

Abstract
It has been shown that 2,4-diamino-6-arylmethylpteridines and 2,4-diamino-5-arylmethylpyrimidines containing a O-carboxylalkyloxy group in the aryl moiety are potent and selective inhibitors of the dihydrofolate reductase (DHFR) from such opportunistic pathogens as Pneumocystis carinii, the causative agent of Pneumocystis pneumonia in HIV AIDS patients. In order to understand the structure-activity profile observed for a series of substituted dibenz[b,f]azepine antifolates, the crystal structures of mouse (m) DHFR, a mammalian homologue, holo and ternary complexes with NADPH and the inhibitor 2,4-diamino-6-(2′-hydroxydibenz[b,f]azepin-5-yl)methylpteridine were determined to 1.9Ǻ and 1.4Ǻ resolution, respectively. Structural data for the ternary complex with the potent O-(3-carboxypropyl) inhibitor PT684 revealed no electron density for the O-carboxylalkyloxy side chain. The side chain either was cleaved or was completely disordered. The electron density fit the less potent hydroxyl compound, PT684a. Additionally, co-crystallization of mDHFR with NADPH and the less potent 2′-(4-carboxybenzyl) inhibitor PT682 showed no electron density for the inhibitor and resulted in the first report of a holo enzyme complex despite several attempts at crystallization of a ternary complex. Modeling data of PT682 in the active site of mDHFR and pcDHFR indicate binding would require ligand-induced conformational changes to the enzyme for the inhibitor to fit in the active site or that the inhibitor side chain would have to adopt an alternative binding mode than that observed for other carboxyalkyloxy inhibitors. These data also show that the mDHFR complexes have a decreased active site volume as reflected in the relative shift of helix C (residues 59-64) by 0.6Ǻ, compared to Pneumocystis carinii DHFR ternary complexes. These data are consistent with the greater inhibitory potency against pcDHFR.
Introduction
Antifolates have been shown effective against dihydrofolate reductase (DHFR) from such opportunistic pathogens as Toxoplasma gondii (tg) and Pneumocystis carinii (pc), the causative agent in Pneumocystis pneumonia (PcP) that is still a cause of mortality among immuno compromised patients such as those with HIV AIDS (Kovacs et al, 2002; Thomas & Limper, 2004; Wakefield, 2002). The antifolate trimethoprim (TMP, Fig. 1), when used in combination with sulfamethoxazole, synergistically targets the folate synthesis of these pathogens and is currently the preferred treatment against PcP (Stringer et al., 2002; Cushion, Keely & Stringer, 2004). However, TMP has limited efficacy and drug resistance to TMP treatment is becoming more prevalent (Medrano et al, 2005). These data illustrate the need for continued efforts to design more selective and potent inhibitors against these opportunistic infectious pathogens.
Fig. 1.

Schematic representation of antifolates under study.
Recent structure activity data have shown that dibenz[b,f]azepines such as PT653 (Fig. 1) is moderately selective against pcDHFR and has a 100-fold increased binding preference for tgDHFR (Rosowsky et al, 1999). These studies led to the structure-based design of a series of 2,4-diamino-6-[(ω-carboxyalkyl)oxy]dibenz[b,f]azepin-5-yl]pteridines that were more potent and selective than trimethoprim as inhibitors of DHFR from such opportunistic pathogens as Pneumocystis carinii, Toxoplasma gondii or Mycobacterium avium (Rosowsky et al, 2004; Chan et al., 2005). As illustrated in Table 1, these data revealed that the 2′-O-(3-carboxypropyl) analogue (PT684, Fig. 1) had the greatest inhibitory potency against pcDHFR with an IC50 of 1.1 nM and a selectivity ratio of 1363 when compared with rat DHFR. Similarly, the 2′-O-(4-carboxybenzyl) analogue (PT682, Fig. 1) showed significant potency with an IC50 of 1.0 nM against pcDHFR, but had lower selectivity against the pathogenic DHFR and showed a selectivity ratio of 580 when compared to rat DHFR (Rosowsky et al., 2004). These data suggest that modification of the 2′-position of the dibenz[b,f]azepine ring contributes significantly to defining selectivity for the pathogenic DHFR enzymes. Computational models of the binding of the potent dibenz[b,f]azepine PT684 in human and pcDHFR has been carried out to better understand the selectivity profiles observed for this class of inhibitors (Rosowsky et al., 2004). The crystal structures of the parent antifolate, PT653 (Fig. 1), bound to pcDHFR (Cody et al, 2002), and the tight binding inhibitor PT523 (Fig. 1) (Cody, et al., 1997) were used as starting models for these calculations. The results revealed a model in which the inhibitor carboxylate interacted with the conserved Arg in the active site, similar to that observed for other O-carboxyalkyloxy inhibitors (Cody et al, 2006). These data suggested that differences in the interactions between the carboxylate of the inhibitor and Lys37 in pcDHFR and Gln35 in hDHFR could contribute to their enhanced selectivity for pcDHFR.
