell M, Sanchez-Valle R, Vilaseca I, Lomeña F, Vilas D, Lladó A, Gaig C, Santamaria J. Neurodegenerative disease status and post-mortem pathology in idiopathic rapid-eye-movement sleep behaviour disorder: an observational cohort study. Lancet Neurol. 2013;12:443–53. doi: 10.1016/S1474-4422(13)70056-5. [DOI] [PubMed] [Google Scholar]
- Jack CR, Holtzman DM. Biomarker modeling of Alzheimer’s disease. Neuron. 2013;80:1347–58. doi: 10.1016/j.neuron.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju YE, Lucey BP, Holtzman DM. Sleep and Alzheimer disease pathology–a bidirectional relationship. Nat Rev Neurol. 2014;10:115–9. doi: 10.1038/nrneurol.2013.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju YE, McLeland JS, Toedebusch CD, Xiong C, Fagan AM, Duntley SP, Morris JC, Holtzman DM. Sleep quality and preclinical Alzheimer disease. JAMA Neurol. 2013;70:587–93. doi: 10.1001/jamaneurol.2013.2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jyoti A, Plano A, Riedel G, Platt B. Progressive age-related changes in sleep and EEG profiles in the PLB1Triple mouse model of Alzheimer’s disease. Neurobiol Aging. 2015;36:2768–84. doi: 10.1016/j.neurobiolaging.2015.07.001. [DOI] [PubMed] [Google Scholar]
- Kang JE, Lim MM, Bateman RJ, Lee JJ, Smyth LP, Cirrito JR, Fujiki N, Nishino S, Holtzman DM. Amyloid-beta dynamics are regulated by orexin and the sleep-wake cycle. Science. 2009;326:1005–7. doi: 10.1126/science.1180962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerbler GM, Fripp J, Rowe CC, Villemagne VL, Salvado O, Rose S, Coulson EJ, A. s. D. N. Initiative Basal forebrain atrophy correlates with amyloid β burden in Alzheimer’s disease. Neuroimage Clin. 2015;7:105–13. doi: 10.1016/j.nicl.2014.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koss DJ, Robinson L, Drever BD, Plucińska K, Stoppelkamp S, Veselcic P, Riedel G, Platt B. Mutant Tau knock-in mice display frontotemporal dementia relevant behaviour and histopathology. Neurobiol Dis. 2016;91:105–23. doi: 10.1016/j.nbd.2016.03.002. [DOI] [PubMed] [Google Scholar]
- Krenzer M, Anaclet C, Vetrivelan R, Wang N, Vong L, Lowell BB, Fuller PM, Lu J. Brainstem and spinal cord circuitry regulating REM sleep and muscle atonia. PLoS One. 2011;6:e24998. doi: 10.1371/journal.pone.0024998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundermann B, Thum A, Rocamora R, Haag A, Krieg JC, Hemmeter U. Comparison of polysomnographic variables and their relationship to cognitive impairment in patients with Alzheimer’s disease and frontotemporal dementia. J Psychiatr Res. 2011;45:1585–92. doi: 10.1016/j.jpsychires.2011.07.008. [DOI] [PubMed] [Google Scholar]
- Lessig S, Ubhi K, Galasko D, Adame A, Pham E, Remidios K, Chang M, Hansen LA, Masliah E. Reduced hypocretin (orexin) levels in dementia with Lewy bodies. Neuroreport. 2010;21:756–60. doi: 10.1097/WNR.0b013e32833bfb7c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leverenz JB, Fishel MA, Peskind ER, Montine TJ, Nochlin D, Steinbart E, Raskind MA, Schellenberg GD, Bird TD, Tsuang D. Lewy body pathology in familial Alzheimer disease: evidence for disease- and mutation-specific pathologic phenotype. Arch Neurol. 2006;63:370–6. doi: 10.1001/archneur.63.3.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liguori C, Romigi A, Mercuri NB, Nuccetelli M, Izzi F, Albanese M, Sancesario G, Martorana A, Sancesario GM, Bernardini S, Marciani MG, Placidi F. Cerebrospinal-fluid orexin levels and daytime somnolence in frontotemporal dementia. J Neurol. 2014a;261:1832–6. doi: 10.1007/s00415-014-7455-z. [DOI] [PubMed] [Google Scholar]
