

Abstract
Neural stem/progenitor cells (NPSCs) express receptors for many inflammatory cytokines, with varying effects on differentiation and proliferation depending on the stage of development and the milieu of inflammatory mediators. In primary neurons and astrocytes, we recently showed that interferon gamma (IFNγ), a potent antiviral cytokine that is required for the control and clearance of many central nervous system (CNS) infections, can differentially affect cell survival and cell cycle progression depending upon the cell type and the profile of activated intracellular signaling molecules. Here, we show that IFNγ inhibits proliferation of primary NSPCs through dephosphorylation of the tumor suppressor Retinoblastoma protein (pRb), which is dependent on activation of Signal Transducers and Activators of Transcription-1 (STAT1) signaling pathways. Our results show i) IFNγ inhibits neurosphere growth and proliferation rate in a dosedependent manner; ii) IFNγ blocks cell cycle progression through a late-stage G1/S phase restriction; iii) IFNγ induces phosphorylation and expression of STAT1 and STAT3; iv) IFNγ decreases cyclin E/cdk2 expression and reduces phosphorylation of cyclin D1 and pRb on serine residue 795; and v) the effects of IFNγ on NSPC proliferation, cell cycle protein expression, and pRb phosphorylation are STAT1-dependent. These data define a mechanism by which IFNγ could contribute to a reduction in NSPC proliferation in inflammatory conditions. Further delineation of the effects of inflammatory cytokines on NSPC growth could improve our understanding of how CNS infections and other inflammatory events disrupt brain development and NSPC function.
Keywords: Interferon-gamma, neural stem/progenitor cell, cell cycle, retinoblastoma protein, Stat1
Graphical Abstract
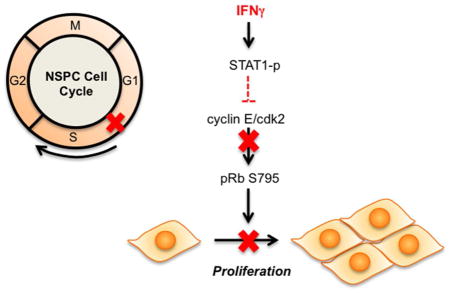
The anti-viral cytokine interferon-gamma (IFNγ) inhibits neural stem/progenitor (NSPC) progression through the cell cycle by reducing phosphorylation of the retinoblastoma protein (pRb) in a STAT1-dependent manner. NSPCs are restricted in the late G1 phase with reduced cyclin E/cdk2 expression and a loss of phosphorylation at serine 795 on pRb.
Introduction
Inflammatory events in the central nervous system can impact on the development of the brain and the function of immature neural cells. In particular, viruses induce neuroinflammatory disease that often results in negative neurological outcomes in neonatal or pediatric infections (Bonthius 2009; Douvoyiannis et al. 2009; Schleede et al. 2013; Tardieu et al. 2000). As part of the antiviral immune response, a milieu of cytokines and chemokines mediate viral clearance in the brain parenchyma. Interferon-gamma (IFNγ), a pluripotent cytokine expressed by activated immune cells, is necessary for the control of many neurotropic viruses (Binder and Griffin 2001; Burdeinick-Kerr and Griffin 2005; Cantin et al. 1999; Fiette et al. 1995; Geiger et al. 1997; Patterson et al. 2002; Pearce et al. 1994). However, the effect of IFNγ on individual neural cells, and the resultant changes in neurodevelopment or differentiation, is largely undefined.
IFNγ activates diverse signaling pathways, which coordinate the expression of IFNγ-responsive genes that are differentially regulated depending upon the cell type and the inflammatory milieu (reviewed in (van Boxel-Dezaire and Stark 2007)). The general paradigm of IFNγ signaling begins when IFNγ engages the IFNγ receptor (IFNGR), which includes two ligand-binding subunits (IFNGR1) and two intracellular subunits (IFNGR2). Upon ligand binding, Janus activated kinases (JAK)-1 and -2 are activated at the receptor, leading to recruitment and phosphorylation of STAT1. STAT1 then translocates to the nucleus to initiate transcription of IFN-responsive genes. Though hundreds of IFN-responsive genes are known, the profile of antiviral gene expression, and the subsequent effects on cellular function and survival, is dependent partially upon cell type. Within the CNS, specific neural cells display unique signaling profiles and cellular responses upon IFNγ stimulation, dependent upon the endogenous expression of the JAK-STAT1 proteins (Burdeinick-Kerr et al. 2009; O’Donnell et al. 2012; Podolsky et al. 2012; Rose et al. 2007; Zhang et al. 2005).
Neural stem/progenitor cells (NSPCs) are capable of self-renewal and differentiation into neurons, astrocytes or oligodendrocytes. During CNS insults (viral infection, stroke, protein misfolding stress), the NSPC pool can expand or contract (Chucair-Elliott et al. 2014; Das and Basu 2011; Das et al. 2011; Deierborg et al. 2010; Gonzalez-Sanchez et al. 2015; Hoglinger et al. 2004; Lee et al. 2011; Sun et al. 2014). During many neurotropic infections, NSPC proliferation and neurogenesis declines (Mutnal et al. 2011a; Ruller et al. 2012; Sharma et al. 2002). These cytostatic responses by NSPCs may be attributed to direct infection by the virus and/or the anti-proliferative effect of inflammatory cytokines (Makela et al. 2010; Mutnal et al. 2011a; Zheng et al. 2014). For example, activated CD8+ lymphocytes, which persist during inflammatory responses to certain CNS viruses, inhibit NSPC proliferation through IFNγ release, suggesting that a single cytokine can impact on NSPC renewal (Hu et al. 2014; Mutnal et al. 2011b).
Our previous work demonstrated that neural cells display tailored cellular responses to IFNγ, depending upon the availability of JAK/STAT signaling molecules. We have observed that IFNγ inhibits proliferation of primary astrocytes in a STAT1-dependent manner, but protects primary neurons from inflammatory insults in STAT1-independent manner (O’Donnell et al. 2015). Here, we examined IFNγ-mediated signaling pathways in primary NSPCs and the consequences on proliferation and survival. Our results demonstrate that IFNγ reduces NSPC proliferation by restricting cell cycle progression in the late G1 phase with site-specific dephosphorylation of the Retinoblastoma protein (pRb), which is dependent upon STAT1 signaling. This study demonstrates that IFNγ profoundly affects NSPC growth through alterations of cell cycle regulatory proteins, and suggests that antiviral cytokine responses may influence the course of neurodevelopment.
Materials and Methods
Culture of neural stem/progenitor cells
Animal use protocols were reviewed and approved by the Duquesne University Institutional Animal Care and Use Committee. STAT1-knockout (STAT1-KO) mice were a generous gift from Dr. Glenn Rall (Fox Chase Cancer Center) (Meraz et al. 1996). The genotype of the STAT1-KO mice used in these experiments was confirmed by PCR analysis of tail biopsy DNA (O’Donnell et al. 2012). Primary neural stem/progenitor cell (NSPC) cultures were prepared from wildtype C57BL/6 mice (WT) or STAT1-KO mouse embryos on embryonic day 12.5 (E12.5) as described previously with modifications (Currle et al. 2007). Briefly, cortical tissue was digested using trypsin-EDTA solution (0.05%, Mediatech, Inc., Manassas, VA) containing bovine serum albumin (0.02%) to obtain a cell suspension. The trypsin was neutralized using an equal volume of soybean trypsin inhibitor (1mg/mL, Sigma-Aldrich, St. Louis, MO), and the cell suspension was passed through a 70μm cell strainer (Thermo Fisher, New York, NY). Cells were centrifuged (1150 rpm for 5 min) and the cell pellet was resuspended in 1 mL NSPC culture medium consisting of DMEM (Mediatech, Inc., Manassas, VA), 1X B27 supplement without vitamin A (Life Technologies, New York, NY), 1X N2 supplement (Thermo Fisher, New York, NY), heparin (2μg/mL, Sigma-Aldrich, St. Louis, MO), epidermal growth factor (20ng/mL, Peprotech, Rocky Hill, NJ) and fibroblast growth factor-2 (10ng/mL, Peprotech, Rocky Hill, NJ). The NSPCs were cultured in suspension at 2*106 cells in 5 mL NSPC culture medium and maintained at 37°C/5% CO2. After 3 days in vitro (DIV), neurospheres were disassociated with trypsin, counted, and used for subsequent experiments.
Neurosphere Assay
NSPCs (passage 1) were cultured in T-25 flasks at 20,000 cells/mL with different concentrations of interferon-gamma (IFNγ; 1–1000 U/ml). IFNγ solutions were prepared by making 10X solutions in NSPC culture medium from a 1000 U/μL stock solution (BD Biosciences, San Jose, CA). As a negative control, heat-inactivated IFNγ (ΔH-IFNγ) was prepared by boiling IFNγ (1000U/mL) at 100°C for 5 min. After IFNγ treatment, the neurospheres were imaged after 3, 5, or 7 DIV using an EVOS FL microscope (Life Technologies, New York, NY). Five fields were taken from each flask at 2X magnification by an observer blinded to experimental conditions. The neurosphere diameter (μm) and area (pixel2) was measured for each neurosphere using Image J software (Version 1.6.0_65). The average diameter for each treatment group was calculated and graphed as percent of the untreated group. Neurosphere area was graphed as a histogram plot using pixel2/neurosphere for each treatment condition.
Bromodeoxyuridine (BrdU) Assay
WT and STAT1-KO NSPCs (100,000 cells/mL) were cultured in T-25 flasks and treated with IFNγ (1–1000 U/ml) for 3 DIV. BrdU staining was performed following manufacturer’s instructions using the BD Pharmingen™-FITC BrdU Flow Kit (BD Biosciences, San Jose, CA). On DIV 3, the neurospheres were treated with BrdU (25μM) solution for 90 min. at 37°C. The neurospheres were dissociated using 0.05% trypsin solution (3 min at 37°C) into a cell suspension. BrdU content was determined by incubating the cells with a FITC-conjugated anti-BrdU antibody (1:50; 559619, BD Biosciences, San Jose, CA) for 30 min. A FITC-conjugated isotype control antibody (1:50; 556649, BD Biosciences, San Jose, CA) was used as a negative control for BrdU staining. The cells were counterstained with 7-Aminoactinomycin D (25μM) for 15 min at 20°C. Single cells (100,000 events per sample) were analyzed by flow cytometry (Accuri C6, BD Biosciences, San Jose, CA) using an initial gate to exclude debris (Supplementary Fig. 1A). Signals for BrdU-FITC (FL1) and 7-AAD (FL3) were compensated by subtracting FL3 from FL1 (2%) and FL1 from FL3 (10%) as per manufacturer’s guidelines prior to analyses. Co-labeling with BrdU and 7-AAD staining allowed for discrimination of cell cycle phases: G0/G1 (7-AAD low, BrdU low), S-phase (BrdU high, 7-AAD intermediate), and G2/M-phase (BrdU low, 7-AAD high).
