

Abstract
Autism spectrum disorder (ASD) is a complex neurodevelopmental disorder often accompanied by intellectual disability, language impairment and medical co-morbidities. The heritability of autism is high and multiple genes have been implicated as causal. However, most of these genes have been identified in de novo cases. To further the understanding of familial autism, we performed whole-exome sequencing on five families in which second and third degree relatives were affected. By focusing on novel and protein-altering variants, we identified a small set of candidate genes. Among these, a novel private missense C1143F variant in the second intracellular loop of the voltage-gated sodium channel NaV1.7, encoded by the SCN9A gene, was identified in one family. Through electrophysiological analysis, we show that NaV1.7C1143F exhibits partial loss-of-function effects, resulting in slower recovery from inactivation and decreased excitability in cultured cortical neurons. Furthermore, for the same intracellular loop of NaV1.7, we found an excess of rare variants in a case-control variant-burden study. Functional analysis of one of these variants, M932L/V991L, also demonstrated reduced firing in cortical neurons. However, while this variant is rare in Caucasians, it is frequent in Latino population, suggesting that genetic background can alter its effects on phenotype. While the involvement of the SCN1A and SCN2A genes encoding NaV1.1 and NaV1.2 channels in de novo ASD has previously been demonstrated, our study indicates the involvement of inherited SCN9A variants and partial loss of function of NaV1.7 channels in the etiology of rare familial ASD.
INTRODUCTION
Autism Spectrum Disorder (ASD) is a behaviorally defined, childhood-onset complex neurodevelopmental disorder with a broad spectrum of symptoms and a wide range of severity. The core symptoms of ASD include deficits in social communication and social interactions, along with restrictive, repetitive patterns of behaviors, interests or activities (DSM5)1. A recent report by the Center for Disease Control and Prevention (CDC) puts the incidence of autism as 1 in 68 children, making it a growing public health concern. Autism has a strong genetic component that is further complicated by overlap with other neurodevelopmental disorders such as intellectual disability, epilepsy and schizophrenia2, 3. Advances in array and sequencing technologies facilitated elucidation of the contribution of de novo copy number variations (CNVs) and single nucleotide mutations to the molecular basis of sporadic ASD4, 5. From recent CNV and sequencing studies, it is estimated that there are hundreds to thousands of ASD risk loci3, 4 and de novo events account for 10–40% of cases6. The analysis of genes with de novo variants using systems biology approaches such as pathway enrichment and gene co-expression profiling implicates neuronal signaling, synaptic transmission, chromatin biology and transcription regulation4, 5, 7 in the etiology of de novo ASD. However, in familial ASD much of heritability remains unexplained8. Traditional linkage studies have implicated regions cumulatively covering more than 10% of the genome and only a few loci have been replicated at genome-wide significance level9. To identify genes involved in familial autism, new sequencing methods were initially applied in consanguineous affected individuals in order to detect recessive genes10, 11. More recently whole-exome sequencing (WES) has been used in multiplex families to identify heterozygous shared variants that are associated with ASD risk12, 13. Such studies identified additional candidate genes for autism but lacked statistical power and did not include replication14. Although evidence is lacking for the involvement of specific genes, some prior studies indicated an association between voltage-gated sodium channels and familial ASD15.
The alpha subunits of voltage-gated sodium (NaV) channels are evolutionarily conserved and responsible for initiation and propagation of action potentials in nerve and muscle16. Nine pore-forming alpha subunits of NaV channels have been identified in mammals, with different channel subtypes showing specific expression profiles in various excitable tissues. Mutations in the primary central nervous system (CNS) NaV channels (encoded by SCN1A, SCN2A, SCN3A and SCN8A) are associated with neurological, psychiatric, and neurodevelopmental disorders including epilepsy, autism and cognitive impairment17-19. In the peripheral nervous system, NaV1.7 channels encoded by SCN9A are important for the excitability of sensory neurons and pain perception. Gain-of-function mutations in NaV1.7 lead to primary erythermalgia20, paroxysmal extreme pain disorder21, small nerve fiber neuropathy22 and increased pain perception23, while homozygous complete loss-of-function mutations cause congenital insensitivity to pain24-27. The absence of other neurological symptoms in these sensory disorders suggested that NaV1.7 channels are crucial for relaying sensory information in humans, but their roles outside the nociceptive system are not well understood.
The traditional family-based design for gene discovery uses large extended families with multiple affected family members and combines linkage and sequencing analyses to limit the search space for variant identification and to obtain statistical evidence for causality. For study of genetically heterogeneous traits, use of extended families in this context is particularly useful28. As large multigenerational families with autism are rare, we applied a modified approach. We focused on families with affected distant relatives such as 2nd and 3rd cousins and used stringent frequency filtering of the exome data to identify putatively causal variants under the assumption that for each family the causal variant is private and sufficient to cause the phenotype. Our focus on protein-altering variants further narrowed the number of candidate genes in each family. This approach resulted in the identification of a small number of candidate genes in each family including SCN9A. Functional analysis of the SCN9A variant, NaV1.7C1143F, revealed partial loss-of-function effects on excitability of cultured neurons. Further case-control analysis provided additional evidence for association of NaV1.7 mutations in the second intracellular loop with ASD, and functional analysis of one of these variants, NaV1.7M932L/V991L, also demonstrated reduced firing of cortical neurons. Together, our results suggest that SCN9A is important for normal brain function and that rare variants in this gene, encoding partial loss-of-function mutations in the second intracellular loop of NaV1.7, are involved in familial autism.
MATERIALS AND METHODS
Study sample
For the family based exome sequencing study we identified three second-cousin and two third-cousin families in the NIMH repository (https://www.nimhgenetics.org/) and selected two affected cousins from each family for exome sequencing (N=10). All families were of Caucasian background.
For the association study, we selected 855 familial Caucasian cases and 960 Caucasian controls from the NIMH repository. An additional unrelated 288 familial Caucasian cases were included from a University of Washington (UW) multiplex autism collection and 208 Caucasian subjects from a UW study on dyslexia served as control samples. For both NIMH and UW ASD subjects, the diagnostic status was determined by gold-standard ADOS and ADI-R assessment followed by expert clinical judgment using all available information. The NIMH controls sample consists of adults who completed an online short self-report assessment to exclude severe psychiatric disorders, but were not specifically screened for autism. The UW controls were assessed for dyslexia by comprehensive testing, and screened for absence of ASD with a questionnaire. Samples from NIMH participating institutions and University of Washington were utilized in concordance with Institutional Review Boards approvals for the participation in the NIMH repository, consenting of subjects and data sharing.
Whole exome and targeted capture sequencing, variant identification and annotation
Exome capture using the Nimblegen SeqCap EZ Human Exome Library v2.0 (Roche, Basel, Switzerland), paired-end 50bp sequencing on an Illumina HiSeq2000 sequencing platform, and variant calling and annotation were performed in the UW Genome Sciences Center for Mendelian Genomics as described before29. For targeted sequencing we designed 117 single-molecule molecular inversion probes (smMIPs) for the protein-coding region of four candidate genes and performed sequencing and analysis as described before30. Sequence reads were aligned to NCBI human reference genome GRCh37 (hg19) using Burrows-Wheeler Aligner31 (BWA v0.7.10) and Genome analysis toolkit32 (GATK) was used for variant calling. Single nucleotide variants were annotated as nonsynonymous, splice, stop gain, stop lost, or synonymous using ANNOVAR33 and further classified as rare or private based on their frequencies in variant databases. We defined variants with frequency < 0.01 in 1000 Genomes (1KG) “European” samples as rare and variants not present in any of the data sets (dbSNP132, 1KG (2012April) and Exome Sequencing Project (ESP) European American data sets) as private. Detailed methods are provided in the Supplementary Information.
Primary cultures of mouse cortical neurons, transient expression of NaV channels and electrophysiology
Primary cultures of cortical neurons were prepared from C57BL/6 mice on postnatal days 0–3 as described 34. The cerebral cortices were dissected, digested, triturated and plated on coverslips. Cortical neurons were transfected with NaV1.7WT, NaV1.7C1143F and NaV1.7M932L/V991L using electroporation (Neon, Life Technologies) or Lipofectamine 2000 (Life Technologies). Co-electroporation of cDNA encoding GFP allowed identification of the transfected cells and all the recordings were performed on GFP labeled cells only. Recordings were made 3-5 days after transfections. Detailed methods are described in the Supplementary Information.
Quantitative real time PCR
Cerebral cortices and hippocampi were isolated from P21-P24 mice and used for quantitative real time PCR (comparative CT method) using TaqMan primers and probes (Applied Biosystems). See Supplementary Information for full description.