Table 1.
Enzyme inhibition (IC50 nM) against DHFR for selected inhibitors (Fig. 1) (Chan et al, 2005; Rosowsky et al, 2004)
| Inhibitor | pcDHFR | rat DHFR | rat/pcDHFR Selectivity Ratio |
|---|---|---|---|
| TMP | 13000 | 180000 | 13.8 |
| PTX | 13 | 3.3 | 0.26 |
| PT684 | 1.1 | 1500 | 1363 |
| PT682 | 1.0 | 580 | 580 |
| PT684a | 31 | 14 | 1.3 |
| PT653 | 79 | 3000 | 37.9 |
Based on the observation that the active site residues of the mammalian DHFRs are highly conserved and that mouse DHFR was more facile to crystallize than human DHFR, we carried out structural studies using mouse DHFR. To better understand the mechanism of selectivity in binding to pcDHFR, the crystal structures of PT684 and PT682 were carried out in complex with mouse DHFR and are compared with the parent benz[b,f]azepine, PT653 (Fig. 1) (Cody, et al, 2002). Efforts are also underway to crystallize these inhibitors with pcDHFR.
Methods
Expression of Wild-type mDHFR
Recombinant mouse DHFR was expressed and purified as described previously (Pineda et al., 2003). A single colony of JM105 E. coli cells containing the pPH70D plasmid that harbors the fusion product of E. coli L54F DHFR and mouse DHFR linked with thrombin, was used to inoculate a 10 mL culture of Luria-Bertani (LB) broth (25g per 1L) containing 50μg/mL ampicillin. After incubation at 37°C overnight with shaking, one 10 mL culture was used to inoculate 1 L culture of LB broth containing 50μg/mL ampicillin. Bacteria were grown to an OD600 of 0.4-0.6, after which expression of mDHFR was induced by the addition of isopropyl-β-D-thiogalactoside (IPTG) at a final concentration of 1mM. After an induction time of 3 hours, the cells were harvested by centrifugation at 4°C, 7000 × g for 30 minutes. The cell pellets were resuspended in 12.5 mL lysis buffer A (50mM Tris, 5mM EDTA, 50μg NaN3/mL, 10mL protease inhibitor cocktail, pH 8.0) per gram wet cells. The solution was left to incubate for 10 minutes at room temperature on a stir plate. Then 1.5mL lysis buffer B (1.5M sodium chloride, 0.1M calcium chloride, 20 μg DNase/mL, 1mM PMSF) per gram of wet cells was added. The solution was incubated for 10 minutes at room temperature on a stir plate. Dithiothreitol (DTT) was added to a final concentration of 5mM. The sample was then subjected to 6 cycles of ultra-sonic cell disruption for 15 seconds with intermittent cooling periods of 30 seconds in ice. The clarified supernatant was obtained by centrifugation at 4° C, 12,000 rpm for 30 minutes.
Purification of mDHFR
The supernatant was then dialyzed into PE buffer (20mM potassium phosphate, 1mM EDTA, 1mM DTT, pH 7.4). The protein was loaded at 0.5mL/min onto a DEAE column equilibrated with PE buffer. The column was washed with 50mL PE buffer with 1mM DTT. The fusion protein, containing both E. coli DHFR L54F and mDHFR joined by a thrombin linker, was eluted with an 800mL gradient of 0-0.5M KCl. Fractions were monitored (Blakley, 1960) and those containing DHFR activity were pooled, concentrated using a YM-30 membrane, and dialyzed into thrombin cleavage buffer (50mM Tris, 0.1M NaCl, 2.5mM CaCl2, pH 8.0). Cleavage of the fusion protein was initiated by the addition of 5 units of thrombin per mg of fusion protein. The sample was left to incubate overnight (15-18 hours) at 4°C. The next day, the protein was dialyzed for >3hours back into PE buffer containing freshly added DTT at 1mM final concentration. The protein was then loaded onto a DEAE (GE Bioscience) column pre-equilibrated with PE buffer containing 1mM DTT at 0.5mL/minute. After washing the column with 50mL of PE buffer containing 1mM DTT, the cleaved mDHFR was eluted from the column using a 600mL gradient of 0-0.1M KCl. Fractions containing DHFR activity were pooled and concentrated using a YM10 membrane.