For the BrdU pulse-chase assay, NSPCs were grown in T-25 flasks with or without IFNγ (100 or 1000 U/mL) treatment as described for the BrdU assay. On DIV 3, the neurospheres were pulsed with BrdU for one hour and washed with 1X PBS. Neurospheres were harvested at 0, 3, 6 and 9 hours post-BrdU washout. Neurospheres for the zero-hour time point were harvested immediately post-BrdU washout. For the other time points, the neurospheres were resuspended in NSPC culture medium and harvested at the respective time points. Following harvest, the protocol for the BrdU assay described above was followed.
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay
The TUNEL assay was performed using the Trivigen Flow TACS Apoptosis kit (R&D Systems, Minneapolis, MN) according to manufacturer’s protocol. Briefly, 50,000 cells/mL NSPCs (WT or STAT1-KO) were cultured with or without IFNγ (100 and 1000 U/ml) for 3 DIV. Neurospheres were collected by centrifuging at 400 rpm and trypsinized as described above. The cells were fixed in 3.7% PFA for 10 min, permeabilized using 100 μL cytonin solution (Trevigen, Gaithersburg, MD, #4876-60-01) for 30 min, and treated with 25μL/sample TdT labeling mixture for 1 hr at 37°C. The cells were then treated with 25μL/sample of strep-fluorescein for 10 min in the dark and washed 1x in PBS. Neurosphere cells (1×10^5 cells/sample) were analyzed by flow cytometry to quantify TUNEL positive cells (FL1 channel) with gating to exclude cellular debris. DNase-treated and TdT enzyme-omitted groups were used as positive and negative controls for the assay.
Western Blot
NSPCs were cultured in T-75 flasks by seeding 50,000 cells/mL in NSPC culture medium. The cells were treated with IFNγ (100 U/mL) or left untreated. On DIV 2, 3, or 5, the neurospheres were collected by centrifugation at 400 rpm and lysed using 200μL 1x Cell Lysis Buffer (Cell Signaling Technology, Danvers) with protease inhibitor cocktail (10 uL/106 cells Sigma-Aldrich, St. Louis, MO). The lysate was centrifuged at 11,000 rpm and the supernatant was stored at −80°C until analysis. The protein concentration of each lysate was measured using the Pierce BCA Protein Assay Kit (Thermo Fisher, New York, NY). For each sample, 20μg of lysate was subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis on NuPAGE 7% Tris-acetate gels (Life Technologies, Grand Island, NY). The gel was blotted onto Immobilon-FL Membrane (Millipore, Billerica, MA) and the membranes were blocked using a 1:1 mixture of 1X phosphate buffered saline/Tween-20 solution (Sigma-Aldrich, St. Louis, MO) and Odyssey blocking buffer (Licor Biosciences, Lincoln, NE) for 60 min at 20°C. The membranes were treated with primary antibody solutions diluted in odyssey blocking buffer (Licor Biosciences, Lincoln, NE) overnight at 4°C on a rocker. The membranes were washed thrice PBS-Tween for 10 min each) and incubated in secondary antibody solutions (goat anti-rabbit 680 or donkey anti-mouse 800 (1:10000); Licor Biosciences, Lincoln, NE) for 60 min at 20°C. The membranes were washed thrice in PBS-Tween and imaged on the Odyssey Infrared Imaging System (Licor Biosciences, Lincoln, NE). Individual bands were quantified using Image Studio software (Licor Biosciences, Lincoln, NE, version 4.0.21). The signal from each band(s) was normalized against the GAPDH signal as a loading control. Primary antibodies used were as follows: anti-phospho STAT1 (Y701, #612133), anti-STAT1 (N-terminus, #610120), anti-STAT3 (610190) from BD Biosciences; anti-phospho STAT1 (S727, #9177) anti-Cyclin D1 (#2978), anti-Cyclin D3 (#2936); pRb S780 (#8180), pRb S807/811 (#8516), pRb S780 (#9307), Rb-total (#9313) anti-phospho STAT3 (#9131) form Cell Signaling Technology; pRb S795 (1:500, ab47474) from Abcam; and anti-cdk4 (MAB8879), anti-cdk2 (#07-631) anti-cyclin E (#07-687) from Millipore. All primary antibody concentrations were 1:1000 unless otherwise stated. Antibody research resource identification numbers (RRIDs) and characterization details are described in Table 1.
Table 1.
Characterization of antibodies used in flow cytometry and western blot analyses.
| Antigen | Molecular weight (kDa) | Immunogen | Source (RRID) | Concentration | Characterization |
|---|---|---|---|---|---|
| anti-nestin PE conjugated antibody | 200 | E. coli-derived recombinant rat Nestin: Met544-Glu820 (Gly756Asp, Ile758Met, Arg572Lys, Ala574Pro, Ile802Met, Arg816Lys) | R and D Systems Cat# IC2736P (AB_2151131) | 1:50 | Mutnal et al. Murine cytomegalovirus infection of neural stem cells alters neurogenesis in the developing brain. Nestin was detected in murine NSPCs using flow cytometry |
| anti-doublecortin antibody | 40 | Amino acids 162–441 at the C-terminus of doublecortin of human origin | Santa Cruz Biotechnology Cat# sc-28939, (AB_2088488) | 1:50 | Maire et al. Pten Loss in Olig2 Expressing Neural Progenitor Cells and Oligodendrocytes Leads to Interneuron Dysplasia and Leukodystrophy. Doublecortin was detected in paraffin frozen sections of mouse brain using immunofluorescence |
| anti-phospho STAT1 | 91,84 | Phosphorylated Human Stat1 (pY701) Peptide | BD Biosciences Cat# 612133 Clone 14/P-STAT1 (AB_399504) | 1:1000 | Lin et al. Distinct Roles of Type I and Type III Interferons in Intestinal Immunity to Homologous and Heterologous Rotavirus Infections. phospho STAT1 was detected in mouse lung, kidney and liver using western blot |
| anti-STAT1-total | 91,84 | STAT1 aa 1–94 | BD Biosciences Cat# 610120 (AB_397526) | 1:1000 | Grant et. al. Zika Virus Targets Human STAT2 to Inhibit Type I Interferon Signaling. STAT1 was detected in 293T cells using western blot |
| anti-STAT3-total, Two isoforms, alpha(85kDa) beta(79 kDa) | 86,79 | Stat3 aa. 1–175 | BD Biosciences Cat# 610190 (AB_397589) | 1:1000 | Ye et al. Wnt/β-catenin and LIF-Stat3 signaling pathways converge on Sp5 to promote mouse embryonic stem cell self-renewal. STAT3 was detected in mouse embryonic stem cells derived from preimplantation embryos using western blot |
| anti-phospho STAT3 (Two isoforms, alpha and beta | 86,79 | Synthetic phosphopeptide corresponding to residues surrounding Tyr705 of mouse Stat3 | Cell Signaling Cat# 9131 (AB_331587) | 1:1000 | Fukuda et al. Potentiation of Astrogliogenesis by STAT3-Mediated Activation of Bone Morphogenetic Protein-Smad Signalling in Neural Stem Cells. phospho STAT3 was detected in mouse neuroepitherlial cells and differentiating cells derived from E.14 embryos using western blot |
| anti-phospho STAT1 (S727) | 91,84 | Synthetic phosphopeptide corresponding to residues surrounding Ser727 of human Stat1 | Cell Signaling Cat# 9177 (AB_2197983) | 1:1000 | Rosowski et al. Toxoplasma gondii Inhibits Gamma Interferon (IFN-γ)- and IFN-β-Induced Host Cell STAT1 Transcriptional Activity by Increasing the Association of STAT1 with DNA. Phospho STAT1 (S727) was detected in 293 FT and HEK cells using western blot. |
| anti-cyclin D1 | 36 | Synthetic peptide corresponding to the carboxy-terminus of human cyclin D1. | Cell Signaling Cat# 2978 (AB_2259616) | 1:1000 | Kosmac et al. Glucocortiocoid Treatment of MCMV Infected Newborn Mice Attenuates CNS Inflammation and Limits Deficits in Cerebellar Development. Cyclin D1 was detected in mouse cerebellar tissue using western blot |
| anti-cyclin D3 | 31 | Recombinant human cyclin D3 corresponding to residues 241–260 | Cell Signaling Cat# 2936 (AB_2070801) | 1:1000 | Barton et al. PD-0332991, a CDK4/6 inhibitor, significantly prolongs survival in a genetically engineered mouse model of brainstem glioma. Cyclin D3 and pRb S780 were detected in glioblasotoma cells using western blot. |
| anti-pRb phospho S780 | 110 | Synthetic peptide corresponding to residues surrounding Ser780 of human Rb protein | Cell Signaling Cat# 8180 (AB_10950972) | 1:1000 | |
| anti-pRb phospho S795 | 110 | Human Rb around the phosphorylation site of serine 795 (P-S-SP-P-L) | Cell Signaling Cat# ab47474 (AB_882295) | 1:500 | Ali et al. The development of a selective cyclin-dependent kinase inhibitor which demonstrates anti-tumor activity. pRb S795 was detected in MCF-7 cells using western blot |
| anti-pRb phospho S807/811 anti-pRb Total |
110 110 |
Synthetic peptide corresponding to residues surrounding Ser807/811 of human Rb protein. Synthetic peptide corresponding to central residues of mouse Rb. |
Cell Signaling Cat# 8516 (AB_11179075) Cell Signaling Cat# 9313(AB_1904119) |
1:1000 1:1000 |
Mo et al. MicroRNA-610 suppresses the proliferation of human glioblastoma cells by repressing CCND2 and AKT3. pRb S807/811 and pRb total were detected in glioblasotoma cells using western blot. |
| anti-cdk4 | 34 | Purified recombinant cdk4 protein | Millipore Cat# MAB8879, Clone DCS-35 (AB_2078833) | 1:1000 | Xu et al. Lin28 modulates cell growth and associates with a subset of cell cycle regulator mRNAs in mouse embryonic stem cells. Cdk4 was detected in mouse embryonic stem cells using western blot. |
| anti-cdk2 | 33 | Peptide corresponding to amino acids 287–298 (C-QDVTKPVPHLRL) of human cdk2 | Millipore Cat# 07-631 (AB_2078833) | 1:1000 | Rus et al. Sublytic Complement Attack Induces Cell Cycle in Oligodendrocytes. Cdk2 was detected in murine oligodendrocytes and oligodendrocyte progenitor cells using western blot. |
| anti-cyclin E | 52 | Synthetic peptide corresponding to amino acids 378–396 (GVLTPPHSSKKQSSEQETE) of rat Cyclin E | Millipore Cat# 07-687 (AB_11213792) | 1:1000 | Ikenishi et al. Cell cycle regulation in mouse heart during embryonic and postnatal stages. Cyclin E was detected in murine embryonic cardiomyocytes using western blot. |
Blots for STAT1 and STAT3 displayed two bands, owing to the presence of two isoforms of each of these proteins. For the STAT proteins, both bands were quantified together. Blots for cyclin D1 also showed two bands, which correspond to phosphorylated (upper band) and unphosphorylated (lower band) forms. These bands were quantified together (for total cyclin D1 levels) and individually (for levels of phosphorylated and unphosphorylated cyclin D1, respectively). All values were STAT proteins and cyclin D1 were normalized to GAPDH for analysis.
