

Conspectus
The physical, biological, and materials properties of organic compounds are determined by their three-dimensional molecular shape. The development of methods to dictate the stereochemistry of organic reactions has consequently emerged as one of the central themes of contemporary synthetic chemistry. Over the past several decades, chiral catalysts have been developed to control the enantioselectivity of almost every class of synthetically useful transformation. Photochemical reactions, however, are a conspicuous exception. Relatively few examples of highly enantioselective catalytic photoreactions have been reported to date, despite almost a century of research in this field. The development of robust strategies for photochemical enantiocontrol has thus proven to be a long-standing and surprisingly difficult challenge.
For the past decade, our laboratory has been studying the application of transition metal photocatalysts to a variety of problems in synthetic organic chemistry. These efforts have recently culminated in the discovery of an effective system in which the activity of a visible light absorbing transition metal photoredox catalyst is combined with a second stereocontrolling chiral Lewis acid catalyst. This dual catalyst strategy has been applied to a diverse range of photochemical reactions; these have included highly enantioselective photocatalytic [2+2] cycloadditions, [3+2] cycloadditions, and radical conjugate addition reactions.
This Account describes the development of the tandem Lewis acid photoredox catalysis strategy utilized in our laboratory. It provides an analysis of the factors that we believe to be particularly important to the success of this seemingly robust approach to photocatalytic stereocontrol. (1) The photocatalysts utilized in our systems are activated by wavelengths of visible light where the organic substrates are transparent, which minimizes the possibility of competitive racemic background photoreactions. (2) The high degree of tolerance that Ru(bpy)3 2+ and similar octahedral metal polypyridine complexes exhibit towards Lewis acids affords great flexibility in tuning the structure of the stereocontrolling chiral catalyst without perturbing the photoredox properties of the photocatalyst. (3) Synthetic chemists have amassed a substantial understanding of the features that are common in highly successful chiral Lewis acid catalyzed reactions, and these deep, well-validated insights are readily applied to the reactions of a variety of photogenerated intermediates. We hope that the recent success of this and similar dual catalytic systems will provide a useful foundation for the further development of powerful, stereocontrolled photochemical reactions.
Graphical abstract
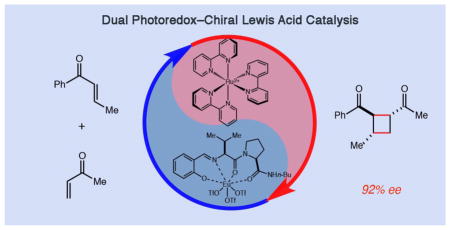
Introduction
The ability to construct organic compounds with control over their molecular shape has been critical to progress in multiple contemporary research areas. The importance of stereochemistry to such fields as drug discovery and materials science has motivated the development of strategies to control stereochemistry in almost every major class of chemical transformation. Photochemical reactions, however, represent a conspicuous exception to this general trend.
Early speculations concerning the possibility of enantioselective photoreactions can be found in publications by Le Bel and van’t Hoff in the late 19th century;1,2 the first experimental studies probing these theories appeared several decades later.3 By the beginning of the 21st century, efforts towards the development of stereoselective photochemical reactions4, 5 had culminated in several successful methods, including chiral auxiliary approaches, 6,7 the use of chiral supramolecular assemblies,8,9,10 and photoreactions in chiral crystalline matrices. 11,12 Although high levels of stereocontrol have been achieved using each of these tactics, the generality of the resulting reactions and their corresponding impact on the broader field of organic synthesis have unfortunately been relatively modest.
The use of substoichiometric chiral catalysts in enantioselective photochemistry has proven to be an even more elusive goal. The first highly enantioselective photoreactions were not developed until the early 2000s,13 and only a handful of successful of alternative strategies have emerged since then.14,15,16 The slow rate of development in this field is particularly surprising considering the rapid rate at which asymmetric catalytic methods were developed for so many other, nonphotochemical reaction types in the last decades of the 20th century.
Our laboratory became interested in this challenge for several reasons. First, photochemical reactions provide a distinctive synthetic capability that can produce regiochemical and stereochemical outcomes that differ from their non-photochemical, thermal counterparts. Structural motifs such as cyclobutanes and oxetanes that are especially well-suited to photochemical synthesis remain challenging to prepare in stereochemically pure form,17 and the biological and materials properties of compounds in this region of chemical diversity space remain relatively unexplored.
There is also an interesting fundamental question underlying the slow rate of progress in this field: Why have asymmetric catalytic photoreactions proven to be more challenging to develop than other classes of enantioselective catalytic reactions? Is there some intrinsic characteristic of photochemical activation that has made the development of highly enantioselective catalytic photoreactions particularly difficult?
This Account offers one analysis of this question that has informed much of my laboratory’s research in photocatalysis. We believe that our investigations have resulted in a strategy that addresses several central issues that have hindered progress in asymmetric photochemistry, and we hope it will provide a general conceptual framework to address the long-standing challenge of stereocontrolled photochemical synthesis.
Why Are Enantioselective Photoreactions Difficult?
The evolution of asymmetric photochemistry as a field has been the subject of several excellent reviews.4,5 Many researchers in this area have offered their own thoughts on why catalytic enantioselective photochemical reactions are relatively difficult to develop. Among the most common explanations one finds in this literature are various formulations of the “reactivity– selectivity principle”. These arguments begin with the observation that the absorption of a photon results in the formation of highly energetic reactive intermediates: for instance, the reactive triplet-state enones involved in classical [2+2] photocycloadditions are ~60–70 kcal/mol higher in energy than their ground-state configurations.18 One might reasonably imagine that such high-energy intermediates would be short-lived, difficult to intercept with chiral catalysts, and have a propensity to react indiscriminately to provide low selectivity among multiple possible stereo- and regioisomers.
As intuitively satisfying as the reactivity–selectivity principle is, however, there is little theoretical justification for it except in specific, specialized contexts.19 To the extent that the reactivity–selectivity principle has predictive value at all, it cannot be used to make any broad generalizations about a class of chemical processes as diverse as photochemical reactions.
