

Abstract
Oxidized LDL (oxLDL) is known to activate inflammatory responses in a variety of cells, especially macrophages and dendritic cells (DCs). Interestingly, much of the oxLDL in circulation is complexed to antibodies and these resulting immune complexes (ICs) are a prominent feature of chronic inflammatory disease such as atherosclerosis, Type-2 diabetes, systemic lupus erythematosus and rheumatoid arthritis. Levels of oxLDL-ICs often correlate with disease severity, and past studies have demonstrated that oxLDL-ICs elicit potent inflammatory responses in macrophages. Here we show that bone marrow-derived dendritic cells (BMDCs) incubated with oxLDL-ICs for 24 hrs secrete significantly more IL-1β compared to BMDCs treated with free oxLDL, while there was no difference in levels of TNFα or IL-6. Treatment of BMDCs with oxLDL-ICs increased expression of inflammasome-related genes Il1a, Il1b, and Nlrp3; and pre-treatment with a caspase 1 inhibitor decreased IL-1β secretion in response to oxLDL-ICs. This inflammasome priming was due to oxLDL-IC signaling via multiple receptors as inhibition of CD36, TLR4 and FcγR significantly decreased IL-1β secretion in response to oxLDL-ICs. Signaling through these receptors converged on the adaptor protein CARD9, a component of the CARD9-Bcl10-MALT1 signalosome complex involved in NF-κB translocation. Finally, oxLDL-IC-mediated IL-1β production resulted in increased Th17 polarization and cytokine secretion. Collectively, these data demonstrate that oxLDL-ICs induce inflammasome activation through a separate and more robust mechanism than oxLDL alone, and that these ICs may be immunomodulatory in chronic disease and not just biomarkers of severity.
Introduction
Immune complexes (ICs) are formed by specific antibody binding to its soluble antigen. Many sterile inflammatory disorders are characterized by increased serum titers of disease-specific ICs, which can have mechanistic roles in pathogenesis (1, 2). In systemic lupus erythematosus, anti-nuclear and anti-double stranded DNA ICs bind to glomerular basement membranes and capillary walls resulting in glomerular nephritis (3). ICs precipitated from the serum of rheumatoid arthritis patients increase the production of tumor necrosis factor alpha (TNFα) from peripheral blood mononuclear cells (4). Atherosclerosis is another disease associated with increased titers of ICs containing antibodies directed to oxidized low-density lipoprotein (oxLDL) (5). In fact, up to 90% of circulating oxLDL can be found in oxLDL-ICs (6). While the majority of studies in atherosclerosis focus on free oxLDL, much less is known about responses to oxLDL-ICs. In vitro studies have demonstrated that treatment of the human macrophage cell line THP-1 with oxLDL-ICs results in increased cell activation, inflammatory cytokine production, and foam cell formation (7). In vivo evidence using Fcγ receptor (FcγR)-deficient mice supports the role of IC-mediated modulation of atherosclerosis (8–10). Understanding how these ICs modulate immune responses is important to identify potential therapeutic targets and provide mechanistic insight into IC-associated diseases.
Interestingly, the majority of sterile inflammatory disorders characterized by high serum titers of ICs also have inflammasome hyperactivation (11–13). The inflammasome is a multi-protein oligomer that, when activated, results in secretion of robust levels of the pro-inflammatory cytokine IL-1β (14). This innate immune mechanism is important and necessary in the clearance of many bacterial and fungal pathogens (15, 16). However, over-activation of the inflammasome exacerbates or even drives many inflammatory diseases (17). IL-1β blockade is currently used clinically to treat many IC-related diseases such as rheumatoid arthritis and juvenile systemic lupus erythematosus (18, 19). Additionally, it is known that knocking out the inflammasome-related gene Nlrp3 in mice completely abolishes atherosclerosis (20). Yet, while diseases of sterile inflammation are characterized by both increased serum IC levels and inflammasome activation, a direct connection has not been made between these two factors.
The current study shows that oxLDL-ICs prime the inflammasome in dendritic cells via FcγRs, TLR4 and CD36. This inflammasome activation is independent of previously established mechanisms such as cholesterol crystal formation (20). Taken together these findings identify a novel and important immunomodulatory role for oxLDL-ICs and provide a link between TLR-ligand containing ICs and the inflammasome in sterile inflammatory disorders.
Materials and Methods
Mice
C57BL/6J (B6), B6N.129-Nlrp3tm1Hhf/J (Nlrp3−/−) and B6.129P2 (SJL)-Myd88tm1Defr/J (Myd88−/−) mice and B6.Cg-Tg (TcraTcrb) 425Cbn/J (OT-II) mice were originally obtained from the Jackson Laboratory (Bar Harbor, ME) and maintained and housed at Vanderbilt University. All mice used in these studies were on the C57BL/6J background. Procedures were approved by the Vanderbilt University Institutional Animal Care and Use Committee.
OxLDL and oxLDL-ICs
Human native LDL was purchased from Intracel (Frederick, MD) or Sigma-Aldrich (St. Louis, MO). OxLDL was made by dialyzing human LDL for 24 hrs against 0.9 M NaCl at 4°C with two buffer changes, followed by dialysis against 0.9 M NaCl containing 20 μM CuSO4 for 4 hrs at room temperature. Oxidation was terminated by dialysis against 1 mM EDTA in 1X PBS for 16 hrs with two buffer changes. Extent of oxidation was determined by TBARS assay (Cell Biolabs, Inc., San Diego, CA). OxLDL-ICs were generated by incubating polyclonal rabbit anti-human apoB-100 (Alfa Aesar, Ward Hill, MA) with oxLDL at a ratio of 10:1 (500 μg of antibody, 50 μg of oxLDL) overnight at 37°C. Unbound antibody and antigen were removed by size exclusion filtration. For all experiments, immune complex concentrations were normalized based on oxLDL concentration to ensure that equal amounts of oxLDL were used in both the oxLDL and oxLDL-IC conditions. Fab2 fragments were made using the Pierce Fab Fragmentation Kit (Thermo Fisher Scientific, Waltham, MA) according to manufacturer’s protocol. OxLDL enriched immune complexes were obtained from the serum of Apoe−/− mice fed Western diet (21% saturated fat, 0.15% cholesterol) for 12 weeks. Whole blood was obtained by retro-orbital bleeding. Serum was incubated with protein G beads for 1 hr at room temperature. Immune complexes were eluted from protein G beads and protein concentration was calculated by BCA assay according to manufacturer’s instructions (Thermo Fisher Scientific).
Cell Culture
BMDCs were generated as previously described (21). Briefly, bone marrow from hind legs was flushed with RPMI-1640 (Corning, Corning, MA) supplemented with 10% FBS (Gibco, Grand Island, NY), 10 mM HEPES (Corning), and 1× Penicillin/Streptomycin/L-glutamine (Sigma-Aldrich) (hereafter referred to as TCM). Cells were plated in 100 mm2 petri dishes at 2×105 cells/mL in TCM containing 20 ng/mL recombinant GM-CSF (R&D Systems, Minneapolis, MN). Media was replaced on days 3 and 6 and cells were harvested on day 9. To make BMDCs from various transgenic strains femurs were shipped overnight. Femurs from Cd36−/− mice were obtained from Dr. Kathryn Moore (New York University, New York, NY). Cd11ccre/Sykflox/flox and Il1b−/− femurs were obtained from Dr. John Lukens (University of Virginia, Charlottesville, VA). Femurs from Card9−/− mice were received from Dr. Thirumala Kanneganti (St. Jude Children’s Research Hospital, Memphis, TN) (22).
ELISA and Western Blotting
IL-1β, IL-6, and TNFα (BD Biosciences, San Jose, CA) ELISAs were performed according to manufacturer’s instructions. For Western blotting experiments, 1×106 BMDCs were treated with indicated stimuli for 24 hrs. Cells were lysed with 1× RIPA buffer and lysates were separated by 4%–20% reducing SDS-PAGE. Blots were incubated with anti-mouse caspase-1 monoclonal antibody (Adipogen, San Diego, CA) or anti-mouse NFκB p65 antibody (Cell Signaling Technology, Danvers, MA), overnight at 4°C followed by IRDye 680RD goat anti-mouse or goat anti-rabbit (LI-COR, Lincoln, NE) for 30 min at room temperature. Bands were visualized using the LI-COR Odyssey System.