- Liguori C, Romigi A, Nuccetelli M, Zannino S, Sancesario G, Martorana A, Albanese M, Mercuri NB, Izzi F, Bernardini S, Nitti A, Sancesario GM, Sica F, Marciani MG, Placidi F. Orexinergic system dysregulation, sleep impairment, and cognitive decline in Alzheimer disease. JAMA Neurol. 2014b;71:1498–505. doi: 10.1001/jamaneurol.2014.2510. [DOI] [PubMed] [Google Scholar]
- Lim AS, Ellison BA, Wang JL, Yu L, Schneider JA, Buchman AS, Bennett DA, Saper CB. Sleep is related to neuron numbers in the ventrolateral preoptic/intermediate nucleus in older adults with and without Alzheimer’s disease. Brain. 2014a;137:2847–61. doi: 10.1093/brain/awu222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim AS, Kowgier M, Yu L, Buchman AS, Bennett DA. Sleep Fragmentation and the Risk of Incident Alzheimer’s Disease and Cognitive Decline in Older Persons. Sleep. 2013;36:1027–1032. doi: 10.5665/sleep.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim MM, Gerstner JR, Holtzman DM. The sleep-wake cycle and Alzheimer’s disease: what do we know? Neurodegener Dis Manag. 2014b;4:351–62. doi: 10.2217/nmt.14.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippa CF, Fujiwara H, Mann DM, Giasson B, Baba M, Schmidt ML, Nee LE, O’Connell B, Pollen DA, St George-Hyslop P, Ghetti B, Nochlin D, Bird TD, Cairns NJ, Lee VM, Iwatsubo T, Trojanowski JQ. Lewy bodies contain altered alpha-synuclein in brains of many familial Alzheimer’s disease patients with mutations in presenilin and amyloid precursor protein genes. Am J Pathol. 1998;153:1365–70. doi: 10.1016/s0002-9440(10)65722-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippa CF, Knopman DS. Dementia: many roads, but not built in a day. Neurology. 2007;69:2193–4. doi: 10.1212/01.wnl.0000287144.13726.3e. [DOI] [PubMed] [Google Scholar]
- Lu J, Sherman D, Devor M, Saper CB. A putative flip-flop switch for control of REM sleep. Nature. 2006;441:589–94. doi: 10.1038/nature04767. [DOI] [PubMed] [Google Scholar]
- Mattsson N, Zetterberg H, Hansson O, Andreasen N, Parnetti L, Jonsson M, Herukka SK, van der Flier WM, Blankenstein MA, Ewers M, Rich K, Kaiser E, Verbeek M, Tsolaki M, Mulugeta E, Rosén E, Aarsland D, Visser PJ, Schröder J, Marcusson J, de Leon M, Hampel H, Scheltens P, Pirttilä T, Wallin A, Jönhagen ME, Minthon L, Winblad B, Blennow K. CSF biomarkers and incipient Alzheimer disease in patients with mild cognitive impairment. JAMA. 2009;302:385–93. doi: 10.1001/jama.2009.1064. [DOI] [PubMed] [Google Scholar]
- McKeith IG. Spectrum of Parkinson’s disease, Parkinson’s dementia, and Lewy body dementia. Neurol Clin. 2000;18:865–902. doi: 10.1016/s0733-8619(05)70230-9. [DOI] [PubMed] [Google Scholar]
- McKeith IG, Dickson DW, Lowe J, Emre M, O’Brien JT, Feldman H, Cummings J, Duda JE, Lippa C, Perry EK, Aarsland D, Arai H, Ballard CG, Boeve B, Burn DJ, Costa D, Del Ser T, Dubois B, Galasko D, Gauthier S, Goetz CG, Gomez-Tortosa E, Halliday G, Hansen LA, Hardy J, Iwatsubo T, Kalaria RN, Kaufer D, Kenny RA, Korczyn A, Kosaka K, Lee VM, Lees A, Litvan I, Londos E, Lopez OL, Minoshima S, Mizuno Y, Molina JA, Mukaetova-Ladinska EB, Pasquier F, Perry RH, Schulz JB, Trojanowski JQ, Yamada M, C. o. DLB Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology. 2005;65:1863–72. doi: 10.1212/01.wnl.0000187889.17253.b1. [DOI] [PubMed] [Google Scholar]