Genetic association analysis
For exome-sequenced cases, the level of relatedness was confirmed using KING35. We used PLINK36 to identify potentially duplicated samples in case-control analysis as some UW cases were also present in the NIMH sample. Pairs with PI_HAT (Proportion IBD) more than 0.9 were considered identical/duplicates. For such pairs only one sample was considered for further analysis. For unrelated subjects we performed gene-based variant burden and single-variant association analyses with a chi-squared test (1-df, Yates correction) and calculated a P value for each gene/region. For the association study we included protein-altering variations (nonsynonymous, splice, stop gain, stop loss) with a frequency <0.01 in 1KG EA. For the SCN9A LII-III region that was associated with autism in our case-control study we performed Transmission Disequilibrium Test (TDT) analysis to control for spurious association that might be due to population substructure. TDT uses chi-squared goodness of fit statistics to determine if there is a preferential transmission of a risk allele to the affected cases. In the TDT analysis we used affected siblings of cases with rare protein-altering variants in the SCN9A LII-III region, under the assumption that if SCN9A LII-III variants contribute to the phenotype we should see over-transmission of risk variants to affected siblings. This analysis incorporated a rare-variant extension of the TDT as described37. In a manner equivalent to our case control study we used a TDT-Burden of rare variants (BRV) that counts the number of minor-allele-transmission events to affected siblings.
Variant validation and evaluation in family members
Selected private variants identified by exome sequencing that were shared between affected cousins were independently validated using Sanger sequencing and tested for co-segregation in additional available family members. Rare variants identified in case-control targeted sequencing that code for the second intracellular loop of SCN9A were also confirmed with Sanger sequencing. For each proband, available family members were genotyped to determine if they carry the variant allele. Primers for PCR amplification and sequencing were designed using Primer3 (v 4.0.0) and bidirectional sequencing on an ABI 3730 DNA analyzer (Applied Biosystems) was done as previously described13.
Bioinformatics analysis of gene expression, co-expression and biological function enrichment
Expression data based on RNA sequencing of 53 different tissues, including 13 brain regions, were downloaded from the GTEx portal38. To identify genes with a brain expression pattern that correlates with that of SCN9A, we calculated the Pearson's correlation coefficient for each gene and SCN9A. Genes with strong correlation (−0.5≥r≥0.5) with p<0.05 were retained. Pearson correlations and p values were calculated using the rcorr function of library Hmisc in R. To identify biological implications for this set of genes we performed Gene Ontology (GO) Biological Processes (BP) enrichment analysis using Database for Annotation, Visualizations and Integration Discovery (DAVID)39. For DAVID analysis Benjamini-Hochberg adjusted p< 0.01 and Fold Enrichment > 2 were considered to be significant.
Genotype-phenotype correlation analysis
To investigate whether SCN9A LII-III variants associate with a phenotypic subtype of ASD we compared available phenotypes in 29 subjects who carried LII-III variants to a comparison sample of 2334 individuals from the full NIMH sample. We examined demographics, cognitive abilities (nonverbal IQ and receptive vocabulary), parental report of autism symptoms domains as assessed with the Autism Diagnostic Interview40, milestones (age of walking, use of first words, and phrases), as well as specific clinical characteristics assessed with parental questionnaire (regression, aggression, self injury and history of seizures). To detect differences between groups, ANOVA was used for the quantitative variables age, cognition and parental report of autism symptom domains, whereas a chi-square nonparametric tests were used for gender and rates of specific phenotypic characteristics (e.g., regression). All analyses were performed using SPSS version 23.0.
RESULTS
Family exome analysis
We sequenced ten exomes from five families with cousins affected with autism, which are deposited in NDAR (https://ndar.nih.gov/edit_collection.html?id=1919). On average, we generated 3.8 Gb of mapped sequence data per individual and >92% of bases had >8x coverage across all samples. After quality filtering, on average 23,685 +/− (429) variants were identified in each individual. After frequency and function filtering was performed, each subject on average had 104 +/− (26) gene disrupting (splice, frameshift, stop) and nonsynonymous private variants (Supplementary Table 1). The affected cases in each family shared between one to four private variants that were all nonsynonymous. We evaluated the impact of missense variants on the structure and function of the proteins using physical and comparative considerations as implemented in SIFT, Polyhen2 and GERP to identify variants more likely to have effects on the protein function (Table 1). Variants were confirmed and evaluated if they were present in additional affected family members. In this way we identified four candidate genes SCN9A, PLEC, KANK3 and CCDC73, which we further evaluated for association with autism. Given the strong evidence for involvement of voltage-gated sodium channels in ASD, and the high impact of the SCN9AC1143F variant based on bioinformatics analysis, we performed functional studies on NaV1.7C1143F.
Table 1. Private variants shared by the affected cousins of each exome sequenced family. Genes selected for case-control study are noted in bold.
SIFT (Sorting Intolerant From Tolerant) predicts impact of amino acid substitutions based on the degree of conservation in sequence alignments derived from closely related sequences. Scores <0.05 are considered deleterious. PolyPhen-2 (Polymorphism Phenotyping v2) predicts impact of a variant on the structure and function of a human protein using eight sequence-based and three structure-based predictive features. Scores >0.95 are considered probably damaging. GERP (Genomic Evolutionary Rate Profiling) identifies functional constraint of a sequence variant by quantifying substitution deficits in multiple alignments. Substitution deficits represent a natural measure of constraint that reflects the strength of past purifying selection. Higher GERP scores are more deleterious.
| Family Id | Position | Gene | Nucleotide/ AA Change | SIFT | Polyphen2 | GERP |
|---|---|---|---|---|---|---|
| 74-0668 | chr14:104029378 | APOPT1 | c.C79A:p.P27T | 0.01 | 0 | −1.81 |
| chr19:35850022 | FFAR3 | c.A230G:p.N77S | 0.88 | 0.003 | −6.53 | |
| 152MM0304 | chr1:3328298 | PRDM16 | c.G1537A:p.G513S | 0.75 | 0.852 | 2.22 |
| chr2:167108286 | SCN9A | c.G3428T:p.C1143F | 0 | 1 | 5.82 | |
| 156-3860 | chr6:167594182 | TCP10L2 | c.A831T:p.E277D | 1 | 0 | −1.58 |
| chr8:144991583 | PLEC | c.G12364C:p.V4122L | 0.99 | 1 | 5.08 | |
| chr11:32636075 | CCDC73 | c.G1789C:p.E597Q | 0.41 | 0.047 | 1.63 | |
| chr19:55998317 | NAT14 | c.C615G:p.D205E | 0.1 | 0.009 | 0.636 | |
| 156-3897 | chr19:8399390 | KANK3 | c.C1321G:p.P441A | 0.23 | 0.131 | 3.36 |
| chr19:58117083 | ZNF530 | c.A190T:p.T64S | 0.79 | 0.001 | −3.39 | |
| 152MM0122 | chr2:233712266 | GIGYF2 | c.G3651C:p.Q1217H | 0.06 | 0.22 | −2.51 |
Functional analysis of the NaVC1143F variant
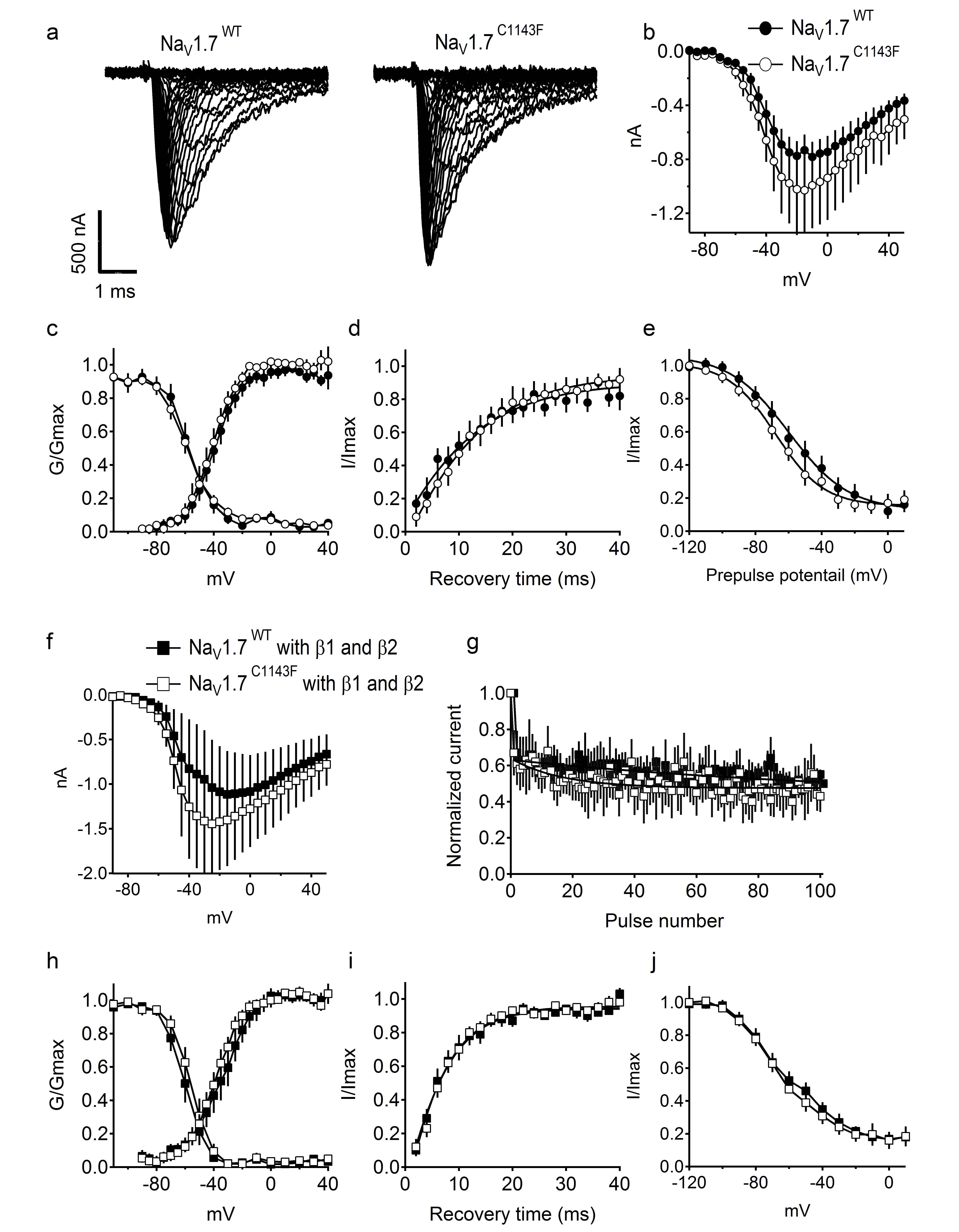
To investigate the biophysical ramifications of this mutation, we first expressed NaV1.7WT and NaV1.7C1143F in human embryonic kidney tsA-201 cells and examined the biophysical properties of the sodium currents. When NaV1.7 channels were expressed without auxiliary β subunits, the voltage dependence and kinetics of activation and fast inactivation were unchanged by the mutation (Supplementary Figure 1a-d), but we observed a trend toward more negative voltage dependence of slow inactivation (Supplementary Figure 1e). Activation, fast inactivation and slow inactivation were unaffected by the mutation when NaV1.7 channels were with co-expressed with β1 and β2 subunits (Supplementary Figure 1f-j).