Moreover, the effects at play in enantioselective reactions can be quite subtle in magnitude. A difference in activation energy of only 1.8 kcal/mol is sufficient to produce highly enantioenriched products (>90% ee) for an idealized enantioselective reaction at room temperature. This energetic difference is smaller even than the barrier for rotation about the carbon–carbon bond of ethane. Thus the high intrinsic reactivity of any particular organic intermediate is unlikely to be an insurmountable obstacle for effective asymmetric catalysis. For example, organoradical chemistry also involves highly energetic intermediates that can react at extremely fast rates and have short lifetimes, and yet there have been enough examples of highly enantioselective radical additions reported in the literature to date that these methods, while still not commonplace, are no longer as astonishing as they once seemed.20 Thus the high reactivity of photogenerated reactive intermediates seems insufficient to rationalize the slow pace of development in asymmetric photochemistry.
We posit that a more central issue has been the challenge of managing racemic background photoreactions. As a specific example to frame this discussion, consider the experiment shown in Figure 1A, which is the first, seminal example of a highly enantioselective stoichiometric [2+2] photocycloaddition in solution reported by Bach in 2000.8 This reaction exploits the propensity of quinolinone 1 to associate with chiral controller 2 into a hydrogen-bonded complex (4); irradiation of this well-organized assembly with UV light affords the [2+2] cycloadduct 3 in 93% ee. However, 2.6 equiv of 2 are required for optimal enantioselectivity; the reaction is markedly less selective at lower concentrations of the chiral controller. The requirement for high loadings of 2 results from the fact that binding of the controller does not significantly impact the photophysical properties of the quinolinone, and photoreaction of free and bound substrate occurs at similar rates (Figure 2B). At lower concentrations of 2, therefore, product formation via the uncatalyzed background reaction becomes competitive, resulting in dramatically lower ee’s of 3 despite the high intrinsic stereocontrol afforded by the controller. The only way to achieve high enantioselectivity at low catalyst loadings, therefore, would be to develop a strategy for selective activation of catalyst-bound substrate under conditions where the free substrate is unreactive.
Figure 1.

(A) First example of a highly enantioselective solution-phase photocycloaddition reaction using a non-covalent controller reported by Bach. (B) The enantioselectivity of a catalytic photocycloaddition is compromised by the participation of any uncatalyzed racemic background processes.
Figure 2.

(A) Initial report of a visible light induced [2+2] cycloaddition reaction using a Ru(bpy)3 2+ photocatalyst. (B) Examples of substrate scope in the [2+2] cycloaddition. (C) Proposed mechanism for the photocatalytic [2+2] cycloaddition of 5. (D) Gram-scale photocycloaddition conducted using ambient sunlight as the irradiation source.
Recognizing this challenge, Bach subsequently reported a strategy to limit the racemic background reaction by using a monochromatic UV LED source specifically tuned to excite a sensitizing moiety in a similar chiral controller without exciting the free substrate.21 This elegant solution has enabled enantioselectivities up to 92% using 10 mol% of a chiral xanthone-containing host. Nevertheless, the generality of this strategy is limited by partial overlap between the UV spectra of the sensitizer and substrate; in Bach’s report, there is an attenuated but still noticeable dependence of product ee on sensitizer loading.
My laboratory’s interest in visible light photochemistry has been heavily influenced by this analysis. Most simple organic substrates are colorless and transparent to visible light. On the other hand, there are many transition metal chromophores that absorb intensely in the visible region of the electromagnetic spectrum and have interesting photochemistry in this regime. Our goal has been to use these complexes to develop photochemical reactions in which no background photoreaction is possible because the photocatalyst is activated by wavelengths of visible light where the free substrate is photochemically inactive.
Lewis acid catalysis of photoredox reactions
Our initial investigations in visible light photochemistry focused on the [2+2] photocycloaddition reaction depicted in Figure 2.22 Based on our collected observations of this reaction over many years, we have proposed the mechanism depicted in Figure 2C. Photoexcitation of Ru(bpy)3 2+ with visible light affords a redox-active excited state (Ru*(bpy)3 2+) that is reduced by tertiary amines to produce a Ru(I) complex. The redox potential of this complex (−1.3 V vs SCE) is insufficient to reduce aryl enone 5 directly (−1.4 V). Successful electron transfer to 5 requires coordination of the enone to a Li+ additive that facilitates electron transfer and stabilizes the resulting radical anion (5•−). Subsequent stepwise [2+2] cycloaddition can occur in both intra-and intermolecular23,24 fashions to afford a wide range of structurally varied cyclobutanes (e.g., 6–9). Importantly, because the photophysical properties of the Ru(bpy)3 2+ chromophore are well-suited for solar energy applications, we also find that this reaction can be conducted using essentially any easily accessible source of visible light, including ambient sunlight (Figure 2D).
This reaction is an example of dual photoredox/Lewis acid catalysis.25 The role of the Ru photocatalyst is to absorb visible light and convert its energy into usable electrochemical potential. In the absence of the photocatalyst, there is no reaction because no other component of this reaction mixture absorbs visible light, and thus no uncatalyzed direct photochemistry can occur. The Lewis acid co-catalyst, in contrast, does not impact the photophysical properties of the photocatalyst; rather, it facilitates an otherwise unfavorable electron transfer event.26 Again, in the absence of Li+ or some other Lewis acidic cation, no background reaction occurs.