- Mesulam M, Shaw P, Mash D, Weintraub S. Cholinergic nucleus basalis tauopathy emerges early in the aging-MCI-AD continuum. Ann Neurol. 2004;55:815–28. doi: 10.1002/ana.20100. [DOI] [PubMed] [Google Scholar]
- Moe KE, Vitiello MV, Larsen LH, Prinz PN. Symposium: Cognitive processes and sleep disturbances: Sleep/wake patterns in Alzheimer’s disease: relationships with cognition and function. J Sleep Res. 1995;4:15–20. doi: 10.1111/j.1365-2869.1995.tb00145.x. [DOI] [PubMed] [Google Scholar]
- Montplaisir J, Petit D, Décary A, Masson H, Bédard MA, Panisset M, Rémillard G, Gauthier S. Sleep and quantitative EEG in patients with progressive supranuclear palsy. Neurology. 1997;49:999–1003. doi: 10.1212/wnl.49.4.999. [DOI] [PubMed] [Google Scholar]
- Moran M, Lynch CA, Walsh C, Coen R, Coakley D, Lawlor BA. Sleep disturbance in mild to moderate Alzheimer’s disease. Sleep Med. 2005;6:347–52. doi: 10.1016/j.sleep.2004.12.005. [DOI] [PubMed] [Google Scholar]
- Musiek ES, Xiong DD, Holtzman DM. Sleep, circadian rhythms, and the pathogenesis of Alzheimer disease. Exp Mol Med. 2015;47:e148. doi: 10.1038/emm.2014.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura S, Takemura M, Ohnishi K, Suenaga T, Nishimura M, Akiguchi I, Kimura J, Kimura T. Loss of large neurons and occurrence of neurofibrillary tangles in the tuberomammillary nucleus of patients with Alzheimer’s disease. Neurosci Lett. 1993;151:196–9. doi: 10.1016/0304-3940(93)90019-h. [DOI] [PubMed] [Google Scholar]
- Ooms S, Overeem S, Besse K, Rikkert MO, Verbeek M, Claassen JA. Effect of 1 night of total sleep deprivation on cerebrospinal fluid β-amyloid 42 in healthy middle-aged men: a randomized clinical trial. JAMA Neurol. 2014;71:971–7. doi: 10.1001/jamaneurol.2014.1173. [DOI] [PubMed] [Google Scholar]
- Osorio RS, Ducca EL, Wohlleber ME, Tanzi EB, Gumb T, Twumasi A, Tweardy S, Lewis C, Fischer E, Koushyk V, Cuartero-Toledo M, Sheikh MO, Pirraglia E, Zetterberg H, Blennow K, Lu SE, Mosconi L, Glodzik L, Schuetz S, Varga AW, Ayappa I, Rapoport DM, de Leon MJ. Orexin-A is Associated With Increases in Cerebrospinal Fluid Phosphorylated-Tau in Cognitively Normal Elderly Subjects. Sleep. 2016 doi: 10.5665/sleep.5846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan XD, Chen XC. Clinic, neuropathology and molecular genetics of frontotemporal dementia: a mini-review. Transl Neurodegener. 2013;2:8. doi: 10.1186/2047-9158-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parvizi J, Van Hoesen GW, Damasio A. Selective pathological changes of the periaqueductal gray matter in Alzheimer’s disease. Ann Neurol. 2000;48:344–53. [PubMed] [Google Scholar]
- Parvizi J, Van Hoesen GW, Damasio A. The selective vulnerability of brainstem nuclei to Alzheimer’s disease. Ann Neurol. 2001;49:53–66. doi: 10.1002/1531-8249(200101)49:1<53::aid-ana30>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- Peter-Derex L, Yammine P, Bastuji H, Croisile B. Sleep and Alzheimer’s disease. Sleep Med Rev. 2015;19:29–38. doi: 10.1016/j.smrv.2014.03.007. [DOI] [PubMed] [Google Scholar]