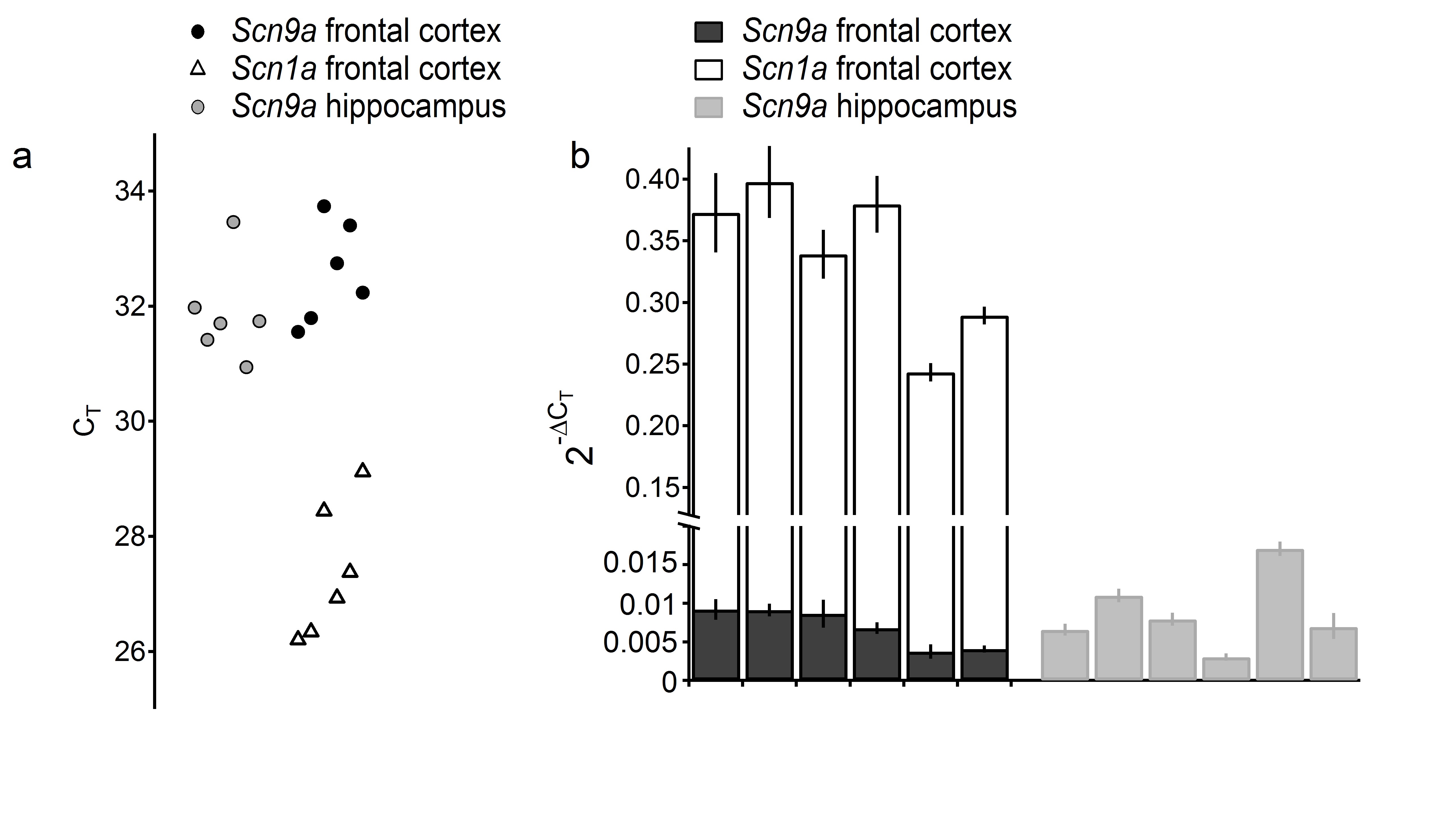
In neurons, NaV1.7 channels have many additional interacting partners, including multiple combinations of β subunits41, fibroblast growth factor homology factors42, protein kinases43, and potentially other regulatory proteins. Therefore, further characterization of the NaV1.7C1143F variant was carried out in mouse cultured cortical neurons. We confirmed that SCN9A is expressed in frontal cortex and hippocampus of mice using quantitative PCR. SCN9A expression is clearly detectable although 52.6 ± 5.5 fold lower compared to the expression of SCN1A, a gene with a well-demonstrated role and expression in CNS (Supplementary Figure 2). Our results of SCN9A expression are in full agreement with recently published results of mouse cortex and hippocampus single cell RNA sequencing studies 44, as well as with previous results from monkeys45. In addition evidence for SCN9A brain expression in mice has been documented, although not published (Mouse Genome Database (MGD), MGI: 3631994, (URL: http://www.informatics.jax.org/) (April 2016)).
NaV1.7 currents recorded in voltage clamp mode with endogenous sodium channels blocked revealed that the NaV1.7C1143F mutation had no effect on peak current amplitude, voltage dependence of activation or voltage dependence of fast inactivation (Figure 1a-c). However, the rate of recovery from fast inactivation was 2-fold slower in NaV1.7C1143F (Figure 1d). Sodium channel availability, during repetitive depolarization at 20 Hz and 50 Hz, declined ~20% in amplitude at both stimulation frequencies for NaV1.7WT. Notably, for NaV1.7C1143F, the decline was significantly increased, with a 35% reduction in current amplitude at 20 Hz and a 67% reduction at 50 Hz (Figures 1e, f).
Figure 1. Properties of NaV1.7WT and NaV1.7C1143F expressed in cultured cortical neurons.

