

Abstract
CLN2 neuronal ceroid lipofuscinosis is a hereditary lysosomal storage disease with primarily neurological signs that results from mutations in TPP1 which encodes the lysosomal enzyme tripeptidyl peptidase-1 (TPP1). Studies using a canine model for this disorder demonstrated that delivery of TPP1 enzyme to the cerebrospinal fluid (CSF) by intracerebroventricular administration of an AAV-TPP1 vector resulted in substantial delays in the onset and progression of neurological signs and prolongation of lifespan. We hypothesized that the treatment may not deliver therapeutic levels of this protein to tissues outside the central nervous system that also require TPP1 for normal lysosomal function. To test this hypothesis, dogs treated with CSF administration of AAV-TPP1 were evaluated for development of non-neuronal pathology. Affected treated dogs exhibited progressive cardiac pathology reflected by elevated plasma cardiac troponin-1, impaired cardiac function, and development of histopathological myocardial lesions. Progressive increases in plasma activity levels of alanine aminotransferase and creatine kinase indicated development of pathology in the liver and muscles. The treatment also did not prevent disease-related accumulation of lysosomal storage bodies in heart or liver. These studies indicate that optimal treatment outcomes for CLN2 disease may require delivery of TPP1 systemically as well as directly to the central nervous system.
Introduction
Hereditary lysosomal storage diseases known as neuronal ceroid lipofuscinoses (NCLs) are characterized by apparently normal development followed by progressive degeneration of the central nervous system and usually the retina as well 1. Clinical signs of the NCLs include progressive cognitive decline and loss of motor functions, seizures, and vision loss usually culminating in blindness. Ultimately the severe neurological declines associated with these disorders lead to premature death. The clinical disease signs are accompanied by massive accumulations of autofluorescent lysosomal storage material in cells of most tissues. Mutations in at least 13 different genes underlie the different forms of NCL 2, with the ages of disease onset and rates of progression varying depending on the gene involved and the specific mutations within that gene. Most of the NCLs are autosomal recessive in inheritance.
One form of NCL, known as CLN2 disease, results from mutations in TPP1, a gene that encodes the soluble lysosomal enzyme tripeptidyl peptidase-1 (TPP1) 3. In most cases of CLN2 disease the onset of signs occurs between 2 and 4 years of age and progresses to death by the teenage years 1. A canine form of CLN2 disease that results from a TPP1 null mutation in Longhaired Dachshunds has been well characterized 4–10. Onset of neurological signs in affected dogs occurs at about 4 months of age and progresses to end-stage disease at approximately 11 months of age. In the canine model, delivery of functional TPP1 enzyme to the cerebrospinal fluid (CSF), either by periodic infusion of the recombinant protein or by introduction of a TPP1 expression construct into the cells lining the brain ventricles, results in significant delays in the onset and progression of neurological signs and prolongation of life span 6, 10. Based on the efficacy of recombinant TPP1 infusion into the CSF in the canine model, this approach to therapy for CLN2 disease is currently undergoing late-stage clinical trials in children (ClinicalTrials.gov Identifiers: NCT01907087, NCT02485899 and NCT02678689).
Although the NCLs are considered primarily neurological diseases, a lack of adequate functional TPP1 in tissues outside of the central nervous system (CNS) could potentially be associated with functional impairment and tissue pathology. This possibility has been largely overlooked because of the predominance of the severe neurological signs to which ultimate death has been attributed. Indeed, accumulation of lysosomal storage material occurs in many tissues and organs outside of the CNS in CLN2 disease and other NCLs both in both human patients 1, 11–14 and in canine models 4, 5, 15–18. Progressive development of cardiac pathology has also been reported in at least some forms of NCL, including CLN2 19–22. Once CSF administration of recombinant TPP1 or TPP1 gene therapy administered via the CSF is adopted as the treatment for CLN2 disease, it appears quite possible that when neurological signs are ameliorated by these treatments and survival is extended, pathology in other tissues and organs will become apparent. This is particularly important because in the current clinical trials children with CLN2 disease are being treated with administration of recombinant TPP1 only to the CNS. To evaluate the possibility that these children may eventually exhibit extra-neuronal pathology, TPP1−/− dogs with delayed CLN2 disease progression due to CSF administration of a TPP1 gene therapy vector 10 were evaluated for indications of pathology in the heart and other tissues.