Crystallization
The protein was washed in a centricon-10 with 10 mM HEPES buffer, pH 7.4 and concentrated to 27 mg/mL and incubated with NADPH and a 10:1 molar excess of the inhibitors, 2,4-diamino-6-(2′-O-(3-carboxylpropyl)oxydibenz[b,f]azepin-5-yl)methylpteridine (PT684) or 2,4-diamino-6-[2′O-(4-carboxybenzyl)oxydibenz[b,f] azepin-5-yl]methylpteridine (PT682), for one hour over ice prior to crystallization using the hanging drop vapor diffusion method. Protein droplets contained 0.15M Tris, pH 8.3, 75 mM Na cacodylate, 21% PEG 4K for the PT684 complex and, 10 mM HEPES, pH 7.4, 17 mM Na acetate, pH 6.5, 85 mM Tris-HCl and 25% PEG 4K for the PT682 complex. Crystals grew over several weeks time and were treated in 15% glycerol as a cryo protectant prior to mounting in the cold stream. The crystals of both complexes are monoclinic, space group P21, and diffracted to 1.4Ǻ and 1.9Ǻ resolution for PT684 and PT682, respectively. Data for the complex with PT684 was collected on beamline 9-1 at the Stanford Synchrotron Resource Laboratory (SSRL) using the remote access protocol (McPhillips et al., 2002; Cohen et al, 2002; Gonzalez et al., 2008) and the PT682 complex was collected on a Rigaku RaxisIV imaging plate system with MaxFlux optics. Data were processed with using both Denzo (Otwinowki & Minor, 1997) and Mosflm (CCP4, 1994). Diffraction statistics are shown in Table 2 for both complexes.
Table 2.
Data collection and refinement statistics
| Data collection | PT 684a-NADPH | NADPH |
| PDB number | 3d80 | 3d84 |
| Space group | P21 | P21 |
| Cell dimensions (Ǻ) | 41.48 61.30 43.59 | 41.23 61.17 43.15 |
| β = 117.22 | β = 118.26 | |
| Beamline | SSRL 9-1 | RaxisIV |
| Resolution (Ǻ) | 1.00 | 1.90 |
| Wavelength (Ǻ) | 1.00 | 1.5418 |
| Rmerge | 0.083 (1.08) | 0.034 (0.076) |
| Rsym (%) a,b | 0.100 (1.49) | 0.051 (0.48) |
| Completeness (%) a | 98.3 (97.4) | 90.7(50.5) |
| Observed reflections | 38237 | 14951 |
| Unique reflections | 35455 | 13551 |
| I/σ(I) | 7.3 (0.7) | 13.3 (2.5) |
| Multiplicity a | 3.4 (2.0) | 15.1 (3.4) |
| Refinement and model quality | ||
| Resolution range (Ǻ) | 31.61 - 1.40 | 23.83 - 1.90 |
| No. of reflections | 35455 | 12865 |
| R-factor c | 19.8 | 18.6 |
| Rfree-factor d | 21.3 | 21.0 |
| Total protein atoms | 1665 | 1665 |
| Total ligand atoms | 76 | 60 |
| Total water atoms | 319 | 82 |
| Average B-factor (Ǻ2) | 19.7 | 19.0 |
| Rms deviation from ideal | ||
| Bond lengths (Ǻ) | 0.011 | 0.018 |
| Bond angles (°) | 1.682 | 2.060 |
| Ramachandran plot | ||
| Residues in most favored regions (%) | 91.8 | 93.7 |
| Residues in additional allowed regions (%) | 8.2 | 6.3 |
| Residues in generously allowed regions (%) | 0.6 | 1.1 |
| Residues in disallowed regions (%) | 0.0 | 0.0 |
The values in parentheses refer to data in the highest resolution shell.
Rsym = ∑h∑i|Ih,i - <Ih>| / ∑h∑i|Ih,i|, where <Ih> is the mean intensity of a set of equivalent reflections.
R-factor = ∑|Fobs - Fcalc|/∑Fobs, where Fobs and Fcalc are observed and calculated structure factor amplitudes.
Rfree-factor was calculated for R-factor for a random 5% subset of all reflections.
Structure Determination
The structures were solved by molecular replacement methods using the coordinates for mouse DHFR (2fzj) (Cody et al, 2006) in the program Molrep (CCP4, 1994). To monitor the refinement, a random subset of all reflections was set aside for the calculation of Rfree (5%). Inspection of the resulting difference electron density maps were made using the program COOT (Emsley & Cowtan, 2004) running on a Mac G5 workstation. The structures of the inhibitors were modeled based on those of PT653 (Fig. 1) reported in the structure of pcDHFR (Cody, et al. 2002) using the builder function in Sybyl (Tripos, St. Louis, MO) and the parameter file for the inhibitors were prepared using the Dundee PRODGR2 Server website (http://davapc1.bioch.dundee.ac.uk/programs/prodrg) (Schuettelkopf & van Aalten, 2004). The final cycles of refinement were carried out using the program Refmac5 in the CCP4 suite of programs (CCP4, 1994). The Ramachandran conformational parameters from the last cycle of refinement generated by PROCHECK (Laskowski et al, 1993) showed that more than 90% of the residues in both mDHFR complexes have the most favored conformation and none are in the disallowed regions. Coordinates for this structure have been deposited with the Protein Data Bank (PDB code 3d80 and 3d84). Figures were prepared using the modeling program PyMol (DeLano, 2006).