Immunoprecipitation
CBM complex formation was assessed in whole cell lysates from BMDCs stimulated for 2 hrs with oxLDL or oxLDL-ICs. Cells were lysed in 1× RIPA buffer followed by immunoprecipitation with antibody to MALT1, CARD9, or Bcl10 (Santa Cruz Biotechnology, Dallas, TX). Western blot analysis was performed as described above with anti-CARD9, anti-Bcl-10, and anti-MALT1 (Cell Signaling Technologies).
Real-Time Quantitative PCR
BMDCs were treated with indicated stimuli for two hrs. Total RNA was isolated from cells using Norgen Total RNA Isolation Kits (Norgen Biotek Corporation, Thorold, Ontario, Canada). RNA concentrations were normalized and RNA was reversed transcribed with a high capacity RNA to cDNA reverse transcription kit (Applied Biosystems, Grand Island, NY). The reverse transcription product was used for detecting mRNA expression by quantitative real time PCR using the QuantStudio 6-flex System (Life Technologies, Grand Island, NY). The cycling-threshold (CT) value for each gene was normalized to that of the house keeping gene Ppia, and relative expression calculated by the change in cycling threshold method (ΔΔCT).
Flow Cytometry
To measure FcγR expression, BMDCs were then stained on ice for 30 min with CD16.2-APC, CD16/32-FITC, CD32-Alexa Fluor 488, or CD64-APC in the absence of Fc-block. The CD16.2, CD16/32, and CD64 antibodies were purchased from BD Bioscience and diluted 1:200. Antibodies were diluted 1:200 in FACS buffer containing HBSS, 1% BSA, 4.17mM sodium bicarbonate, and 3.08mM sodium azide. The CD32 antibody, a gift from Dr. Jeffrey Ravetch (The Rockefeller University, New York, NY), was labeled using an Alexa Fluor 488 Antibody Labeling Kit (Thermo Fisher Scientific). Cells were washed and re-suspended in 2% PFA for analysis on a MACSQuant seven color flow cytometer (Miltenyi Biotech) and data were analyzed using FlowJo Single Cell Analysis Version 7.6.5. To measure pSyk and pErk, cells were stimulated with LPS, oxLDL, oxLDL-Fab2 or oxLDL-IC for 5 or 15min. Cells were then fixed for 10minf in 1× lyse/fix buffer (BD Bioscience) and permeabilized for 30min using Perm Buffer III (BD Bioscience). After permeabilization, cells were Fc blocked for 15min followed by staining with either CD11b-V450 (BD Bioscience), CD11c-FITC (BD Bioscience) and pSyk Y525/526- PE (Cell Signaling Technology); or, CD11b-V450 (BD Biosciences), CD11c-PeCy7 (BD Biosciences) and pERK1/2-FITC (BD Biosciences).
Statistical Analyses
Where appropriate statistical significance was determined using a Student’s t test. If more than two groups were compared, a one way Analysis of Variance (ANOVA) was used. In all cases p<0.05 was considered statistically significant.
Results
OxLDL-ICs act as a priming signal for the inflammasome
It has been demonstrated that ICs containing TLR ligands can enhance inflammatory responses in DCs and macrophages (4, 23). To determine whether the cytokine response to oxLDL-ICs was different than that generated with oxLDL alone, we incubated BMDCs with either stimuli for 24hrs. While there were no differences in TNFα or IL-6 production between the 2 treatment groups, oxLDL-ICs induced robust IL-1β production compared to free oxLDL (Figure 1A). An additional control of oxLDL-enriched ICs isolated from hyperlipidemic ApoE deficient mice (ApoE-IC) was added to validate our prepared oxLDL-ICs. Similar results were seen with bone marrow-derived macrophages (BMDMs) indicating that this was not a DC-specific response but likely represented a fundamental difference in signaling between oxLDL and oxLDL-ICs (Supplementary Figure 1). Figure 1A demonstrates that enhanced IL-1β production to lab-prepped oxLDL-ICs is similar to ApoE-ICs and thus likely to be physiologic.
Figure 1. OxLDL-ICs prime the inflammasome.
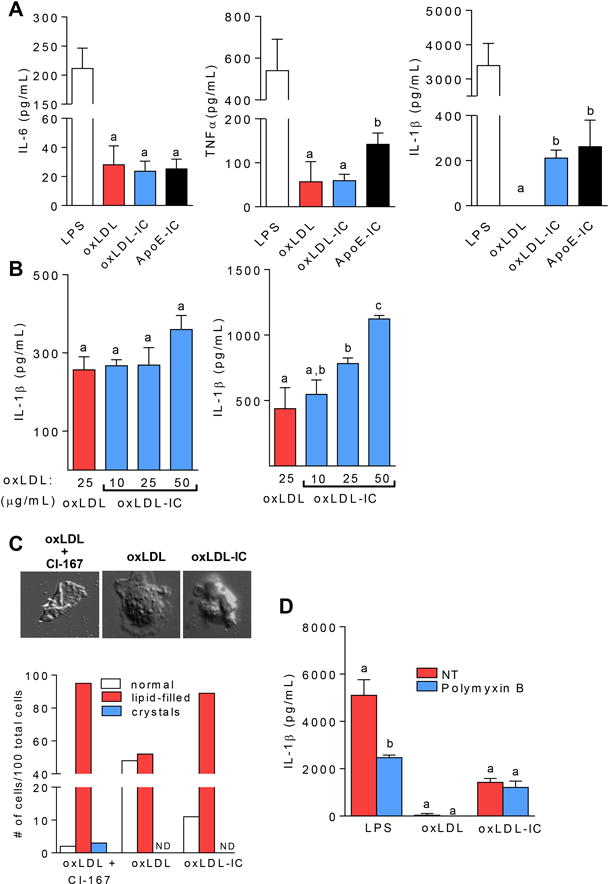
BMDCs were treated for 24 hrs with oxLDL or oxLDL-ICs. (A) Cytokine levels in culture supernatants were measured by ELISA. Shown are representative experiments where n= at least 3 biological and technical replicates. Error bars indicate SEM. Unlike letters denote significance (p<0.01) by Student’s t test. (B) OxLDL-ICs were tested for their ability to act as an activating (left) or priming (right) signal for the inflammasome. Briefly, BMDCs were treated for 3 hrs with 20 ng/mL LPS followed by oxLDL or increasing concentrations of oxLDL-ICs (based on oxLDL concentration) for an additional 3 hrs (left). For priming experiments (right panel) BMDCS were treated for 3 hrs with oxLDL or increasing concentrations of oxLDL-ICs followed by 5mM ATP for 1 hr. Culture supernatants were tested for IL-1β by ELISA. Shown is one representative of 3 experiments with 3 mice/experiment. Unlike letters denote significance (p<0.05) by Student’s t test and error bars represent the SEM. (C) BMDCs were treated with oxLDL or oxLDL-ICs for 3 hrs or with oxLDL in the presence of the ACAT inhibitor CLI-067 (positive control) for 24 hrs and analyzed by polarizing light microscopy for crystal formation. Lipid filled cells and crystal formation is quantified and representative images are depicted. Shown is one representative of two experiments. (D) BMDCs were treated with oxLDL-ICs in the presence of polymyxin B. Shown is one representative of 2 experiments. IL-1β in culture supernatants was measured by ELISA. Unlike letters denote significance (p<0.01) by One-way ANOVA with a Bonferroni post-test. Error bars represent SD.