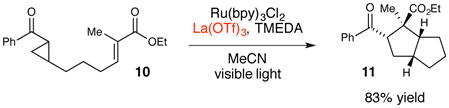
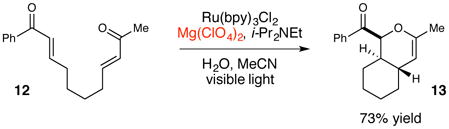
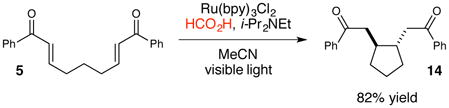
Many of the photoredox reactions subsequently developed in our laboratories similarly utilize Lewis or Brønsted acids as co-catalysts for analogous photoinduced electron transfer events, and the identity of the co-catalyst has proven to be an important variable for optimization. For example, in the photocatalytic [3+2] cycloaddition shown in eq 1, we observed that stronger, trivalent Lewis acids such as La(OTf)3 were required to compensate for the more negative intrinsic reduction potential of the aryl cyclopentyl ketone.27 Similarly, in the photocatalytic radical anion Diels–Alder cycloaddition shown in eq 2, we found that Mg(ClO4)2 was the optimal Lewis acid co-catalyst that balanced the rate of productive cycloaddition against the rate of an undesired competitive overreduction and α-cleavage process.28 Finally, we also found that replacing a Lewis acid co-catalyst with a Brønsted acid can change the course of a reaction entirely (eq 3). The photocatalytic reaction of 5 using formic acid in place of LiBF4 results in a reductive cyclization instead of [2+2] cycloaddition.29
 |
(1) |
 |
(2) |
 |
(3) |
Other investigators have similarly observed that the identity of a Lewis30 or Brønsted acid31 cocatalyst can have a substantial impact on the success and product distribution of photoredox reactions. This underscores an important characteristic of photoredox catalysts that distinguishes them from other, more common modes of catalytic activation. Although many structurally varied photoredox catalysts are known, with correspondingly diverse electrochemical and photophysical properties,32 photoredox catalysts all fundamentally operate by using the energy of an absorbed photon to create redox potential. Subsequent bimolecular, outer-sphere electron transfer events with organic substrates result in the formation of odd-electron reactive intermediates, which need not react in the environment of the photocatalytic species. Non-photoactive co-catalysts, then, can have a dramatic impact not only by catalyzing electron transfer but also by interacting directly with these odd-electron organic intermediates and modulating their reactivity.
This analysis has guided our thinking about how best to develop a general strategy for asymmetric catalytic photoredox transformations. While enantiopure, chiral Ru(II) polypyridyl photocatalysts have been studied for their ability to participate in enantioselective electron transfer processes,33 we suspected that subsequent bond-forming reactions would have little propensity to occur within the proximity of the chiral photocatalyst. We also suspected that these reactions, like many other radical reactions, might be dominated by chain processes,34 in which case a chiral photocatalyst would be involved only in the initiating turnover and not in the chain propagating steps. It seemed to us more logical to study systems using racemic photocatalysts in combination with chiral Lewis acid co-catalysts. We imagined that the heteroatom-containing radical anion intermediates involved in these transformations could bind relatively tightly to an appropriate Lewis acidic scaffold and thus be strongly influenced by the spatial disposition of stereogenic substituents. Control experiments also showed no product formation in the absence of a Lewis acid, suggesting that racemic background processes would not be competitive. Finally, we were attracted to the use of chiral Lewis acids because of the large number of privileged chiral structures available35 and because of the sophisticated understanding that synthetic chemists have developed of the features common to successful enantioselective Lewis acid catalysts.
Chiral Lewis acid co-catalysis
Our search for an effective chiral Lewis acid for the photocatalytic [2+2] cycloaddition reaction began with a screen of simple achiral salts to identify a promising metal center with good catalytic activity at substoichiometric loading. Many main group and transition metal complexes were effective Lewis acids for this process and resulted in the formation of varying amounts of [2+2] cycloadduct. The most effective Lewis acids in this screen, however, proved to be lanthanide triflates (e.g., Gd(OTf)3, Eu(OTf)3). We do not yet have evidence supporting any hypotheses for this empirical observation, but it could be consistent with the high kinetic lability of lanthanide Lewis acids,36 a feature that would facilitate catalyst turnover in this reaction. We consequently focused our subsequent efforts on examining chiral lanthanide-based Lewis acids.
Several families of chiral lanthanide Lewis acids were screened for their ability to control the stereoselectivity of the model cycloaddition shown in Table 1. Many chiral lanthanide Lewis acids are known and have previously been shown to impart high enantioselectivities in non-photochemical cycloaddition reactions;37 however, among the selection that we screened, none of these provided selectivities greater than ~25% ee. While the low ee’s were disappointing, these experiments nevertheless validated our hypothesis that the Lewis acid would be involved in the stereochemistry-determining bond-forming steps.
Table 1.
Screen of chiral Lewis acids for enantioselective [2+2] photocycloaddition.
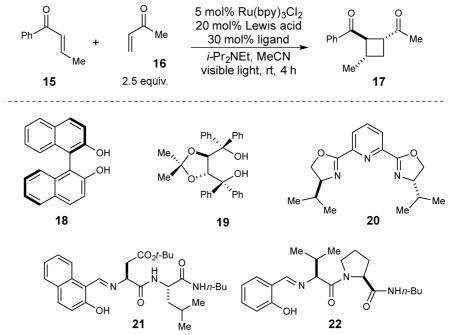
| ||||
|---|---|---|---|---|
| Entry | Lewis acid | Ligand | Yielda | ee |
| 1 | Gd(OTf)3 | -- | 32% | -- |
| 2 | Gd(OTf)3 | 18 | 0% | -- |
| 3 | Gd(OTf)3 | 19 | 27% | 0% |
| 4 | Gd(OTf)3 | 20 | 32% | −6% |
| 5 | Gd(OTf)3 | 21 | 56% | 21% |
| 6 | Gd(OTf)3 | 22 | 43% | 56% |
| 7 | Eu(OTf)3 | 22 | 41% | 78% |
| 8b | Eu(OTf)3 | 22 | 74% | 85% |
| 9b,c | Eu(OTf)3 | 22 | 71% | 92% |
Yield determined by 1H NMR using an internal standard.
Using 5 equiv. 16, 5 mol% Ru(bpy)3Cl2, 10 mol% Eu(OTf)3, and 20 mol% ligand 22.
Reaction conducted at −20 °C.