Results
Mouse DHFR Ternary Complex
Inspection of the difference electron density map for the ternary complex of mDHFR with NADPH and what was initially assumed to be the carboxylic acid PT684 revealed no electron density for the 4-carboxypropyl side chain (Fig. 2). It is unclear whether the side chain is completely disordered in this region, or whether the sample had decomposed on storage or on crystallization. One other possibility is that intramolecular catalysis had resulted in spontaneous cleavage of the carboxypropyl side chain during storage or under the crystallization conditions used, so that the molecule actually in the active site was the 2-hydroxydibenzazepine PT684a (Fig. 1). The side chains of Phe31 and Gln35 were also observed in two alternate conformations in this complex. There is a partial water molecule present in one of the Gln35 conformers that also contacts Arg 70 when the partially occupied Gln35 is not present.
Fig. 2.

(right) Difference electron density (2Fo-Fc, 1.0 σ) showing the inhibitor PT684a as a ternary complex with NADPH in mouse DHFR. Also shown are two alternate conformations for Phe31. (left) Difference electron density (Fo-Fc, 3σ) from omit map calculated from the final refinement without inhibitor present. There is no indication of a partially occupied inhibitor side chain.
The more surprising observation for these data is the lack of involvement of the putative hydroxyl group of PT684a in any hydrophilic interactions. The closest contact of the hydroxyl group of PT684a is with a partially occupied conformer of Phe31 (2.9Ǻ), while the other contact distances range from 3.7-4.7Ǻ to the partially occupied water/Gln35 and the side chains of Asn64 and Leu60 (Fig. 4).
Fig. 4.
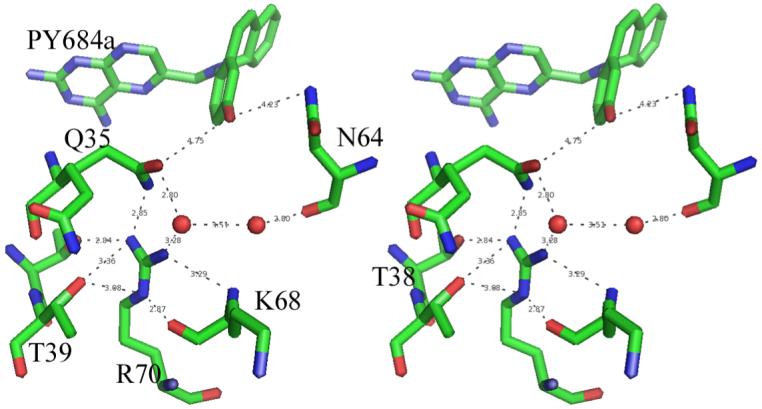
Stereo view of the conserved Arg70 interactions with their contact distances shown for mDHFR PT684a NADPH ternary complex. Figure drawn with PyMol.
As previously described in the structures of mDHFR inhibitor complexes (Cody et. al., 2006; Cody, Luft & Pangborn, 2005; Cody & Schwalbe, 2006), the orientation of the conserved Arg70 is held in place by a network of hydrogen bonds with the conserved Thr38 and Thr39 and the backbone functional groups of Lys68, and through water, to the backbone carbonyl of Asn64. There are also contacts to other structural water molecules in the active pocket (Fig. 4).
Mouse DHFR Holo Enzyme Complex
When the difference density maps were analyzed for the complex with PT682, it was shown that the final structure is a holo enzyme complex containing only the cofactor NADPH (Fig. 5). Although diffraction data from three separate crystallization trials were collected, all crystals were found to be holo enzyme complexes. This is the first report of a mammalian DHFR holo complex structure. The active site is occupied by a molecule of glycerol used as a cryo protection agent during diffraction data collection (Fig. 6).
Fig. 5.

Difference electron density (2Fo-Fc, 1σ) for the holo complex with cofactor, NADPH in mouse DHFR. The density in the active site was fit to glycerol from the cryo protection in the buffer.
Fig. 6.

Stereo view of mouse DHFR with NADPH and a glycerol molecule in the active site of the holo structure. Residues Glu30, Gln35, Asn64 and Arg70 are shown. Figure drawn with PyMol.