Statistical Analysis
Data are presented as the mean ± SEM from 3–4 technical replicates for all experiments. For the neurosphere assay, a one-way ANOVA was performed. To compare the IFNγ-treated groups with the untreated group, a Dunnett’s multiple comparison test was applied. For the BrdU assay and western blot analyses, a one-way ANOVA was performed and a Bonferroni multiple comparisons post-hoc test was applied for pair-wise comparisons between the untreated and IFNγ-treated groups. For the BrdU pulse-chase assay, a two-way ANOVA was performed to compare the differences between the different treatment groups across different time points, with Bonferroni multiple comparisons post-hoc analysis. For the TUNEL assays, a two-tailed Student’s t-test was applied for statistical comparisons. For all other assays, a one-way ANOVA was performed. Differences were deemed significant when p ≤ 0.05. For p values between 0.0001 and 0.05, actual p values are reported. For any values <0.0001, Graphpad software reports the values as “p<0.0001”, which we list as appropriate. All statistical analysis was performed using Graphpad Prism (Version 6.0b).
Outliers were identified using the Grubb’s method and the alpha level was set to 0.05 using Graphpad Prism. In the neurosphere assay for WT NSPCs, five outlier data points were reported corresponding to individual neurospheres. Upon statistical analysis of the cleaned data, the comparisons retained significance. Technical replicates were obtained from at least 3 separate set of dissections. Each replicate was defined as embryonic cortical NSPCs from obtained from one female dam. Typically, each replicate involved NSPCs derived from 6–8 embryos.
Results
IFNγ inhibits neurosphere growth
Our previous work has shown that IFNγ can reduce the growth of mitotically active cells, such as primary fibroblasts and astrocytes, while promoting the survival of primary neurons, depending on the signaling pathways that are invoked (O’Donnell et al. 2015). In NSPCs, we hypothesized that IFNγ would impair NSPC growth due to the availability of STAT1. To assess how IFNγ impacts NSPC proliferation, we measured the diameter and area of primary murine NSPCs grown as neurospheres in suspension culture. The neurospheres were comprised of 95.3% nestin+ cells, with 81.6% of cells in the neurospheres expressing the IFNGR1 subunit of the receptor, as measured by flow cytometry (Supplementary Fig. 1). Neurospheres were exposed to a range of IFNγ concentrations (1–1000 U/ml) for 3, 5, or 7 days in vitro (DIV) (Fig. 1). Neurosphere diameter was significantly smaller in IFNγ-treated cultures in comparison to untreated cells or to cells treated with heat-inactivated IFNγ (ΔH IFNγ; 1000 U/ml) at all concentrations tested (Fig. 1A). At DIV 3, IFNγ limited neurosphere diameter in comparison to untreated controls at both low (1 U/ml IFNγ) and high (1000 U/ml IFNγ) concentrations of IFNγ. Neurospheres were restricted to 89.5%±3.3 of untreated controls at 1 U/ml IFNγ (n=3, p=0.0063) and 59.4%±3.0 of untreated controls at 1000 U/ml (n=3, p<0.0001) (Fig. 1B, left panel). By 7 days post-IFNγ treatment, neurosphere diameter was less than half of the untreated cells at 100 and 1000 U/ml of IFNγ (44.6%±3.2 of untreated; n=3, p<0.0001 and 43.7%±3.2 of untreated; n=3, p<0.0001, respectively). These results show that IFNγ treatment was associated with a prolonged reduction in neurosphere proliferation.
Figure 1. IFNγ inhibits neurosphere growth in a concentration-dependent manner.

Wildtype NSPCs (WT/NSPCs) were treated with IFNγ (1–1000 U/ml) or with heat-inactivated IFNγ (ΔH IFNγ; 1000 U/ml) as a negative control for 3, 5, or 7 days in vitro (DIV). (A) Representative images of neurospheres imaged 5 days post-IFNγ treatment at 2x magnification for different concentrations of IFNγ (Scale bar=250 μm). (B) Quantitation of neurosphere diameter at 3, 5, and 7 days post-IFNγ treatment. The longest diameter of each neurosphere was measured using Image J software. Data was collected from neurospheres in five fields/condition from three biological replicates and graphed as a percentage of untreated controls. Statistical analysis was applied by one-way ANOVA (** p<0.01, **** p<0.0001). (C) Histogram plots of neurosphere area for different concentrations of IFNγ on DIV 5 as measured by pixel2 for each neurosphere. For each condition, five fields/condition were measured using cells from three biological replicates. Frequency distribution of the number of neurospheres corresponding to the indicated neurosphere area in pixel2 was plotted for each condition.
We next determined the distribution of neurosphere sizes during IFNγ treatment using a histogram analysis of neurosphere area. We observed a decrease in median neurosphere area with IFNγ treatment (100 U/mL, DIV 5) as measured by the number of pixels2 in each neurosphere (Fig. 1C). The median neurosphere area was reduced 3-fold from 2054.4 pixel2 in untreated cells to 656.5 pixel2 in IFNγ-treated NSPCs (100 U/ml). Furthermore, the distribution of neurosphere sizes shifted toward a smaller-sized population of neurospheres with the addition of IFNγ, as shown by the leftward shift of the curve in the IFNγ-treated (100 U/ml) group (Fig. 1C, right panel) versus untreated cells (left panel) or cells treated with 1 U/ml IFNγ (middle panel).
NSPCs proliferation is restricted at the G1/S checkpoint in response to IFNγ
Based on the inhibition of neurosphere growth that we observed with IFNγ treatment, we reasoned that cell death and/or changes in the cell cycle could contribute to the reduction in neurosphere size. IFNγ can induce apoptotic or pro-survival pathways depending on the cell type and on the context of other inflammatory mediators in the system (Chawla-Sarkar et al. 2003; Dai and Krantz 1999; Medina-Echeverz et al. 2014; Wall et al. 2003). To determine if apoptosis contributed to the IFNγ-mediated restriction in neurosphere size, we quantified the percentage of apoptotic cells in the neurospheres using the TUNEL assay (Fig. 2A. A significant but modest increase in apoptosis was seen at the highest concentration of IFNγ used in the study (0.45%±0.08 TUNEL+ cells in untreated versus 3%±0.5 TUNEL+ cells at 1000 U/mL IFNγ (n=4, p=0.0037), demonstrating that IFNγ can lead to limited cell death.
Figure 2. IFNγ restricts cell cycle progression in NSPCs and induces minimal apoptosis.

(A) The TUNEL assay was performed on untreated and IFNγ-treated (100 and 1000 U/ml; 72h) NSPCs. The average percentage of total TUNEL+ cells from 3 independent experiments is plotted with SEM (one-way ANOVA, ** p<0.01). (B) WT/NSPCs were treated with IFNγ (1, 100, 1000 U/ml) for 72h and labeled with BrdU and 7-AAD. The intensity of BrdU and 7-AAD staining per cell was assayed by flow cytometry. Cell populations were gated in different phases of cell cycle (S = synthesis phase, G1= gap phase 1, M= mitosis phase, G2= gap phase 2). Representative plots for untreated and IFNγ-treated NSPCs (1000 U/ml) are shown. (C) Quantitation of NSPCs in each cell cycle phase. The average percentage of cells in each cell cycle was plotted with SEM (n=3). Statistical analysis was applied using one-way ANOVA (****p<0.0001; ***p<0.001). (D) Cells in the S-phase gates were analyzed for mean fluorescence intensity (MFI, arbitrary units, A.U.) of the BrdU signal for all treatment groups. The MFI for the BrdU signal of IFNγ-treated cells was compared with untreated controls using a one-way ANOVA (*p<0.05).
We next performed BrdU/7-AAD staining in IFNγ-treated neurospheres in order to identify the stages of the cell cycle. The thymidine analog BrdU is incorporated into newly synthesized DNA, which marks cells in the S (synthesis) phase of the cell cycle. The cells are also counterstained with 7-AAD to measure the total DNA content (Rothaeusler and Baumgarth 2007). We analyzed BrdU/7-AAD-stained NSPCs by flow cytometry, and gated single cells by cell cycle stage (representative plots are shown in Fig. 2B). With IFNγ treatment, there was a dose-dependent decrease in the percentage of cells in the S-phase (48.8%±1.0 untreated versus 33.9%±0.5 IFNγ-treated (1000 U/ml; n=4 p<0.0001)), suggesting that fewer cells were actively synthesizing DNA (Fig. 2C, left panel). Moreover, we observed a dose-dependent increase in the number of cells in the G0/G1 phase (38.7%±1.0 untreated versus 48.9%±1.0 IFNγ-treated (1000 U/ml; n=4, p<0.0001)) and G2/M phase (0.36%±0.1 untreated versus 3.7%±0.6 IFNγ-treated (1000 U/ml; n=4, p=0.001)) (Fig. 2C, middle and right panels respectively). These findings indicate that IFNγ induces a major restriction in cell cycle progression at the G1/S checkpoint, and a significant, but relatively minor, restriction at the G2/M checkpoint.