We next examined complexes bearing several other classes of chiral ligands that had not previously been combined with lanthanide catalysts, again observing only modest enantioselectivities. We were intrigued, however, by the observation that good ee’s were obtained using Gd complexes in conjunction with 21, part of a class of a dipeptide Schiff base ligand originally reported by Inoue38 and extensively studied by Snapper and Hoveyda39,40 because of the ease with which modified ligands could be synthesized and screened to optimize for high ee. Upon performing such a combinatorial screen, we ultimately discovered that the optimal Lewis acid for the [2+2] photocycloaddition was a Eu(OTf)3 complex of proline-valine Schiff base ligand 22. Using this combination, a diverse assortment of unsymmetrically substituted cyclobutane cycloadducts could be obtained in good yields and excellent enantioselectivity (Figure 3A, 17–27).41
Figure 3.

Scope and diastereocontrol in the enantioselective [2+2] photocycloaddition. (A) Using trans-selective imine ligand 22. (B) Using cis-selective amine ligand 28.
Notably, although the concentration of the Lewis acid co-catalyst affected the overall rate of the photocycloaddition, there was no significant impact on enantioselectivity (Figure 4). This is an important observation that suggests that there is indeed no racemic background reaction, neither via direct photoexcitation of the enone nor a Lewis-acid-independent pathway. The insensitivity of ee to Lewis acid concentration indicates that the chiral product is formed exclusively through a Lewis acid mediated pathway.
Figure 4.

The ee of cycloadduct 17 is independent of chiral Lewis acid catalyst concentration.
A second unexpected observation was that even though the identity of the dipeptide ligand had a large effect on the ee of the reaction, the overall reaction rate proved relatively insensitive to its structure. The process of optimizing the catalyst structure for good ee involved the screening of a library of 70 modified dipeptide ligands, and while there was a marked effect on enantioselectivity, most of these complexes gave similar reaction rates.
This observation highlights an important strategic departure from the majority of prior approaches to enantioselective catalysis of photochemical reactions. In general, previous attempts to control the stereochemistry of photocatalytic reactions relied upon a single chiral photocatalyst to simultaneously mediate the photochemical processes as well as control stereochemistry in the product-forming steps. The optimization of a single structure capable of fulfilling these two distinct roles is relatively challenging, as the small structural perturbations usually required to achieve an effective stereocontrolling environment could simultaneously have a large deleterious impact on the photophysical properties important for efficient photocatalysis (e.g., absorption, excited state lifetime, intersystem crossing efficiency, etc.). By separating the photochemical and stereocontrolling functions into two discrete co-catalysts, the structure of the chiral Lewis acid could be independently tuned for optimal enantioselectivity without perturbing the photoelectrochemistry of the photoredox catalyst.
One practical outcome of this flexibility has been the discovery that both relative and absolute stereochemistry of the [2+2] cycloaddition can be controlled through modifications of the structure of the chiral Lewis acid (Figure 3B). Amine ligand 28 is readily available by reduction of the parent imine ligand. The Eu(III) complex of this reduced ligand still provides crossed [2+2] cycloadducts with excellent ee; however, the 1,2-cis diastereoisomer is favored instead of the trans isomer formed using imine ligand 22.
Thus the broad range of chiral environments that can be brought to bear by straightforward modifications of the chiral Lewis acid can be used to alter the stereochemical course of a particular photoreaction. The mutual compatibility of the co-catalysts is crucial; in attempts to apply a similar dual-catalyst strategy to organic photosensitizers, we have observed that the presence of strong oxophilic Lewis acids can interfere with the photochemistry of common ketone sensitizers such as benzophenones. The lack of available Lewis basic sites on the octahedrally saturated, chemically stable Ru(bpy)3 2+ chromophore, however, endows it with an insensitivity to the presence of strong Lewis acids that has proven to be powerfully enabling in our studies.
Generality of the tandem photoredox and chiral Lewis acid catalysis system
We hypothesize that the tunability of the stereocontrolling catalyst independent of the photoredox catalyst will be beneficial in a range of mechanistically distinct reactions. Consider for example, our proposed mechanism for the photocatalytic [3+2] cycloaddition of aryl cyclopropyl ketones (Scheme 1). This reaction shares several features with the [2+2] cycloaddition: (1) it is an overall redox-neutral process initiated by electron transfer; (2) a Lewis acid co-catalyst accelerates the photoreduction step; (3) no background reaction occurs in the absence of the Lewis acid. This reaction presented an ideal opportunity to test the generality of dual catalysis for enantiocontrolled photoreactions.
Scheme 1.

Mechanistic proposal for photocatalytic [3+2] cycloaddition.
There were, however, several unattractive features that we wished to address first. Unlike the analogous racemic [2+2] cycloaddition, we found that only intermolecular reactions were high-yielding. More problematically, optimal reaction conditions required stoichiometric La(OTf)3 as the Lewis acid co-catalyst as well as 5 equiv of TMEDA as both a ligand for La3+ and a reductive quencher of the excited-state photocatalyst.
The strategy that emerged from our optimization studies are depicted in Figure 5.42 We continued to utilize Ru(bpy)3 2+ as the photocatalyst for this reaction, but the chiral Lewis acid co-catalyst that proved to provide the highest yields and enantioselectivities was chiral Gd(III)(pybox) complex 35. Thus, the two-catalyst approach readily accommodated both a different Lewis acidic metal center as well as a different class of chiral ligands from the dipeptide Schiff base ligand that proved to be optimal for the [2+2] photocycloaddition reaction. In particular, the 4- dimethylamino substituent on the central pyridine ring of 3543 proved to be crucial. The unsubstituted parent complex slowly formed an unreactive Lewis acid-base complex with the reductive quencher used in this reaction (i-Pr2NEt). The electron-releasing dimethylamino substituent was incorporated based on our hypothesis that a more strongly donating ligand might attenuate the occurrence of this catalyst-deactivating process. The fact that such a large electronic perturbation in the structure of the Lewis acid catalyst is readily tolerated without deleterious effect on the photophysical properties of the photocatalyst further highlights the flexibility of the two-catalyst approach.