Overall Structure
There are no significant changes in the overall structure of the holo and ternary mDHFR complexes (RMSD = 0.38Ǻ between all residues in these two structures), which also are similar to previously reported mDHFR structures (Cody et al., 2006; Cody, Luft & Pangborn, 2005). The interactions of the 2,4-diaminopteridine ring of PT684a preserves the overall pattern of contacts with invariant residues in the active site as observed in other DHFR-inhibitor complexes (Cody & Schwalbe, 2006). The intermolecular interactions of PT684a are also similar to those observed for the parent compound PT653 reported in the crystal structure with pcDHFR (Cody, et al, 2002) (Fig. 7), and shows the 4-amino group makes a number of hydrogen bond contacts with the backbone functional groups of Ile7 (2.9Ǻ) and Val115 (3.1Ǻ) and the side chains of Tyr121 (3.4Ǻ) and the nicotinamide of NADPH (3.5Ǻ) that contribute to the tight binding of antifolates. There is an invariant hydrogen bond network involving structural water, the conserved residues Thr136, Glu30 and Trp24 and the N1 nitrogen and 2-amino group of PT684a. The pteridine ring N8 makes contact to Glu30 and Trp24 through a structural water molecule that is observed in most DHFR structures.
Fig. 7.
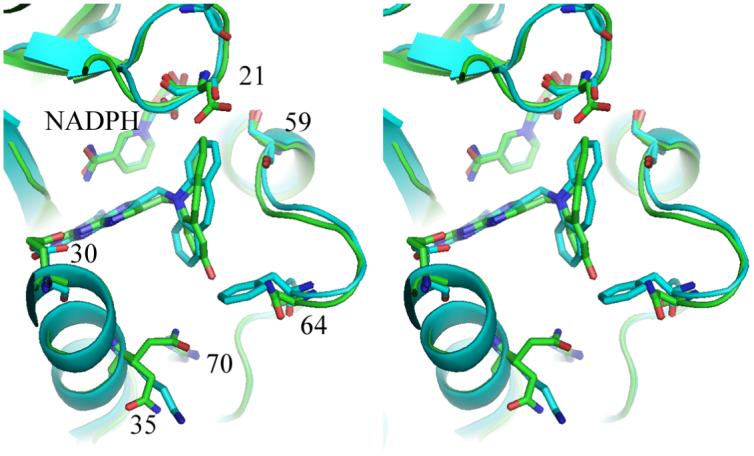
Stereo superposition of mouse DHFR ternary complex with PT684a (green) and pcDHFR (cyan) ternary complex with NADPH and inhibitor PT653 (Cody, et al, 2002). The mouse DHFR sequence numbers are shown for the active site residues. Figure is drawn with PyMol.
Comparison of these mDHFR complexes with those of the parent PT653 bound to pcDHFR (Cody, et al., 2002) and with methotrexate (MTX) bound to mouse and human DHFR (Cody et al., 2006) reveals that the backbone of helix C (encompassing residues 59-64, human numbering) is displaced by 0.2-1.3Ǻ depending on the ligand bound and reflects ligand-induced conformational changes (Cody et al, 1999). Measurements of the distances between the alpha carbon of residue Glu30 to the alpha carbon of residues Asp21, Leu22, Ser59 and Leu60 (Table 3) describe the relative size of the active site among these structures. As illustrated, the largest differences measured at positions 59 and 60 are between the human DHFR MTX complex (Cody et al., 2005) and that of pcDHFR PT653 (Cody et al., 2002) which reflects a shift of 1.3Ǻ in the relative position of helix C making the size of the pcDHFR active site larger as a result of ligand-induced conformational changes (Cody et al., 1999) (Fig. 8). The difference between the helix C positions of mDHFR PT684a and hDHFR MTX is 0.7-1.3Ǻ. It is somewhat surprising to note that there is little change in these contact distances between the holo and PT684a ternary complex with mDHFR (Table 3). These data suggest that the shifts in helix C of the MTX ternary complexes are more tightly bound and that the human enzyme has the smallest active site volume.
Table 3.
Contact distances between active site residues for DHFR complexes
| DHFR Species Ligand | Residue Cα 30-22 | Residue Cα 30...21 | Residue Cα 30...59 | Residue Cα 30...60 |
|---|---|---|---|---|
| m-PT684a | 12.2Ǻ | 15.1Ǻ | 17.5Ǻ | 15.3Ǻ |
| m-holo | 12.3 | 15.3 | 17.2 | 15.3 |
| mL22R-MTX Cody et al, 2005 | 12.1 | 14.5 | 16.5 | 14.8 |
| h-MTX Cody et al, 2005 | 12.2 | 14.7 | 16.2 | 14.6 |
| pc-PT653 Cody et al, 2002 | 12.3 | 15.1 | 17.9 | 15.9 |
| pc-MTX Cody et al, 1999 | 11.7 | 15.0 | 16.5 | 14.7 |
Fig. 8.