(a-f) Biophysical characterization of NaV1.7WT and NaV1.7C1143F in cultured cortical neurons. Because cortical neurons express endogenous NaV1.7 channels, as well as many other types of sodium channels, we used a tetrodotoxin-resistant form of NaV1.7 (See Supplementary Information, NaV1.7Y362S ) and blocked endogenous channels with 500 nM TTX. (a) Representative set of sodium current traces from cortical neurons expressing NaV1.7WT and NaV1.7C1143F. (b) Mean current-voltage (I-V) relationships of peak currents. (c) Voltage dependence of activation (right curves, the V1/2 of channel activation was 19.7±4.4 mV for NaV1.7WT and 19.3±9.3 mV for NaV1.7C1143F, p>0.05), or the voltage dependence for steady-state fast inactivation (left curves, V1/2 for NaV1.7WT was −62.6 ± 1.6 mV and for NaV1.7C1143F −69.7 ± 3.62 mV, p>0.05). (d) Recovery from fast inactivation (τ = 3.72±0.6 ms in NaV1.7WT and 7.63±1.35 ms in NaV1.7C1143F, p=0.017). (e-f) Mean normalized currents during 100 depolarizations to 0 mV at 20 Hz (e) and 50 Hz (f). NaV1.7WT, n= 7, NaVC1143F, n=5. (g-j) Firing of cortical neurons expressing NaV1.7WT and NaV1.7C1143F. (g) Representative firing in response to 160 pA depolarizing current injection. (h) Membrane input resistance. (i) Rheobase, the minimal current needed to evoke AP (j) Average number of AP in response to 1 s depolarizing current injection at the indicated intensity. The resting membrane potential was unaffected by the C1143F mutation (−65.8±2.7 mV in NaV1.7WT and −60.7±3.6 mV in NaV1.7C1143F,p>0.05). Similarly, the threshold for action potential (−32.7±1.7 mV for NaV1.7WT and −27.68±5.4 mV for NaV1.7C1143F p>0.05) and cell capacitance (95.6±22 pF in NaV1.7WT and 85.3±37 pF in NaV1.7C1143F, p>0.05) were unchanged. *, p<0.05. NaV1.7WT, n= 7, NaV1.7C1143F, n=5.
The measures of cortical neuron membrane capacitance, resting membrane potential, and threshold for action potential generation, were comparable between neurons expressing NaV1.7WT and NaV1.7C1143F (Figure 1g, j). For neurons expressing NaV1.7C1143F, the input resistance was lower and rheobase increased, requiring larger current injection to evoke action potentials (Figures 1h, i; p<0.05). In neurons expressing NaV1.7C1143F the number of action potentials was decreased during trains evoked by a broad range of injected current intensities (Figure 1j).
Genetic association analysis and functional analysis of the NaV1.7M932L/V991L variant
To further test the significance of mutations in SCN9A in autism, we conducted extensive genetic association analyses. After removal of duplicated and poorly captured samples, 1004 unrelated familial cases (NIMH cases = 800, UW cases = 204) and 1127 controls (NIMH controls = 924, UW controls = 203) remained. Gene-based variant-burden analysis did not identify significant results for any of the 4 evaluated genes (Table 2). However, single variant analysis was significant for the SCN9AV991L rs4369876 variant (p=0.00004) that is located in the same cytoplasmic loop (LII-III) as the C1143F variant that we observed in a single ASD exome sequenced family. The V991L variant was previously reported to be in complete linkage disequilibrium with an M932L variant46. In our analysis the M932L variant was initially filtered out due to poor quality of reads for that position. Capillary sequencing confirmed the presence of M932L in all subjects with the V991L variant. Eight additional rare protein-altering variants were observed in LII-III and none in controls (p=0.0083). For LII-III, we identified a total of 29 rare variants in cases and 2 in controls for a combined variant-burden LII-III, p=5.1×10−7 (Table 3). Variants identified in all 4 genes are reported in Supplementary Table 2.
Table 2. Gene based variant-burden association study for SCN9A, PLEC, KANK3 and CCDC73 in 1004 familial cases and 1127 unscreened controls.
For PLEC only 1kb coding region surrounding original variant identified in affected family was sequenced. Association for the second intracellular loop (LII-III) of SCN9A gene is shown in bolds. Case: Number of ASD case samples with variant / Number of ASD case samples without variant. Control: Number of control samples with variant / Number of control samples without variant.
| Gene | Case | Control | Chi-Square test (Yates’ P value) |
|---|---|---|---|
| SCN9A | 67/936 | 60/1067 | 0.18 |
| SCN9A (LII-III) | 29/975 | 2/1119 | 5.1×10−7 |
| KANK3 | 5/999 | 8/1119 | 0.72 |
| CCDC73 | 1/1003 | 5/1122 | 0.27 |
| PLEC | 7/997 | 5/1122 | 0.62 |
Table 3. All variants identified in second intracellular loop (LII-III) of SCN9A.
ESP and 1000 Genome frequencies are only for “European American” and “European” population respectively. Case: Number of ASD case samples with variant/ Number of ASD case samples without variant. Control: Number of control samples with variant/ Number of control samples without variant. dbSNP: database of Short Genetic Variation. NA indicates that the variant is not present. SIFT, Polyphen-2 and GERP are described in Table 1.
| Position | Nucleotide change | AA change | ESP6500 EU | 1000Genome EUR | dbSNP132 | Case | Control | SIFT | Polyphen2 | GERP |
|---|---|---|---|---|---|---|---|---|---|---|
| chr2:167099083 | c.T3523A | p.Y1175N | 0 | 0 | NA | 1/1003 | 0/1127 | 0 | 0.969 | 5.01 |
| chr2:167099157 | c.G3449A | p.W1150X | 0 | 0 | NA | 1/1003 | 0/1126 | 0 | 0 | 1.74 |
| chr2:167108345 | c.G3369T | p.L1123F | 0.000364 | 0.0013 | NA | 3/1001 | 0/1126 | 0.05 | 0.13 | −2.18 |
| chr2:167108386 | c.C3328T | p.R1110W | 0.000854 | 0.0026 | NA | 1/901 | 0/1091 | 0.01 | 0.6 | 3.92 |
| chr2:167129091 | c.G3136A | p.D1046N | 0.000121 | 0 | NA | 1/1003 | 0/1126 | 1 | 0.003 | 5.42 |
| chr2:167129135 | c.C3092T | p.T1031I | 0 | 0 | NA | 1/1001 | 0/1110 | 0.23 | 0.054 | 5.42 |
| chr2:167129256 | c.G2971T | p.V991L | 0.004177 | 0 | rs4369876 | 21/976 | 2/1119 | 0.08 | 0.004 | −0.949 |
| #chr2:167133540 | c.A2794C | p.M932L | 0.004070 | 0 | rs12478318 | 21/976 | 2/1119 | 0.53 | 0.043 | 5.65 |
This variant was initially filtered out due to poor quality of reads for that position. Capillary sequencing confirmed the presence of M932L in all subjects with the V991L variant.
The TDT was performed on 32 affected siblings of cases that had rare gene disrupting variants in the SCN9A LII-III region. For 25 affected siblings proband had SCN9AM932L/V991L variant and for 7 siblings proband had the other rare LII-III variants. We found over-transmission of rare alleles to affected siblings for SCN9AM932L/V991L variant (17 minor alleles, 8 major alleles, Chi-square 3.24, p=0.071) and for other LII-III variants (5 minor alleles, 2 major alleles, Chi-square 1.28, p=0.256). Combining SCN9AM932L/V991L and other rare LII-III variants the over-transmission was statistically significant (22 minor alleles, 10 major alleles, Chi-square 4.5, p = 0.033).
As NaV1.7M932L/V991L variant, which was present in 21 cases and 2 controls, carried the bulk of genetic association, we performed functional analysis of the NaV1.7M932L/V991L variant in cultured cortical neurons (Figure 2). NaV1.7M932L/V991L variant did not affect peak current amplitude, voltage dependence of activation, voltage dependence of fast inactivation or channel availability during repetitive depolarization at 50 Hz (Figure 2a-d). However, neurons expressing NaV1.7 M932L/V991L fired less action potentials during 1 sec long depolarizing current injection, indicating reduced excitability (Figure 2e-f).
Figure 2. Properties of NaV1.7WT and NaV1.7M932L/V991L expressed in cultured cortical neurons.

(a) Representative set of sodium current traces from cortical neurons expressing NaV1.7WT and NaV1.7M932L/V991L. (b) Mean current-voltage (I-V) relationships of peak currents. (c) Voltage dependence of activation (right curves) and the voltage dependence of steady-state fast inactivation. (d) Mean normalized currents during 100 depolarizations to 0 mV at 50 Hz. NaV1.7WT, n= 4, NaV1.7M932L/V991L, n=5. (e-f) Firing of cortical neurons expressing NaV1.7WT and NaV1.7M932L/V991L NaV1.7M932L/V991L. (e) Representative firing in response to 100 pA depolarizing current injection. (f) Average number of action potentials (AP) in response to 1 s depolarizing current injection at the indicated intensity. NaV1.7WT, n= 7, NaV1.7M932L/V991L, n=7.
Phenotype analysis
Available phenotypic information for the exome-sequenced families is presented in Supplementary Information. Genotype-phenotype correlation analysis for 29 subjects that had variants in SCN9A LII-III did not reveal any specific phenotype characteristics for available measures. Phenotype information is shown in Supplementary Table 3.