Figure 5.

Dual catalyst strategy for enantioselective [3+2] photocycloadditions.
From a synthetic perspective, this enantioselective [3+2] cycloaddition reaction provides an interesting, complementary capability compared to previous asymmetric reactions of cyclopropyl ketones. Although several different strategies for highly enantioselective cycloadditions of cyclopropanes have been reported, all of these have involved highly activated donor–acceptor cyclopropanes44,45,46 or vinylcyclopropanes. 47,48 The method developed in our laboratory, however, works well for a variety of electronically dissimilar cyclopropyl ketones, affording cycloadducts arising from cyclopropanes bearing either alkyl or ester β-substituents (38–41). Finally, Figure 6 depicts a third enantioselective photocatalytic method developed in our laboratory that exploits a two-catalyst strategy in the context of a conjugate addition reaction of an α-silylamine pronucleophile (43) to a β-substituted Michael acceptor (44).49 The key intermediate in this reaction is an α-amino radical, a nucleophilic radical that is readily available via photosensitized oxidation of amines, α-amino acids, and α-silylamines.50,51,52 Prior to our investigations, however, control over the stereochemistry of α-amino radical reactions had remained challenging; the only prior example of an enantioselective photocatalytic reaction involving α-amino radical intermediates was an intramolecular conjugate addition reported by Bach using a chiral hydrogen-bonding photosensitizer.53
Figure 6.

Dual catalyst strategy for photochemical desilylative conjugate addition.
Our solution to this challenge bears a superficial similarity to the previous examples of tandem photoredox/chiral Lewis acid catalysis; Ru(bpy)3 2+ serves as a photoredox catalyst that oxidatively triggers desilylation of 43 to afford a nucleophilie α-amino radical, and the subsequent stereoselectivity of the conjugate addition process is controlled by a chiral Sc(pybox) Lewis acid co-catalyst (42). However, closer inspection reveals an important mechanistic difference: a variety of photocatalytic methods for the production of α-amino radicals using Ru(bpy)3 2+ and similar transition metal photoredox catalysts are known,54 and no co-catalyst is typically required to facilitate the electron transfer step. Indeed, when the Lewis acid is omitted from our optimized reaction conditions, we observe a slower but still significant rate of formation of conjugate adduct 45, albeit in racemic form. The role of the Lewis acid co-catalyst, then, is to accelerate the rate-limiting carbon–carbon bond-forming conjugate addition step55 and to impart high enantiofacial control in this process.
These results show that a Lewis acid co-catalyst can influence enantioselectivity of a photocatalytic transformation even when it does not act directly upon steps involving the photocatalyst. They can instead impact the rate of secondary, downstream events. Lewis acids can influence organic reactions in a multitude of ways, and given the increasing number of reactive intermediates that are being shown to be easily accessible using photoredox catalysis, our studies suggest that the combination of chiral Lewis acid and photoredox catalysts might offer a robust, flexible strategy to control the reactions of many of them.
Our optimization studies drew upon an extensive literature describing successful Lewis acid catalysts for other enantioselective transformations. For instance, our initial screen of common Michael acceptors afforded generally low to modest ee’s in Lewis acid catalyzed reactions of photogenerated α-amino radicals. For our optimal solution, we utilized Sibi’s chiral relay auxiliary, which we selected because it has been shown to afford excellent stereoinduction in other radical conjugate additions using bis(oxazoline)-based chiral Lewis acids.56 Thus the ability to use chiral Lewis acids in photochemical applications offers a final advantage in enantioselective photochemistry. Synthetic chemists’ understanding of the factors that are common in chiral Lewis acid catalyzed reactions is quite deep. The ability to apply these insights broadly to photoredox reactions offers a significant conceptual advantage that we have found powerfully enabling in our continuing studies of enantioselective photochemical reactions.
Conclusion
Our laboratory’s studies in enantioselective photochemistry have occurred contemporaneously with analogous efforts in this area from several other groups. Many examples of highly enantioselective photoreactions have recently been reported, providing conclusive evidence that catalytic photochemical stereocontrol is feasible. Interestingly, most of these examples have also involved transition metal photoredox catalysts operating in tandem with chiral, non-photoactive co-catalysts such as Brønsted acids57,58 and organocatalysts.59,60 One could argue that this is not coincidental, and that the features of dual photocatalytic systems outlined above are indeed advantageous in the design of asymmetric photoreactions.
There have also been several reports involving successful single-catalyst solutions, notably from Bach,61 Meggers,62 and Sivaguru.63 Thus the recent renewal of interest in stereocontrolled photochemical synthesis has also involved approaches that differ strategically from the dualcatalyst systems favored in our laboratory. There is a fascinating comparison among all of these methods to be made, and it remains to be seen whether any of them will prove to be as general as we all might hope. Regardless, the pace of progress in the field of asymmetric photochemistry is clearly accelerating, and it seems reasonable to hope that this long-standing, conspicuous gap in synthetic capability is finally being filled.
Acknowledgments
I am deeply indebted to the coworkers whose work is mentioned here, and particularly to Michael Ischay, Juana Du, Kaz Skubi, Dani Schultz, Adrian Amador, Evan Sherbrook, Laura Ruiz Espelt, Iain McPherson, and Eric Wiensch.
Funding Sources
Our enantioselective photochemistry research is funded by the NIH (GM095666).
Biography
Tehshik P. Yoon received his M.S. and Ph.D. from Caltech under Erick Carreira and David MacMillan, respectively. After postdoctoral research at Harvard with Eric Jacobsen, Tehshik began his independent career at the University of Wisconsin–Madison, where he has continued to be fascinated by stereocontrolled synthesis, particularly as it applies to radical and photochemical reactions.