Previous studies showed that oxLDL activates the inflammasome through the formation of cholesterol crystals (20). Given that oxLDL-ICs caused enhanced IL-1β production from BMDCs, we hypothesized that oxLDL-ICs activate the inflammasome by a similar mechanism. Canonical inflammasome activation is a two-step process that requires a priming signal, typically a pathogen associated molecular pattern, and an activating signal that can be cell damage, ATP or cholesterol or uric acid crystals (24). The first signal leads to production of pro-IL1β and the second signal cleaves pro-caspase 1 to active caspase 1, allowing it to convert pro-IL1β to its mature secreted form (14). To determine whether oxLDL-ICs served as signal 2, BMDCs were primed with LPS for 3 hrs followed by oxLDL (25 μg/ml) or increasing concentrations of oxLDL-ICs (containing 10, 25 or 50 μg/mL total oxLDL) for an additional 3 hrs. As an activating signal, oxLDL-ICs elicited IL-1β levels similar to that of oxLDL (Figure 1B, left). To test oxLDL-ICs as inflammasome priming signal 1, BMDCs were incubated with oxLDL or oxLDL-ICs in increasing concentration for 3 hrs followed by ATP for an additional hr. OxLDL-ICs elicited significantly more IL-1β than free oxLDL (Figure 1B, right panel). OxLDL-ICs did not promote IL-1β through formation of cholesterol crystals, as incubation of BMDCs with oxLDL or oxLDL-ICs for 3 hrs was not sufficient for crystal formation (Figure 1C). Likewise pretreatment of BMDCs with the LPS inhibitor polymyxin B prior to exposure to oxLDL or oxLDL-ICs had no effect on elicited IL-1β production in BMDCs treated with oxLDL or oxLDL-ICs, ruling out the possibility of endotoxin contamination in our IC preparations (Figure 1D).
OxLDL-IC priming of the inflammasome is Nlrp3 and caspase-1 dependent
qPCR analysis of RNA from BMDCs treated with oxLDL-ICs for 2 hrs resulted in increased transcription of inflammasome-related genes Il1a, Il1b, and Nlrp3 with no change in inflammasome-related genes Aim2, Nlrc4, or Il18 (Figure 2A). These data indicate that oxLDL-ICs induce Nlrp3 mRNA levels. To support this finding, wild-type and Nlrp3−/− BMDCs were treated with oxLDL-ICs for 3 hrs followed by ATP for an additional hr and IL-1β was measured in culture supernatants by ELISA. As expected, absence of Nlrp3 completely abolished mature IL-1β production (Figure 2B). Western blot analysis for cleaved caspase-1 and measurement of IL-1β production from BMDCs pre-treated with a caspase-1 and pan-caspase inhibitor demonstrate that oxLDL-IC mediated inflammasome activation was capsase-1 dependent (Supplemental Figure 2 and Figure 2C). Taken together, these data show that oxLDL-ICs elicit robust IL-1β production from BMDCs by inducing production of pro-IL-1β and Nlrp3.
Figure 2. Inflammasome priming is NLRP3 and capase-1 dependent.
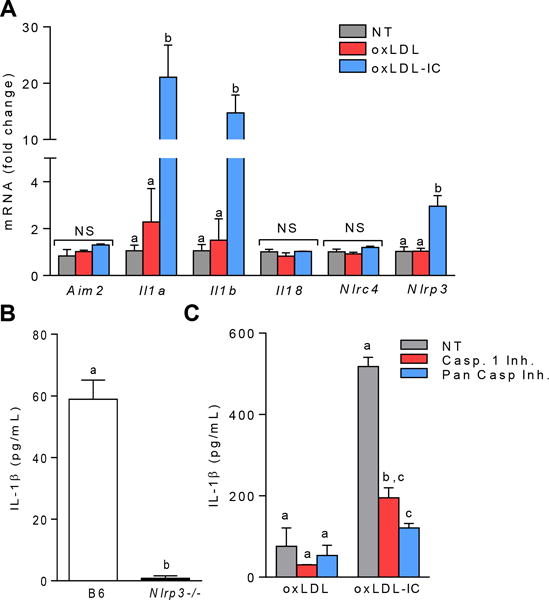
(A) BMDCs were stimulated for 2 hrs with oxLDL or oxLDL-ICs. Expression of inflammasome-related genes was measured using real-time RT PCR and expressed as the 2−ΔΔCT method compared to the no treatment group (n=6 mice). Unlike letters denote significance (p<0.01) by one-way ANOVA with a Bonferroni post-test. (B) B6 and Nlrp3−/− BMDCs were treated for 3 hrs with oxLDL or oxLDL-ICs followed by 1 hr with ATP. IL-1β production in culture supernatants was measured by ELISA. Shown is one of 3 experiments with 3 mice/experiment. Unlike letters indicate significance (p<0.01) by Student’s t test. Error bars represent SEM. (C) BMDCs were treated as in (B) in the presence or absence of a caspase-1 (Z-VAD-FMK) or pan-caspase inhibitor (Z-YVAD-FMK). IL-1β production in culture supernatants was measured by ELISA. Unlike letters denote significance (p<0.01) by One-way ANOVA with a Bonferroni post-test. Shown is one of 3 experiments with 3 mice/experiment. Error bars represent SD.
OxLDL-ICs prime the inflammasome via FcγR, TLR4, and CD36
FcγRs are the canonical receptors for IgG containing-ICs, however it is known that oxLDL activates the inflammasome via TLRs and scavenger receptors (25, 26). To test the role of these receptors in oxLDL-IC mediated inflammasome activation, we first measured the baseline expression of FcγRs on BMDCs and found that BMDCs mainly express the activating receptors FcγRI and FcγRIV (Figure 3A). Next, BMDCs were treated with oxLDL-ICs or oxLDL-Fab2 complex (lacking the Fc portion of the antibody and inhibiting interaction with FcγRs) in the presence or absence of the TLR-4 inhibitor CLI-095 for 3 hrs followed by ATP for an additional hr. Treatment of BMDCs with the Fab2 complex or the TLR4 inhibitor decreased IL-1β production by approximately 50% (Figure 3B). Interestingly, treatment of BMDCs with both the TLR4 inhibitor and oxLDL-Fab2 further decreased IL-1β suggesting a cooperative role for these receptors (Figure 3B). The contribution of TLR signaling to oxLDL-IC mediated inflammasome activation was confirmed using Myd88−/− and Cd36−/− BMDCs, (Figure 3C and D). These data show that oxLDL-IC priming of IL-1β responses in BMDCs is likely a collaboration between FcγRs, TLRs and CD36.
Figure 3. OxLDL-ICs prime the inflammasome using multiple receptors.
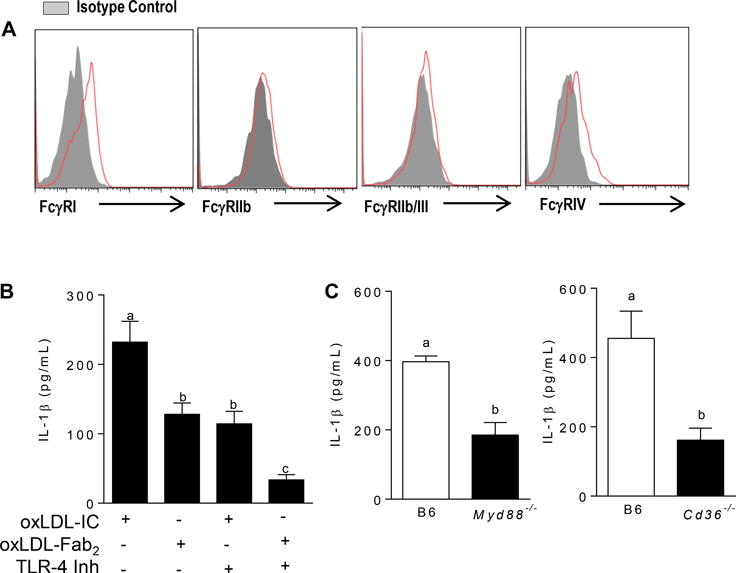
(A) Surface expression of FcγRs on BMDCs was measured by flow cytometry. Shown is one representative of 3 experiments. (B) BMDCs were pre-treated with the TLR-4 inhibitor CLI-095 prior to treatment with oxLDL-IC or oxLDL-Fab2 for 3 hrs and ATP for an additional hr. Culture supernatants were tested for IL-1β by ELISA. Shown is one of 3 experiments with similar results. Unlike letters denote significance (p<0.01) by One-way ANOVA with a Bonferroni post-test. (C) BMDCs from Myd88−/− (left) and Cd36−/− (right) mice (n=3 per group) were treated with oxLDL or oxLDL-ICs for 3 hrs followed by ATP for an additional hr. IL-1β in culture supernatants was measured by ELISA. Unlike letters denote significance (p<0.01) by Student’s t test. Error bars represent SEM.