Bioinformatics analysis of SCN9A expression, gene co-expression and biological function enrichment
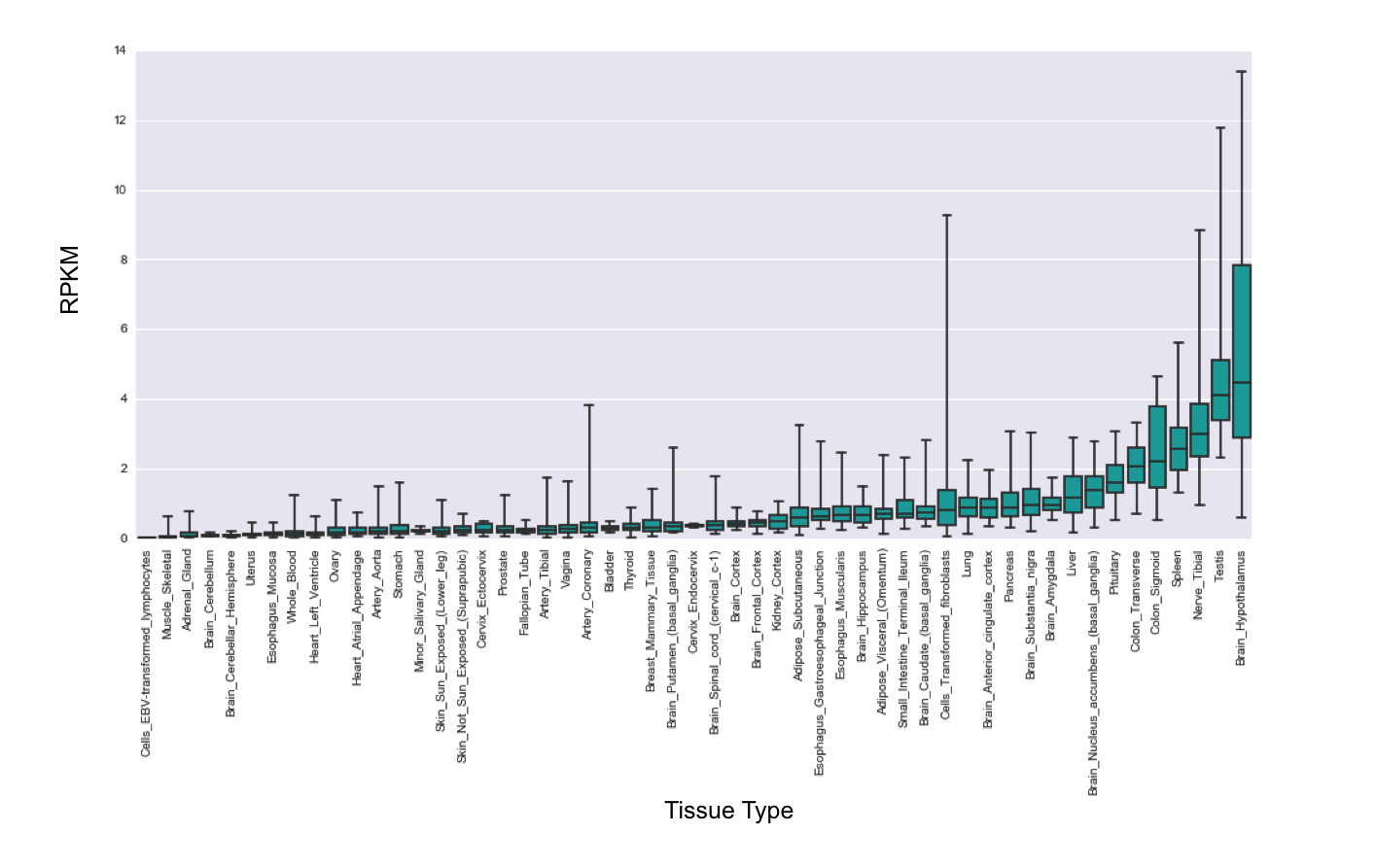
In humans, the GTeX data38 also confirms the expression of SCN9A in the brain (Supplementary Figure 3). Brain co-expression analysis of GTeX data identified 538 genes positively co-expressed with SCN9A. DAVID analysis identified 7 enriched biological processes for SCN9A co-expressed genes: forebrain development and neuronal differentiation, regulation of secretion and hormone levels, cell-cell signaling, and feeding and adult behavior (Supplementary Table 4). The most significant enrichment was observed for feeding behaviors (p= 8.8×10−6, 7.91 fold enrichment, 15 genes) and forebrain differentiation (p=0.002, 4.06 fold enrichment, 17 genes).
DISCUSSION
While recent studies have identified multiple genetic factors involved in sporadic, de novo autism, genetic factors contributing to familial autism are less well characterized. Here, to identify genes that harbor rare variants that contribute to familial autism, we adopted a three step design that combines family based whole-exome sequencing for candidate gene identification, followed by electrophysiology studies to show functional relevance of identified variants, and additional genetic analysis to provide statistical evidence for the association with ASD. For the exome family analysis, we narrowed the number of identified genes by focusing on variants that are both private and protein disrupting. In this way we identified 11 candidate variants in 5 families. Bioinformatics analysis suggested that four of these variants were more likely to have functional effects. A variant-burden analysis of four genes was significant for rare protein disrupting variants in the second intracellular loop of SCN9A. This is the same LII-III loop that harbored a SCN9AC1143F variant in one family that was exome sequenced. In further support of the association with autism, for LII-III variants we show preferential transmission to affected siblings as well. SCN9A is in tight linkage disequilibrium with a cluster of sodium channel genes on chromosome 2q24.3 (SCN1A, SCN2A, SCN3A, SCN7A) and SCN1A is less then 200 kb away. This raises the possibility that the association we have identified with SCN9A might be due to mutations in SCN1A and SCN2A genes that are implicated in autism as well. Although we were not able to directly sequence SCN1A and SCN2A genes, we feel that this scenario is very unlikely as the main phenotype associated with SCN1A (OMIM: 182389) and SCN2A (OMIM *182390) mutations are seizures, which are not reported in our subjects carrying SCN9A variants. However we cannot exclude the possibility of SCN1A involvement as mutations in this gene were associated with autism without seizures as well47, 48. In addition to genetic association, both NaV1.7C1143F variant identified in a single exome-sequenced family, and NaV1.7M932L/V991L variant identified in association analysis, demonstrated a partial loss-of-function effect when expressed in cortical neurons.
We did not find genetic evidence for association in the four other families studied by exome sequencing. This is not surprising as ASD is highly heterogeneous. In these families other genetic mechanisms, that we have not evaluated, might be contributory. Such mechanisms include disruptions of noncoding regulatory DNA49, genetic heterogeneity within the family50 and polygenic inheritance51.
Functional consequences of the NaV1.7C1143F and NaV1.7M932L/V991L variants
Electrophysiological analysis of NaV1.7C1143F expressed in tsA-201 human embryonic kidney cell lines with and without auxiliary β1 and β2 subunits suggests, at most, a small effect on basic sodium channel functions. In neurons, NaV1.7 channels have many additional interacting partners, and indeed, in cortical neurons the rate of recovery from fast inactivation was 2-fold slower for NaV1.7C1143F channels. Such slowed recovery is expected to result in longer periods of reduced sodium channel availability during and following trains of stimuli. In agreement with this expectation, sodium current amplitude conducted by NaV1.7C1143F expressing neurons declined significantly more compared to NaV1.7WT during repetitive depolarization at 20 Hz and 50 Hz. These biophysical alterations are both expected to result in reduced firing and excitability of cortical neurons. The experiments in current-clamp mode revealed that neurons expressing NaV1.7C1143F had lower input resistance and increased rheobase, so larger current injection was needed to evoke action potentials, resulting in decreased action potential firing (Figure 1).
NaV1.7M932L/V991L variant has been implicated before in neuropathic pain syndromes22 and is associated with partial deletion of pain perception52, suggesting both increased and decreased neuronal firing. Here, electrophysiological analysis of NaV1.7M932L/V991L did not detect changes in the biophysical properties of the currents. However, similarly to NaV1.7C1143F, cortical neurons expressing NaV1.7M932L/V991L fired less action potentials, indicating reduced excitability (Figure 2). While we do not completely understand the causes for reduced firing in the NaV1.7M932L/V991L variant, it might be related to changes in cellular localization or neuronal maturation. LII-III contains binding sites for ankyrin-G that are crucial for the localization of sodium channels at the axon initial segment43. Neuronal excitability was therefore examined in mature neurons (after the formation of axon and dendrites). In contrast, biophysical characterization was performed on immature cultures (three to four days in-vitro) in order to avoid space clamp problems, but potentially before the manifestation of functional alterations. Additionally, unlike the C1143F variant, which was found to be highly conserved and predicted to be damaging with 3 bioinformatics tools (Table 1), strong evidence for conservation and potential impact on protein function is lacking for the M932L and V991L variants (Table 3). Furthermore, functional implication of M932L/V991L variant might strongly depend on genetic background, as indicated by its frequency in different populations. Based on ExAC database that aggregates multiple studies (www.ExAC.broadinstitute) M932L and V991L variants are rare in European (MAF=0.00236) and African American (MAF=0.0029) populations with frequencies that are comparable to our Caucasian controls (MAF=0.0016). However, M932L and V991L variants have higher frequency in East (MAF=0.061) and South Asians (MAF=0.012), and in Latino population variant frequency (MAF=0.23) far exceeds the frequency we have observed in our Caucasian cases (MAF=0.022). However, for sodium channel genes, genetic background is known to play a role in seizure phenotype expression53 and in mouse model SCN1A mutations result in severe epilepsy and impaired cognitive abilities in the C57BL/6 strain and in extremely rare spontaneous seizures with normal cognitive function on the 129/SvJ background54.