Results
No consistent abnormalities observed in most blood cytology and chemistry profiles of treated affected dogs
The concentrations of red blood cells, white blood cell types and platelets remained within the reference intervals throughout the lifespans of the affected treated dogs. A few isolated abnormalities were seen in one or two dogs at some time points, but no consistent or persistent abnormalities were observed.
Plasma biochemistry abnormalities were defined as values outside the corresponding reference interval. Likewise, the blood chemistry profiles remained within or close to the normal range for all parameters measured throughout the disease progression. The occasional abnormalities were not consistently observed between dogs of the same age or in the same dog over time. Even when values were outside of the normal range, the deviations from normal values were relatively small.
Blood activity levels of 5 enzymes (ALT, CK, ALP, and GGT) and concentration of cTn1 were used as biomarkers of specific types of tissue damage and were monitored over the courses of the lifespans of the treated dogs. Of these, ALP and GGT remained within the normal reference intervals for dogs throughout the lifespans of all of the affected treated Dachshunds. Deviations from the normal range for ALT, CK and cTn1 are described below.
Elevation of blood levels of cardiac troponin-1, alanine aminotransferase and creatine kinase
Plasma cTn1 levels in 10 normal Dachshunds across the age range of 15 to 80 weeks of age were within an established reference interval for dogs of 0.0 to 0.1 ng/ml 23, 24. Among the affected treated dogs, the cTn1 levels were within the reference interval when first measured at 16 to 17 weeks of age. However, all affected dogs exhibited progressive increases in serum troponin-1 levels with advancing age (Figure 1), despite the fact that the CSF administration of the TPP1 gene therapy vector resulted in sustained presence of TPP1 protein in the CSF and slowed the progression of neurological disease signs 10. By end stage disease all affected treated dogs had substantially elevated cTn1 levels (as high as 4.62 ng/ml).
Figure 1.
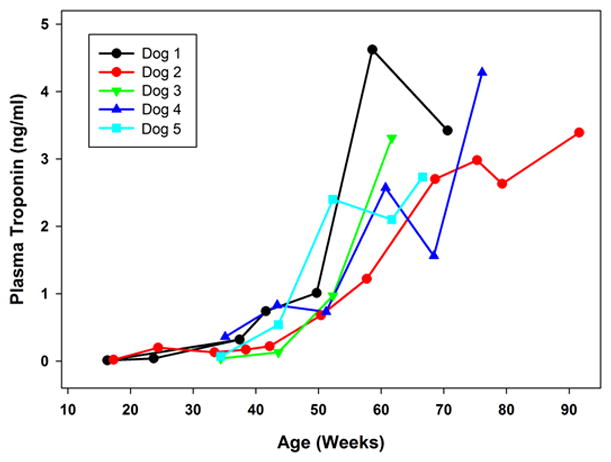
Change over time in serum cTn1 concentrations in TPP1−/− Dachshunds that received a TPP1 gene therapy vector via intracerebroventricular injection at 10 to11 weeks of age. In healthy normal dogs, including TPP1+/− Dachshunds from our colony, cTn1 levels are less than 0.1 ng/ml.
Our reference interval for ALT in dogs is 9 to 58 units/liter. Among the affected treated dogs, plasma ALT activities were all within the reference interval when the dogs were fist evaluated at 8 to 10 weeks of age. All of the affected treated dogs exhibited progressive increases in plasma ALT over time and in all but one dog these levels exceeded the maximum of the reference interval at or prior to reaching end-stage disease (Figure 2).
Figure 2.