References
- 1.Le Bel JA. Sur les relations qui existent entre les formules atomiques des corps organiques et le pouvoir rotatoire de leurs dissolutions. Bull Soc Chim (Paris) 1874;22:337–347. [Google Scholar]
- 2.van t’Hoff JH. Die Lagerung der Atome in Raume. 2. Vieweg: Braumschweig; 1894. p. 30. [Google Scholar]
- 3.Kuhn W, Knopf E. Photochemische Erzeugung optisch aktiver Stoffe. Naturwissenschaften. 1930;18:183. [Google Scholar]
- 4.Inoue Y. Asymmetric Photochemical Reactions in Solution. Chem Rev. 1992;92:741–770. [Google Scholar]
- 5.Brimioulle R, Lenhart D, Maturi MM, Bach T. Enantioselective Catalysis of Photochemical Reactions. Angew Chem Int Ed. 2015;54:3872–3890. doi: 10.1002/anie.201411409. [DOI] [PubMed] [Google Scholar]
- 6.Green BS, Hagler AT, Rabinsohn Y, Rejtõ M. Photochemical Asymmetric Synthesis. Irradiation of Ring and Open-chain Derivatives of L-Erythritol 1,4-Dicinnamate. Isr J Chem. 1976/77;15:124–130. [Google Scholar]
- 7.Tolbert LM, Ali MB. High Optical Yields in a Photochemical Cycloaddition. Lack of Cooperativity as a Clue to Mechanism. J Am Chem Soc. 1982;104:1742–1744. [Google Scholar]
- 8.Bach T, Bergmann H, Harms K. Enantioselective Intramolecular [2 + 2]-Photocycloaddition Reactions in Solution. Angew Chem Int Ed. 2000;39:2302–2304. [PubMed] [Google Scholar]
- 9.Joy A, Scheffer JR, Ramamurthy V. Chirally Modified Zeolites as Reaction Media: Photochemistry of an Achiral Tropolone Ether. Org Lett. 2000;2:119–121. doi: 10.1021/ol9903335. [DOI] [PubMed] [Google Scholar]
- 10.Nishijima M, Wada T, Mori T, Pace TCS, Bohne C, Inoue Y. Highly Enantiomeric Supramolecular [4 + 4] Photocyclodimerization of 2-Anthracenecarboxylate Mediated by Human Serum Albumin. J Am Chem Soc. 2007;129:3478–3479. doi: 10.1021/ja068475z. [DOI] [PubMed] [Google Scholar]
- 11.Addadi L, van Mil J, Lahav M. Photopolymerization in Chiral Crystals. 4. Engineering of Chiral Crystals for Asymmetric (2π + 2 π) Photopolymerization. Execution of an ‘Absolute’ Asymmetric Synthesis with Quantitative Enantiomeric Yield. J Am Chem Soc. 1982;104:3422–3429. [Google Scholar]
- 12.Evans SV, Garcia-Garibay M, Omkaram N, Scheffer JR, Trotter J, Wireko F. Use of Chiral Single Crystals to Convert Achiral Reactants to Chiral Products in High Optical Yield: Application to the Di-π-Methane and Norrish Type II Photorearrangements. J Am Chem Soc. 1986;108:5648–5650. [Google Scholar]
- 13.Bauer A, Westkämper W, Grimme S, Bach T. Catalytic enantioselective reactions driven by photoinduced electron transfer. Nature. 2005;436:1139–1140. doi: 10.1038/nature03955. [DOI] [PubMed] [Google Scholar]
- 14.Guo H, Herdtweck E, Bach T. Enantioselective Lewis Acid Catalysis in Intramolecular [2 + 2] Photocycloaddition Reactions of Coumarins. Angew Chem Int Ed. 2010;49:7782–7785. doi: 10.1002/anie.201003619. [DOI] [PubMed] [Google Scholar]
- 15.Vallavoju N, Selvakumar S, Jockusch S, Sibi MP, Sivaguru J. Enantioselective Organo- Photocatalysis Mediated by Atropisomeric Thioruea Derivatives. Angew Chem Int Ed. 2014;53:5604–5608. doi: 10.1002/anie.201310940. [DOI] [PubMed] [Google Scholar]
- 16.Huo H, Shen X, Wang C, Zhang L, Röse P, Chen LA, Harms K, Marsch M, Hilt G, Meggers E. Asymmetric photoredox transition-metal catalysis activated by visible light. Nature. 2014;515:100–103. doi: 10.1038/nature13892. [DOI] [PubMed] [Google Scholar]
- 17.Lee-Ruff E, Mladenova G. Enantiomerically Pure Cyclobutane Derivatives and Their Use in Organic Synthesis. Chem Rev. 2003;103:1449–1483. doi: 10.1021/cr010013a. [DOI] [PubMed] [Google Scholar]
- 18.Schuster DI, Dunn DA, Heibel GE, Brown PB, Rao JM, Woning J, Bonneau R. Enone Photochemistry. Dynamic Properties of Triplet Excited States of Cyclic Conjugated Enones as Revealed by Transient Absorption Spectroscopy. J Am Chem Soc. 1991;113:6245– 6255. [Google Scholar]
- 19.Mayr H, Ofial AR. The Reactivity–Selectivity Principle: An Imperishable Myth in Organic Chemistry. Angew Chem Int Ed. 2006;45:1844–1854. doi: 10.1002/anie.200503273. [DOI] [PubMed] [Google Scholar]
- 20.Sibi MP, Manyem S, Zimmerman J. Enantioselective Radical Processes. Chem Rev. 2003;103:3263–3295. doi: 10.1021/cr020044l. [DOI] [PubMed] [Google Scholar]
- 21.Müller C, Bauer A, Bach T. Light-Driven Enantioselective Organocatalysis. Angew Chem Int Ed. 2009;48:6640–6642. doi: 10.1002/anie.200901603. [DOI] [PubMed] [Google Scholar]
- 22.Ischay MA, Anzovino ME, Du J, Yoon TP. Efficient Visible Light Photocatalysis of [2+2] Enone Cycloadditions. J Am Chem Soc. 2008;130:12886–12887. doi: 10.1021/ja805387f. [DOI] [PubMed] [Google Scholar]