OxLDL-IC signaling through FcγRs induces Syk phosphorylation
Given that BMDCs express high levels of activating FcγRs, and that treatment of BMDCs with oxLDL-Fab2 complex did not result in enhanced levels of IL-1β (Figure 3B), we hypothesized that oxLDL-ICs induce phosphorylation of Syk downstream of activating FcγRs. To test this hypothesis, BMDCs were treated with oxLDL or oxLDL-ICs for 15 minutes and Syk phosphorylation was measured by phosphoroflow cytometry. OxLDL-IC treatment of BMDCs increased pSyk levels; whereas oxLDL did not induce pSyk (Figure 4A right). To demonstrate that phosphorylation of Syk was due to ligation of FcγRs and not some off-target effect of oxLDL, BMDCs were treated with oxLDL-Fab2 or oxLDL-ICs. Again, only oxLDL-ICs increased levels of pSyk (Figure 4A middle). As an additional control, BMDCs were treated with OVA-containing ICs (ova-ICs) which caused Syk phosphorylation, although not quite as robustly as oxLDL-ICs (Figure 4A right). Interestingly, however, while ova-ICs induced pSyk, they did not elicit the increased IL-1β production seen with oxLDL-ICs (Figure 4B), indicating that pSyk is not sufficient to increase production of mature IL-1β. Unbound anti-oxLDL also did not elicit IL-1β production from BMDCs, further confirming that both the antigen and antibody must be present for this response to occur (Figure 4B). Because FcγRs can also signal through ERK in order to promote anti-inflammatory responses such as IL-10 secretion, we wanted to confirm that oxLDL-ICs did not just generally induce FcγR-associated kinases, but that the response was more specifically associated with pSyk. BMDCs treated with oxLDL-ICs for 15 minutes did not result in detectable pERK (Figure 4C). This was supported by the significant decrease in IL-1β production from oxLDL-IC treated BMDCs from Syk−/− mice and in the presence of a Syk inhibitor (Figure 4D). Collectively, these data support the hypothesis that signaling via pSyk is necessary but not sufficient to induce enhanced inflammasome priming by oxLDL-ICs and that concomitant ligation of FcγRs, TLRs, and CD36 is required.
Figure 4. OxLDL-ICs enhance IL-1β production via pSyk.
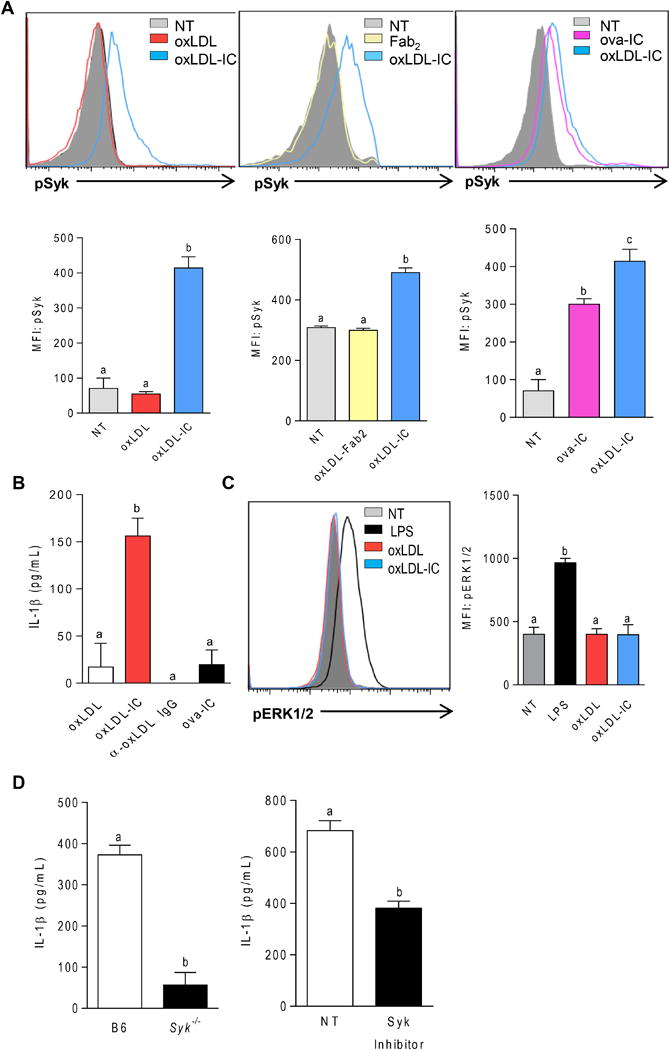
(A) BMDCs were incubated with oxLDL or oxLDL-ICs, oxLDL-Fab2, oxLDL-ICs, or ova-ICs for 15 minutes. Syk phosphorylation was measured by phosphoro-flow cytometry. Representative histograms are shown (top panels). n=3 mice per group and error bars represent SEM. Unlike letters indicate significance p<0.001 by One-way ANOVA with Bonferroni post-test. (B) Cells were treated with oxLDL, anti-oxLDL, oxLDL-ICs or ova-ICs for 3 hrs followed by ATP for an additional hr. IL-1β in culture supernatants was measured by ELISA. Error bars indicate SD. Shown is one representative of 3 separate experiments. Unlike letters denote significance by One-way ANOVA with a Bonferroni post-test. (C) BMDCs were incubated with LPS oxLDL or oxLDL-ICs, for 15 minutes. Erk phosphorylation was measured by phosphoro-flow cytometry. Representative histogram is shown (left) and results are quantified by MFI (right). N=3 separate experiments and error bars represent SEM. Unlike letters indicate significance p<0.001 by One-way ANOVA with Bonferroni post-test. (D) B6 or Syk−/− BMDCs (right) or B6 BMDCs plus or minus the Syk inhibitor Bay61-3606 (left) were incubated with oxLDL or oxLDL-ICs for 3hrs followed by ATP for 1hr. n=3 separate experiments and error bars represent SEM. Unlike letters indicate significance (p<0.05) by Student’s t test.
OxLDL-IC-mediated inflammasome priming is CARD9 dependent
CARD9 is an adaptor protein component of the CARD9-Bcl10-MALT1 (CBM) complex, a signalosome involved in the translocation of NF-κB to the nucleus and subsequent production of pro-inflammatory cytokines (27). Previous studies showed that ITAM and TLR signaling pathways converge on CARD9, and CARD9-dependent inflammasome activation and IL-1β production are critical in models of fungal pathogenesis (28–30). To determine whether CARD9 is necessary for oxLDL-IC-mediated inflammasome responses, wild type and Card9−/− BMDCs were treated with oxLDL or oxLDL-ICs for 3 hrs followed by ATP for an additional hr in the presence or absence of a TLR4 or Syk inhibitor. CARD9 deficiency had no effect on IL-1β responses to LPS priming (Figure 5A, left panel) or oxLDL-mediated IL-1β production (Figure 5A). However, absence of CARD9 resulted in significant decreases in IL-1β secretion in response to oxLDL-IC priming (Figure 5A, right panel). As seen previously, treatment of B6 BMDCs with a TLR-4 or Syk inhibitor reduced IL-1β levels to that of oxLDL alone, indicating that both TLR-4 and FcγRs are necessary for IC-enhanced cytokine production. Supporting the convergence of these signals on CARD9, pre-treatment of Card9−/− BMDCs with either TLR4 or Syk inhibitors completely abolished IL-1β production. In addition, CARD9 deficiency had no effect on oxLDL-induced expression of mRNA for Il1a, Il1b or Nlrp3 (Figure 5B, left panel) but decreased levels of mRNA for these genes in response to oxLDL-ICs (Figure 5B, right panel). Absence of CARD9 had no effect on levels of IL6 or TNFα (Figure 5C). Taken together, these data indicate that oxLDL-ICs prime the IL-1β response by signaling through multiple receptors and converging on the adaptor protein CARD9.
