

Abstract
Prime-boost vaccination with recombinant (r) Vaccinia(V)-CEA(6D)-TRICOM (triad of co-stimulatory molecules B7.1, ICAM-1 and LFA-3) and rFowlpox(F)-CEA(6D)-TRICOM, infect antigen presenting cells and direct expression of co-stimulatory molecules. We hypothesized that co-administration of vaccine with GM-CSF and interferon-alpha (IFN-α) would have efficacy in CEA-expressing cancers. Patients with CEA-expressing cancers received the rV-CEA(6D)-TRICOM vaccine subcutaneously (s.c.) on day 1 followed by GM-CSF s.c. to the injection site on days 1–4. In Cycle 1, patients received thrice weekly s.c. injections of IFN-α-2b the week after rV-CEA(6D)-TRICOM. In Cycles 2–4, patients received thrice weekly s.c. injections of IFN-α-2b the same week that rF-CEA(6D)-TRICOM was given. The first cohort received no IFN followed by dose escalation of IFN-α in subsequent cohorts. Thirty-three patients were accrued (mean 59.8 years). Grade ≥3 toxicities included fatigue and hyperglycemia. Grade 4–5 adverse events (unrelated to treatment) were confusion (1), elevated aspartate transaminase (AST)/alanine transaminase (ALT) (1), and sudden death (1). No patients had a partial response, and eight patients exhibited stable disease of >3 months. Median progression-free survival and overall survival (OS) were 1.8 and 6.3 months, respectively. Significantly higher serum CD27 levels were observed after vaccine therapy (p=0.006 post 1–2 cycles, p=0.003 post 3 cycles, p=0.03 post 4–7 cycles) and 42% of patients assayed developed CEA-specific T-cell responses. Pre-treatment levels of myeloid derived suppressor cells correlated with overall survival (p=0.04). Administration of IFN-α led to significantly increased OS (p=0.02) compared to vaccine alone. While the vaccine regimen produced no clinical responses, IFN-α administration was associated with improved survival.
Keywords: Vaccines, Carcinoembryonic Antigen (CEA), Colorectal Cancer, Immunotherapy, Interferon alpha-2b
INTRODUCTION
Colorectal cancers are the third leading cause of cancer-related mortality in the United States [1]. Locally advanced tumors and tumors with lymph node involvement frequently recur even after successful surgery and adjuvant chemotherapy [2]. The 5-year survival rate for patients with metastatic disease is typically around 20%. However, improvements in systemic therapy are leading to increased survival rates. 5-fluorouracil (5-FU) has served as the cornerstone of therapy for patients with metastatic or locally-advanced colorectal cancer [3]. Newer chemotherapeutic drugs have been approved (e.g., irinotecan and oxaliplatin) and when utilized in combination with 5-FU have demonstrated improved response rates and overall survival [4]. Despite these advances, the search for novel therapies utilizing alternate anti-tumor mechanisms is ongoing [5].
Carcinoembryonic antigen (CEA) is a glycoprotein normally found at low levels in the gut crypts and healing intestinal mucosa [6]. However, CEA over-expression can be found in virtually all adenocarcinomas of the colon and rectum, [7–9]. In colorectal carcinomas, CEA is produced at high levels and can be detected in the circulation [10]. This over-expression of CEA in colorectal cancers has led to intensive research on vaccine strategies targeting this protein.
The immunologic destruction of tumors necessitates the presentation of antigenic peptides on antigen presenting cells and the acquisition of costimulatory signals by the T cell. Recombinant (r) vaccinia-based and recombinant avipox-based vectors containing the transgenes encoding for the costimulatory molecules B7.1, ICAM-1, and LFA-3 were created (designated TRICOM). These TRICOM constructs were shown in pre-clinical studies to be superior to constructs that contained only one or two of the costimulatory molecules [11]. Additionally, a unique CEA agonist epitope, CAP1-6D, was included in the vaccine in an attempt to increase the immune response directed against “self”. CAP1-6D, a mutated 9-mer peptide (YLSGANLNL) of the CEA protein that binds to HLA-A2, appears to be more effective in the induction of an immune response [12, 13]. Marshall et al. evaluated four sequential vaccinations with rF-CEA(6D)-TRICOM alone, in combination with rV-CEA(6D)-TRICOM and with GM-CSF. These vaccines were administered to patients with CEA-expressing carcinomas demonstrating the feasibility of this approach [14].
The effects of IFN-α on the expression of tumor antigens such as CEA have been evaluated by several investigators. Monoclonal antibody-based treatments are significantly enhanced by IFN-α pre-treatment in tumor models and the mechanism for this seems to depend on the ability of IFN-α to enhance the expression of tumor antigens [15, 16]. IFN-α, IFN-β, and IFN-γ can all mediate the increased expression of CEA on human cell lines [17–20]. The actions of IFN-α occur at concentrations easily attainable in the clinic via infusion/injection of IFN-α. Moreover, these effects continue well past the time of exposure to IFN [21]. It has also been shown that combining IFN-α-2b and GM-CSF induces a rapid maturation of monocytes into dendritic cells that are functionally superior to those induced by treatment with IL-4 plus GM-CSF [22].
It was hypothesized that upregulation of carcinoma cell CEA expression by IFN-α and the potential for increased dendritic cell activity would lead to enhanced activation of antigen-specific T cells in subjects receiving IFN-α in combination with the vaccine regimen. The purpose of this trial was to evaluate the safety of adding IFN-α to rV-CEA (6D)-TRICOM and rF-CEA(6D)-TRICOM in combination with GM-CSF.
METHODS
Eligibility Criteria
A National Cancer Institute (NCI) sponsored phase I trial of prime-boost vaccinations with rV-CEA(6D)-TRICOM and rF-CEA(6D)-TRICOM in combination with GM-CSF and IFN-α-2b in patients with CEA-expressing carcinomas was conducted at the Ohio State University Comprehensive Cancer Center under an Institutional Review Board-approved protocol (OSU-0312, NCI-5633). Eligible patients had histologically confirmed metastatic or locally-advanced cancer that was shown to express CEA. Patients must have received at least one previous systemic regimen and 4 weeks must have passed since any previous treatment (6 weeks for patients receiving nitrosoureas or mitomycin C). Patients also met the following criteria: age ≥18 years, life expectancy ≥6 months, Eastern Cooperative Oncology Group (ECOG) status ≤2, normal organ function, negative pregnancy test, and ability to provide informed consent. Patients must not have had a history of allergy or untoward reaction to a vaccinia vaccine or any of its components. Patients with known brain metastases or allergy to eggs were excluded. Patients with concurrent steroid use, except topical and inhaled steroids, and those who were immunocompromised (HIV positive or Hepatitis B/C positive) were excluded. Patients who had close contact with immunocompromised persons were excluded. Patients were required to exhibit acceptable renal function and must not have had splenectomy. Pregnant women and those with any recent cardiovascular events were excluded.