Nevertheless, although the evidence for the disease-causing role of NaV1.7M932L/V991L variant is tempered due to high population frequency of the variant in Asian and Latino populations and lack of strong evolutionary conservation, we have identified 6 additional rare LII-III variants that support association with ASD. Overall our findings indicate that ASD associated SCN9A variants results in partial loss-of-function of NaV1.7 channel, causing decreased action potential firing and altering the input-output relationships in circuits whose neurons express SCN9A.
How might NaV1.7 channels contribute to autism?
Although traditionally considered as a gene that is important for the peripheral nervous system, there is increased understanding that SCN9A has a role in the CNS as well. Missense mutations in SCN9A are associated with familial febrile seizures55, a benign form of epilepsy. Similarly, mutations in SCN9A can modify the severity of Dravet Syndrome, a rare genetic intractable epilepsy syndrome caused by complete loss-of-function (LOF) mutations in NaV1.1 channels, which are prominent in interneurons of the brain55, 56. One emerging concept from studies of mouse models of autism spectrum disorders, including idiopathic autism57, Rett Syndrome58, Fragile X Syndrome59, and Dravet Syndrome60, is that core autistic-like behaviors are associated with an increased ratio of excitatory to inhibitory neurotransmission in the brain. Most of the experimental evidence suggests increased excitation. In contrast, in Dravet Syndrome mice, autistic-like behaviors are caused by loss-of-function mutations in NaV1.1 channels and consequent selective impairment of action potential firing in GABAergic inhibitory neurons 60, 61. These autistic-like behaviors are rescued by enhancement of inhibitory neurotransmission by treatment with low doses of clonazepam, a positive allosteric modulator of GABAA receptors60. Data presented here (Supplementary Figure 2) confirm the expression of NaV1.7 in the cortex. The single cell neuronal gene transcription analysis by Zeisel et al.44 demonstrated that the NaV1.7 channel is expressed in both GABAergic and glutamatergic neurons, but with higher levels of expression in interneurons44. Thus, by analogy to loss of function mutations in NaV1.1, a hypothesis can be made that rare variants in NaV1.7 decrease the firing of a specific set of GABAergic neurons that are important in control of social behaviors. Furthermore, using GTeX data we show that SCN9A is co-expressed with genes that are enriched for forebrain neuronal differentiation. These genes include transcription factors DLX1 and DLX2 that are essential for the production of forebrain GABAergic interneurons during embryonic development62 and have been implicated in autism in a genetic association study63. These observations support a hypothesis that functional variants in SCN9A channels contribute to autism phenotype by affecting a specific set of GABAergic neurons that are important in control of social behaviors.
Our analyses of GTeX data indicate that SCN9A is highly expressed in the hypothalamus, a brain region that has been implicated in autism64, 65. Among other roles, the hypothalamus is responsible for synthesizing the behaviorally important hormone oxytocin that has a role in social bonding behaviors and has been implicated in autism as well66. Additionally, the hypothalamus is a crucial regulator of feeding behavior67. We have found that genes co-expressed with SCN9A are enriched for functions related to feeding behaviors and the function of NaV1.7 channels in hypothalamic neurons was shown to be important for body weight regulation68. This indicates that, in addition to the role in GABAergic neurotransmission, the role of SCN9A in autism might be mediated through changes in hypothalamic functions, which in turn can affect multiple hormonally regulated processes that are frequently disrupted in autism such as oxytocin mediated social interactions69 and feeding behaviors70.
Further directions
Until now, truncating mutations in NaV1.7 with full loss-of-function effects that completely abolish NaV1.7 currents were found to be responsible for rare autosomal-recessive congenital insensitivity to pain (CIP)24-27. Fewer than 100 individuals with CIP were reported in the literature and phenotype description in these reports indicate that beyond CIP other sensory modalities are unaffected. Interestingly, extensive neuropsychological testing was reported for a single 9 year old girl with partial CIP, caused by compound heterozygous partial loss of function SCN9A variant. Based on parental report this individual had impaired social skills and below average levels of empathy71. Although in this report presence of ASD was not evaluated, presence of deficits in social skills and empathy indicate the need for more detailed evaluation for ASD in individuals with CIP. In addition, our study has shown the association of heterozygous partial loss of function variants with ASD. This indicates that carriers of SCN9A CIP variants might be at increased risk for autism as well, and formal ASD screening and evaluation in patients with CIP will allow to establish the diagnosis, which in turn, will improve the care and treatments such individuals are receiving.
In our sample, we were unable to detect any specific autism subtype based on demographic, ADOS variables and clinical characteristics. However, our analysis of SCN9A brain co-expressed genes indicates that feeding behaviors might be a prominent component of the SCN9A associated phenotype. We did not have information about feeding behaviors for our cohort. Our results indicate that ASD subjects with deregulated feeding behaviors might be enriched for SCN9A variants. In addition dysregulated-feeding behaviors could be a phenotype that is suitable for genetic studies and it might be valuable to add such phenotype to genetic studies of autism.
The physiological role of LII-III region in voltage gated sodium channels is not well understood. Evidence from human genetic studies implicates variants in LII-III in a growing number of hereditary disorders including cardiac arrhythmias72-74 and epilepsy 56; and we add the association with ASD. Functional analysis demonstrating effects on channel inactivation72-75, recovery from fast inactivation and neuronal excitability (Figure 1). Further functional studies on SCN9A and other brain expressed NaV family genes that examine electrophysiological properties and subcellular localization in specific cell subtypes like GABAergic neurons are warranted to further elucidate the mechanism by which NaV channels LII-III variants increase the risk for ASD. In addition, pharmacological interventions in SCN1A-null mice that reverse ASD-like symptoms6060 indicate that for individuals with ASD and NaV mutations ASD, symptoms might be responsive to pharmacological interventions as well.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
ACKNOWLEDGEMENTS
We acknowledge the clinicians, organizations and families that contributed to data and samples used in this study (https://www.nimhgenetics.org/acknowledgements.php). DNA samples and phenotype information were obtained from NIMH Center for Collaborative Genomic Research on Mental Disorders. AP acknowledges Dr. Fernando Gelin for his help in generating Supplementary Figure 3. MR and WAC acknowledge Dr. Gilbert Martinez for assistance with molecular biology. Research reported in this publication was supported by funding from the National Institute of Mental Health and the National Institute of Neurological Disorders and Stroke under award numbers R01 MH092367, R01 NS25704 from the National Institutes of Health and research grant number 240243 from the Simons Foundation. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the Simons Foundation.
Footnotes
CONFILCT OF INTEREST
The authors declare no conflict of interest.