Figure 5. OxLDL-IC inflammasome priming is CARD9 dependent.
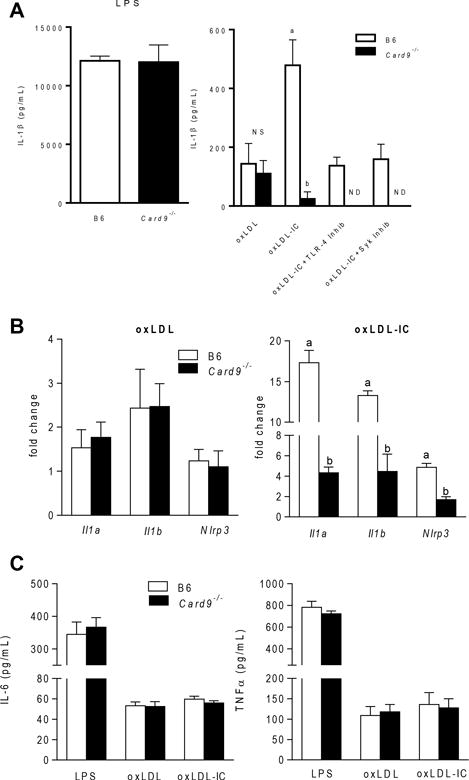
(A) B6 and Card9−/− BMDCs were incubated with LPS, oxLDL, or oxLDL-ICs in the presence or absence of a TLR4 or Syk inhibitor for 3 hrs followed by ATP for an additional hr. IL-1β in culture supernatants was measured by ELISA. Error bars indicate SEM (n=3 mice per group). Unlike letters denote significance (P<0.001) by Student’s t test. (B) B6 and Card9−/− BMDCs were incubated with oxLDL (left) or oxLDL-ICs (right) for 2 hrs, and expression of inflammasome-related genes was measured using real-time RT PCR and expressed as the 2−ΔΔCT compared to the no treatment group (n=3 mice per group). Unlike letters denote significance (p<0.01) by one-way ANOVA with a Bonferroni post-test. (C) BMDCs from B6 and Card9−/− mice were incubated with LPS, oxLDL or oxLDL-ICs for 24 hrs. TNFα and IL-6 in culture supernatants was measured by ELISA. N=3 mice per group and error bars represent SEM.
OxLDL-ICs induce CBM complex formation and NF-kB translocation
To test if oxLDL-ICs elicited transcription of Nlrp3 genes and IL-1β production by promoting formation of the CBM complex, BMDCs were treated for 2 hrs with oxLDL or oxLDL-ICs. Immunoprecipitation experiments with antibodies to MALT1 and CARD9 showed that the entire CBM complex was only pulled down when cells were treated with oxLDL-ICs, and not oxLDL alone (Figure 6A, left and middle). When lysates were immunoprecipitated with an antibody to Bcl10, the entire complex was pulled down in both treatment groups; however there were much higher levels of MALT1 and CARD9 in the BMDCs treated with oxLDL-ICs (Figure 6A, right). Because CBM complex formation has been found to promote nuclear translocation of NF-κB, we then measured NF-κB p65 levels in the cytosol and nucleus of BMDCs treated with oxLDL or oxLDL-ICs. OxLDL-ICs induced nuclear translocation of NF-κB, and this translocation did not occur when the experiment was repeated in Card9−/− BMDCs (Figure 6B). These results support Nlrp3 gene transcription through formation of the CBM complex.
Figure 6. OxLDL-ICs cause CBM formation.
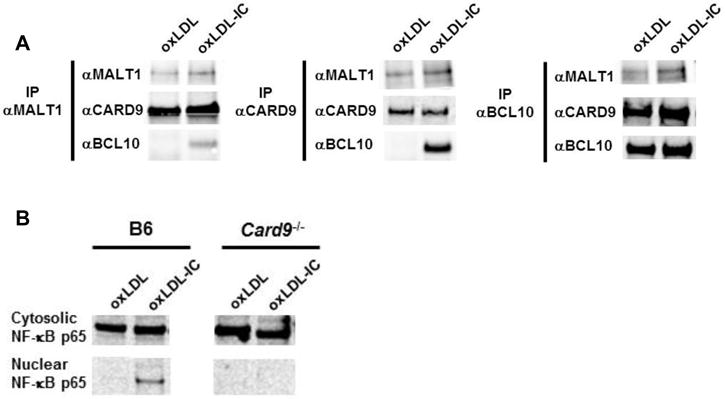
(A) BMDCs were treated with oxLDL or oxLDL-ICs for 2 hrs. Immunoprecipitation using antibodies to MALT1, CARD9, and BCL10 was performed on whole cell lysates followed by Western blot analysis. Shown is one representative of 3 similar experiments (B) BMDCs were treated with oxLDL or oxLDL-ICs for 2 hrs. Lysates were separated into nuclear and cytosolic fractions followed by Western blotting for NF-κB p65. Shown is one of three representative experiments.
OxLDL-IC-mediated IL-1β enhances IL-17 production from T cells
Given that IL-1β is an important cytokine involved in TH17 polarization, we hypothesized that oxLDL-IC-mediated inflammasome activation would modulate T cell responses. To test whether increased oxLDL-IC-induced IL-1β production by BMDCs modulated antigen-specific T cell responses, BMDCs were treated for 24 hrs with oxLDL or oxLDL-ICs then co-cultured with bead-purified splenic OT-II CD4+ T cells in the presence of OVA323–339 peptide for an additional 72 hrs. While there were no differences in T cell activation (Figure 7A) or proliferation (Figure 7B), analysis of cytokines in culture supernatants by ELISA showed that oxLDL-IC treatment of BMDCs induced increased production of IL-17 from T cells compared to oxLDL alone (Figure 7C). When this experiment was repeated using Nlrp3 or Il1b deficient BMDCs, IL-17 production was levels obtained with oxLDL alone, confirming that oxLDL-ICs enhance both the innate and adaptive immune response (Figure 7D and E).
Figure 7. OxLDL-mediated IL-1β production by BMDCs enhances antigen-specific TH17 responses.
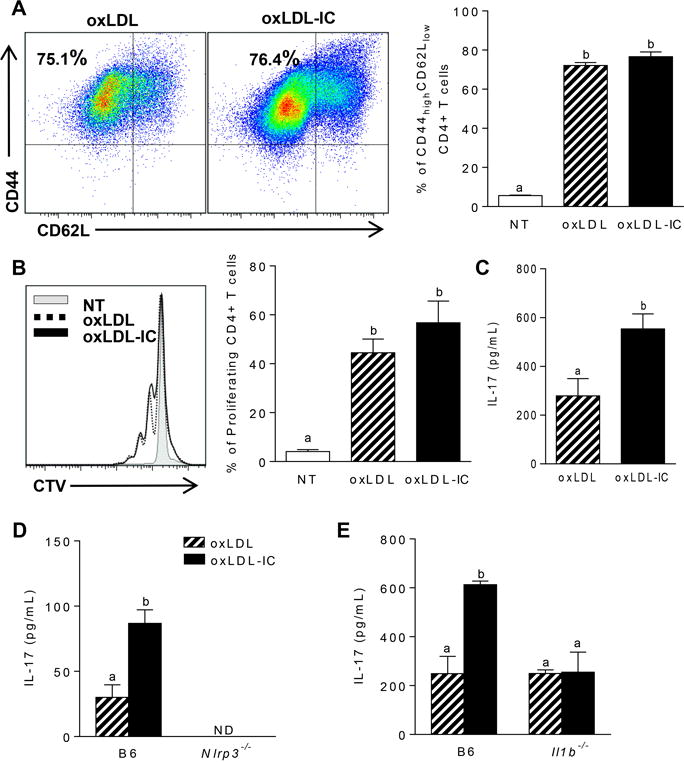
(A–C) 104 BMDCs were treated with oxLDL or oxLDL-ICs for 24 hrs followed by co-culture with 105 MACs sorted OT-II CD4+ T cells and OVA peptide (50μg/mL) for 72hrs. (A) T cell activation was measured by expression of CD44 and CD62L on CD4+TCRβ+ cells using flow cytometry. Representative dot plots are shown (left) and percent of CD44hiCD62Llo cells are quantified (right). (B) T cell proliferation was determined by Cell Trace Violet dilution. Shown is a representative histogram (left) and percent of proliferating cells quantified (right). (C) IL-17 was measured in T cell culture supernatants by ELISA. 3 mice per group were used and the experiment was repeated 3 times. Unlike letters denote significance p<0.05 by Student’s t test. Error bars represent SEM. The experiment described was repeated using (D) Nlrp3−/− and (E) Il1b−/− BMDCs. IL-17 in culture supernatants was measured by ELISA. Unlike letters denote significance (p<0.01) by one way ANOVA with Bonferroni post-test. Error bars represent SEM of 3 mice per group.