The changes in cell cycle progression could be explained by a longer G0/G1 phase and/or by a shorter S phase with IFNγ treatment. To determine if IFNγ treatment also altered the duration of the cell cycle phases, we first compared the mean fluorescence intensities (MFI) of NSPCs in S phase (Fig. 2D). The MFI of IFNγ-treated NSPCs in S-phase was higher than the untreated group (n=4; p=0.0491 for 100 U/ml and p=0.0357 for 1000 U/ml). Moreover, when comparing the number of BrdU+ cells in the G2/M phase, we found that the IFNγ-treated groups (100 and 1000 U/ml) had higher numbers of BrdU+ cells than the untreated control (Supplementary Fig. 2), suggesting that more NSPCs were transitioning from the S phase to G2/M with IFNγ treatment. Together, these findings support the notion that IFNγ induces a shorter S phase in NSPCs.
To further investigate the effect of IFNγ on cell cycle progression, we also performed a BrdU pulse chase experiment. IFNγ-treated neurospheres (72h) were pulsed with BrdU for one hour and cell cycle progression was tracked in cells for nine hours post-BrdU washout (Fig. 3A). At all time points, IFNγ treatment decreased the proportion of cells transitioning from G1 to S phase (G1/S) and from S-phase to G2 (S/G2) (Fig. 3B). However, the number of cells BrdU+ cells in the G0/G1 and G2/M phases increased with IFNγ treatment. This observation corroborates the increased MFI of BrdU+ S-phase cells seen with IFNγ treatment (Fig. 2D), suggesting that IFNγ leads to a shortened S-phase. These studies also suggest that IFNγ treatment lengthens the G0/G1 phase, as the percentage of cells increased in G0/G1 in the presence of IFNγ.
Figure 3. IFNγ decreases NSPC cell cycle progression.

WT NSPCs were treated with IFNγ (100 U/ml) 3 days or left untreated. On day 3, the cells were pulsed with BrdU for one hour. The BrdU was washed out and the cells were harvested at 0, 3, 6, and 9h post-washout. Cells were processed as described in materials and methods for BrdU/7-AAD staining. (A) Representative plots for untreated (top row) and IFNγ-treated (1000 U/mL, bottom row) NSPCs with gates for G0/G1, G1/S, S/G2 and G2/M phases at different time points. (B) Averages of cell percentages in the each of cell cycle gate were quantified for untreated, and IFNγ-treated (100 and 1000 U/mL) NSPCs. Error bars represent SEM. Statistical analysis was applied using two-way ANOVA with Bonferroni post-hoc analysis (n=3), * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001.
IFNγ alters activation and expression of STAT1 and STAT3 in NSPCs
IFNγ primarily signals through the JAK/STAT1 pathway. However, alternative signaling via STAT3 has also been reported (Qing and Stark 2004), and activation of both STAT1 and STAT3 has been demonstrated in primary hippocampal neurons after IFNγ stimulation (Rose et al. 2007). STAT1 and STAT3 also are associated with inhibition and activation of cell proliferation, respectively (Nakashima et al. 2000; Sherry et al. 2009; Tu et al. 2012). Thus, based upon the cell cycle blockade observed in IFNγ-treated NSPCs, we next analyzed the activation of STAT1 and STAT3. Both STAT1 and STAT3 were phosphorylated upon IFNγ treatment (Fig. 4A and B). Phosphorylation of STAT1 on tyrosine 701 (Y701) and serine 727 (S727) was elevated in all IFNγ-treated groups, with phosphorylation of Y701 and S727 peaking at 3 days post-treatment when normalized to GAPDH (348-fold increase above untreated for Y701 and 184-fold above untreated for S727; n=3, p<0.0001). Phosphorylation of STAT3 was less pronounced than STAT1, with a 2.4-fold increase in phosphorylation versus untreated NSPCs at DIV 2 (n=4, p=0.0018), followed by a decline in phosphorylation below the untreated group by DIV 5. Basal levels of phosphorylated STAT3 also increased over time in the untreated groups (10-fold increase in phosphorylated STAT3 in the untreated group at day 5 versus day 0). We also observed an increase in the total expression levels of STAT1 and STAT3, regardless of phosphorylation status, in all IFNγ-treated groups. These findings indicate that NSPCs respond to IFNγ with a sustained activation of STAT1 and a transient, but significant, activation of STAT3. Normalization of phosphorylated STAT1 and 3 with their respective total protein levels was also performed (Fig. 4C). The protein ratio of STAT1-P/STAT1 at both phosphorylation sites was consistent with that seen with GAPDH normalization. However, the protein ratio of STAT3-P/STAT3 did not show increased activation with IFNγ at DIV 2 or 3. These data indicate that increased STAT1 activation is not only attributed to greater total STAT1 expression but also to increased activation of the STAT1 pathway. However, increased STAT3 phosphorylation corresponded with greater expression of total STAT3 protein levels post-IFNγ treatment.
Figure 4. NSPCs activate STAT1 and STAT3 upon IFNγ stimulation.

IFNγ-treated NSPCs (100U/ml) were collected on DIV 2, 3, and 5, and lysed for western blot analysis. (A) Representative blots are shown for phosphorylated STAT1 (Y701 and S727), total STAT1, phosphorylated STAT3 (Y705), and total STAT3. GAPDH is shown as the loading control. (B) The fluorescence signal for each protein was quantified and normalized to GAPDH. (C) For the phosphorylated STAT1 and STAT3 bands, the fluorescence signals were also normalized to the levels of the total STAT1 and STAT3, respectively. The average of 3–5 biological replicates is plotted with SEM. Statistical analysis was applied using repeated measures one-way ANOVA with Bonferroni multiple comparisons post-hoc analysis (****p<0.0001, *** p<0.001, ** p<0.01 *p<0.5).
IFNγ decreases site-specific phosphorylation of pRb and expression of late G1/S cyclin/cdk complexes
Growth arrest is mediated by decreased expression of positive regulators of the cell cycle, including particular cyclins and cyclin-dependent kinases (cdks) depending on the cell cycle checkpoint. In order to progress through the G1/S checkpoint, both cyclin D1/cdk4 and cyclin E/cdk2 phosphorylate the retinoblastoma protein (pRb) at serine 795 (S795), which is a rate-limiting step in the progression from the G1 phase to the S phase (Connell-Crowley et al. 1997; Zarkowska and Mittnacht 1997). Phosphorylation at S795 is critical for pRb inactivation and dissociation from the transcription factor, E2F1 (Rubin et al. 2005); whereas, phosphorylation at serine 807/811 (S807/811) is not required for progression to the S phase (Knudsen and Wang 1996). Because we observed that IFNγ restricted cell cycle progression at G1/S checkpoint, we next analyzed the protein expression of cyclin D/cdk4 (early G1 phase) and cyclin E/cdk2 (late G1 phase) complexes in NSPCs post-IFNγ treatment (Fig. 5A). Cyclin D1 expression did not significantly change after IFNγ treatment until DIV 5, where cyclin D1 levels increased transiently (n=4, p=0.0011). Cyclin D1 also undergoes cyclical changes in phosphorylation and subcellular localization (Yang et al. 2006). This includes nuclear to cytoplasmic redistribution and phosphorylation, with the highest levels of phosphorylation occurring after the G1/S transition (Alt et al. 2000; Diehl et al. 1997). Therefore, if IFNγ treatment slowed the transition of NSPCs into the S-phase, we would expect a decrease in the phosphorylated form of cyclin D1. In Figure 5, the upper cyclin D band (open arrowhead) corresponds to the phosphorylated form of the protein; the lower band corresponds to the unphosphorylated form (closed arrowhead) (Fig. 5A). When quantified separately, the phosphorylated form of cyclin D1 decreases in the IFNγ-treated groups at DIV 2 and 3, consistent with a G0/G1 restriction. Cdk4 expression was also unchanged in the presence of IFNγ (data not shown). Expression of cyclin D3 decreased with IFNγ treatment only at DIV 3, suggesting that the regulation of expression of D-type cyclins do not play a major role in IFNγ-mediated control of cell cycle progression. In contrast, the expression of both cyclin E and cdk2 decreased at all time points post-IFNγ treatment, suggesting that IFNγ acts at the late G1 phase.
Figure 5. IFNγ modulates the expression of cell cycle checkpoint proteins and the phosphorylation of pRb in NSPCs.

(A) Expression of cyclins D1, D2, D3, E and cdk2 were measured using western blot and fluorescence signals were normalized to GAPDH as a loading control. For cyclin D1, the top band (open arrowhead) corresponds to the phosphorylated form of cyclin D1 and the bottom band (closed arrowhead) corresponds to the unphosphorylated form of cyclin D1. (B) Expression of total retinoblastoma protein (pRb) and associated pRb phosphorylation at different serine residues (S780, S795, and S807/811) was measured. The fluorescence signal for each band was normalized to GAPDH as a loading control. For pRb S795, normalization was also performed against total pRb. Quantitation of samples is shown as the average with SEM. Statistical analysis was applied using repeated measures one-way ANOVA with Bonferroni multiple comparisons post-hoc analysis (****p<0.0001, *** p<0.001, ** p<0.01 *p<0.5; n=3–5).
We also examined multiple phosphorylation sites on pRb (Fig. 5B). Expression levels of total pRb did change with IFNγ treatment. However, phosphorylation of pRb at S795 was decreased at both DIV 2 (2.25-fold decrease versus untreated) and DIV 3 (2-fold decrease versus untreated) with IFNγ treatment (n=4, p=0.0002), while S780 and S807/811 were not affected by IFNγ. In untreated NSPCs, pRb phosphorylation at each of the phosphorylation sites increased at DIV 2 and 3, and declined by DIV 5, suggesting that the untreated neurospheres undergo a period of sustained growth early in culture that slows by DIV 5. Together, these results show that IFNγ interferes with G1/S cell cycle progression by decreasing the expression of cyclin/cdk complexes that control the late G1 phase transition, and ultimately decrease pRb phosphorylation. However, the expression of early G1 phase cyclin/cdk complexes are unaffected by IFNγ.