Study Design
The clinical trial employed two novel anti-CEA vaccines: rV-CEA(6D)-TRICOM and rF-CEA(6D)-TRICOM. The vaccinia vaccine was administered s.c. on day 1 followed by GM-CSF administered to the same site on days 1–4. In Cycle 1, patients received thrice weekly injections of IFN-α-2b s.c. the week after the rV-CEA(6D)-TRICOM vaccine was administered. This schema was chosen so that 7 days had passed since vaccination, as the administration of IFN-α-2b could inhibit replication of the vaccinia virus. In Cycles 2–4, patients received thrice weekly s.c. injections of IFN-α-2b simultaneously with the administration of rF-CEA(6D)-TRICOM vaccine and GM-CSF, since IFN-α-2b can augment dendritic cell maturation and should not inhibit the activity of the non-replicating fowlpox vector. An initial six-patient cohort (Cohort 0) received no IFN-α-2b. Dose escalation of IFN-α-2b (1×106, 3×106, 6×106, 9×106 U) therapy took place in cohorts of three patients following a standard 3+3 design based on the number of dose-limiting toxicities (DLT) observed during the first cycle. The maximum tolerated dose (MTD) was defined as the highest dose level at which fewer than two patients experienced first course DLT. Six additional patients were treated at the MTD and were all HLA-A2 positive. There was no intra-patient dose escalation of IFN-α-2b. IFN-α-2b was administered s.c. at rotating sites away from the vaccination site during Cycle 1. In Cycles 2–4, IFN-α-2b was given along with GM-CSF at the vaccination site. There were at least three patients in each of the four cohorts. HLA-A2 status was determined for most patients, but was not a requirement.
Patients who did not progress or have unacceptable toxicity after the initial phase of vaccinations were offered additional vaccinations of rF-CEA(6D)-TRICOM (with GM-CSF and IFN-α-2b). Patients received the same dose of vaccine and IFN-α-2b as the treatment arm on which they were enrolled every 28 days through vaccination number 6 and every 3 months thereafter for up to 2 years. Off-study criteria and clinical/immunologic monitoring continued for these patients. Patients were re-staged every 2 months.
Vaccine Preparation
rV-CEA(6D)-TRICOM and rF-CEA(6D)-TRICOM were manufactured by Therion Biologics Corp. (Cambridge, MA) and supplied by the Cancer Therapy Evaluation Program, National Cancer Institute (Bethesda, MD). 0.2 mL (=1.2×108 pfu) of rV-CEA(6D)/TRICOM was administered by subcutaneous injection. 0.2 mL of rF-CEA(6D)-TRICOM was withdrawn to a sterile glass vial containing 1.96 mL of 0.9% sodium chloride for injection, and vortexed at high power for at least 10 seconds. 1 mL (=4×108 pfu) of the diluted vaccine was then withdrawn for subcutaneous administration.
Measurement of Serum sCD27 and sCD40L
Serum levels of sCD27 were determined using the Human sCD27 Instant ELISA Kit (eBioscience, San Diego, CA) according to the manufacturer’s instructions. The standard range of this kit is 15.6-1000 U/ml. The serum levels of sCD40L were determined using the Human sCD40L Platinum ELISA Kit (eBioscience) according to the manufacturer’s instructions. The standard range of this kit is 0–10 ng/ml.
Assessment of Antigen Specific Responses
Antigen specific responses were assessed by intracellular cytokine staining (ICS) following a period of in vitro stimulation with overlapping 15-mer peptide pools encoding the tumor-associated antigen (TAA) CEA as previously described [23]. The TAA peptide pool was designed to contain an agonist epitope (CAP1-6D), which was also present in the vaccines [24]; peptide pools encoding for HLA and CEFT (a mixture of cytomegalovirus, Epstein-Barr virus, influenza, and tetanus toxin) served as negative and positive controls, respectively. Peptide mixes were purchased from JPT (Berlin, Germany), reconstituted in DMSO, and utilized immediately. Cryopreserved PBMCs were defrosted, rested overnight, and then stimulated for 7 days with peptide pools (0.1ug/mL) with cytokines (IL-7 and IL-15, 10ng/mL, PeproTech, Rocky Hill, NJ) supplemented on days 3 and 5. On day 7 cultures were rested (with the removal of cytokine and peptide), and 4 days later (day 11), 1×106 cells were restimulated for 24 hours with peptide pools (0.1ug/mL) in the presence of anti-CD107a-APC (clone H4A3, BD Biosciences, San Jose, CA); brefeldin A (1ul/mL) and monensin (0.7ul/mL) (BD Biosciences) were added to cultures 2 hours after the start of the restimulation and incubated for the remaining 22 hours. PBMCs were then stained with anti-CD4-PerCP-Cy5.5 (clone OKT4, Biolegend, San Diego, CA), anti-CD8-AF700 (clone OKT8, eBioscience), and anti-TNF-PE (clone MAb11), anti-IFNγ-PE-Cy7 (clone 4SB3), and anti-IL-2-BV521 (clone 5344.111) (BD Biosciences). At least 3×105 events in the live gate were acquired with a BD LSR-II flow cytometer and analyzed with FlowJo software (TreeStar, Ashland, OR). The absolute number of CD4+ or CD8+ lymphocytes producing cytokine or positive for CD107a was calculated per 1×106 cells plated at the start of the in vitro stimulation. The background signal (obtained with the HLA peptide pool), and values obtained prior to therapy were subtracted from those obtained post-therapy. Values >250 were scored as positive for TAA-specific immune response following therapy.
Flow Cytometry for Myeloid-Derived Suppressor Cells
PBMC from each patient at the pre-treatment and post-Cycle 1 time points were analyzed for myeloid derived suppressor cells (MDSC) as previously described [25]. Specific antibodies included CD15-FITC (Beckman Coulter), CD33-APC (Beckman Coulter), HLA-DR-PC7 (Beckman Coulter), CD11b-PE (Beckman Coulter), and CD14-V450 (BD Biosciences). Single color staining was performed for compensation. All samples were run on a BD LSR II flow cytometer and were subsequently analyzed with FlowJo software (TreeStar). MDSC were defined as cells positive for CD33 and CD11b and lacking HLA-DR with subsets expressing CD15 or CD14 representing granulocytic and monocytic MDSC, respectively, as discussed in figure legends.
Statistical Analysis
Patient demographic and clinical characteristics were summarized by number and percentage for categorical data, and mean or median as well as range for each continuous variable. The changes in serum sCD27 and sCD40L levels before and after treatment were compared using the Wilcoxon signed rank test and MDSC percentages were compared using a paired t-test. Response and progression were evaluated using the international criteria proposed by the Response Evaluation Criteria in Solid Tumors (RECIST) Committee [26]. Progression free survival (PFS) and overall survival (OS), defined as the time from initiation of study drug to disease progression or death, censored at last evaluation date for PFS and last known survival data for OS, were estimated using the Kaplan-Meier method. Results were also summarized by dose cohort, and compared using log rank test. All statistical analyses were performed using SAS 9.4 (SAS Inc., Cary, NC).