Discussion
Most studies of the immune response to oxLDL have looked at either free oxLDL or antibody to oxLDL; however, only a few have examined the importance of oxLDL-containing ICs (31–33). While direct evidence of their role in cardiovascular disease pathogenesis is lacking, a number of studies have provided data suggesting that oxLDL-ICs have disease-modifying potential. For example, Apoe−/− and Ldlr−/− mice deficient in activating FcγRI/III have decreased atherosclerosis, while atherosclerosis-susceptible mice lacking the inhibitory FcγRIIb show strain-dependent increases or decreases atherosclerosis (8, 9, 34). Interestingly, Kyaw et al. elegantly showed that IgG plays an inflammatory role, whereas IgM inhibits the development of atherosclerosis (35, 36). Although these studies support pathogenic potential for oxLDL-ICs, a direct mechanistic understanding of how these ICs may modulate disease progression is lacking. Furthermore, aside from work showing that oxLDL-ICs bind FcγRI on a human macrophage cell line and induce the secretion of pro-inflammatory cytokines, very little is understood about how oxLDL-ICs affect immune responses in potential inflammatory cells (33). Our studies show that oxLDL-ICs act directly on dendritic cells (and BMDMs) leading to up-regulation of activation markers and differential cytokine production compared to oxLDL alone. This finding is novel and important as dendritic cells provide a link between the innate and adaptive immune response.
Our current investigation shows that oxLDL-ICs act as priming signals for IL-1β production and Nlrp3 inflammasome activation via FcγR, CD36 and TLR4. Studies by Sheedy et al. demonstrated that oxLDL can be a second, activating signal for the inflammasome through a TLR4/TLR6/CD36 heterotrimer complex (37). This was largely facilitate by formation of cholesterol crystals and resulting lysosomal disruption. The authors went on to suggest that oxLDL could act as both a priming and activating signal for the inflammasome via TLR ligation and cholesterol crystal formation, respectively. Given that high levels of IL-1β were observed following 24 hrs of treatment with oxLDL-ICs (Figure 1A), it is likely that oxLDL-ICs are able to act as both a priming and activating signal for the inflammasome even more robustly than free oxLDL. OxLDL-IC priming of the inflammasome appears to be due to receptor-specific signaling leading to production of pro-IL-1β but the primary mechanism does not involve cholesterol crystal formation. These data are supported by the observations that 1) oxLDL-ICs enhance IL-1β production above oxLDL when used as a priming signal for the inflammasome; 2) IL-1β production is partially decreased by removing CD36, TLR4, or FcγR; and 3) oxLDL-ICs increase transcription of the inflammasome-related genes Il1a, Il1b, and Nlrp3. It is possible that oxLDL-ICs also act as an activating signal in vivo both by inducing cell death via pyroptosis resulting in the release of cellular contents including ATP and by cholesterol crystal formation and lysosomal disruption following uptake via FcγRs.
The aforementioned study and others have shown that oxLDL induces inflammasome mediated IL-1β production from BMDM’s; however, we were not able to detect IL-1β in BMDM supernatants under our treatment conditions (Supplemental Figure 1)(37–39). This discrepancy is likely both a time and dose dependent issue. Jiang et al. observed IL-1β production from BMDMs treated with increasing concentrations of oxLDL (25–200 μg/mL) for 12 hrs, choosing to do the majority of the experiments with 200 μg/mL of oxLDL (38). Similarly, Liu et al. used high concentrations of oxLDL (50–200 μg) for 24 hrs (39). While these high concentrations of oxLDL elicit robust responses from innate immune cells, they are at the extreme upper limit of being physiologically relevant. We chose to use 10 μg/mL oxLDL for our studies in order to more closely mimic conditions in vivo. In addition, the longer incubation periods utilized during these studies allowed time for the formation of cholesterol crystals which is the primary mechanism by which oxLDL activates the inflammasome. Our studies used a much shorter 3 hr incubation with the antigens in order to tease apart the different mechanisms by which oxLDL and oxLDL-ICs induced IL-1β production.
A recent study from Duffy et al. showed that IgG-opsonized inactivated Franciscella tularensis was able to activate the inflammasome in an FcγR/TLR dependent fashion (40). The ability of ICs to enable crosstalk between these two receptors is likely facilitated by the tight clustering of TLRs and FcγRs in glycoprotein microdomains (41). In addition, fungal antigens, such as those from Candida albicans (C. albicans), have been found to activate the inflammasome by binding to several ITAM associated C-type lectins including dectin-1, dectin-2, and mincle (28). Binding of these antigens leads to recruitment and phosphorylation of Syk and further signal propagation resulting in the formation of a CARD9/Bcl10/MALT1 complex and nuclear translocation of NF-κB (28). Our current study shows that, similar to septic inflammation, oxLDL-ICs utilize the CBM signaling pathway during sterile inflammation to enhance IL-1β responses in BMDCs, and by extension, IL-17 production by antigen-specific T cells. Surprisingly, increased CARD9-mediated NF-κB translocation did not result in increased production of TNFα or IL6, both of which are known transcriptional targets of NF-κB. There have been a handful of studies implicating CARD9 in increased TNFα production in models of fungal pathogenesis, and no reports connecting CARD9 signaling and IL-6 production to date (42–44). The studies examining CARD9 mediated TNFα production all required dectin-1 ligation. Perhaps the FcγR-CARD9 pathway is distinct from the dectin-1-CARD9 pathway and involves a phosphorylation or ubiquitination event that gives NF-κB greater affinity for the IL-1 promoter. Because we only examined TNFα levels at 24 hrs, it is also possible that levels are increased at an earlier time point. Greater understanding of the FcγR-CARD9 pathway in sterile inflammation is an area of continued interested and warrants further study.
In stark contrast to our study, Janczy et al. recently published that IgG ICs inhibit inflammasome activation by LPS in bone marrow derived macrophages (BMDMs). Specifically, BMDMs primed with LPS in the presence of ICs containing sheep’s red blood cell, OVA, or C. albicans resulted in decreased IL-1β production compared to priming with LPS alone (45). We observed that, similar to BMDCs, BMDMs also exhibit enhanced IL-1β secretion in response to oxLDL-ICs (Supplementary Figure 1). One possible explanation for the discrepancy between this work and our current study is that the specific antigen contained in the immune complex plays an important role in the immune response. Our data show that oxLDL-ICs activate the inflammasome by binding to multiple receptors on DCs including TLR4, CD36, and FcγR. There is no precedent for sheep’s red blood cell or OVA binding to pattern recognition receptors and, although C. albicans can bind both TLR2 and TLR4, LPS has been shown to upregulate the expression of the inhibitory receptor FcγRIIb on the surface of cells downregulating inflammation (46). Therefore, it is possible that pre-treatment with LPS decreases IC-mediated inflammatory cytokines due to increased FcγRIIb expression. Given that high levels of LPS are not found in sterile inflammation, our experimental system may be more clinically relevant to diseases such as atherosclerosis, systemic lupus erythematosus and rheumatoid arthritis associated with ICs containing molecules that can signal through both TLRs and FcγRs.