STAT1 is crucial for IFNγ-mediated inhibition of NSPC proliferation
IFNγ induced an anti-proliferative phenotype in NSPCs that was characterized by strong, persistent STAT1 activation. To study the role of STAT1 in IFNγ-induced growth arrest in NSPCs, we analyzed primary NSPCs from STAT1-KO mice, which express a truncated form of STAT1 that is devoid of the first three translated exons and is functionally deficient in the ability to bind to GAS elements (Meraz et al. 1996). The impact of IFNγ on NSPC proliferation was abrogated by the loss of functional STAT1 (Fig. 6). We first determined the diameter and area of STAT1-KO neurospheres treated with IFNγ. Neurosphere diameter was decreased at the highest concentration of IFNγ (1000 U/ml) (Fig. 6A and 6B), but not at the other concentrations of IFNγ that had an impact on the growth of wildtype neurospheres (1, 10, and 100 U/ml, Fig. 1B). At 1000 U/ml IFNγ, there was a 27% decrease in neurosphere diameter in STAT1-KO NSPCs (n=3, p<0.0001) as compared to a 53% decrease in the WT NSPCs at the same time point (DIV 5). Histogram plots of the area of the STAT1-KO neurospheres showed that IFNγ treatment (1 and 100 U/ml, Fig. 6C) reduced neurosphere area to a lesser extent than in wildtype NSPCs. The median area decreased from 2547.5 pixel2 in untreated STAT1-KO neurospheres to 2247 pixel2 in neurospheres treated with 100 U/ml IFNγ; a 300 pixel2 decrease as compared to a 1400 pixel2 decrease in WT NSPCs. Notably, the area of the untreated STAT1-KO neurospheres (2547.5 pixel2, DIV 5) was greater than untreated WT neurospheres (2054 pixel2) at the same time point. Moreover, the diameter in all treatment groups of the STAT1-KO NSPCs was greater than in the corresponding treatment groups of WT NSPCs (Supplementary Fig. 3), suggesting that deletion of STAT1 permitted greater growth of the neurospheres regardless of IFNγ treatment. The higher growth rate of the STAT1-KO NSPCs limited data collection at DIV 7, as the larger neurospheres began to clump and settle at the bottom of the flask, with the appearance of a darker center by phase-contrast microscopy. We suspect that the larger size of the STAT1-KO neurospheres prevented necessary growth factors and nutrients from reaching the cells located in the core at later time points.
Figure 6. STAT1 is required for IFNγ-mediated inhibition of neurosphere growth.

STAT1-KO NSPCs were treated with IFNγ (1–1000 U/ml) or with heat-inactivated IFNγ (ΔH IFNγ; 1000 U/ml) as a negative control for DIV 3 and 5 post-IFNγ treatment (A) Representative images of neurospheres imaged 5 days post-IFNγ treatment at 2x magnification for different concentrations of IFNγ (Scale bar=250 μm). (B) Quantitation of neurosphere diameter at 3 and 5 days post-IFNγ treatment. The longest diameter of each neurosphere was measured using Image J software. Data was collected from neurospheres in five fields/condition from three biological replicates and graphed as a percentage of untreated controls. Statistical analysis was applied by one-way ANOVA (*** p<0.001, **** p<0.0001). (C) Histogram plots of neurosphere area for different concentrations IFNγ on DIV 5 as measured by pixel2 for each neurosphere. Frequency distribution of the number of neurospheres corresponding to the indicated neurosphere area in pixel2 was plotted for each condition. Note that the range of the x-axis is greater than in Figure 1C.
To further identify the role of STAT1 in inhibiting NSPC proliferation, we also measured the cell cycle progression in STAT1-KO NSPCs during IFNγ treatment. We found that the cytostatic effect in WT NSPCs was lost at all but the highest concentration of IFNγ (1000 U/ml) (Fig. 7B). There was no change in the number of cells in the S, G0/G1, and G2/M phases with 1–100 U/mL IFNγ treatment (Fig. 7A and B) at any time point. A modest effect was observed with 1000U/mL IFNγ, with only a 4%±0.9 decrease (n= 4, p=0.005) in S phase cells in STAT1-KO NSPCs as compared to 15%±0.5 decrease for WT NSPCs at the same concentration (Fig. 2B and 5B). These findings demonstrate that STAT1 mediates the cytostatic effect of IFNγ on NSPCs, though STAT1-independent pathways may also contribute.
Figure 7. IFNγ-mediated regulation of cell cycle progression is STAT1-dependent in NSPCs.

(A) STAT1-KO NSPCs were treated with IFNγ (1, 100, 1000 U/ml) for 72h and labeled with BrdU and 7-AAD. The BrdU and 7-AAD intensities per cell were assayed by flow cytometry. Cell populations were gated in different phases of cell cycle (S = synthesis phase, G1= gap phase 1, M= mitosis phase, G2= gap phase 2). Representative plots for untreated and IFNγ-treated NSPCs (1000 U/ml) are shown. (B) Quantitation of NSPCs in each cell cycle phase. The average percentage of cells in each cell cycle phase was plotted with SEM (n=3). Statistical analysis was applied using one-way ANOVA (** p<0.01).
IFNγ-treated WT NSPCs transitioned faster into the G2/M phase than untreated cells (Fig. 2). This effect was not observed in the STAT1-KO NSPCs (Fig. 7B; right panel), where IFNγ did not affect the percentage of cells in G2/M. However, when comparing the untreated controls of WT and STAT1-KO NSPCs, the latter had significantly higher cells in the G2/M phase (SupplementaryFig. 2C). Collectively, these data indicate that STAT1 is responsible for mediating much of the antiproliferative effects of IFNγ on NSPCs. However, STAT1-KO NSPCs may also be inherently cycling a faster rate than WT NSPCs, as indicated by the greater neurosphere sizes of untreated STAT1-KO cells.
STAT1 mediates the effects of IFNγ on pRb phosphorylation and cyclin/cdk expression
We further explored whether the absence of STAT1 abrogated changes in the expression of cell cycle regulatory proteins. Western blot analysis on STAT1-KO NSPCs showed that STAT3 activation increased at DIV 2 (Fig. 8A; n=3, p<0.0001). Interestingly, IFNγ-mediated STAT3 activation was greater in the absence of STAT1 than in WT NSPCs (10-fold vs. 2.4-fold induction, respectively, on DIV 2). Cyclin E expression was unaffected by IFNγ treatment at DIV 2 and 3, but increased at DIV 5, in contrast to WT NSPCs where cyclin E expression is reduced at all time points. Cdk2 expression did not change at any time point. IFNγ treatment also did not alter the expression of cyclin D1 and D3, similar to WT NSPCs (Fig. 8B). The dephosphorylation of pRb at S795 seen in WT NSPCs was not observed with IFNγ treatment in STAT1-KO cells. Rather, a significant increase in pRb S795 phosphorylation was observed on DIV 3 (Fig. 8C; n=4, p=0.0406). As with WT NSPCs, the phosphorylation of pRb at S780 and S807/811 was unaffected in STAT1-KO cells during IFNγ treatment (Fig. 8C). Together, these studies further suggest that functional STAT1 is required for IFNγ-mediated modulation of cyclin E/cdk2 expression and pRb dephosphorylation during inhibition of cell cycle progression.
Figure 8. IFNγ activates STAT3, but does not inhibit cyclin E expression or pRb phosphorylation at S795, in the absence of STAT1.

STAT1-KO NSPCs were treated with IFNγ (100U/ml) and collected on DIV 2, 3, and 5 for western blot analysis. Representative blots for phosphorylated and total STAT3 (A); cyclins D1, D2, D3, E and cdk2, (B); and for total pRb and associated pRb phosphorylation sites (C) are shown. Signal intensity for each band was normalized to GAPDH as a loading control. Quantitation of signal intensity is shown as the average from 3 independent biological replicates with SEM. Statistical analysis was applied using repeated measures one-way ANOVA with Bonferroni multiple comparisons posthoc analysis (*p<0.5, **** p<0.0001).
Discussion
Given the significant role of IFNγ in anti-viral immunity in the brain, we explored the impact of IFNγ on NSPC growth and survival through analysis of cell cycle regulatory proteins. We found that IFNγ inhibited NSPC proliferation as characterized by a decrease in neurosphere diameter and median neurosphere area (Fig. 1). Cell cycle analysis showed that IFNγ restricted the transition of NSPCs into the S-phase and shortened the transition from S to the G2/M-phase (Fig 2). We also observed a modest but significant increase in apoptosis at the highest concentration of IFNγ tested (1000U/ml). Our results contrast with studies on E14 NSPCs from C57BL/6J mice, where IFNγ substantially activated caspase-3/7 and only slightly decreased proliferation (Walter et al. 2011). However, studies on striatal NSPCs from post-natal rat showed a similar increase in TUNEL+ cells with IFNγ treatment (~2-fold increase above untreated controls), albeit with a higher basal level of apoptosis in untreated neurospheres (15% TUNEL+ cells versus 0.5% TUNEL+ cells in our experiments, Fig. 2D) (Ben-Hur et al. 2003). The cytostatic effects we observed are also consistent with other studies on E14 NSPCs from BALB/c mice and on post-natal day 2 and adult murine NSPCs from the subventricular zone, where IFNγ was shown to inhibit NSPC proliferation (Cheeran et al. 2008; Li et al. 2010). Thus, whether IFNγ induce a cytotoxic or cytostatic effect on NSPCs may be dependent, at least in part, on the stage of development during which the NSPCs are harvested or on differential susceptibility of NSPCs in different brain regions.