- 23.Du J, Yoon TP. Crossed Intermolecular [2 + 2] Cycloadditions of Acyclic Enones via Visible Light Photocatalysis. J Am Chem Soc. 2009;131:14604–14605. doi: 10.1021/ja903732v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tyson EL, Farney EP, Yoon TP. Photocatalytic [2 + 2] Cycloadditions of Enones with Cleavable Redox Auxiliaries. Org Lett. 2012;14:1110–1113. doi: 10.1021/ol3000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Skubi KL, Blum TR, Yoon TP. Dual Catalysis Strategies in Photochemical Synthesis. Chem Rev. 2016:116. doi: 10.1021/acs.chemrev.6b00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fournier F, Fournier M. Transferts d’électrons assistés par les métaux de transition: influence de la nature du cation métallique sur la réduction de composés carbonylés en milieu aprotique. Can J Chem. 1986;64:881–890. [Google Scholar]
- 27.Lu Z, Shen M, Yoon TP. [3 + 2] Cycloadditions of Aryl Cyclopropyl Ketones by Visible Light Photocatalysis. J Am Chem Soc. 2011;133:1162–1164. doi: 10.1021/ja107849y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hurtley AE, Cismesia MA, Ischay MA, Yoon TP. Visible Light Photocatalysis of Radical Anion Hetero-Diels-Alder Cycloadditions. Tetrahedron. 2011;67:4442–4448. doi: 10.1016/j.tet.2011.02.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Du J, Ruiz Espelt L, Guzei IA, Yoon TP. Photocatalytic Reductive Cyclizations of Enones: Divergent Reactivity of Photogenerated Radical and Radical Anion Intermediates. Chem Sci. 2011;2:2115–2119. doi: 10.1039/C1SC00357G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao G, Yang C, Guo L, Sun H, Lin R, Xia W. Reactivity Insight into Reductive Coupling and Aldol Cyclization of Chalcones by Visible Light Photocatalysis. J Org Chem. 2012;77:6302–6306. doi: 10.1021/jo300796j. [DOI] [PubMed] [Google Scholar]
- 31.Tarantino KT, Liu P, Knowles RR. Catalytic Ketyl-Olefin Cyclizations Enabled by Proton-Coupled Electron Transfer. J Am Chem Soc. 2013;135:10022–10025. doi: 10.1021/ja404342j. [DOI] [PubMed] [Google Scholar]
- 32.Juris A, Balzani V, Barigelletti F, Campagna S, Belser P, von Zelewsky A. Ru(II) Polypyridine Complexes: Photophysics, Photochemistry, Electrochemistry, and Chemiluminescence. Coord Chem Rev. 1988;84:85–277. [Google Scholar]
- 33.Ohkubo K, Hamada T, Ishida H. Novel Enantioselective Photocatalysis by Chiral, Helical Ruthenium(II) Complexes. J Chem Soc, Chem Commun. 1993:1423–1425. [Google Scholar]
- 34.Cismesia MA, Yoon TP. Characterizing Chain Processes in Visible Light Photoredox Catalysis. Chem Sci. 2015;6:5426–5434. doi: 10.1039/c5sc02185e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yoon TP, Jacobsen EN. Privileged Chiral Catalysts. Science. 2003;299:1691–1693. doi: 10.1126/science.1083622. [DOI] [PubMed] [Google Scholar]
- 36.Anwander R. Principles in Organolanthanide Chemistry. In: Kobayashi S, editor. Lanthanides: Chemistry and Use in Organic Synthesis. Springer; Berlin: 1999. pp. 11–61. [Google Scholar]
- 37.Mikami K, Terada M, Matsuzawa H. Asymmetric Catalysis by Lanthanide Complexes. Angew Chem Int Ed. 2002;41:3555–3571. doi: 10.1002/1521-3773(20021004)41:19<3554::AID-ANIE3554>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 38.Nitta H, Yu D, Kudo M, Mori A, Inoue S. Peptide-Titanium Complex as Catalyst for Asymmetric Addition of Hydrogen Cyanide to Aldehydes. J Am Chem Soc. 1992;114:7969– 7975. [Google Scholar]
- 39.Cole BM, Shimizu KD, Krueger CA, Harrity JPA, Snapper ML, Hoveyda AH. Discovery of Chiral Catalysts through Ligand Diversity: Ti-Catalyzed Enantioselective Addition of TMSCN to meso Epoxides. Angew Chem Int Ed Eng. 1996;35:1668–1671. [Google Scholar]
- 40.Hoveyda AH. Catalyst Discovery Through Combinatorial Chemistry. Chemistry & Biology. 1998;5:R187–R191. doi: 10.1016/s1074-5521(98)90155-7. [DOI] [PubMed] [Google Scholar]
- 41.Du J, Skubi KL, Schultz DM, Yoon TP. A Dual-Catalysis Approach to Enantioselective [2 + 2] Photocycloadditions Using Visible Light. Science. 2014;344:392–396. doi: 10.1126/science.1251511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Amador AG, Sherbrook EM, Yoon TP. Enantioselective Photocatalytic [3 + 2] Cycloadditions of Aryl Cyclopropyl Ketones. J Am Chem Soc. 2016;138:4722–4725. doi: 10.1021/jacs.6b01728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nishiyama H, Yamaguchi S, Kondo M, Itoh K. Electronic Substituent Effect of Nitrogen Ligands in Catalytic Asymmetric Hydrosilylation of Ketones: Chiral 4-Substituted Bis(oxazolinyl)pyridines. J Org Chem. 1992;57:4306–4309. [Google Scholar]
- 44.Parsons AT, Johnson JS. Catalytic Enantioselective Synthesis of Tetrahydrofurans: A Dynamic Kinetic Asymmetric [3 + 2] Cycloaddition of Racemic Cyclopropanes and Aldehydes. J Am Chem Soc. 2009;131:3122–3123. doi: 10.1021/ja809873u. [DOI] [PubMed] [Google Scholar]