In conclusion, the current study demonstrates that oxLDL-ICs have the potential to enhance inflammation by priming the Nlrp3 inflammasome, and molecular mechanisms share signaling pathways with pathogens and/or ICs formed during bacterial infections (28, 40). Collectively, the data suggest that while such responses may be beneficial during acute septic inflammation, IC-mediated production of cytokines such as IL-1β during chronic sterile inflammation are likely pathogenic. These findings identify an important contribution of oxLDL-ICs to both innate and adaptive immune responses that go beyond its previous recognition as a biomarker for atherosclerosis disease severity.
Supplementary Material
Footnotes
This work was supported by grants from the Lupus Research Institute, the NIH (R21AR066971) and the Veterans Administration (I01BX002968) to A.S.M. J.P.R. was supported by the NIH (T32 AR059039 and F31 HL128040).
Literature Cited
- 1.Fernandez-Madrid F, Mattioli M. Antinuclear antibodies (ANA): Immunologic and clinical significance. Semin Arthritis Rheum. 1976;6:83–124. doi: 10.1016/0049-0172(76)90018-4. [DOI] [PubMed] [Google Scholar]
- 2.Zhao X, Okeke NL, Sharpe O, Batliwalla FM, Lee AT, Ho PP, Tomooka BH, Gregersen PK, Robinson WH. Circulating immune complexes contain citrullinated fibrinogen in rheumatoid arthritis. Arthritis Res Ther. 2008;10:R94. doi: 10.1186/ar2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Krishnan MR, Wang C, Marion TN. Anti-DNA autoantibodies initiate experimental lupus nephritis by binding directly to the glomerular basement membrane in mice. Kidney Int. 2012;82:184–92. doi: 10.1038/ki.2011.484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sokolove J, Zhao X, Chandra PE, Robinson WH. Immune complexes containing citrullinated fibrinogen costimulate macrophages via toll-like receptor 4 and Fc?? receptor. Arthritis Rheum. 2011;63:53–62. doi: 10.1002/art.30081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Salonen JT, Korpela H, Salonen R, Nyyssonen K, Yla-Herttuala S, Yamamoto R, Butler S, Palinski W, Witztum JL. Autoantibody against oxidised LDL and progression of carotid atherosclerosis. Lancet. 1992;339:883–887. doi: 10.1016/0140-6736(92)90926-t. [DOI] [PubMed] [Google Scholar]
- 6.Lopes-Virella MF, Virella G, Orchard TJ, Koskinen S, Evans RW, Becker DJ, Forrest KY. Antibodies to oxidized LDL and LDL-containing immune complexes as risk factors for coronary artery disease in diabetes mellitus. Clin Immunol. 1999;90:165–172. doi: 10.1006/clim.1998.4631. [DOI] [PubMed] [Google Scholar]
- 7.Virella G, Atchley D, Koskinen S, Zheng D, Lopes-Virella MF. Proatherogenic and proinflammatory properties of immune complexes prepared with purified human oxLDL antibodies and human oxLDL. Clin Immunol. 2002;105:81–92. doi: 10.1006/clim.2002.5269. [DOI] [PubMed] [Google Scholar]
- 8.Mendez-Fernandez YV, Stevenson BG, Diehl CJ, Braun NA, Wade NS, Covarrubias R, van Leuven S, Witztum JL, Major AS. The inhibitory Fc??RIIb modulates the inflammatory response and influences atherosclerosis in male apoE−/− mice. Atherosclerosis. 2011;214:73–80. doi: 10.1016/j.atherosclerosis.2010.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hernandez-Vargas P, Ortiz-Munoz G, Lopez-Franco O, Suzuki Y, Gallego-Delgado J, Sanjuan G, Lazaro A, Lopez-Parra V, Ortega L, Egido J, Gomez-Guerrero C. Fc Receptor Deficiency Confers Protection Against Atherosclerosis in Apolipoprotein E Knockout Mice. Circ Res. 2006;99:1188–1196. doi: 10.1161/01.RES.0000250556.07796.6c. [DOI] [PubMed] [Google Scholar]
- 10.Zhao M, Wigren M, Dunér P, Kolbus D, Olofsson KE, Björkbacka H, Nilsson J, Fredrikson GN, Bjo H. Fc{gamma}RIIB Inhibits the Development of Atherosclerosis in Low-Density Lipoprotein Receptor-Deficient Mice. 2010 doi: 10.4049/jimmunol.0902654. [DOI] [PubMed] [Google Scholar]
- 11.Ruscitti P, Cipriani P, Di Benedetto P, Liakouli V, Berardicurti O, Carubbi F. Monocytes from patients with rheumatoid arthritis and type 2 diabetes mellitus display an increased production of interleukin (IL)-1 b via the nucleotide-binding domain and leucine-rich repeat containing family pyrin 3 (NLRP3) -inflammasome activatio. 2015:35–44. doi: 10.1111/cei.12667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Choulaki C, Papadaki G, Repa A, Kampouraki E, Kambas K, Ritis K, Bertsias G, Boumpas DT, Sidiropoulos P. Enhanced activity of NLRP3 inflammasome in peripheral blood cells of patients with active rheumatoid arthritis. Arthritis Res Ther. 2015:1–11. doi: 10.1186/s13075-015-0775-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pontillo A, Reis EC, Liphaus BL, Silva CA, Carneiro-sampaio M, Pontillo A, Reis EC, Liphaus BL, Silva CA, Pontillo A, Reis EC, Liphaus BL, Silva CA, Carneiro-sampaio M. Inflammasome polymorphisms in juvenile systemic lupus erythematosus Inflammasome polymorphisms in juvenile systemic lupus erythematosus. 2016:6934. [Google Scholar]
- 14.Schroder K, Tschopp J. The Inflammasomes. Cell. 2010;140:821–832. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 15.Hise AG, Tomalka J, Ganesan S, Patel K, Hall BA, Brown GD, Fitzgerald KA. Article An Essential Role for the NLRP3 Inflammasome in Host Defense against the Human Fungal Pathogen Candida albicans. Cell Host Microbe. 2009;5:487–497. doi: 10.1016/j.chom.2009.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Body-malapel M, Amer A, Park J, Kanneganti T, Nesrin O, Franchi L, Whitfield J, Barchet W, Colonna M, Vandenabeele P, Bertin J, Coyle A, Grant EP, Akira S, Nu G. Bacterial RNA and small antiviral compounds. 2006;440:233–236. doi: 10.1038/nature04517. [DOI] [PubMed] [Google Scholar]
- 17.Yang C, Chiang B. Inflammasomes and human autoimmunity : A comprehensive review. 2015:61. doi: 10.1016/j.jaut.2015.05.001. [DOI] [PubMed] [Google Scholar]
- 18.2016. juvenile idiopathic arthritis and systemic lupus erythematosus.