BrdU-labeling demonstrated a major restriction in the G1 phase of the cell cycle in the presence of IFNγ (Fig. 2). In the G1 phase, hypophosphorylated pRb binds to the transcription factor E2F1, which prevents nuclear translocation of E2F1 and in turn inhibits E2F1-mediated gene expression of other cell cycle regulators (Johnson and Walker 1999). The C-terminal domain of pRb is sequentially and differentially phosphorylated at specific amino acid residues by cyclin/cdk complexes, which then regulate pRb-E2F1 binding. We observed only transient changes in cyclin D1 and D3 expression and no changes in cdk4 expression at all time points following IFNγ treatment (Fig. 5A). These cyclin/cdk complexes control the early G1 phase checkpoint (Ezhevsky et al. 2001), which suggests that IFNγ does not restrict cell cycle progression at the early G1 phase. In contrast, the cyclin E/cdk2 complex controls the late G1 phase checkpoint (Ezhevsky et al. 2001). IFNγ reduced cyclin E and cdk2 expression at all time points examined (Fig. 5A), suggesting that IFNγ acts at the late G1 phase in NSPCs, prior to entry into S phase. IFNγ also caused a minor restriction in G2/M phase (Fig 2B), which may be due to early exit from S phase or delayed exit from G2/M. In conjunction with the moderate increase in TUNEL+ cells (Fig. 2D), these data suggest that DNA damage signaling pathways may be activated at high concentrations of IFNγ. Induction of DNA damage signaling has been noted in other cell types during IFNγ treatment, with similar effects on apoptosis and G2/M arrest (Hubackova et al. 2016; Kim et al. 2009).
pRb phosphorylation at S795 is necessary for disruption of pRb-E2F1 binding (Connell-Crowley et al. 1997; Rubin et al. 2005). Consistent with reports in human fibrosarcoma cells, we found that IFNγ led to dephosphorylation of pRb at S795 (Dimco et al. 2010). In primary cortical neurons, phosphorylation of pRb at S795 is also associated with cell death in a model of HIV-induced neurotoxicity (Akay et al. 2011). Thus, the low levels of apoptosis (~3%) seen in IFNγ-treated neurospheres may have been limited because of the loss of phosphorylation at S795. We did not observe any effect on other pRb C-terminal phosphorylation sites (S780 and S807/811) (Fig. 5B). This is consistent with previous studies that show the S807/811 residues are important for binding to the proto-oncogene c-Abl, but do not implicate them in affecting pRb binding with E2F1 (Wall et al. 2003). pRb is also phosphorylated at S780 by the cyclin D1/cdk4 complex but not by the cyclin E/cdk2 complex (Futatsugi et al. 2012; Kitagawa et al. 1996). Therefore, the lack of change in phosphorylation of S780 by IFNγ at any time point further reinforces the role of cyclin E/cdk2 in mediating effects of IFNγ.
IFNγ mediates unique signaling pathways in neural cells depending upon the availability of intracellular signaling molecules. Our previous work has shown that astrocytes express relatively high levels of endogenous STAT1 and respond to IFNγ with rapid but transient STAT1 phosphorylation (O’Donnell et al. 2012). IFNγ also blocks cell cycle progression in the G0/G1 phase in astrocytes in a STAT1-dependent manner, with minimal induction of apoptosis (O’Donnell et al. 2015). In contrast, primary hippocampal neurons express low endogenous levels of STAT1, which is associated with delayed but sustained engagement of JAKs with IFNGR and prolonged STAT1 phosphorylation (Podolsky et al. 2012; Rose et al. 2007). IFNγ also induces protective signaling pathways in neurons that are STAT1-independent. Similar to neurons, NSPCs express low basal levels of STAT1, and STAT1 expression and activation is sustained (over 72h) after IFNγ treatment. However, IFNγ induces similar cytostatic effects in astrocytes and NSPCs that are STAT1-dependent, despite different kinetics of STAT1 expression and activation (Fig 3). Thus, STAT1 is central to the anti-proliferative effects of IFNγ in neural cells, although the profile of STAT1 activation is not predictive of changes in cell cycle progression. Interestingly, IFNγ also inhibits neuronal differentiation through the JAK/STAT1 pathway, suggesting that STAT1 activation is likely to impact on multiple aspects of NSPC function (Ahn et al. 2015).
STAT1-KO NSPCs show a minor but significant decrease in neurosphere diameter and BrdU incorporation at the highest concentration of IFNγ tested (1000 U/mL) (Fig. 7). This result could be attributed to residual STAT1 activity in the STAT1-KO mouse model (the truncated STAT1 protein retains 2% of DNA binding activity compared to full length STAT1) (Meraz et al. 1996). Nevertheless, we observed that a functional loss of STAT1 abrogated most of the cytostatic effects of IFNγ. In cells lacking STAT1, IFNγ caused increased phosphorylation of pRb on S795 as opposed to the dephosphorylation at S795 seen in the WT NSPCs. STAT1-KO NSPCs also did not reduce cyclin E/cdk2 expression in response to IFNγ, but instead increased expression of cyclin E. These observations indicate that the effects of IFNγ on proliferation, pRb phosphorylation, and cyclin/cdk expression are dependent on STAT1.
In the absence of STAT1, IFNγ induced greater STAT3 activation as compared to WT NSPCs. STAT3 can function as an alternative signaling pathway in response to IFNγ, as has been observed in embryonic fibroblasts (Qing and Stark 2004). Moreover, STAT1 and STAT3 show opposing effects on cell proliferation; STAT1 being anti-proliferative and STAT3 being pro-proliferative (Nakashima et al. 2000; Tu et al. 2012). STAT1 inhibits cell proliferation in epithelial carcinoma cells through the upregulation of cyclin-dependent kinase inhibitors such as p21 and p27, and primary rat NSPCs upregulate p21 in response to IFNγ (Dimberg et al. 2003; Egwuagu et al. 2006; Kominsky et al. 2000; Makela et al. 2010). Conversely, heightened STAT3 expression and activation has been observed in B-cell lymphomas, which increases proliferation and survival (Ding et al. 2008). The substantial increase in STAT3 activation in STAT1-KO NSPCs could also explain the enhanced neurosphere growth compared to WT NSPCs (Supplementary Fig. 3). Therefore, it is possible that the absence of STAT1 leads to a loss of cell cycle inhibitory proteins, such as p21, as well as compensatory changes in other signaling pathways (e.g. STAT3), leading to greater cell cycle progression and more rapid neurosphere growth.
Besides IFNγ, other cytokines signal through the JAK/STAT pathway and are expressed during neurodevelopment and immune activation. Cytokines such as ciliary neurotrophic factor (CNTF) and leukemia inhibitory factor (LIF) maintain the NSPC pool and initiate glial differentiation in vivo through the STAT3 pathway (Fukuda et al. 2007; Shimazaki et al. 2001). Acute maternal administration of IL-6 in pregnant mice increased NSPC number in the forebrains of adult offspring and resulted in perturbations in the localization of NSPCs in the SVZ and cortex (Gallagher et al. 2013). In this study, we describe the STAT1-dependent role of IFNγ in controlling NSPC proliferation, which not only supports recent studies on NSPCs in the adult subventricular zone (Li et al. 2010; Pereira et al. 2015), but also identifies a mechanism for mediating NSPC cell cycle control. However, an outstanding question is how the inflammatory cytokine milieu would affect NSPC function. Since IFNγ, IL-6, and other inflammatory cytokines may be expressed concurrently during an anti-viral immune response, NSPC proliferation may be dictated by multiple signals. Moreover, IFNγ itself induces expression of multiple chemokines in adult NSPCs in vitro with downstream effects on differentiation (Turbic et al. 2011). Further studies to explore the combinatory effects of multiple cytokines will be necessary to fully understand how NSPCs respond to an inflammatory environment.
This was the first study that describes STAT1-mediated control of pRb in the NSPC cell cycle. Our data show that IFNγ-mediated activation of STAT1 may directly or indirectly control pRb activity in NSPCs, and suggests that immune mediators can affect NSPC growth and the phosphorylation status of pRb. It will be interesting to determine whether IFNγ mediates similar effects on NSPCs in the context of an anti-viral immune response in vivo, and whether communication between NSPCs and other brain cells by exchange of IFNγ-rich extracellular vesicles influences the anti-proliferative response (Cossetti et al. 2014). Moreover, the long-term impact of alterations in NSPC proliferation on the infected host remains to be explored. Our studies, and others like them, underscore the importance of considering the influence of the immune response in developing CNS tissue and whether inflammatory mediators bear upon neurodevelopmental disorders.
Supplementary Material
Significance Statement.
Neural stem/progenitor cells (NSPCs) are responsible for fetal neural development and for maintenance of limited neurogenesis in adulthood. Immune responses to brain injury, including that resulting from viral infections, affects the proliferation of NSPCs, potentially due to the signaling pathways induced by inflammatory cytokines. Here, we define cytokine-mediated intracellular signaling mechanisms that result in decreased NSPC proliferation. We show that the anti-viral cytokine interferon-gamma induces site-specific dephosphorylation of the Retinoblastoma protein, which is dependent upon the upstream activation of STAT1. Understanding these mechanisms is necessary for discovering new therapeutic targets to protect NSPCs from potentially deleterious effects of inflammatory damage.
Acknowledgments
Grant information: This work was supported by the following sources: R15 NS087606-01A1 (LOD), a grant from the Samuel and Emma Winters Foundation (LOD), and the Hunkele Dreaded Disease Award and the Faculty Development Fund from Duquesne University (LOD).
We are grateful to Dr. Kelly Jordan Sciutto and Dr. Cagla Akay (University of Pennsylvania), Dr. Michel Modo (University of Pittsburgh), and Dr. John Pollock (Duquesne University) for their advice and feedback on this project. We thank Mrs. Deborah Willson, Mary Caruso, and Jackie Farrer for their excellent administrative support. We also thank Dr. David Draper and Dr. Alan M. Watson from BD Biosciences for their valuable inputs on acquisition and analysis of flow cytometry data.
Abbreviations
- IFNγ
Interferon-gamma
- NSPC
Neural stem/progenitor cell
- pRb
retinoblastoma protein
- IFNGR
Interferon gamma receptor
- Jak
Janus activated kinase
- Stat
Signal Transducers and Activators of Transcription
- cdk
Cyclin dependent kinase
Footnotes
Conflict of Interest
The authors do not have any conflicts of interest to report.
Role of authors
All authors had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. Study concept and design: AK and LOD. Acquisition of data: AK and TS. Analysis and interpretation of data: AK and LOD. Drafting of the manuscript: AK and LOD. Critical revision of the manuscript for important intellectual content: AK and LOD. Statistical analysis: AK. Obtained funding: LOD. Study supervision: LOD.