REFERENCES
- 1.Association AP. Diagnostic and Statistical Manual of Mental Disorders: Dsm-5. Amer Psychiatric Pub Incorporated; 2013. [Google Scholar]
- 2.Tuchman R, Rapin I. Epilepsy in autism. The Lancet Neurology. 2002;1(6):352–358. doi: 10.1016/s1474-4422(02)00160-6. [DOI] [PubMed] [Google Scholar]
- 3.Hoischen A, Krumm N, Eichler EE. Prioritization of neurodevelopmental disease genes by discovery of new mutations. Nature neuroscience. 2014;17(6):764–772. doi: 10.1038/nn.3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Iossifov I, O'Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014;515(7526):216–221. doi: 10.1038/nature13908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pinto D, Delaby E, Merico D, Barbosa M, Merikangas A, Klei L, et al. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am J Hum Genet. 2014;94(5):677–694. doi: 10.1016/j.ajhg.2014.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berg JM, Geschwind DH. Autism genetics: searching for specificity and convergence. Genome biology. 2012;13(7):247. doi: 10.1186/gb-2012-13-7-247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. 2014;515(7526):209–215. doi: 10.1038/nature13772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Devlin B, Scherer SW. Genetic architecture in autism spectrum disorder. Current opinion in genetics & development. 2012;22(3):229–237. doi: 10.1016/j.gde.2012.03.002. [DOI] [PubMed] [Google Scholar]
- 9.Freitag CM. The genetics of autistic disorders and its clinical relevance: a review of the literature. Mol Psychiatry. 2007;12(1):2–22. doi: 10.1038/sj.mp.4001896. [DOI] [PubMed] [Google Scholar]
- 10.Shi L, Zhang X, Golhar R, Otieno FG, He M, Hou C, et al. Whole-genome sequencing in an autism multiplex family. Molecular autism. 2013;4(1):8. doi: 10.1186/2040-2392-4-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chahrour MH, Yu TW, Lim ET, Ataman B, Coulter ME, Hill RS, et al. Whole-exome sequencing and homozygosity analysis implicate depolarization-regulated neuronal genes in autism. PLoS genetics. 2012;8(4):e1002635. doi: 10.1371/journal.pgen.1002635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Toma C, Torrico B, Hervas A, Valdes-Mas R, Tristan-Noguero A, Padillo V, et al. Exome sequencing in multiplex autism families suggests a major role for heterozygous truncating mutations. Mol Psychiatry. 2014;19(7):784–790. doi: 10.1038/mp.2013.106. [DOI] [PubMed] [Google Scholar]
- 13.Chapman NH, Nato AQ, Jr., Bernier R, Ankenman K, Sohi H, Munson J, et al. Whole exome sequencing in extended families with autism spectrum disorder implicates four candidate genes. Human genetics. 2015;134(10):1055–1068. doi: 10.1007/s00439-015-1585-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sham PC, Purcell SM. Statistical power and significance testing in large-scale genetic studies. Nature reviews Genetics. 2014;15(5):335–346. doi: 10.1038/nrg3706. [DOI] [PubMed] [Google Scholar]
- 15.Weiss LA, Escayg A, Kearney JA, Trudeau M, MacDonald BT, Mori M, et al. Sodium channels SCN1A, SCN2A and SCN3A in familial autism. Mol Psychiatry. 2003;8(2):186–194. doi: 10.1038/sj.mp.4001241. [DOI] [PubMed] [Google Scholar]
- 16.Catterall WA. Voltage-gated sodium channels at 60: structure, function and pathophysiology. The Journal of physiology. 2012;590(Pt 11):2577–2589. doi: 10.1113/jphysiol.2011.224204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Catterall WA, Kalume F, Oakley JC. NaV1.1 channels and epilepsy. The Journal of physiology. 2010;588(Pt 11):1849–1859. doi: 10.1113/jphysiol.2010.187484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Imbrici P, Camerino DC, Tricarico D. Major channels involved in neuropsychiatric disorders and therapeutic perspectives. Frontiers in genetics. 2013;4:76. doi: 10.3389/fgene.2013.00076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.O'Brien JE, Meisler MH. Sodium channel SCN8A (Nav1.6): properties and de novo mutations in epileptic encephalopathy and intellectual disability. Frontiers in genetics. 2013;4:213. doi: 10.3389/fgene.2013.00213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Drenth JP, te Morsche RH, Guillet G, Taieb A, Kirby RL, Jansen JB. SCN9A mutations define primary erythermalgia as a neuropathic disorder of voltage gated sodium channels. The Journal of investigative dermatology. 2005;124(6):1333–1338. doi: 10.1111/j.0022-202X.2005.23737.x. [DOI] [PubMed] [Google Scholar]
- 21.Fertleman CR, Baker MD, Parker KA, Moffatt S, Elmslie FV, Abrahamsen B, et al. SCN9A mutations in paroxysmal extreme pain disorder: allelic variants underlie distinct channel defects and phenotypes. Neuron. 2006;52(5):767–774. doi: 10.1016/j.neuron.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 22.Faber CG, Hoeijmakers JG, Ahn HS, Cheng X, Han C, Choi JS, et al. Gain of function Nanu1.7 mutations in idiopathic small fiber neuropathy. Annals of neurology. 2012;71(1):26–39. doi: 10.1002/ana.22485. [DOI] [PubMed] [Google Scholar]
- 23.Reimann F, Cox JJ, Belfer I, Diatchenko L, Zaykin DV, McHale DP, et al. Pain perception is altered by a nucleotide polymorphism in SCN9A. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(11):5148–5153. doi: 10.1073/pnas.0913181107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kurban M, Wajid M, Shimomura Y, Christiano AM. A nonsense mutation in the SCN9A gene in congenital insensitivity to pain. Dermatology (Basel, Switzerland) 2010;221(2):179–183. doi: 10.1159/000314692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nilsen KB, Nicholas AK, Woods CG, Mellgren SI, Nebuchennykh M, Aasly J. Two novel SCN9A mutations causing insensitivity to pain. Pain. 2009;143(1-2):155–158. doi: 10.1016/j.pain.2009.02.016. [DOI] [PubMed] [Google Scholar]
- 26.Goldberg YP, MacFarlane J, MacDonald ML, Thompson J, Dube MP, Mattice M, et al. Loss-of-function mutations in the Nav1.7 gene underlie congenital indifference to pain in multiple human populations. Clinical genetics. 2007;71(4):311–319. doi: 10.1111/j.1399-0004.2007.00790.x. [DOI] [PubMed] [Google Scholar]
- 27.Ahmad S, Dahllund L, Eriksson AB, Hellgren D, Karlsson U, Lund PE, et al. A stop codon mutation in SCN9A causes lack of pain sensation. Human molecular genetics. 2007;16(17):2114–2121. doi: 10.1093/hmg/ddm160. [DOI] [PubMed] [Google Scholar]
- 28.Wijsman EM. The role of large pedigrees in an era of high-throughput sequencing. Human genetics. 2012;131(10):1555–1563. doi: 10.1007/s00439-012-1190-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.O'Roak BJ, Deriziotis P, Lee C, Vives L, Schwartz JJ, Girirajan S, et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet. 2011;43(6):585–589. doi: 10.1038/ng.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boyle EA, O'Roak BJ, Martin BK, Kumar A, Shendure J. MIPgen: optimized modeling and design of molecular inversion probes for targeted resequencing. Bioinformatics. 2014;30(18):2670–2672. doi: 10.1093/bioinformatics/btu353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic acids research. 2010;38(16):e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Slutsky I, Sadeghpour S, Li B, Liu G. Enhancement of synaptic plasticity through chronically reduced Ca2+ flux during uncorrelated activity. Neuron. 2004;44(5):835–849. doi: 10.1016/j.neuron.2004.11.013. [DOI] [PubMed] [Google Scholar]
- 35.Manichaikul A, Mychaleckyj JC, Rich SS, Daly K, Sale M, Chen WM. Robust relationship inference in genome-wide association studies. Bioinformatics. 2010;26(22):2867–2873. doi: 10.1093/bioinformatics/btq559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.He Z, O'Roak BJ, Smith JD, Wang G, Hooker S, Santos-Cortez RL, et al. Rare-variant extensions of the transmission disequilibrium test: application to autism exome sequence data. Am J Hum Genet. 2014;94(1):33–46. doi: 10.1016/j.ajhg.2013.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Human genomics The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015;348(6235):648–660. doi: 10.1126/science.1262110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 40.Michael Rutter ALC, Catherine Lord . ADI-R Autism Diagnostic Interview-Revised, vol. Manual. Western Psychological Services; Los Angeles: 2003. [Google Scholar]
- 41.Ho C, Zhao J, Malinowski S, Chahine M, O'Leary ME. Differential expression of sodium channel beta subunits in dorsal root ganglion sensory neurons. The Journal of biological chemistry. 2012;287(18):15044–15053. doi: 10.1074/jbc.M111.333740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Goldfarb M, Schoorlemmer J, Williams A, Diwakar S, Wang Q, Huang X, et al. Fibroblast growth factor homologous factors control neuronal excitability through modulation of voltage-gated sodium channels. Neuron. 2007;55(3):449–463. doi: 10.1016/j.neuron.2007.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Leterrier C, Brachet A, Fache MP, Dargent B. Voltage-gated sodium channel organization in neurons: protein interactions and trafficking pathways. Neuroscience letters. 2010;486(2):92–100. doi: 10.1016/j.neulet.2010.08.079. [DOI] [PubMed] [Google Scholar]