RESULTS
Patient Demographics
Thirty-three patients were enrolled on this study (20 male, 13 female). Their mean age was 59.8 years (range 38–71). All patients had metastatic disease at trial entry (Table 1). Most patients had colorectal cancers (n=21, 64%). Other cancer types included lung (4), breast (3), pancreas (2), appendix (1), esophagus (1) and bladder (1). Sites of metastasis included lung (16), liver (16), lymph node (8) kidney (3), abdomen (3), pelvis (2), stomach (2), spine (2), brain (2), bone (1), pancreas (1), and small intestine (1). Fourteen patients were HLA-A2 positive, 11 patients were HLA-A2 negative, and eight patients had unknown HLA status. Patients were heavily pre-treated. Thirty-one out of 33 patients (94%) had received at least two prior chemotherapy regimens and 29 out of 33 patients (88%) had greater than two prior chemotherapy regimens (range 3–8). Twenty-five patients (76%) had prior surgery, while only 12 (36%) had received radiation treatment.
Table 1.
Patient Demographics and Outcomes
| No. of patients (n) | |
|---|---|
|
| |
| Sex | |
| Male | 20 |
| Female | 13 |
|
| |
| Age | |
| Mean = 59.79 | |
| Median = 63 | |
| Range = 38–71 | |
|
| |
| ECOG performance status | |
| 0 | 13 |
| 1 | 20 |
|
| |
| Primary Site | |
| Colon | 14 |
| Rectum | 7 |
| Lung | 4 |
| Breast | 3 |
| Pancreas | 2 |
| Appendix | 1 |
| Esophagus | 1 |
| Bladder | 1 |
|
| |
| Location of Metastatic Site(s) | |
| Lung | 16 |
| Liver | 16 |
| Lymph node | 8 |
| Kidney/Adrenal | 3 |
| Abdomen | 3 |
| Pelvis | 2 |
| Stomach | 2 |
| Spinal | 2 |
| Brain | 2 |
| Other | 3 |
|
| |
| HLA-A2 | |
| Positive | 14 |
| Negative | 11 |
| Unknown | 8 |
|
| |
| Prior Treatments Received | |
| Surgery | 25 |
| Radiation | 12 |
| Chemotherapy - 0 prior regimens | 0 |
| Chemotherapy - 1 prior regimen | 2 |
| Chemotherapy - 2 prior regimens | 31 |
| Chemotherapy - > 2 prior regimens | 29 |
|
| |
| Response to Therapy | |
| Partial Response | 0 |
| Stable Disease | 8 |
| Progressive Disease | 21 |
| Not evaluable | 4 |
|
| |
| Progression-free Survival (mo) | |
| Median | 1.8 |
| Range | 0.8 – 15.7 |
|
| |
| Overall Survival (mo) | |
| Median | 6.3 |
| Range | 1.2 – 98.8 |
Toxicities
Toxicities are listed in Table 2. In general, the regimen was well tolerated. Common grade 3 toxicities included fatigue, hyperglycemia and abdominal pain and were felt to be disease related. Other notable toxicities were encephalopathy (1), troponin elevation (1) and disseminated intravascular coagulation (DIC) (1). In each case, the adverse event was felt to be related to disease progression and not the experimental therapy. Grade 4 and 5 toxicities included confusion, elevated aspartate transaminase to alanine transaminase (AST/ALT) ratio and sudden death. The elevated liver enzymes were felt to be related to metastatic disease in the liver. The patient with confusion also had uncontrolled hypertension and underwent a brain MRI for further evaluation. The imaging was consistent with posterior reversible encephalopathy syndrome, which was deemed to be related, in part, to the hypertension and possibly the IFN-α treatment. However, the patient was removed from the trial shortly afterwards recovered fully. The only patient who expired while on trial was noted to have breast adenocarcinoma metastatic to liver, lung and bone. She had a recurrent pleural effusion and had received seven different systemic regimens for her metastatic disease along with surgery and radiation to the lung. She developed an upper respiratory infection (likely viral) on cycle 2 and refused further supportive therapy. On day 8 of cycle 2 she presented to the emergency department with a chief complaint of progressive dyspnea. She was found to be hypoxic and imaging revealed a large pleural effusion. She expired within hours of presentation without having had any supportive interventions.
Table 2.
Toxicities
| Type of Toxicity | Grade 3, n (%) | Grade 4 n (%) | Grade 5 n (%) |
|---|---|---|---|
|
| |||
| Cardiovascular/Pulmonary | |||
| DIC | 1 (3.0) | ||
| Dyspnea | 2 (6.0) | ||
| Hypertension | 1 (3.0) | ||
| Plueral effusion | 1 (3.0) | ||
| Pulmonary/Upper respiratory- Other | 1 (3.0) | ||
| Troponin 1 | 1 (3.0) | ||
| Gastrointestinal | |||
| Anorexia | 2 (6.0) | ||
| Nausea | 3 (9.0) | ||
| Vomiting | 2 (6.0) | ||
| Diarrhea | 1 (3.0) | ||
| Constitutional | |||
| Fatigue | 5 (15.2) | ||
| Neurologic | |||
| Confusion | 1 (3.0) | ||
| Encephalopathy | 1 (3.0) | ||
| Laboratory Abnormalities | |||
| Alkaline phosphatase | 3 (9.0) | ||
| Anemia | 2 (6.0) | ||
| AST/ALT | 1 (3.0) | ||
| Bilirubin | 2 (6.0) | ||
| Hemoglobin | 1 (3.0) | ||
| Hyperglycemia | 4 (12.1) | ||
| Hypoalbuminemia | 2 (6.0) | ||
| Hyponatremia | 1 (3.0) | ||
| Leukocytes | 1 (3.0) | ||
| Lymphopenia | 2 (6.0) | ||
| Neutrophils | 1 (3.0) | ||
| Pain | |||
| Anthralgia/Myalgia | 2 (6.0) | ||
| Back Pain | 1 (3.0) | ||
| Headache | 1 (3.0) | ||
| Pelvic/Abdominal pain | 3 (9.0) | ||
| Other | |||
| Glaucoma | 1 (3.0) | ||
| Hypoxia | 1 (3.0) | ||
| Infection without neutropenia | 1 (3.0) | ||
| Sudden death | 1 (3.0) | ||
Response to Therapy
Outcome data are presented in Table 1. No patients had a partial response (PR). Eight patients (24%) exhibited stable disease (SD) that lasted 3 months or more (16, 16, 16, 17, 22, 26, 28, and 67 weeks). Twenty-one patients (64%) had progressive disease prior to 3 months. Complete information was unavailable for four patients (12%) who were removed from the study at early time points due to toxicities (2), refusal to continue treatment (1) or death (1). The median number of treatment cycles received was 2 (range, 1–8). The median PFS was 1.8 months and the median OS was 6.3 months. While those patients who received any dose of IFN-α-2b (cohorts 1–4) had significantly improved OS compared to those who did not receive IFN-α-2b (6.40 vs. 3.94 months, p=0.02) (Fig. 1a and b), PFS did not differ significantly between these two groups (1.80 vs. 1.87 months, p=0.54). However, Cohorts 3 and 4 had marginally better PFS than Cohort 0–2 (p=0.07). Due to the small sample size in each cohort, there were no significant differences in PFS or OS among dose cohorts (Fig. 1c and d).