Literature Cited
- Akay C, Lindl KA, Wang Y, White MG, Isaacman-Beck J, Kolson DL, Jordan-Sciutto KL. Site-specific hyperphosphorylation of pRb in HIV-induced neurotoxicity. Molecular and cellular neurosciences. 2011;47(2):154–165. doi: 10.1016/j.mcn.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Hur T, Ben-Menachem O, Furer V, Einstein O, Mizrachi-Kol R, Grigoriadis N. Effects of proinflammatory cytokines on the growth, fate, and motility of multipotential neural precursor cells. Mol Cell Neurosci. 2003;24(3):623–631. doi: 10.1016/s1044-7431(03)00218-5. [DOI] [PubMed] [Google Scholar]
- Binder GK, Griffin DE. Interferon-gamma-mediated site-specific clearance of alphavirus from CNS neurons. Science. 2001;293(5528):303–306. doi: 10.1126/science.1059742. [DOI] [PubMed] [Google Scholar]
- Bonthius DJ. Lymphocytic choriomeningitis virus: a prenatal and postnatal threat. Adv Pediatr. 2009;56:75–86. doi: 10.1016/j.yapd.2009.08.007. [DOI] [PubMed] [Google Scholar]
- Burdeinick-Kerr R, Govindarajan D, Griffin DE. Noncytolytic clearance of sindbis virus infection from neurons by gamma interferon is dependent on Jak/STAT signaling. J Virol. 2009;83(8):3429–3435. doi: 10.1128/JVI.02381-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdeinick-Kerr R, Griffin DE. Gamma interferon-dependent, noncytolytic clearance of sindbis virus infection from neurons in vitro. J Virol. 2005;79(9):5374–5385. doi: 10.1128/JVI.79.9.5374-5385.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantin E, Tanamachi B, Openshaw H. Role for gamma interferon in control of herpes simplex virus type 1 reactivation. J Virol. 1999;73(4):3418–3423. doi: 10.1128/jvi.73.4.3418-3423.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chawla-Sarkar M, Lindner DJ, Liu YF, Williams BR, Sen GC, Silverman RH, Borden EC. Apoptosis and interferons: role of interferon-stimulated genes as mediators of apoptosis. Apoptosis : an international journal on programmed cell death. 2003;8(3):237–249. doi: 10.1023/a:1023668705040. [DOI] [PubMed] [Google Scholar]
- Cheeran MC, Jiang Z, Hu S, Ni HT, Palmquist JM, Lokensgard JR. Cytomegalovirus infection and interferon-gamma modulate major histocompatibility complex class I expression on neural stem cells. J Neurovirol. 2008;14(5):437–447. doi: 10.1080/13550280802356845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chucair-Elliott AJ, Conrady C, Zheng M, Kroll CM, Lane TE, Carr DJ. Microglia-induced IL-6 protects against neuronal loss following HSV-1 infection of neural progenitor cells. Glia. 2014;62(9):1418–1434. doi: 10.1002/glia.22689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connell-Crowley L, Harper JW, Goodrich DW. Cyclin D1/Cdk4 regulates retinoblastoma protein-mediated cell cycle arrest by site-specific phosphorylation. Molecular biology of the cell. 1997;8(2):287–301. doi: 10.1091/mbc.8.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Currle DS, Hu JS, Kolski-Andreaco A, Monuki ES. Culture of mouse neural stem cell precursors. Journal of visualized experiments : JoVE. 2007;(2):152. doi: 10.3791/152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai C, Krantz SB. Interferon gamma induces upregulation and activation of caspases 1, 3, and 8 to produce apoptosis in human erythroid progenitor cells. Blood. 1999;93(10):3309–3316. [PubMed] [Google Scholar]
- Das S, Basu A. Viral infection and neural stem/progenitor cell’s fate: implications in brain development and neurological disorders. Neurochem Int. 2011;59(3):357–366. doi: 10.1016/j.neuint.2011.02.020. [DOI] [PubMed] [Google Scholar]
- Das S, Dutta K, Kumawat KL, Ghoshal A, Adhya D, Basu A. Abrogated inflammatory response promotes neurogenesis in a murine model of Japanese encephalitis. PloS one. 2011;6(3):e17225. doi: 10.1371/journal.pone.0017225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deierborg T, Roybon L, Inacio AR, Pesic J, Brundin P. Brain injury activates microglia that induce neural stem cell proliferation ex vivo and promote differentiation of neurosphere-derived cells into neurons and oligodendrocytes. Neuroscience. 2010;171(4):1386–1396. doi: 10.1016/j.neuroscience.2010.09.045. [DOI] [PubMed] [Google Scholar]
- Dimberg A, Karlberg I, Nilsson K, Oberg F. Ser727/Tyr701-phosphorylated Stat1 is required for the regulation of c-Myc, cyclins, and p27Kip1 associated with ATRA-induced G0/G1 arrest of U-937 cells. Blood. 2003;102(1):254–261. doi: 10.1182/blood-2002-10-3149. [DOI] [PubMed] [Google Scholar]
- Dimco G, Knight RA, Latchman DS, Stephanou A. STAT1 interacts directly with cyclin D1/Cdk4 and mediates cell cycle arrest. Cell cycle. 2010;9(23):4638–4649. doi: 10.4161/cc.9.23.13955. [DOI] [PubMed] [Google Scholar]
- Ding BB, Yu JJ, Yu RY, Mendez LM, Shaknovich R, Zhang Y, Cattoretti G, Ye BH. Constitutively activated STAT3 promotes cell proliferation and survival in the activated B-cell subtype of diffuse large B-cell lymphomas. Blood. 2008;111(3):1515–1523. doi: 10.1182/blood-2007-04-087734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douvoyiannis M, Litman N, Goldman DL. Neurologic manifestations associated with parvovirus B19 infection. Clin Infect Dis. 2009;48(12):1713–1723. doi: 10.1086/599042. [DOI] [PubMed] [Google Scholar]
- Egwuagu CE, Li W, Yu CR, Che Mei Lin M, Chan CC, Nakamura T, Chepelinsky AB. Interferon-gamma induces regression of epithelial cell carcinoma: critical roles of IRF-1 and ICSBP transcription factors. Oncogene. 2006;25(26):3670–3679. doi: 10.1038/sj.onc.1209402. [DOI] [PubMed] [Google Scholar]
- Ezhevsky SA, Ho A, Becker-Hapak M, Davis PK, Dowdy SF. Differential regulation of retinoblastoma tumor suppressor protein by G(1) cyclin-dependent kinase complexes in vivo. Mol Cell Biol. 2001;21(14):4773–4784. doi: 10.1128/MCB.21.14.4773-4784.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiette L, Aubert C, Muller U, Huang S, Aguet M, Brahic M, Bureau JF. Theiler’s virus infection of 129Sv mice that lack the interferon alpha/beta or interferon gamma receptors. J Exp Med. 1995;181(6):2069–2076. doi: 10.1084/jem.181.6.2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda S, Abematsu M, Mori H, Yanagisawa M, Kagawa T, Nakashima K, Yoshimura A, Taga T. Potentiation of astrogliogenesis by STAT3-mediated activation of bone morphogenetic protein-Smad signaling in neural stem cells. Mol Cell Biol. 2007;27(13):4931–4937. doi: 10.1128/MCB.02435-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futatsugi A, Utreras E, Rudrabhatla P, Jaffe H, Pant HC, Kulkarni AB. Cyclin-dependent kinase 5 regulates E2F transcription factor through phosphorylation of Rb protein in neurons. Cell cycle. 2012;11(8):1603–1610. doi: 10.4161/cc.20009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher D, Norman AA, Woodard CL, Yang G, Gauthier-Fisher A, Fujitani M, Vessey JP, Cancino GI, Sachewsky N, Woltjen K, Fatt MP, Morshead CM, Kaplan DR, Miller FD. Transient maternal IL-6 mediates long-lasting changes in neural stem cell pools by deregulating an endogenous self-renewal pathway. Cell stem cell. 2013;13(5):564–576. doi: 10.1016/j.stem.2013.10.002. [DOI] [PubMed] [Google Scholar]
- Geiger KD, Nash TC, Sawyer S, Krahl T, Patstone G, Reed JC, Krajewski S, Dalton D, Buchmeier MJ, Sarvetnick N. Interferon-gamma protects against herpes simplex virus type 1-mediated neuronal death. Virology. 1997;238(2):189–197. doi: 10.1006/viro.1997.8841. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Sanchez HM, Monsivais-Urenda A, Salazar-Aldrete CA, Hernandez-Salinas A, Noyola DE, Jimenez-Capdeville ME, Martinez-Serrano A, Castillo CG. Effects of cytomegalovirus infection in human neural precursor cells depend on their differentiation state. J Neurovirol. 2015;21(4):346–357. doi: 10.1007/s13365-015-0315-5. [DOI] [PubMed] [Google Scholar]
- Guarascio AJ, Faust AC, Sheperd L, O’Donnell LA. Ebola virus disease: roles and considerations for pharmacists. Ann Pharmacother. 2015;49(2):247–249. doi: 10.1177/1060028014561782. [DOI] [PubMed] [Google Scholar]
- Hoglinger GU, Rizk P, Muriel MP, Duyckaerts C, Oertel WH, Caille I, Hirsch EC. Dopamine depletion impairs precursor cell proliferation in Parkinson disease. Nat Neurosci. 2004;7(7):726–735. doi: 10.1038/nn1265. [DOI] [PubMed] [Google Scholar]
- Hu S, Rotschafer JH, Lokensgard JR, Cheeran MC. Activated CD8+ T lymphocytes inhibit neural stem/progenitor cell proliferation: role of interferon-gamma. PloS one. 2014;9(8):e105219. doi: 10.1371/journal.pone.0105219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubackova S, Kucerova A, Michlits G, Kyjacova L, Reinis M, Korolov O, Bartek J, Hodny Z. IFNgamma induces oxidative stress, DNA damage and tumor cell senescence via TGFbeta/SMAD signaling-dependent induction of Nox4 and suppression of ANT2. Oncogene. 2016;35(10):1236–1249. doi: 10.1038/onc.2015.162. [DOI] [PubMed] [Google Scholar]
- Johnson DG, Walker CL. Cyclins and cell cycle checkpoints. Annual review of pharmacology and toxicology. 1999;39:295–312. doi: 10.1146/annurev.pharmtox.39.1.295. [DOI] [PubMed] [Google Scholar]
- Kim KS, Kang KW, Seu YB, Baek SH, Kim JR. Interferon-gamma induces cellular senescence through p53-dependent DNA damage signaling in human endothelial cells. Mech Ageing Dev. 2009;130(3):179–188. doi: 10.1016/j.mad.2008.11.004. [DOI] [PubMed] [Google Scholar]
- Kitagawa M, Higashi H, Jung HK, Suzuki-Takahashi I, Ikeda M, Tamai K, Kato J, Segawa K, Yoshida E, Nishimura S, Taya Y. The consensus motif for phosphorylation by cyclin D1-Cdk4 is different from that for phosphorylation by cyclin A/E-Cdk2. EMBO J. 1996;15(24):7060–7069. [PMC free article] [PubMed] [Google Scholar]