- 19.Fleischmann RM, Tesser J, Schiff MH, Schechtman J, Burmester G, Bennett R, Modafferi D, Zhou L, Bell D, Appleton B. Safety of extended treatment with anakinra in patients with rheumatoid arthritis. 2006:1006–1012. doi: 10.1136/ard.2005.048371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, Schnurr M, Espevik T, Lien E, Fitzgerald KA, Abela GS, Franchi L, Nun G. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. 2010;464:1357–1362. doi: 10.1038/nature08938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lutz MB, Kukutsch N, Ogilvie AL, Rößner S, Koch F, Romani N, Schuler G. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J Immunol Methods. 1999;223:77–92. doi: 10.1016/s0022-1759(98)00204-x. [DOI] [PubMed] [Google Scholar]
- 22.Shornick LP, De Togni P, Mariathasan S, Goellner J, Strauss-Schoenberger J, Karr RW, a Ferguson T, Chaplin DD. Mice deficient in IL-1beta manifest impaired contact hypersensitivity to trinitrochlorobenzone. J Exp Med. 1996;183:1427–36. doi: 10.1084/jem.183.4.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Means TK, Latz E, Hayashi F, Murali MR, Golenbock DT, Luster AD. Human lupus autoantibody-DNA complexes activate DCs through cooperation of CD32 and TLR9. J Clin Invest. 2005;115:407–417. doi: 10.1172/JCI23025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stutz A, Golenbock DT, Latz E. Inflammasomes: Too big to miss. J Clin Invest. 2009;119:3502–3511. doi: 10.1172/JCI40599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nimmerjahn F, Ravetch JV. Fc?? receptors: Old friends and new family members. Immunity. 2006;24:19–28. doi: 10.1016/j.immuni.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 26.Perrin-Cocon L, Coutant F, Agaugué S, Deforges S, André P, Lotteau V. Oxidized Low-Density Lipoprotein Promotes Mature Dendritic Cell Transition from Differentiating Monocyte. J Immunol. 2001;167:3785–3791. doi: 10.4049/jimmunol.167.7.3785. [DOI] [PubMed] [Google Scholar]
- 27.Hara H, Ishihara C, Takeuchi A, Imanishi T, Xue L, Morris SW, Inui M, Takai T, Shibuya A, Saijo S, Iwakura Y, Ohno N, Koseki H, Yoshida H, Penninger JM, Saito T. The adaptor protein CARD9 is essential for the activation of myeloid cells through ITAM-associated and Toll-like receptors. Nat Immunol. 2007;8:619–629. doi: 10.1038/ni1466. [DOI] [PubMed] [Google Scholar]
- 28.Gross O, Poeck H, Bscheider M, Dostert C, Hannesschläger N, Endres S, Hartmann G, Tardivel A, Schweighoffer E, Tybulewicz V, Mocsai A, Tschopp J, Ruland J. Syk kinase signalling couples to the Nlrp3 inflammasome for anti-fungal host defence. Nature. 2009;459:433–436. doi: 10.1038/nature07965. [DOI] [PubMed] [Google Scholar]
- 29.Saijo S, Iwakura Y. Dectin-1 and Dectin-2 in innate immunity against fungi. Int Immunol. 2011;23:467–472. doi: 10.1093/intimm/dxr046. [DOI] [PubMed] [Google Scholar]
- 30.Robinson MJ, Osorio F, Rosas M, Freitas RP, Schweighoffer E, Gross O, Verbeek JS, Ruland J, Tybulewicz V, Brown GD, Moita LF, Taylor PR, Reis e Sousa C. Dectin-2 is a Syk-coupled pattern recognition receptor crucial for Th17 responses to fungal infection. J Exp Med. 2009;206:2037–2051. doi: 10.1084/jem.20082818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lopes-Virella MF, Hunt KJ, Baker NL, Lachin J, Nathan DM, Virella G. Levels of oxidized LDL and advanced glycation end products-modified LDL in circulating immune complexes are strongly associated with increased levels of carotid intima-media thickness and its progression in type 1 diabetes. Diabetes. 2011;60:582–9. doi: 10.2337/db10-0915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lopes-Virella MF, Virella G. Pathogenic role of modified LDL antibodies and immune complexes in atherosclerosis. J Atheroscler Thromb. 2013;20:743–54. doi: 10.5551/jat.19281. [DOI] [PubMed] [Google Scholar]
- 33.Saad AF, Virella G, Chassereau C, Boackle RJ, Lopes-Virella MF. OxLDL immune complexes activate complement and induce cytokine production by MonoMac 6 cells and human macrophages. J Lipid Res. 2006;47:1975–1983. doi: 10.1194/jlr.M600064-JLR200. [DOI] [PubMed] [Google Scholar]
- 34.Ng HP, Zhu X, Harmon EY, Lennartz MR, Nagarajan S. Reduced atherosclerosis in apoE-inhibitory Fc??RIIb-deficient mice is associated with increased anti-inflammatory responses by T cells and macrophages. Arterioscler Thromb Vasc Biol. 2015;35:1101–1112. doi: 10.1161/ATVBAHA.115.305290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kyaw T, Tay C, Krishnamurthi S, Kanellakis P, Agrotis A, Tipping P, Bobik A, Toh BH. B1a B lymphocytes are atheroprotective by secreting natural IgM that increases IgM deposits and reduces necrotic cores in atherosclerotic lesions. Circ Res. 2011;109:830–840. doi: 10.1161/CIRCRESAHA.111.248542. [DOI] [PubMed] [Google Scholar]
- 36.Kyaw T, Tay C, Khan A, Dumouchel V, Cao A, To K, Kehry M, Dunn R, Agrotis A, Tipping P, Bobik A, Toh BH. Conventional B2 B cell depletion ameliorates whereas its adoptive transfer aggravates atherosclerosis. J Immunol. 2010;185:4410–9. doi: 10.4049/jimmunol.1000033. [DOI] [PubMed] [Google Scholar]
- 37.Sheedy FJ, Grebe A, Rayner KJ, Kalantari P, Carpenter SB, Becker CE, Ediriweera HN, Adam E, Golenbock DT, Stuart LM, Latz E, Katherine A, Moore KJ. Ligands in Sterile Inflammation. 2014;14:812–820. doi: 10.1038/ni.2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jiang Y, Wang M, Huang K, Zhang Z, Shao N, Zhang Y, Wang W, Wang S. Biochemical and Biophysical Research Communications Oxidized low-density lipoprotein induces secretion of interleukin-1 b by macrophages via reactive oxygen species-dependent NLRP3 inflammasome activation. Biochem Biophys Res Commun. 2012;425:121–126. doi: 10.1016/j.bbrc.2012.07.011. [DOI] [PubMed] [Google Scholar]
- 39.Liu W, Yin Y, Zhou Z. OxLDL-induced IL-1beta secretion promoting foam cells formation was mainly via CD36 mediated ROS production leading to NLRP3 inflammasome activation. 2014:33–43. doi: 10.1007/s00011-013-0667-3. [DOI] [PubMed] [Google Scholar]
- 40.Duffy EB, Periasamy S, Hunt D, Drake JR, Harton JA. Fc g R mediates TLR2- and Syk-dependent NLRP3 inflammasome activation by inactivated Francisella tularensis LVS immune complexes. 2016:100. doi: 10.1189/jlb.2A1215-555RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shang L, Daubeuf B, Triantafilou M, Olden R, Dépis F, Raby A, Herren S, Dos Santos A, Malinge P, Dunn-siegrist I, Benmkaddem S, Geinoz A, Magistrelli G, Rousseau F, Buatois V, Salgado-pires S, Reith W, Monteiro R, Pugin J, Leger O, Ferlin W, Kosco-vilbois M, Triantafilou K, Elson G. Selective Antibody Intervention of Toll-like Receptor 4 Activation through Fc γ Receptor Tethering. 2014;289:15309–15318. doi: 10.1074/jbc.M113.537936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pedroza LA, Kumar V, Sanborn KB, Mace EM, Niinikoski H. Autoimmune regulator (AIRE) contributes to Dectin-1 – induced TNF- a production and complexes with caspase recruitment domain – containing protein 9 (CARD9), spleen tyrosine kinase (Syk), and Dectin-1. J Allergy Clin Immunol. 129:464–472.e3. doi: 10.1016/j.jaci.2011.08.027. [DOI] [PubMed] [Google Scholar]
- 43.Whibley N, Jaycox JR, Reid D, Garg V, Taylor JA, Clancy CJ, Hong M, Biswas PS, Mcgeachy MJ, Gordon D, Gaffen SL, Clancy CJ, Nguyen MH, Biswas PS, Mcgeachy MJ. Delinking CARD9 and IL-17: CARD9 Protects against Candida tropicalis Infection through a TNF-α − Dependent, IL-17 − Independent Mechanism. 2016 doi: 10.4049/jimmunol.1500870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Goodridge HS, Shimada T, Wolf AJ, Hsu YS, Becker CA, Lin X, Underhill DM. Differential Use of CARD9 by Dectin-1 in Macrophages. 2016 doi: 10.4049/jimmunol.182.2.1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Janczy JR, Ciraci C, Haasken S, Iwakura Y, Olivier AK, Cassel SL, Sutterwala FS. Immune complexes inhibit IL-1 secretion and inflammasome activation. J Immunol. 2014;193:5190–8. doi: 10.4049/jimmunol.1400628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang Y, Liu S, Liu J, Zhang T, Shen Q, Yu Y, Cao X. Immune complex/Ig negatively regulate TLR4-triggered inflammatory response in macrophages through Fc gamma RIIb-dependent PGE2 production. J Immunol. 2009;182:554–562. doi: 10.4049/jimmunol.182.1.554. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.