- Petit D, Gagnon JF, Fantini ML, Ferini-Strambi L, Montplaisir J. Sleep and quantitative EEG in neurodegenerative disorders. J Psychosom Res. 2004;56:487–96. doi: 10.1016/j.jpsychores.2004.02.001. [DOI] [PubMed] [Google Scholar]
- Pistacchi M, Gioulis M, Contin F, Sanson F, Marsala SZ. Sleep disturbance and cognitive disorder: epidemiological analysis in a cohort of 263 patients. Neurol Sci. 2014;35:1955–62. doi: 10.1007/s10072-014-1870-x. [DOI] [PubMed] [Google Scholar]
- Platt B, Drever B, Koss D, Stoppelkamp S, Jyoti A, Plano A, Utan A, Merrick G, Ryan D, Melis V, Wan H, Mingarelli M, Porcu E, Scrocchi L, Welch A, Riedel G. Abnormal cognition, sleep, EEG and brain metabolism in a novel knock-in Alzheimer mouse, PLB1. PLoS One. 2011;6:e27068. doi: 10.1371/journal.pone.0027068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postuma RB, Gagnon JF, Vendette M, Fantini ML, Massicotte-Marquez J, Montplaisir J. Quantifying the risk of neurodegenerative disease in idiopathic REM sleep behavior disorder. Neurology. 2009;72:1296–300. doi: 10.1212/01.wnl.0000340980.19702.6e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pressman PS, Miller BL. Diagnosis and management of behavioral variant frontotemporal dementia. Biol Psychiatry. 2014;75:574–81. doi: 10.1016/j.biopsych.2013.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers JD, Brogan D, Mirra SS. The nucleus basalis of Meynert in neurological disease: a quantitative morphological study. Ann Neurol. 1985;17:163–70. doi: 10.1002/ana.410170210. [DOI] [PubMed] [Google Scholar]
- Roh JH, Huang Y, Bero AW, Kasten T, Stewart FR, Bateman RJ, Holtzman DM. Disruption of the sleep-wake cycle and diurnal fluctuation of β-amyloid in mice with Alzheimer’s disease pathology. Sci Transl Med. 2012;4:150ra122. doi: 10.1126/scitranslmed.3004291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rüb U, Del Tredici K, Schultz C, Thal DR, Braak E, Braak H. The evolution of Alzheimer’s disease-related cytoskeletal pathology in the human raphe nuclei. Neuropathol Appl Neurobiol. 2000;26:553–67. doi: 10.1046/j.0305-1846.2000.00291.x. [DOI] [PubMed] [Google Scholar]
- Sanchez-Espinosa MP, Atienza M, Cantero JL. Sleep deficits in mild cognitive impairment are related to increased levels of plasma amyloid-β and cortical thinning. Neuroimage. 2014;98:395–404. doi: 10.1016/j.neuroimage.2014.05.027. [DOI] [PubMed] [Google Scholar]
- Saper CB, Fuller PM, Pedersen NP, Lu J, Scammell TE. Sleep state switching. Neuron. 2010;68:1023–42. doi: 10.1016/j.neuron.2010.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saper CB, Scammell TE, Lu J. Hypothalamic regulation of sleep and circadian rhythms. Nature. 2005;437:1257–63. doi: 10.1038/nature04284. [DOI] [PubMed] [Google Scholar]
- Schenck CH, Boeve BF, Mahowald MW. Delayed emergence of a parkinsonian disorder or dementia in 81% of older men initially diagnosed with idiopathic rapid eye movement sleep behavior disorder: a 16-year update on a previously reported series. Sleep Med. 2013;14:744–8. doi: 10.1016/j.sleep.2012.10.009. [DOI] [PubMed] [Google Scholar]
- Schenck CH, Bundlie SR, Mahowald MW. Delayed emergence of a parkinsonian disorder in 38% of 29 older men initially diagnosed with idiopathic rapid eye movement sleep behaviour disorder. Neurology. 1996;46:388–93. doi: 10.1212/wnl.46.2.388. [DOI] [PubMed] [Google Scholar]