- Knudsen ES, Wang JY. Differential regulation of retinoblastoma protein function by specific Cdk phosphorylation sites. The Journal of biological chemistry. 1996;271(14):8313–8320. doi: 10.1074/jbc.271.14.8313. [DOI] [PubMed] [Google Scholar]
- Kominsky SL, Hobeika AC, Lake FA, Torres BA, Johnson HM. Down-regulation of neu/HER-2 by interferon-gamma in prostate cancer cells. Cancer research. 2000;60(14):3904–3908. [PubMed] [Google Scholar]
- Lee MH, Wang T, Jang MH, Steiner J, Haughey N, Ming GL, Song H, Nath A, Venkatesan A. Rescue of adult hippocampal neurogenesis in a mouse model of HIV neurologic disease. Neurobiol Dis. 2011;41(3):678–687. doi: 10.1016/j.nbd.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Walker TL, Zhang Y, Mackay EW, Bartlett PF. Endogenous interferon gamma directly regulates neural precursors in the non-inflammatory brain. J Neurosci. 2010;30(27):9038–9050. doi: 10.1523/JNEUROSCI.5691-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makela J, Koivuniemi R, Korhonen L, Lindholm D. Interferon-gamma produced by microglia and the neuropeptide PACAP have opposite effects on the viability of neural progenitor cells. PloS one. 2010;5(6):e11091. doi: 10.1371/journal.pone.0011091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina-Echeverz J, Haile LA, Zhao F, Gamrekelashvili J, Ma C, Metais JY, Dunbar CE, Kapoor V, Manns MP, Korangy F, Greten TF. IFN-gamma regulates survival and function of tumor-induced CD11b+ Gr-1high myeloid derived suppressor cells by modulating the anti-apoptotic molecule Bcl2a1. Eur J Immunol. 2014;44(8):2457–2467. doi: 10.1002/eji.201444497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meraz MA, White JM, Sheehan KC, Bach EA, Rodig SJ, Dighe AS, Kaplan DH, Riley JK, Greenlund AC, Campbell D, Carver-Moore K, DuBois RN, Clark R, Aguet M, Schreiber RD. Targeted disruption of the Stat1 gene in mice reveals unexpected physiologic specificity in the JAK-STAT signaling pathway. Cell. 1996;84(3):431–442. doi: 10.1016/s0092-8674(00)81288-x. [DOI] [PubMed] [Google Scholar]
- Mutnal MB, Cheeran MC, Hu S, Lokensgard JR. Murine cytomegalovirus infection of neural stem cells alters neurogenesis in the developing brain. PloS one. 2011a;6(1):e16211. doi: 10.1371/journal.pone.0016211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutnal MB, Hu S, Little MR, Lokensgard JR. Memory T cells persisting in the brain following MCMV infection induce long-term microglial activation via interferon-gamma. J Neurovirol. 2011b;17(5):424–437. doi: 10.1007/s13365-011-0042-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashima O, Terada Y, Hanada S, Yamamoto K, Kuwahara M, Sasaki S, Marumo F. Activated STAT1 suppresses proliferation of cultured rat mesangial cells. Kidney international. 2000;57(6):2249–2257. doi: 10.1046/j.1523-1755.2000.00085.x. [DOI] [PubMed] [Google Scholar]
- O’Donnell LA, Conway S, Rose RW, Nicolas E, Slifker M, Balachandran S, Rall GF. STAT1-independent control of a neurotropic measles virus challenge in primary neurons and infected mice. J Immunol. 2012;188(4):1915–1923. doi: 10.4049/jimmunol.1101356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell LA, Henkins KM, Kulkarni A, Matullo CM, Balachandran S, Pattisapu AK, Rall GF. Interferon Gamma Induces Protective Non-Canonical Signaling Pathways in Primary Neurons. Journal of neurochemistry. 2015 doi: 10.1111/jnc.13250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson CE, Lawrence DM, Echols LA, Rall GF. Immune-mediated protection from measles virus-induced central nervous system disease is noncytolytic and gamma interferon dependent. J Virol. 2002;76(9):4497–4506. doi: 10.1128/JVI.76.9.4497-4506.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce BD, Hobbs MV, McGraw TS, Buchmeier MJ. Cytokine induction during T-cell-mediated clearance of mouse hepatitis virus from neurons in vivo. J Virol. 1994;68(9):5483–5495. doi: 10.1128/jvi.68.9.5483-5495.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira L, Medina R, Baena M, Planas AM, Pozas E. IFN gamma regulates proliferation and neuronal differentiation by STAT1 in adult SVZ niche. Frontiers in cellular neuroscience. 2015;9:270. doi: 10.3389/fncel.2015.00270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podolsky MA, Solomos AC, Durso LC, Evans SM, Rall GF, Rose RW. Extended JAK activation and delayed STAT1 dephosphorylation contribute to the distinct signaling profile of CNS neurons exposed to interferon-gamma. J Neuroimmunol. 2012;251(1–2):33–38. doi: 10.1016/j.jneuroim.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qing Y, Stark GR. Alternative activation of STAT1 and STAT3 in response to interferon-gamma. The Journal of biological chemistry. 2004;279(40):41679–41685. doi: 10.1074/jbc.M406413200. [DOI] [PubMed] [Google Scholar]
- Rose RW, Vorobyeva AG, Skipworth JD, Nicolas E, Rall GF. Altered levels of STAT1 and STAT3 influence the neuronal response to interferon gamma. J Neuroimmunol. 2007;192(1–2):145–156. doi: 10.1016/j.jneuroim.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothaeusler K, Baumgarth N. Assessment of cell proliferation by 5-bromodeoxyuridine (BrdU) labeling for multicolor flow cytometry. In: Paul Robinson J, et al., editors. Current protocols in cytometry / editorial board. Unit 7. Chapter 7. 2007. p. 31. [DOI] [PubMed] [Google Scholar]
- Rubin SM, Gall AL, Zheng N, Pavletich NP. Structure of the Rb C-terminal domain bound to E2F1-DP1: a mechanism for phosphorylation-induced E2F release. Cell. 2005;123(6):1093–1106. doi: 10.1016/j.cell.2005.09.044. [DOI] [PubMed] [Google Scholar]
- Ruller CM, Tabor-Godwin JM, Van Deren DA, Jr, Robinson SM, Maciejewski S, Gluhm S, Gilbert PE, An N, Gude NA, Sussman MA, Whitton JL, Feuer R. Neural stem cell depletion and CNS developmental defects after enteroviral infection. Am J Pathol. 2012;180(3):1107–1120. doi: 10.1016/j.ajpath.2011.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schleede L, Bueter W, Baumgartner-Sigl S, Opladen T, Weigt-Usinger K, Stephan S, Smitka M, Leiz S, Kaiser O, Kraus V, van Baalen A, Skopnik H, Hartmann H, Rostasy K, Lucke T, Schara U, Hausler M. Pediatric herpes simplex virus encephalitis: a retrospective multicenter experience. J Child Neurol. 2013;28(3):321–331. doi: 10.1177/0883073812471428. [DOI] [PubMed] [Google Scholar]
- Sharma A, Valadi N, Miller AH, Pearce BD. Neonatal viral infection decreases neuronal progenitors and impairs adult neurogenesis in the hippocampus. Neurobiol Dis. 2002;11(2):246–256. doi: 10.1006/nbdi.2002.0531. [DOI] [PubMed] [Google Scholar]
- Sherry MM, Reeves A, Wu JK, Cochran BH. STAT3 is required for proliferation and maintenance of multipotency in glioblastoma stem cells. Stem cells. 2009;27(10):2383–2392. doi: 10.1002/stem.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimazaki T, Shingo T, Weiss S. The ciliary neurotrophic factor/leukemia inhibitory factor/gp130 receptor complex operates in the maintenance of mammalian forebrain neural stem cells. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2001;21(19):7642–7653. doi: 10.1523/JNEUROSCI.21-19-07642.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun T, Vasek MJ, Klein RS. Congenitally acquired persistent lymphocytic choriomeningitis viral infection reduces neuronal progenitor pools in the adult hippocampus and subventricular zone. PLoS One. 2014;9(5):e96442. doi: 10.1371/journal.pone.0096442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tardieu M, Le Chenadec J, Persoz A, Meyer L, Blanche S, Mayaux MJ. HIV-1-related encephalopathy in infants compared with children and adults. French Pediatric HIV Infection Study and the SEROCO Group. Neurology. 2000;54(5):1089–1095. doi: 10.1212/wnl.54.5.1089. [DOI] [PubMed] [Google Scholar]
- Tu B, Du L, Fan QM, Tang Z, Tang TT. STAT3 activation by IL-6 from mesenchymal stem cells promotes the proliferation and metastasis of osteosarcoma. Cancer letters. 2012;325(1):80–88. doi: 10.1016/j.canlet.2012.06.006. [DOI] [PubMed] [Google Scholar]
- van Boxel-Dezaire AH, Stark GR. Cell type-specific signaling in response to interferon-gamma. Curr Top Microbiol Immunol. 2007;316:119–154. doi: 10.1007/978-3-540-71329-6_7. [DOI] [PubMed] [Google Scholar]
- Wall L, Burke F, Smyth JF, Balkwill F. The anti-proliferative activity of interferon-gamma on ovarian cancer: in vitro and in vivo. Gynecologic oncology. 2003;88(1 Pt 2):S149–151. doi: 10.1006/gyno.2002.6707. [DOI] [PubMed] [Google Scholar]
- Walter J, Honsek SD, Illes S, Wellen JM, Hartung HP, Rose CR, Dihne M. A new role for interferon gamma in neural stem/precursor cell dysregulation. Molecular neurodegeneration. 2011;6:18. doi: 10.1186/1750-1326-6-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarkowska T, Mittnacht S. Differential phosphorylation of the retinoblastoma protein by G1/S cyclin-dependent kinases. The Journal of biological chemistry. 1997;272(19):12738–12746. doi: 10.1074/jbc.272.19.12738. [DOI] [PubMed] [Google Scholar]
- Zhang SS, Liu MG, Kano A, Zhang C, Fu XY, Barnstable CJ. STAT3 activation in response to growth factors or cytokines participates in retina precursor proliferation. Exp Eye Res. 2005;81(1):103–115. doi: 10.1016/j.exer.2005.01.016. [DOI] [PubMed] [Google Scholar]
- Zheng LS, Hitoshi S, Kaneko N, Takao K, Miyakawa T, Tanaka Y, Xia H, Kalinke U, Kudo K, Kanba S, Ikenaka K, Sawamoto K. Mechanisms for interferon-alpha-induced depression and neural stem cell dysfunction. Stem Cell Reports. 2014;3(1):73–84. doi: 10.1016/j.stemcr.2014.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.