Fig. 1. Overall survival is increased with the addition of IFN-α-2b.
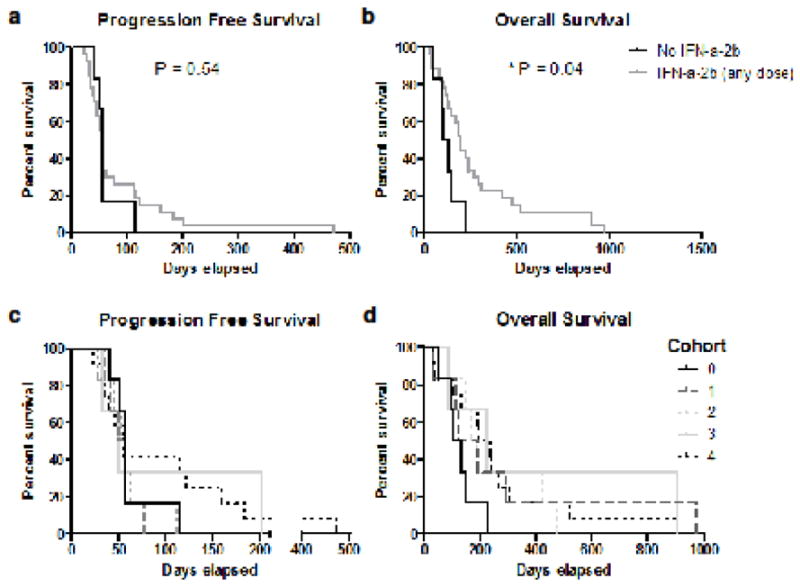
(a) Progression-free survival curves comparing those patients not receiving IFN-α-2b (Cohort 0) to those receiving any dose of IFN-α-2b (Cohorts 1–4). (b) Overall survival curves comparing those patients not receiving IFN-α-2b (cohort 0) to those receiving any dose of IFN-α-2b (Cohorts1–4) (c) Progression-free survival curves and (d) overall survival curves comparing all 5 study arms: Cohort 0, no IFN-α-2b; Cohort 1, IFN-α-2b (1×106 U); Cohort 2, IFN-α-2b (3×106 U); Cohort 3, IFN-α-2b (6×106 U); Cohort 4, IFN-α-2b (9×106 U)
Immune Responses
Serum levels of sCD27 are increased at all time points post-vaccine therapy
Soluble CD27 is a marker of T-cell activation, and previous studies have suggested that elevated levels can be used as a marker for patients responding to immunotherapy [27]. The levels of sCD27 were measured in patient serum at all available time points. The levels of sCD27 before vaccine therapy were compared with those measured after Cycle 1 or 2, Cycle 3 or Cycles 4–7. Six different patients had samples available at two post-therapy time points. As can be seen in Figure 2, the serum levels of sCD27 were significantly increased at all time points after vaccine therapy, with median levels of 103.8 U/ml pre-therapy (n=33), 111.1 U/ml post-Cycles 1–2 (Fig. 2a, n=21, p=0.007), 131.0 U/ml at Cycle 3 (Figure 2b, n=12, p=0.003), and 131.2 at Cycles 4–7 (Fig. 2c, n=6, p=0.03). When the net increase in sCD27 levels from pre-treatment to post-treatment (regardless of cycles) was evaluated in the context of IFN-α-2b dose received, those patients who received any dose of IFN-α-2b had a significant increase in sCD27 levels compared to baseline (p<0.004), while those receiving no IFN-α-2b had no significant change in sCD27 (p=0.065), indicating that the addition of IFN-α-2b as a vaccine adjuvant may have enhanced T-cell activation in this patient subset. A significant correlation with OS was detected when comparing the net sCD27 increase in the IFN-α-2b treated group versus untreated groups (p=0.04). The timespan between the last chemotherapy administration and the first trial vaccination for each patient was evaluated for its relationship to survival and immune response. A trend indicated that increased time between treatments was positively associated with PFS (p=0.071). Additionally, the change in sCD27 from pre-treatment to post-cycle 4 showed a positive, but statistically non-significant association with time between treatments (p=0.072). In addition, serum levels of sCD40L were measured, as they have been shown to have a potential immunosuppressive role [28]. There were no significant changes in sCD40L levels over the time points examined for this group of patients.
Fig. 2. Serum levels of sCD27 increased at all time points post-therapy.
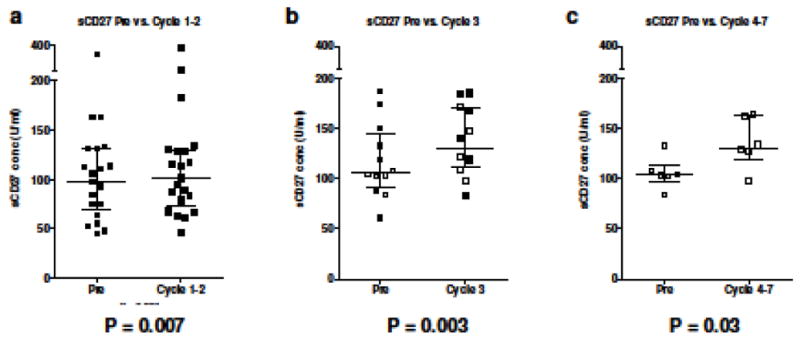
Serum levels of sCD27 were analyzed by ELISA. (a) Patient samples analyzed pre-therapy (day 1 or “Pre”) and after 1–2 cycles (n=21, P=0.007). (b) Patient samples analyzed prior to therapy and after 3 cycles (n=12, P=0.003). Patients who were also assayed at Cycles 4–7 are shown as open circles/squares. (c) Patient samples analyzed pre-therapy and after 4–7 cycles (n=6, P=0.03). Dot plots with medians and interquartile range are shown
Detection of antigen specific CEA responses post-vaccine therapy
Sufficient PBMCs were available from 19 of 33 patients to analyze antigen-specific immune responses by intracellular cytokine staining. These patients were representative of the total patient population. Patients were assessed before vaccination and after Cycle 1 (n=4), Cycle 2 (n=6), and Cycle 3 (n=9) of therapy. Three subjects evaluated at Cycle 3 were also analyzed at Cycle 5 or 6 (all from Cohort 4). As seen in Figure 3 and Supplementary Table S1, 8 of 19 patients (42%) developed T-cell responses to the 15-mer CEA peptide pools during the course of treatment, as evidenced by cytokine production or the induction of CD107a positive CD4+ and/or CD8+ T cells. Of the eight patients developing responses, three were HLA-A2 positive. Notably, all three patients evaluated at cycle 5 or 6 (2/3 HLA-A2 positive and all receiving the highest dose of IFN-α-2b) developed CEA specific T-cell responses. Of these patients, 2/3 also displayed a response at cycle 3 (as indicated by CD107a positivity or IFN-γ production by CD4 T cells). Two patients (B and N, Cohorts 0 and 1, respectively) had pre-existing immune responses to CEA that were further increased following therapy. There were no significant differences in the responses between cohorts. All patients assessed for viral responses to CEFT peptide pools were positive.