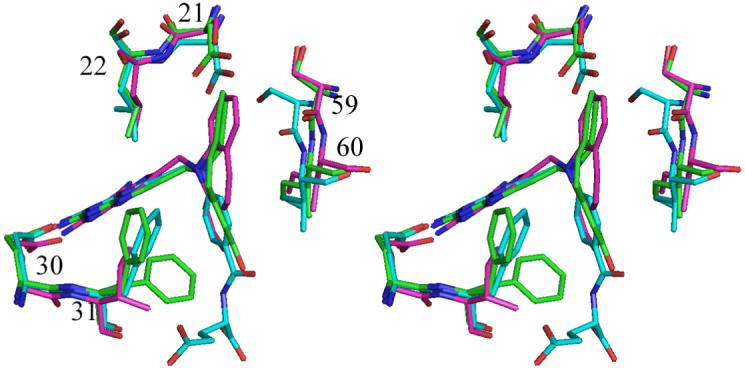
Stereo comparison of mDHFR-NADPH-PY684a (green), hDHFR-MTX-NADPH (Cody et al, 2005) (cyan) and pcDHFR-NADPH-PT653 (Cody, et al, 2002) (violet), highlighting the conformation of loop 21 and in helix C with residues Ser59 and Leu60 in all structures. Residues are numbered for the mammalian enzyme. Position 21 is Asp in the mammalian DHFR and Ser in pcDHFR, 22 is Leu in all structures, 30 is Glu in all three structures, 31 is Phe in mammalian and Ile in pcDHFR. Figure drawn with PyMol.
Discussion
These structural studies describe the binding of a hydrolyzed product PT684a (Fig. 1) as a ternary complex with NADPH and mouse DHFR as no electron density was evident for the carboxyalkyloxy side chain. There was also no evidence of a partially occupied position for the side chain. These observations suggest that the side chain is completely disordered or has been cleaved under the storage or crystallization conditions used, resulting in density that is consistent with the hydroxyl compound PT864a. This product was shown to be less potent (Table 1) than those that contain a carboxylate side chain that can interact with the conserved Arg70 present in all DHFR. Comparison of this complex with the parent dibenz[b,f]azepine, PT653 (Cody et al., 2002) (Fig. 7) reveals only small variations in the buckling of the dibenz[b,f]azepine ring compared to the mDHFR complex.
One of the strategies developed for the design of these dibenz[b,f]azepine antifolates was to probe the binding interactions near the flexible loop near residues 20-24 of the DHFR active site. Thus, by making a rigid group that could occupy this region while still occupying the p-aminobenzoyl glutamate portion of the substrate active site pocket, it would be possible for this rigid group to exploit differences in the active site volume that result from movement of the flexible loop 20-24 between the mammalian and fungal DHFR enzymes (Rosowsky et al, 1999). Comparison of the loop 20-24 positions among the structures of the human, mouse and Pneumocystis carinii DHFR complexes with MTX and the dibenz[b,f]azepine antifolates reveals that the greatest difference is between the hDHFR MTX complex and the mDHFR complex with PT684a, as measured by the differences between this distance from the Cα of Glu30 to that of Asp21 and Leu22 (Table 3) (Fig. 8). These data reveal a progressive increase in the distance for pcDHFR MTX, pcDHFR PT653 and mDHFR PT684a ternary complexes, respectively. It is somewhat surprising that the mDHFR holo enzyme complex has the same contact distances as the PT684a ternary complex.
Similarly, the effects of ligand-induced conformational changes are reflected in the relative movement of helix C (residues 59-64) that indicates an increase in the active site size among these species. However, in this case, the largest shift is observed for the PT653 ternary complex with pcDHFR that has a 1.3Ǻ displacement at residue Leu60 of helix C relative to the human DHFR MTX ternary complex (Table 3). The change for the mDHFR ternary complex with PT684a is 0.7Ǻ relative to the 0.4Ǻ difference for the pcDHFR MTX complex.