- 44.Zeisel A, Munoz-Manchado AB, Codeluppi S, Lonnerberg P, La Manno G, Jureus A, et al. Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science. 2015;347(6226):1138–1142. doi: 10.1126/science.aaa1934. [DOI] [PubMed] [Google Scholar]
- 45.Raymond CK, Castle J, Garrett-Engele P, Armour CD, Kan Z, Tsinoremas N, et al. Expression of alternatively spliced sodium channel alpha-subunit genes. Unique splicing patterns are observed in dorsal root ganglia. The Journal of biological chemistry. 2004;279(44):46234–46241. doi: 10.1074/jbc.M406387200. [DOI] [PubMed] [Google Scholar]
- 46.Li QS, Cheng P, Favis R, Wickenden A, Romano G, Wang H. SCN9A Variants May be Implicated in Neuropathic Pain Associated With Diabetic Peripheral Neuropathy and Pain Severity. The Clinical journal of pain. 2015;31(11):976–982. doi: 10.1097/AJP.0000000000000205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li J, Shi M, Ma Z, Zhao S, Euskirchen G, Ziskin J, et al. Integrated systems analysis reveals a molecular network underlying autism spectrum disorders. Molecular systems biology. 2014;10:774. doi: 10.15252/msb.20145487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Alvarez-Mora MI, Calvo Escalona R, Puig Navarro O, Madrigal I, Quintela I, Amigo J, et al. Comprehensive molecular testing in patients with high functioning autism spectrum disorder. Mutation research. 2016;784-785:46–52. doi: 10.1016/j.mrfmmm.2015.12.006. [DOI] [PubMed] [Google Scholar]
- 49.Turner TN, Hormozdiari F, Duyzend MH, McClymont SA, Hook PW, Iossifov I, et al. Genome Sequencing of Autism-Affected Families Reveals Disruption of Putative Noncoding Regulatory DNA. Am J Hum Genet. 2016;98(1):58–74. doi: 10.1016/j.ajhg.2015.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yuen RK, Thiruvahindrapuram B, Merico D, Walker S, Tammimies K, Hoang N, et al. Whole-genome sequencing of quartet families with autism spectrum disorder. Nature medicine. 2015;21(2):185–191. doi: 10.1038/nm.3792. [DOI] [PubMed] [Google Scholar]
- 51.Talkowski ME, Minikel EV, Gusella JF. Autism spectrum disorder genetics: diverse genes with diverse clinical outcomes. Harvard review of psychiatry. 2014;22(2):65–75. doi: 10.1097/HRP.0000000000000002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yuan R, Zhang X, Deng Q, Si D, Wu Y, Gao F, et al. Two novel SCN9A gene heterozygous mutations may cause partial deletion of pain perception. Pain medicine (Malden, Mass) 2011;12(10):1510–1514. doi: 10.1111/j.1526-4637.2011.01237.x. [DOI] [PubMed] [Google Scholar]
- 53.Ragsdale DS. How do mutant Nav1.1 sodium channels cause epilepsy? Brain research reviews. 2008;58(1):149–159. doi: 10.1016/j.brainresrev.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 54.Rubinstein M, Westenbroek RE, Yu FH, Jones CJ, Scheuer T, Catterall WA. Genetic background modulates impaired excitability of inhibitory neurons in a mouse model of Dravet syndrome. Neurobiology of disease. 2015;73:106–117. doi: 10.1016/j.nbd.2014.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Singh NA, Pappas C, Dahle EJ, Claes LR, Pruess TH, De Jonghe P, et al. A role of SCN9A in human epilepsies, as a cause of febrile seizures and as a potential modifier of Dravet syndrome. PLoS genetics. 2009;5(9):e1000649. doi: 10.1371/journal.pgen.1000649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mulley JC, Hodgson B, McMahon JM, Iona X, Bellows S, Mullen SA, et al. Role of the sodium channel SCN9A in genetic epilepsy with febrile seizures plus and Dravet syndrome. Epilepsia. 2013;54(9):e122–126. doi: 10.1111/epi.12323. [DOI] [PubMed] [Google Scholar]
- 57.Han S, Tai C, Jones CJ, Scheuer T, Catterall WA. Enhancement of inhibitory neurotransmission by GABAA receptors having alpha2,3-subunits ameliorates behavioral deficits in a mouse model of autism. Neuron. 2014;81(6):1282–1289. doi: 10.1016/j.neuron.2014.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dani VS, Chang Q, Maffei A, Turrigiano GG, Jaenisch R, Nelson SB. Reduced cortical activity due to a shift in the balance between excitation and inhibition in a mouse model of Rett syndrome. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(35):12560–12565. doi: 10.1073/pnas.0506071102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.D'Hulst C, Kooy RF. The GABAA receptor: a novel target for treatment of fragile X? Trends Neurosci. 2007;30(8):425–431. doi: 10.1016/j.tins.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 60.Han S, Tai C, Westenbroek RE, Yu FH, Cheah CS, Potter GB, et al. Autistic-like behaviour in Scn1a+/- mice and rescue by enhanced GABA-mediated neurotransmission. Nature. 2012;489(7416):385–390. doi: 10.1038/nature11356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rubinstein M, Han S, Tai C, Westenbroek RE, Hunker A, Scheuer T, et al. Dissecting the phenotypes of Dravet syndrome by gene deletion. Brain. 2015;138(Pt 8):2219–2233. doi: 10.1093/brain/awv142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cobos I, Calcagnotto ME, Vilaythong AJ, Thwin MT, Noebels JL, Baraban SC, et al. Mice lacking Dlx1 show subtype-specific loss of interneurons, reduced inhibition and epilepsy. Nature neuroscience. 2005;8(8):1059–1068. doi: 10.1038/nn1499. [DOI] [PubMed] [Google Scholar]
- 63.Liu X, Novosedlik N, Wang A, Hudson ML, Cohen IL, Chudley AE, et al. The DLX1and DLX2 genes and susceptibility to autism spectrum disorders. European journal of human genetics : EJHG. 2009;17(2):228–235. doi: 10.1038/ejhg.2008.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Biran J, Tahor M, Wircer E, Levkowitz G. Role of developmental factors in hypothalamic function. Frontiers in neuroanatomy. 2015;9:47. doi: 10.3389/fnana.2015.00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jacobson L. Hypothalamic-pituitary-adrenocortical axis: neuropsychiatric aspects. Comprehensive Physiology. 2014;4(2):715–738. doi: 10.1002/cphy.c130036. [DOI] [PubMed] [Google Scholar]
- 66.Guastella AJ, Hickie IB. Oxytocin Treatment, Circuitry and Autism: A Critical Review of the Literature Placing Oxytocin into the Autism Context. Biological psychiatry. 2015 doi: 10.1016/j.biopsych.2015.06.028. [DOI] [PubMed] [Google Scholar]
- 67.Sestan-Pesa M, Horvath TL. Metabolism and Mental Illness. Trends in molecular medicine. 2016;22(2):174–183. doi: 10.1016/j.molmed.2015.12.003. [DOI] [PubMed] [Google Scholar]
- 68.Branco T, Tozer A, Magnus CJ, Sugino K, Tanaka S, Lee AK, et al. Near-Perfect Synaptic Integration by Nav1.7 in Hypothalamic Neurons Regulates Body Weight. Cell. 2016;165(7):1749–1761. doi: 10.1016/j.cell.2016.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Green JJ, Hollander E. Autism and oxytocin: new developments in translational approaches to therapeutics. Neurotherapeutics : the journal of the American Society for Experimental NeuroTherapeutics. 2010;7(3):250–257. doi: 10.1016/j.nurt.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mari-Bauset S, Zazpe I, Mari-Sanchis A, Llopis-Gonzalez A, Morales-Suarez-Varela M. Food selectivity in autism spectrum disorders: a systematic review. Journal of child neurology. 2014;29(11):1554–1561. doi: 10.1177/0883073813498821. [DOI] [PubMed] [Google Scholar]
- 71.Staud R, Price DD, Janicke D, Andrade E, Hadjipanayis AG, Eaton WT, et al. Two novel mutations of SCN9A (Nav1.7) are associated with partial congenital insensitivity to pain. European journal of pain (London, England) 2011;15(3):223–230. doi: 10.1016/j.ejpain.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mohler PJ, Rivolta I, Napolitano C, LeMaillet G, Lambert S, Priori SG, et al. Nav1.5 E1053K mutation causing Brugada syndrome blocks binding to ankyrin-G and expression of Nav1.5 on the surface of cardiomyocytes. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(50):17533–17538. doi: 10.1073/pnas.0403711101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hsueh CH, Chen WP, Lin JL, Tsai CT, Liu YB, Juang JM, et al. Distinct functional defect of three novel Brugada syndrome related cardiac sodium channel mutations. Journal of biomedical science. 2009;16:23. doi: 10.1186/1423-0127-16-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ackerman MJ, Siu BL, Sturner WQ, Tester DJ, Valdivia CR, Makielski JC, et al. Postmortem molecular analysis of SCN5A defects in sudden infant death syndrome. Jama. 2001;286(18):2264–2269. doi: 10.1001/jama.286.18.2264. [DOI] [PubMed] [Google Scholar]
- 75.Kuzmenkin A, Jurkat-Rott K, Lehmann-Horn F, Mitrovic N. Impaired slow inactivation due to a polymorphism and substitutions of Ser-906 in the II-III loop of the human Nav1.4 channel. Pflugers Archiv : European journal of physiology. 2003;447(1):71–77. doi: 10.1007/s00424-003-1137-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.