Fig. 3. CEA-specific T-cell responses post- vs. pre-vaccination.
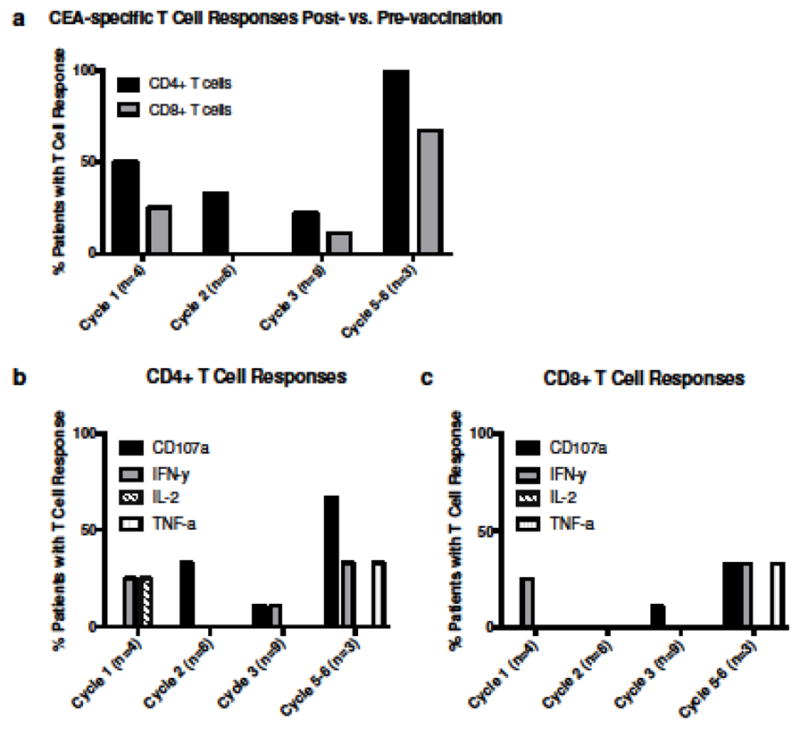
PBMCs from patients were analyzed for antigen-specific immune responses by intracellular cytokine staining. Patients were assessed before vaccination and after Cycle 1 (n=4), Cycle 2 (n=6), Cycle 3 (n=9) and Cycle 5–6 (n=4) of therapy. (a) Percentage of patients with CD4+ or CD8+ T cell responses after indicated cycle. (b) CD4+ or (c) CD8+ T cell responses as evidenced by cytokine production or the induction of CD107a positive T cells
Levels of circulating MDSC correlate with overall survival
Myeloid derived suppressor cells are often found at increased levels in cancer patients and are known to promote disease progression through multiple immunosuppressive mechanisms, including suppression of T-cell function [29]. Cryopreserved, total PBMCs from 19 patients collected prior to the initiation of therapy and following the completion of cycle 1 were stained with fluorochrome-conjugated antibodies targeting CD33, HLA-DR, CD11b, CD14, and CD15. Cells displaying a CD33+/HLA-DR− phenotype were considered to constitute total MDSC, while cells further expressing CD11b, and CD14 or CD15 were considered to be monocytic (M-MDSC) and granulocytic (G-MDSC) subsets, respectively. The mean frequency of total MDSC within patient PBMCs increased slightly following the initial cycle of therapy (from 1.60% to 2.27%, p=0.18), with post-Cycle 1 levels ranging from 1.24–3.30% (Fig. 4a). The absolute change in MDSC percentage from pre-treatment to post-Cycle 1 did not differ significantly by treatment cohort (Fig. 4b). Prior to the initiation of treatment, levels of G-MDSC were higher than the levels of M-MDSC (mean percentage of 0.55% vs. 0.14%, respectively). However, by the end of Cycle 1, levels of G-MDSC and M-MDSC were comparable (0.60 vs. 0.61%). Pre-treatment levels of MDSC were found to have a significant, negative correlation with OS for both the total MDSC (p=0.04) and G-MDSC (p=0.05) populations, but not M-MDSC (Fig. 4c). A similar trend could be seen at the post-Cycle 1 time point, with total MDSC, G-MDSC and M-MDSC all being present at lower percentages in those patients with an OS greater than 150 days. With regard to the T-cell responses, G-MDSC levels following Cycle 1 of treatment were found to have a negative correlation with pre-treatment sCD27 levels (p=0.03), indicating that those patients with higher baseline levels of T-cell activation, as indicated by sCD27 levels, had lower G-MDSC following one cycle of vaccine therapy. Overall, while MDSC levels appeared to be prognostic for survival in this patient population, they did not appear to be significantly affected by treatment, and did not correlate with any other immune responses, including sCD40L levels and antigen-specific T-cell responses.
Fig. 4. MDSC levels are associated with overall survival.

Circulating levels of total (HLADR-,CD33+), granulocytic (HLADR-,CD33+,CD11b+,CD15+) or monocytic (HLADR-,CD33+,CD11b+,CD14+) MDSC were measured by flow cytometry prior to or after completion of cycle 1 of treatment. (a) Patient samples analyzed before (Pre) and after 1 cycle (n=19) and (b) absolute change in MDSC by cohort group, calculated as %MDSC Pre- minus %MDSC Post-Cycle 1. (c) Pre-treatment MDSC and (d) Post-Cycle 1 MDSC levels stratified by overall survival at 150 days. Dot plots with medians and interquartile range are shown
DISCUSSION
A phase I clinical trial evaluating the combination of GM-CSF and IFN-α with the novel anti-CEA vaccines, rV-CEA(6D)-TRICOM and rF-CEA(6D)-TRICOM, was conducted. This study was based on the rationale that GM-CSF has been shown to increase the frequency of antigen presenting cells and enhance the antigen-specific anti-tumor response and that IFN-α can enhance the expression of tumor antigens such as CEA. The present regimen was well-tolerated. The majority of grade 3/4 events were felt to be disease related. In this study, the combination of the rV-CEA(6D)-TRICOM prime and multiple rF-CEA(6D)-TRICOM booster vaccines with GM-CSF and IFN-α-2b resulted in stable disease in 24% of patients and no partial responses. The median PFS and OS were 1.8 and 6.3 months, respectively, and the OS was significantly increased with the addition of IFN-α-2b to the treatment regimen. The sCD27 data indicated that T-cell activation increased with the addition of IFN-α-2b to the regimen. The development of antigen-specific CEA responses to the vaccine therapy was seen in 42% of patients tested, and did not appear to be dependent on treatment cohort. Levels of circulating MDSC measured prior to treatment were found to significantly correlate with overall survival.