- 45.Parsons AT, Smith AG, Neel AJ, Johnson JS. Dynamic Kinetic Asymmetric Synthesis of Substituted Pyrrolidines from Racemic Cyclopropanes and Aldimines: Reaction Development and Mechanistic Insights. J Am Chem Soc. 2010;132:9688–9692. doi: 10.1021/ja1032277. [DOI] [PubMed] [Google Scholar]
- 46.Xiong H, Xu H, Liao S, Xie Z, Tang Y. Copper-Catalyzed Highly Enantioselective Cyclopentannulation of Indoles with Donor–Acceptor Cyclopropanes. J Am Chem Soc. 2013;135:7851–7853. doi: 10.1021/ja4042127. [DOI] [PubMed] [Google Scholar]
- 47.Trost BM, Morris PJ. Palladium-Catalyzed Diastereo- and Enantioselective Synthesis of Substituted Cyclopentanes through a Dynamic Kinetic Asymmetric Formal [3+2]-Cycloaddition of Vinyl Cyclopropanes and Alkylidene Azlactones. Angew Chem Int Ed. 2011;50:6167– 6170. doi: 10.1002/anie.201101684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hashimoto T, Kawamata Y, Maruoka K. An Organic Thiyl Radical Catalyst for Enantioselective Cyclization. Nat Chem. 2014;6:702–705. doi: 10.1038/nchem.1998. [DOI] [PubMed] [Google Scholar]
- 49.Ruiz Espelt L, McPherson IS, Wiensch EM, Yoon TP. Enantioselective Conjugate Additions Of α-Amino Radicals via Cooperative Photoredox and Lewis Acid Catalysis. J Am Chem Soc. 2015;137:2452–2455. doi: 10.1021/ja512746q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yoon UC, Su Z, Mariano PS. The Dynamics and Photochemical Consequences of Aminium Radical Reactions. In: Horspool W, Lenci F, editors. Handbook of Organic Photochemistry and Photobiology. 2. Ch. 101 CRC Press; Boca Raton: 2003. [Google Scholar]
- 51.Brumfield MA, Quillen SL, Yoon UC, Mariano PS. A Novel Method for Heteroatom-Substituted Free Radical Generation by Photochemical Electron-Transfer-Induced Desilylation of RXCH2Me3Si Systems. J Am Chem Soc. 1984;106:6855–6856. [Google Scholar]
- 52.Pandey G, Kumaraswamy G, Bhalerao UT. Photoinduced SET Generation of α-Amineradicals: A Practical Method for the Synthesis of Pyrrolidines and Piperidines. Tetrahedron Lett. 1989;30:6059–6062. [Google Scholar]
- 53.Bauer A, Westkämper F, Grimme S, Bach T. Catalytic Enantioselective Reactions Driven by Photoinduced Electron Transfer. Nature. 2005;436:1139–1140. doi: 10.1038/nature03955. [DOI] [PubMed] [Google Scholar]
- 54.Shi L, Xia W. Photoredox Functionalization of C–H Bonds Adjacent to a Nitrogen. Chem Soc Rev. 2012;41:7687–7697. doi: 10.1039/c2cs35203f. [DOI] [PubMed] [Google Scholar]
- 55.Ruiz Espelt L, Wiensch EM, Yoon TP. Brønsted Acid Cocatalysts in Photocatalytic Radical Addition of α-Amino C–H Bonds Across Michael Acceptors. J Org Chem. 2013;78:4107–4114. doi: 10.1021/jo400428m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sibi MP, Prabagaran N. Chiral Relay in Enantioselective Conjugate Radical Additions Using Pyrazolidinone Templates. How Does Metal Geometry Impact Selectivity. Synlett. 2004:2421–2424. [Google Scholar]
- 57.Rono LJ, Yayla HG, Wang DY, Armstrong MF, Knowles RR. Enantioselective Photoredox Catalysis Enabled by Proton-Coupled Electron Transfer: Development of an Asymmetric Aza-Pinacol Cyclization. J Am Chem Soc. 2013;135:17735–17738. doi: 10.1021/ja4100595. [DOI] [PubMed] [Google Scholar]
- 58.Uraguchi D, Kinoshita N, Kizu T, Ooi T. Synergistic Catalysis of Ionic Brønsted Acid and Photosensitizer for a Redox Neutral Asymmetric α-Coupling of N-Arylaminomethanes with Aldimines. J Am Chem Soc. 2015;137:13768–13771. doi: 10.1021/jacs.5b09329. [DOI] [PubMed] [Google Scholar]
- 59.Nicewicz D, MacMillan DWC. Merging Photoredox Catalysis with Organocatalysis: The Direct Asymmetric Alkylation of Aldehydes. Science. 2008;322:77–80. doi: 10.1126/science.1161976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.DiRocco DA, Rovis T. Catalytic Asymmetric α-Acylation of Tertiary Amines Mediated by a Dual Catalysis Mode: N-Heterocyclic Carbene and Photoredox Catalysis. J Am Chem Soc. 2012;134:8094–8097. doi: 10.1021/ja3030164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Brimioulle R, Bach T. Enantioselective Lewis Acid Catalysis of Intramolecular Enone [2+2] Photocycloaddition Reactions. Science. 2013;342:840–843. doi: 10.1126/science.1244809. [DOI] [PubMed] [Google Scholar]
- 62.Huo H, Shen X, Wang C, Zhang L, Röse P, Chen LA, Harms K, Marsch M, Hilt G, Meggers E. Asymmetric photoredox transition-metal catalysis activated by visible light. Nature. 2014;515:100–103. doi: 10.1038/nature13892. [DOI] [PubMed] [Google Scholar]
- 63.Vallavoju N, Selvakumar S, Jockusch S, Sibi MP, Sivaguru J. Enantioselective Organo- Photocatalysis Mediated by Atropisomeric Thiourea Derivatives. Angew Chem Int Ed. 2014;53:5604–5608. doi: 10.1002/anie.201310940. [DOI] [PubMed] [Google Scholar]