These data also show that the complex with the highly selective inhibitor PT682 resulted in the first report of a mammalian holo mDHFR enzyme complex with cofactor, NADPH. Despite efforts to co-crystallize PT682 as a ternary complex with mDHFR, only the holo enzyme complex was observed. Modeling studies of the binding of PT682 in mouse and pcDHFR indicate that an alternate binding mode is needed for this inhibitor to fit into the active site. If, in both mDHFR and pcDHF, the binding of PT682 is similar to that observed for PT653 in pcDHFR (Cody et al, 2002), then the carboxylate side chain has steric clashes with the conserved Arg in the active site (Fig. 9). The fact that PT684 has significant potency and selectivity in pcDHFR, would suggest that ligand induced conformational changes need to occur for inhibitor binding or that the carboxyalkyloxy side chain can adopt an alterative conformation and forgo interaction with Arg, as observed in other carboxyalkyloxy inhibitors (Cody et al, 2006). These data may indicate that the steric bulk of this antifolate is such that it is prevented from binding to any significant degree in mouse DHFR. These models for the binding of PT682 are in contrast to those derived for the binding of PT684 in which the carboxylate was shown to interact with the conserved Arg in both pc-and hDHFR (Rosowsky et al, 2004). These data are the first to illustrate the lack of binding to mammalian DHFR to explain its loss of potency compared to pcDHFR.
Fig. 9.
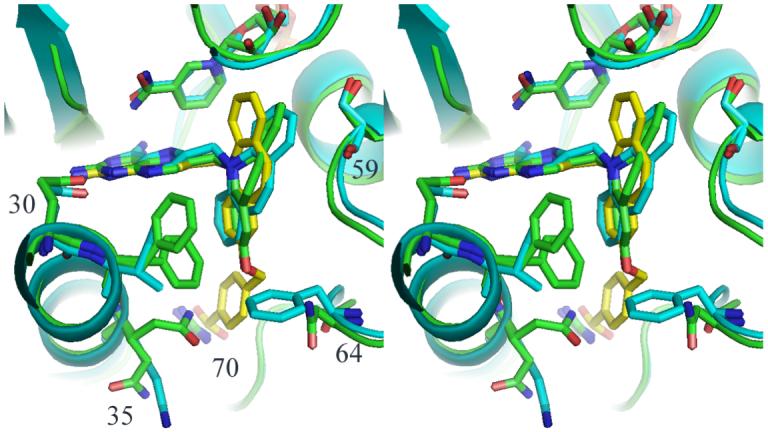
Stereo comparison of the ternary complex of mDHFR with NADPH and PT684a (green), pcDHFR with PT653 (cyan) (Cody et al, 2002) and a model of PT682 (yellow). Note that the carboxylate group clashes with Arg70.
Crystallization screens are underway to obtain complexes with pcDHFR with the potent dibenz[b,f]azepine antifolates PT684 and PT682 to validate the computational models that suggested that interactions of the 2′-(ω-carboxyalkyloxy) or 2′-(4-carboxybenzyloxy) substitutent with the conserved active site Arg70 and the differential interactions with Gln35 in mammalian versus Lys37 in pcDHFR contribute to their high potency and selectivity against the pathogenic DHFR enzymes.
Fig. 3.
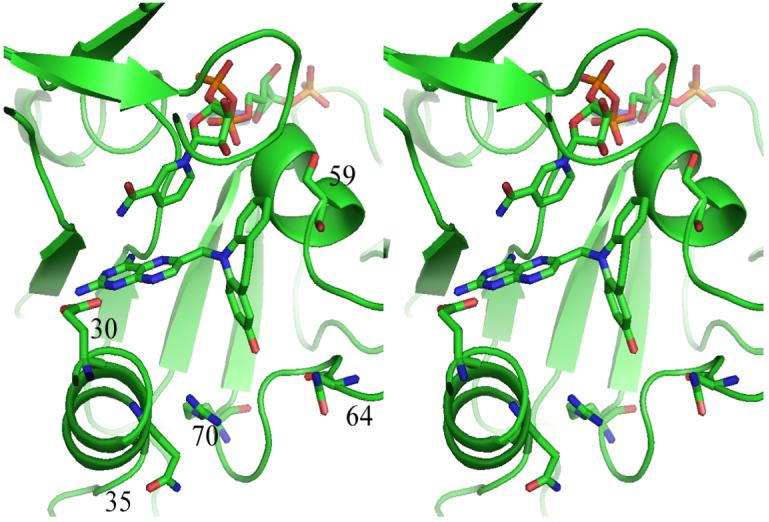
Stereo view of the ternary complex of NADPH and PT684a in mouse DHFR. The active site residues Glu30, Gln35, Ser59, Asn64 and Arg70 are shown. Figure drawn with PyMol.
Acknowledgement
This work was supported in part by grants from the National Institutes of Health GM51670 (VC). The authors thank the beam line 9-1 staff at SSRL for their support. Portions of this research were carried out at the Stanford Synchrotron Radiation Laboratory, a national user facility operated by Stanford University on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program, and the National Institute of General Medical Sciences.