While no patients had a clinical response to therapy, it is important to note that all patients were heavily pre-treated, with 94% having received at least two prior chemotherapy regimens and 76% having undergone prior therapeutic surgeries. The extent of disease and number of prior therapies also likely contributed to the common grade 3 toxicities seen during therapy (fatigue, hyperglycemia and abdominal pain), as IFN-α has been generally well tolerated when administered as an adjuvant in other vaccine trials [30, 31].
Immunotherapy of cancer and the development of cancer vaccines have become an important area of research for the treatment of colorectal cancer, given the predictive value of tumor immune infiltrates with respect to survival [32]. The TRICOM vaccines consist of either a priming vaccinia virus or a booster avipox virus vector, encoding for CEA and three costimulatory molecules. These constructs allow for an enhanced immune response to specific tumor associated antigens, such as CEA [33]. CEA/TRICOM vaccines have shown efficacy in the induction of CEA-specific T-cell responses in other clinical studies [14], and this outcome was confirmed in the current study, with 8 out of 19 (42%) patients tested developing responses to CEA peptide pools, as indicated by the presence of CD4+ and/or CD8+ T cells positive for CD107a or producing the cytokines IFN-γ, IL-2 or TNF-α following peptide stimulation. Additionally, sCD27 levels in all patients tested were found to increase significantly from pre-treatment to post-therapy cycles, indicating that the treatment brought about T-cell activation. Although the development of antigen specific T-cell responses has historically been measured by stimulating PBMCs with a single 9-mer peptide and measuring the production of IFN-γ by ELISPOT, extensive studies have shown a number of advantages to the flow-based intracellular cytokine staining method following a period of in vitro stimulation with overlapping 15-mer peptide pools that span the entire length of a given tumor associated antigen. More recently, a TRICOM vaccine containing transgenes for the MUC1 tumor antigen, in addition to CEA (known as PANVAC), has shown promise when given in combination with GM-CSF and when utilized to modify autologous dendritic cells for injection [34, 35].
The effects of IFN-α administration on CEA and major histocompatibility complex (MHC) expression by tumor cells appear to be fairly robust [36]. Guadagni et al. demonstrated that the cell surface expression of CEA and MHC antigens on freshly isolated tumor cells is significantly enhanced following exposure to type I interferons [37]. The intramuscular administration of IFN-α at a dose of 3 × 106 U on a daily basis significantly increased the localization of an anti-melanoma antibody to tumor deposits in melanoma patients with metastatic disease [38]. Similar results were obtained in a study in which patients with metastatic breast cancer received either IFN-α at a dose of 3 × 106 U daily or no treatment [39]. These results support the applicability of this approach to multiple clinical situations. It has also been observed that many tumors express low levels of MHC molecules [40, 41]. This can lead to impaired recognition by specific immune effector cells. MHC class I and II as well as ICAM-1 are critically important to effector cell recognition of cancer cells [42, 43]. The potentially favorable effects of IFN-α on MHC class I and II expression in adenocarcinoma cells are well-documented [44, 45]. In this trial, patients receiving any dose of IFN-α-2b had significantly improved OS compared to those who did not receive IFN-α, indicating that adding IFN-α as an adjuvant was beneficial. Cohorts 3 and 4 (IFN-α doses 6 × 106 and 9 × 106 U, respectively) also had better PFS than Cohorts 0–2, although this result was not statistically significant (p=0.07). Reflective of the mechanisms previously described, the net sCD27 increase in the IFN-α-2b treated versus untreated groups significantly correlated with OS (p=0.04), further implying that the addition of IFN-α-2b to the therapy could possibly result in increased T-cell activation leading to enhanced survival. This enhancement might have been due to increased expression of MHC and CEA on tumor cells following IFN-α administration.
Our group and others have previously shown that enhanced levels of circulating MDSC correlate with increased disease burden and decreased survival in patients with cancer [46, 47]. Additionally, we have shown that MDSC can function to reduce IFN-α responsiveness of immune cells [25, 48]. In the present trial, circulating levels of MDSC were measurable and pre-treatment MDSC levels were found to negatively correlate (p=0.04) with overall survival. This may be due to the fact that increased MDSC levels could lead to enhanced immunosuppression and diminished response to IFN-α treatment. Moreover, it is noteworthy that G-MDSC levels following one cycle of therapy were negatively correlated with sCD27 levels pre-treatment (p=0.03). This suggests that patients with heightened T-cell activation prior to the initiation of therapy (as indicated by increased serum sCD27) did not experience an influx of immunosuppressive G-MDSC during cycle 1 of therapy. This effect could signal the importance of MDSC levels in determining the responsiveness to a vaccine given with IFN-α. Further studies of patient MDSC levels are warranted and may yield clues for potential combination therapies to target MDSC in the context of immune-based therapies.
This immunotherapy regimen was generally well-tolerated and can be safely administered to patients with CEA-expressing carcinomas. While the vaccine regimen did not produce any clinical responses, the addition of IFN-α to this vaccine was associated with patient immune responses. IFN-α as a vaccine adjuvant is thus worthy of further investigation in future clinical trials.
Supplementary Material
Supplementary table S1. CEA-specific T-cell responses post-vs. pre-vaccination
Acknowledgments
FUNDING
This work was supported by NIH Grants: P01 CA095426 (W. Carson), P30 CA016058 (W. Carson), T32 GM068412 (M. Duggan)
ABBREVIATIONS
- AST/ALT
Aspartate transaminase to alanine transaminase ratio
- CEA
Carcinoembryonic antigen
- CEFT
Mixture of cytomegalovirus, Epstein-Barr virus, influenza, and tetanus toxin
- DIC
Disseminated intravascular coagulation
- DLT
Dose-limiting toxicities
- ECOG
Eastern Cooperative Oncology Group
- G-MDSC
Granulocytic myeloid derived suppressor cell
- M-MDSC
Monocytic myeloid derived suppressor cell
- MDSC
Myeloid derived suppressor cell
- MTD
Maximum tolerated dose
- PFS
Progression free survival
- PR
Partial response
- SD
Stable disease
- TAA
Tumor-associated antigen
Footnotes
CONFLICT OF INTEREST STATEMENT
The authors declare that they have no conflict of interest.