- Schneider F, Baldauf K, Wetzel W, Reymann KG. Behavioral and EEG changes in male 5xFAD mice. Physiol Behav. 2014;135:25–33. doi: 10.1016/j.physbeh.2014.05.041. [DOI] [PubMed] [Google Scholar]
- Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT. Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med. 2011;1:a006189. doi: 10.1101/cshperspect.a006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethi M, Joshi SS, Webb RL, Beckett TL, Donohue KD, Murphy MP, O’Hara BF, Duncan MJ. Increased fragmentation of sleep-wake cycles in the 5XFAD mouse model of Alzheimer’s disease. Neuroscience. 2015;290:80–9. doi: 10.1016/j.neuroscience.2015.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slats D, Claassen JA, Lammers GJ, Melis RJ, Verbeek MM, Overeem S. Association between hypocretin-1 and amyloid-β42 cerebrospinal fluid levels in Alzheimer’s disease and healthy controls. Curr Alzheimer Res. 2012;9:1119–25. doi: 10.2174/156720512804142840. [DOI] [PubMed] [Google Scholar]
- Song H, Moon M, Choe HK, Han DH, Jang C, Kim A, Cho S, Kim K, Mook-Jung I. Aβ-induced degradation of BMAL1 and CBP leads to circadian rhythm disruption in Alzheimer’s disease. Mol Neurodegener. 2015;10:13. doi: 10.1186/s13024-015-0007-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spira AP, Gamaldo AA, An Y, Wu MN, Simonsick EM, Bilgel M, Zhou Y, Wong DF, Ferrucci L, Resnick SM. Self-reported sleep and β-amyloid deposition in community-dwelling older adults. JAMA Neurol. 2013;70:1537–43. doi: 10.1001/jamaneurol.2013.4258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Standaert DG, Lee VM, Greenberg BD, Lowery DE, Trojanowski JQ. Molecular features of hypothalamic plaques in Alzheimer’s disease. Am J Pathol. 1991;139:681–91. [PMC free article] [PubMed] [Google Scholar]
- Stern AL, Naidoo N. Wake-active neurons across aging and neurodegeneration: a potential role for sleep disturbances in promoting disease. Springerplus. 2015;4:25. doi: 10.1186/s40064-014-0777-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterniczuk R, Dyck RH, Laferla FM, Antle MC. Characterization of the 3xTg-AD mouse model of Alzheimer’s disease: part 1. Circadian changes Brain Res. 2010;1348:139–48. doi: 10.1016/j.brainres.2010.05.013. [DOI] [PubMed] [Google Scholar]
- Stopa EG, Volicer L, Kuo-Leblanc V, Harper D, Lathi D, Tate B, Satlin A. Pathologic evaluation of the human suprachiasmatic nucleus in severe dementia. J Neuropathol Exp Neurol. 1999;58:29–39. doi: 10.1097/00005072-199901000-00004. [DOI] [PubMed] [Google Scholar]
- Stratmann K, Heinsen H, Korf HW, Del Turco D, Ghebremedhin E, Seidel K, Bouzrou M, Grinberg LT, Bohl J, Wharton SB, den Dunnen W, Rüb U. Precortical Phase of Alzheimer’s Disease (AD)-Related Tau Cytoskeletal Pathology. Brain Pathol. 2016;26:371–86. doi: 10.1111/bpa.12289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabuchi M, Lone SR, Liu S, Liu Q, Zhang J, Spira AP, Wu MN. Sleep interacts with aβ to modulate intrinsic neuronal excitability. Curr Biol. 2015;25:702–12. doi: 10.1016/j.cub.2015.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terzaghi M, Arnaldi D, Rizzetti MC, Minafra B, Cremascoli R, Rustioni V, Zangaglia R, Pasotti C, Sinforiani E, Pacchetti C, Manni R. Analysis of video-polysomnographic sleep findings in dementia with Lewy bodies. Mov Disord. 2013;28:1416–23. doi: 10.1002/mds.25523. [DOI] [PubMed] [Google Scholar]