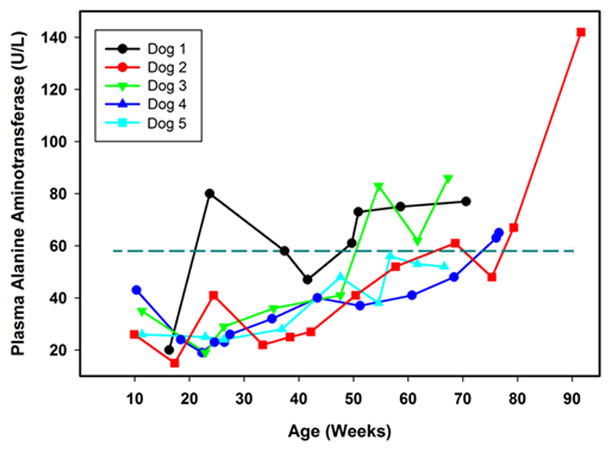
Change over time in plasma ALT activities in TPP1−/− Dachshunds that received a TPP1 gene therapy vector via intracerebroventricular injection at 10 to 11 weeks of age. Dotted horizontal line indicates the maximum of the reference interval.
CK is an enzyme released from skeletal muscle during sarcolemmal damage and has a short half-life in plasma. Our reference interval for CK in dogs is 10 to 274 units/liter. Among the affected treated dogs, there was significant variability between dogs and within each dog over time in plasma CK activities (Figure 3). However, every dog exhibited at least one plasma CK level that was above the maximum of the reference interval, with most of the elevated levels occurring near end stage disease. One dog (Dog 1) exhibited significantly elevated plasma CK levels as early as 37 weeks of age, and the levels remained elevated for most of the rest of its life (Figure 3).
Figure 3.
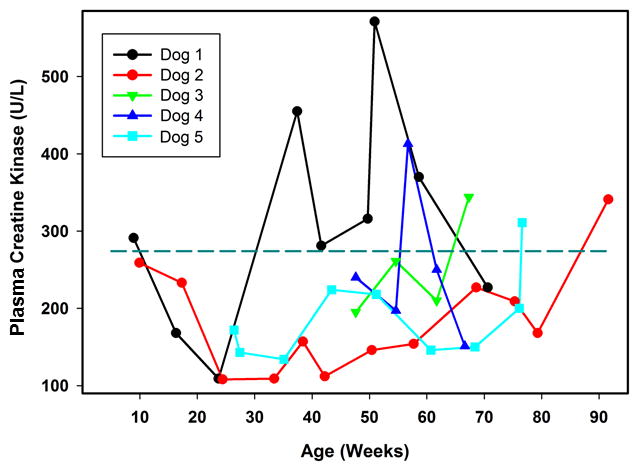
Change over time in plasma CK activities in TPP1−/− Dachshunds that received a TPP1 gene therapy vector via intracerebroventricular injection at 10 to 11 weeks of age. Dotted horizontal line indicates the maximum of the reference interval.
Cardiac functional analyses
No pathologic arrhythmias or conduction disturbances were noted in any of the heterozygous unaffected dogs at any time point. Ventricular premature complexes were noted in one affected treated dog during its final evaluation at 17 months of age. Another affected treated dog had a 2nd degree atrioventricular block, Mobitz type II, noted during the final evaluation at 16 months of age. Among the electrocardiographic measures that were performed on the dogs, the only one that was significantly different between the normal dogs and the affected treated dogs was heart rate. In dogs 12 to 17 months of age, end systolic volume was significantly higher among the affected treated dogs than in the normal control dogs (p<0.01) (Figure 4). No overt structural heart disease was noted in any of the dogs during the initial echocardiographic evaluation at 8.5 months of age. Among the echocardiographic parameters that were assessed in the dogs, the heart rate, ESVI, and LVIDs were significantly elevated in affected treated dogs compared to control dogs over the 12 to 17 month age range (p<0.05) (Supplemental Figures 1 and 2). Over the same age range the ejection fractions were significantly lower in the affected treated dogs than in the normal dogs (p<0.10) (Figure 5). One of the affected treated dogs had a profound reduction in ejection fraction during its final evaluation when compared to control dogs (Figure 6 and Supplementary Videos 1 & 2).
Figure 4.