References
- Blakley RL. Nature (London) 1960;188:231–232. [Google Scholar]
- Collaborative Computational Project, Number 4 Acta Cryst. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Chan DCM, Fu H, Forsch RA, Queener SF, Rosowsky A. J. Med. Chem. 2005;48:4420–4431. doi: 10.1021/jm0581718. [DOI] [PubMed] [Google Scholar]
- Cody V, Galitsky N, Luft JR, Pangborn W, Rosowsky A, Blakley RL. Biochemistry. 1997;36:13897–13903. doi: 10.1021/bi971711l. [DOI] [PubMed] [Google Scholar]
- Cody V, Galitsky N, Luft JR, Pangborn W, Rosowsky A, Queener SF. Acta Cryst. 2002;D58:946–954. doi: 10.1107/s090744490200505x. [DOI] [PubMed] [Google Scholar]
- Cody V, Galitsky N, Rak D, Luft JR, Pangborn W, Queener SF. Biochemistry. 1999;38:4303–4312. doi: 10.1021/bi982728m. [DOI] [PubMed] [Google Scholar]
- Cody V, Luft JR, Pangborn W. Acta Cryst. 2005;D61:147–155. doi: 10.1107/S0907444904030422. [DOI] [PubMed] [Google Scholar]
- Cody V, Pace J, Chisum K, Rosowsky A. Proteins, Structure, Function and Bioinformatics. 2006;65:959–969. doi: 10.1002/prot.21131. [DOI] [PubMed] [Google Scholar]
- Cody V, Schwalbe CH. Cryst. Reviews. 2006;12:301–333. [Google Scholar]
- Cohen AE, Ellis PJ, Miller MD, Deacon AM, Phizackerley RP. J. Appl. Cryst. 2002;35:720–726. doi: 10.1107/s0021889802016709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cushion MT, Keely SP, Stringer JR. Mycologia. 2004;96:429–438. [PubMed] [Google Scholar]
- DeLano Scientific LLC MacPyMOL. 2006 http://www.pymol.org.
- Emsley P, Cowtan K. Acta Cryst. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Gonzalez A, Moorhead P, McPhillips SE, Song J, Sharp K, Taylor JR, Adams PD, Sauter NK, Soltis SM. J. Appl. Cryst. 2008;41:176–184. [Google Scholar]
- Kovacs JA, Gill VJ, Meshnick S, Maur H. J. Am. Med. Assoc. 2002;286:2450–2460. doi: 10.1001/jama.286.19.2450. [DOI] [PubMed] [Google Scholar]
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM. J. Applied Cryst. 1993;26:283–291. [Google Scholar]
- McPhillips TM, McPhillips SE, Chiu HJ, Cohen AE, Deacon AM, Ellis PJ, Garman E, Gonzalez A, Sauter NK, Phizackerley RP, Soltis SM, Kuhn P. J. Synchrotron Rad. 2002;9:401–406. doi: 10.1107/s0909049502015170. [DOI] [PubMed] [Google Scholar]
- Medrano FJ, Montes-Cano M, Conde M, de la Horra C, Respaldiza N, Gasch A, Peres-Lozano M,J, Varela JM, Calderon EJ. Emerging Infect. Dis. 2005;11:245–250. doi: 10.3201/eid1102.040487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. In: Methods in Enzymology. Part A. Carter CW Jr., Sweet RM, editors. Vol. 276. Academic Press; New York: 1997. pp. 224–225. [Google Scholar]
- Pineda P, Kanter A, McIvor RS, Benkovic SJ, Rosowsky A, Wagner CR. J. Med. Chem. 2003;46:2816–2818. doi: 10.1021/jm034057i. [DOI] [PubMed] [Google Scholar]
- Rosowsky A, Cody V, Galitsky N, Fu H, Papoulis AT, Queener SF. J. Med. Chem. 1999;42:4853–4860. doi: 10.1021/jm990331q. [DOI] [PubMed] [Google Scholar]
- Rosowsky A, Fu H, Chan DCM, Queener SF. J. Med. Chem. 2004;47:2475–2485. doi: 10.1021/jm030599o. [DOI] [PubMed] [Google Scholar]
- Schuettelkopf AW, van Aalten DMF. Acta Cryst. 2004;D60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- Stringer JR, Beard CB, Miller RF, Wakefield AE. Emerging Infect. Dis. 2002;8:891–896. doi: 10.3201/eid0809.020096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas CF, Limper AH. N Engl. J. Med. 2004;350:2487–2498. doi: 10.1056/NEJMra032588. [DOI] [PubMed] [Google Scholar]
- Wakefield A. Brit. Med. Bull. 2002;61:175–188. doi: 10.1093/bmb/61.1.175. [DOI] [PubMed] [Google Scholar]