References
- 1.American Cancer Society. Key statistics for colorectal cancer. Am. Cancer Soc; 2016. [Accessed 24 Mar 2016]. http://www.cancer.org/cancer/colonandrectumcancer/detailedguide/colorectal-cancer-key-statistics. [Google Scholar]
- 2.Demols A, Van Laethem J-L. Adjuvant chemotherapy for colorectal cancer. Curr Gastroenterol Rep. 2002;4:420–6. doi: 10.1007/s11894-002-0013-3. [DOI] [PubMed] [Google Scholar]
- 3.Gustavsson B, Carlsson G, Machover D, et al. A review of the evolution of systemic chemotherapy in the management of colorectal cancer. Clin Colorectal Cancer. 2015;14:1–10. doi: 10.1016/j.clcc.2014.11.002. [DOI] [PubMed] [Google Scholar]
- 4.Folprecht G, Hamann S, Schütte K, et al. Dose escalating study of cetuximab and 5-FU/folinic acid (FA)/oxaliplatin/irinotecan (FOLFOXIRI) in first line therapy of patients with metastatic colorectal cancer. BMC Cancer. 2014;14:521. doi: 10.1186/1471-2407-14-521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marques AM, Turner A, de Mello RA. Personalizing medicine for metastatic colorectal cancer: current developments. World J Gastroenterol. 2014;20:10425–31. doi: 10.3748/wjg.v20.i30.10425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rogers GT. Carcinoembryonic antigens and related glycoproteins. Molecular aspects and specificity. Biochim Biophys Acta. 1983;695:227–49. doi: 10.1016/0304-419x(83)90013-6. [DOI] [PubMed] [Google Scholar]
- 7.Muraro R, Wunderlich D, Thor A, et al. Definition by monoclonal antibodies of a repertoire of epitopes on carcinoembryonic antigen differentially expressed in human colon carcinomas versus normal adult tissues. Cancer Res. 1985;45:5769–80. [PubMed] [Google Scholar]
- 8.Steward AM, Nixon D, Zamcheck N, Aisenberg A. Carcinoembryonic antigen in breast cancer patients: serum levels and disease progress. Cancer. 1974;33:1246–52. doi: 10.1002/1097-0142(197405)33:5<1246::aid-cncr2820330509>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 9.Vincent RG, Chu TM. Carcinoembryonic antigen in patients with carcinoma of the lung. J Thorac Cardiovasc Surg. 1973;66:320–8. [PubMed] [Google Scholar]
- 10.Ladenson JH, McDonald JM, Landt M, Schwartz MK. (Washington University Case Conference). Colorectal carcinoma and carcinoembryonic antigen (CEA) Clin Chem. 1980;26:1213–20. [PubMed] [Google Scholar]
- 11.Hodge JW, Sabzevari H, Yafal AG, et al. A triad of costimulatory molecules synergize to amplify T-cell activation. Cancer Res. 1999;59:5800–7. [PubMed] [Google Scholar]
- 12.Zaremba S, Barzaga E, Zhu M, et al. Identification of an enhancer agonist cytotoxic T lymphocyte peptide from human carcinoembryonic antigen. Cancer Res. 1997;57:4570–7. [PubMed] [Google Scholar]
- 13.Salazar E, Zaremba S, Arlen PM, et al. Agonist peptide from a cytotoxic t-lymphocyte epitope of human carcinoembryonic antigen stimulates production of tc1-type cytokines and increases tyrosine phosphorylation more efficiently than cognate peptide. Int J cancer. 2000;85:829–38. doi: 10.1002/(sici)1097-0215(20000315)85:6<829::aid-ijc16>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 14.Marshall JL, Gulley JL, Arlen PM, et al. Phase I study of sequential vaccinations with fowlpox-CEA(6D)-TRICOM alone and sequentially with vaccinia-CEA(6D)-TRICOM, with and without granulocyte-macrophage colony-stimulating factor, in patients with carcinoembryonic antigen-expressing carcinomas. J Clin Oncol. 2005;23:720–31. doi: 10.1200/JCO.2005.10.206. [DOI] [PubMed] [Google Scholar]
- 15.Guadagni F, Schlom J, Pothen S, et al. Parameters involved in the enhancement of monoclonal antibody targeting in vivo with recombinant interferon. Cancer Immunol Immunother. 1988;26:222–30. doi: 10.1007/BF00199933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Greiner JW, Guadagni F, Noguchi P, et al. Recombinant interferon enhances monoclonal antibody-targeting of carcinoma lesions in vivo. Science. 1987;235:895–8. doi: 10.1126/science.3580039. [DOI] [PubMed] [Google Scholar]
- 17.Greiner JW, Fisher PB, Pestka S, Schlom J. Differential effects of recombinant human leukocyte interferons on cell surface antigen expression. Cancer Res. 1986;46:4984–90. [PubMed] [Google Scholar]
- 18.Guadagni F, Witt PL, Robbins PF, et al. Regulation of carcinoembryonic antigen expression in different human colorectal tumor cells by interferon-gamma. Cancer Res. 1990;50:6248–55. [PubMed] [Google Scholar]
- 19.Leon JA, Mesa-Tejada R, Gutierrez MC, et al. Increased surface expression and shedding of tumor associated antigens by human breast carcinoma cells treated with recombinant human interferons or phorbol ester tumor promoters. Anticancer Res. 9:1639–47. [PubMed] [Google Scholar]
- 20.Guadagni F, Roselli M, Schlom J, Greiner JW. In vitro and in vivo regulation of human tumor antigen expression by human recombinant interferons: a review. Int J Biol Markers. 1994;9:53–60. doi: 10.1177/172460089400900111. [DOI] [PubMed] [Google Scholar]
- 21.Greiner JW, Hand PH, Noguchi P, et al. Enhanced expression of surface tumor-associated antigens on human breast and colon tumor cells after recombinant human leukocyte alpha-interferon treatment. Cancer Res. 1984;44:3208–14. [PubMed] [Google Scholar]
- 22.Santini SM, Lapenta C, Logozzi M, et al. Type I interferon as a powerful adjuvant for monocyte-derived dendritic cell development and activity in vitro and in Hu-PBL-SCID mice. J Exp Med. 2000;191:1777–88. doi: 10.1084/jem.191.10.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heery CR, Singh BH, Rauckhorst M, et al. Phase I Trial of a Yeast-Based Therapeutic Cancer Vaccine (GI-6301) Targeting the Transcription Factor Brachyury. Cancer Immunol Res. 2015;3:1248–56. doi: 10.1158/2326-6066.CIR-15-0119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsang KY, Zaremba S, Nieroda CA, et al. Generation of human cytotoxic T cells specific for human carcinoembryonic antigen epitopes from patients immunized with recombinant vaccinia-CEA vaccine. J Natl Cancer Inst. 1995;87:982–90. doi: 10.1093/jnci/87.13.982. [DOI] [PubMed] [Google Scholar]