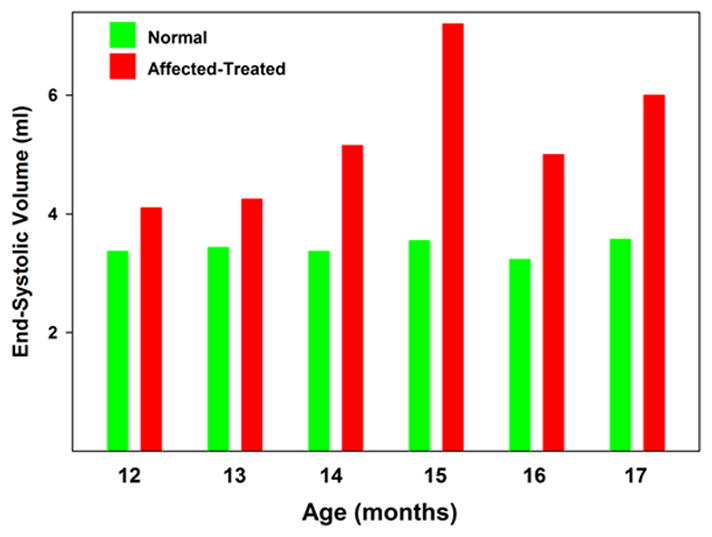
Median heart end systolic volume of normal and affected treated Dachshunds between 12 and 17 months of age. Over this age range the mean end systolic volume of the affected dogs was higher than that of the normal dogs at every age assessed. Comparison of the data using the Friedman repeated measures analysis of variance on ranks test indicated that the end systolic volume in the affected treated dogs was significantly higher than in the normal dogs over this age range (p<0.01).
Figure 5.
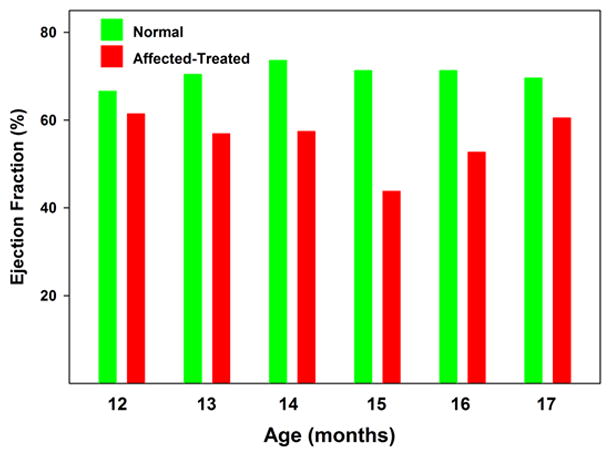
Median ejection fraction of normal and affected treated Dachshunds between 12 and 17 months of age. Over this age range the mean ejection fraction of the affected dogs was lower than that of the normal dogs at every age assessed. Comparison of the data using the Mann-Whitney rank sum test indicated that the ejection volume in the affected treated dogs was significantly lower than in the normal dogs over this age range (p<0.01).
Figure 6.
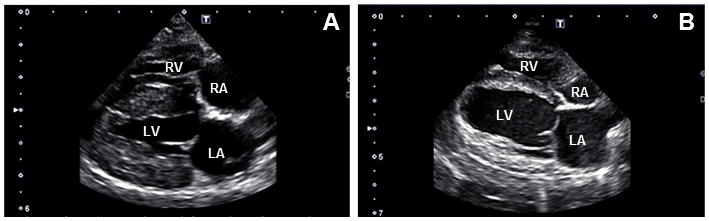
Video stills. Representative two-dimensional right parasternal 4 chamber long axis view of a control dog (A) and affected treated dog (B) at 15 months of age. The affected treated dog has a much larger left ventricular end-systolic diameter and reduced ejection fraction when compared to the control. LA=left atrium, LV=left ventricle, RA=right atrium, RV=right ventricle.