- Tsuboi Y, Dickson DW. Dementia with Lewy bodies and Parkinson’s disease with dementia: are they different? Parkinsonism Relat Disord. 2005;11(Suppl 1):S47–51. doi: 10.1016/j.parkreldis.2004.10.014. [DOI] [PubMed] [Google Scholar]
- Uchiyama M, Isse K, Tanaka K, Yokota N, Hamamoto M, Aida S, Ito Y, Yoshimura M, Okawa M. Incidental Lewy body disease in a patient with REM sleep behavior disorder. Neurology. 1995;45:709–12. doi: 10.1212/wnl.45.4.709. [DOI] [PubMed] [Google Scholar]
- Vitiello MV, Prinz PN, Williams DE, Frommlet MS, Ries RK. Sleep disturbances in patients with mild-stage Alzheimer’s disease. J Gerontol. 1990;45:M131–8. doi: 10.1093/geronj/45.4.m131. [DOI] [PubMed] [Google Scholar]
- Volicer L, Harper DG, Manning BC, Goldstein R, Satlin A. Sundowning and circadian rhythms in Alzheimer’s disease. Am J Psychiatry. 2001;158:704–11. doi: 10.1176/appi.ajp.158.5.704. [DOI] [PubMed] [Google Scholar]
- Walker MP, Ayre GA, Cummings JL, Wesnes K, McKeith IG, O’Brien JT, Ballard CG. Quantifying fluctuation in dementia with Lewy bodies, Alzheimer’s disease, and vascular dementia. Neurology. 2000;54:1616–25. doi: 10.1212/wnl.54.8.1616. [DOI] [PubMed] [Google Scholar]
- Wilson RS, Nag S, Boyle PA, Hizel LP, Yu L, Buchman AS, Schneider JA, Bennett DA. Neural reserve, neuronal density in the locus ceruleus, and cognitive decline. Neurology. 2013;80:1202–8. doi: 10.1212/WNL.0b013e3182897103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisor JP, Edgar DM, Yesavage J, Ryan HS, McCormick CM, Lapustea N, Murphy GM. Sleep and circadian abnormalities in a transgenic mouse model of Alzheimer’s disease: a role for cholinergic transmission. Neuroscience. 2005;131:375–85. doi: 10.1016/j.neuroscience.2004.11.018. [DOI] [PubMed] [Google Scholar]
- Wu YH, Feenstra MG, Zhou JN, Liu RY, Toranõ JS, Van Kan HJ, Fischer DF, Ravid R, Swaab DF. Molecular changes underlying reduced pineal melatonin levels in Alzheimer disease: alterations in preclinical and clinical stages. J Clin Endocrinol Metab. 2003;88:5898–906. doi: 10.1210/jc.2003-030833. [DOI] [PubMed] [Google Scholar]
- Xie L, Kang H, Xu Q, Chen MJ, Liao Y, Thiyagarajan M, O’Donnell J, Christensen DJ, Nicholson C, Iliff JJ, Takano T, Deane R, Nedergaard M. Sleep drives metabolite clearance from the adult brain. Science. 2013;342:373–7. doi: 10.1126/science.1241224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K, Holth JK, Liao F, Stewart FR, Mahan TE, Jiang H, Cirrito JR, Patel TK, Hochgräfe K, Mandelkow EM, Holtzman DM. Neuronal activity regulates extracellular tau in vivo. J Exp Med. 2014;211:387–93. doi: 10.1084/jem.20131685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Veasey SC, Wood MA, Leng LZ, Kaminski C, Leight S, Abel T, Lee VM, Trojanowski JQ. Impaired rapid eye movement sleep in the Tg2576 APP murine model of Alzheimer’s disease with injury to pedunculopontine cholinergic neurons. Am J Pathol. 2005;167:1361–9. doi: 10.1016/S0002-9440(10)61223-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Zhu Y, Zhan G, Fenik P, Panossian L, Wang MM, Reid S, Lai D, Davis JG, Baur JA, Veasey S. Extended wakefulness: compromised metabolics in and degeneration of locus ceruleus neurons. J Neurosci. 2014;34:4418–31. doi: 10.1523/JNEUROSCI.5025-12.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]