- 25.Mundy-Bosse BL, Young GS, Bauer T, et al. Distinct myeloid suppressor cell subsets correlate with plasma IL-6 and IL-10 and reduced interferon-alpha signaling in CD4+ T cells from patients with GI malignancy. Cancer Immunol Immunother. 2011;60:1269–79. doi: 10.1007/s00262-011-1029-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Therasse P, Arbuck SG, Eisenhauer EA, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92:205–16. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- 27.Huang J, Jochems C, Anderson AM, et al. Soluble CD27-pool in humans may contribute to T cell activation and tumor immunity. J Immunol. 2013;190:6250–8. doi: 10.4049/jimmunol.1300022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang J, Jochems C, Talaie T, et al. Elevated serum soluble CD40 ligand in cancer patients may play an immunosuppressive role. Blood. 2012;120:3030–8. doi: 10.1182/blood-2012-05-427799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–74. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Di Pucchio T, Pilla L, Capone I, et al. Immunization of stage IV melanoma patients with Melan-A/MART-1 and gp100 peptides plus IFN-alpha results in the activation of specific CD8(+) T cells and monocyte/dendritic cell precursors. Cancer Res. 2006;66:4943–51. doi: 10.1158/0008-5472.CAN-05-3396. [DOI] [PubMed] [Google Scholar]
- 31.Zeestraten ECM, Speetjens FM, Welters MJP, et al. Addition of interferon-α to the p53-SLP® vaccine results in increased production of interferon-γ in vaccinated colorectal cancer patients: a phase I/II clinical trial. Int J cancer. 2013;132:1581–91. doi: 10.1002/ijc.27819. [DOI] [PubMed] [Google Scholar]
- 32.Singh PP, Sharma PK, Krishnan G, Lockhart AC. Immune checkpoints and immunotherapy for colorectal cancer. Gastroenterol Rep (Oxf) 2015;3:289–97. doi: 10.1093/gastro/gov053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Madan RA, Bilusic M, Heery C, et al. Clinical evaluation of TRICOM vector therapeutic cancer vaccines. Semin Oncol. 2012;39:296–304. doi: 10.1053/j.seminoncol.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mohebtash M, Tsang K-Y, Madan RA, et al. A pilot study of MUC-1/CEA/TRICOM poxviral-based vaccine in patients with metastatic breast and ovarian cancer. Clin Cancer Res. 2011;17:7164–73. doi: 10.1158/1078-0432.CCR-11-0649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morse MA, Niedzwiecki D, Marshall JL, et al. A randomized phase II study of immunization with dendritic cells modified with poxvectors encoding CEA and MUC1 compared with the same poxvectors plus GM-CSF for resected metastatic colorectal cancer. Ann Surg. 2013;258:879–86. doi: 10.1097/SLA.0b013e318292919e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mahvi DM, Madsen JA, Witt PL, Sondel PM. Interferon alpha enhances expression of TAG-72 and carcinoembryonic antigen in patients with primary colorectal cancer. Cancer Immunol Immunother. 1995;40:311–4. doi: 10.1007/BF01519631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guadagni F, Schlom J, Johnston WW, et al. Selective interferon-induced enhancement of tumor-associated antigens on a spectrum of freshly isolated human adenocarcinoma cells. J Natl Cancer Inst. 1989;81:502–12. doi: 10.1093/jnci/81.7.502. [DOI] [PubMed] [Google Scholar]
- 38.Rosenblum MG, Lamki LM, Murray JL, et al. Interferon-induced changes in pharmacokinetics and tumor uptake of 111In-labeled antimelanoma antibody 96.5 in melanoma patients. J Natl Cancer Inst. 1988;80:160–5. doi: 10.1093/jnci/80.3.160. [DOI] [PubMed] [Google Scholar]
- 39.Murray JL, Macey DJ, Grant EJ, et al. Enhanced TAG-72 expression and tumor uptake of radiolabeled monoclonal antibody CC49 in metastatic breast cancer patients following alpha-interferon treatment. Cancer Res. 1995;55:5925s–5928s. [PubMed] [Google Scholar]
- 40.Marincola FM, Jaffee EM, Hicklin DJ, Ferrone S. Escape of human solid tumors from T-cell recognition: molecular mechanisms and functional significance. Adv Immunol. 2000;74:181–273. doi: 10.1016/s0065-2776(08)60911-6. [DOI] [PubMed] [Google Scholar]
- 41.Koopman LA, Corver WE, van der Slik AR, et al. Multiple genetic alterations cause frequent and heterogeneous human histocompatibility leukocyte antigen class I loss in cervical cancer. J Exp Med. 2000;191:961–76. doi: 10.1084/jem.191.6.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van de Stolpe A, van der Saag PT. Intercellular adhesion molecule-1. J Mol Med (Berl) 1996;74:13–33. doi: 10.1007/BF00202069. [DOI] [PubMed] [Google Scholar]
- 43.Imboden M, Murphy KR, Rakhmilevich AL, et al. The level of MHC class I expression on murine adenocarcinoma can change the antitumor effector mechanism of immunocytokine therapy. Cancer Res. 2001;61:1500–7. [PubMed] [Google Scholar]
- 44.Bander NH, Yao D, Liu H, et al. MHC class I and II expression in prostate carcinoma and modulation by interferon-alpha and -gamma. Prostate. 1997;33:233–9. doi: 10.1002/(sici)1097-0045(19971201)33:4<233::aid-pros2>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 45.Makridis C, Juhlin C, Akerström G, et al. MHC class I and II antigen expression and interferon alpha treatment of human midgut carcinoid tumors. World J Surg. 18:481–6. doi: 10.1007/BF00353741. discussion 486–7. [DOI] [PubMed] [Google Scholar]
- 46.Markowitz J, Brooks TR, Duggan MC, et al. Patients with pancreatic adenocarcinoma exhibit elevated levels of myeloid-derived suppressor cells upon progression of disease. Cancer Immunol Immunother. 2015;64:149–59. doi: 10.1007/s00262-014-1618-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Diaz-Montero CM, Salem ML, Nishimura MI, et al. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol Immunother. 2009;58:49–59. doi: 10.1007/s00262-008-0523-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mundy-Bosse BL, Lesinski GB, Jaime-Ramirez AC, et al. Myeloid-derived suppressor cell inhibition of the IFN response in tumor-bearing mice. Cancer Res. 2011;71:5101–10. doi: 10.1158/0008-5472.CAN-10-2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary table S1. CEA-specific T-cell responses post-vs. pre-vaccination