Postmortem evaluation of cardiac pathology
Myocardial pathology was observed grossly and microscopically in all CLN2-affected dogs that we have examined at necropsy, and among the dogs evaluated for this study cardiac measurement abnormalities indicative of biventricular hypertrophy were observed, particularly in the older animals (Table 1). Post mortem cardiac measurements were indicative of heart enlargement at the time the dogs reached neurological end-stage disease. Measurements were made on five affected treated dogs that ranged from 10 to 21 months in age (Table 1). Both right and left ventricular weights were increased relative to total heart weight in 2 dogs euthanized at 10 months of age, reflecting biventricular enlargement relative to the atria. Increased right ventricle to total heart weight ratios were also increased in dogs euthanized at 10 months. Older animals in which lifespan was increased by the ICV gene therapy had an increased right to left ventricular weight ratio and right ventricle to total heart weight ratio compared to normal in dogs 18 to 21 months of age, suggesting relatively more extensive damage to the right ventricle. Abnormal cardiac weight ratios increased with greater survival. Abnormal ventricular and total heart to body weight ratios were present after 18 months with a relative prominence of right ventricular weights, as indicated by decreased LV+S/RV ratios. This prominence is also reflected in residual increase right ventricular weight to total cardiac weight; ratios to total heart weight are usually normal in biventricular hypertrophy.
Table 1.
Anatomical measures in hearts of CLN2-affected dogs that received ICV TPP1 Gene Therapy
| Measurement | Dog 7* 10 Mos |
Dog 6* 10 Mos |
Dog 4 18 Mos |
Dog 1 18 Mos |
Dog 2 21 mos |
Reference Range |
|---|---|---|---|---|---|---|
| H/BW %1 | 0.78% | 0.74% | 1.13% ↑ | 0.93 | 0.94 ↑ | 0.58–0.94% |
| LV+S/RV2 | 2.86 | 3.18 | 2.36 ↓ | 2.4↓ | 2.42 ↓ | 2.76–3.88 |
| RV/BW, %3 | 0.18 | 0.16 | 0.27 ↑ | 0.23 ↑ | 0.24 ↑ | 0.1–0.18 |
| LV+S/BW, %4 | 0.54 | 0.49 | 0.65 ↑ | 0.56 ↑ | 0.59 ↑ | 0.35–0.55 |
| RV/TC, %5 | 23.8 ↑ | 21.0 ↑ | 24% ↑ | 24.9↑ | 25.7 ↑ | 15.3–20.9% |
| LV+S/TC, %6 | 68.2 ↑ | 66.8 ↑ | 56 | 59.7 | 62.3 | 52.5–66.5% |
Total heart weight/body weight
Left ventricle and septum weight/right ventricle weight
Right ventricle weight/body weight
Left ventricle and septum weight/body weight
Right ventricle weight/total cardiac weight
Left ventricle and septum weight/total cardiac weight
A few opalescent pale foci could be observed on the epicardial surface of the heart, and were present as early as 10 months of age in an untreated affected dog Figure 7A. Some foci were observed in each of the older affected treated dogs that were necropsied, and either the right ventricle, the left ventricle or both might be affected in a particular dog. In older dogs the heart had a progressively more flabby appearance Fig 7B. Well defined but irregular opaque foci that correspond to interstitial fibrous tissue were apparent on the epicardial surface in all animals (arrows, Figure 7A,B).
Figure 7.
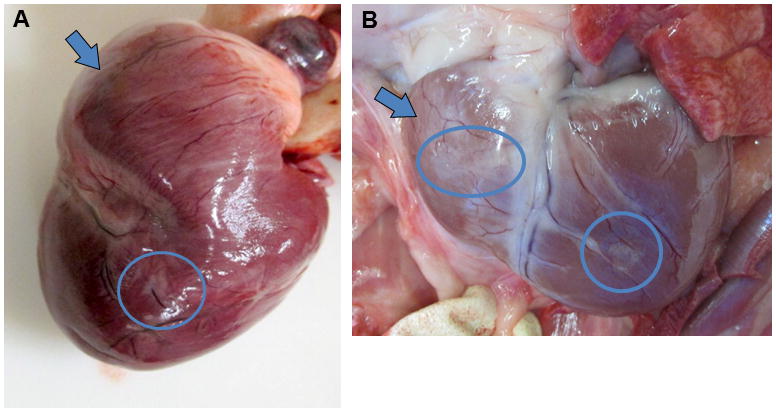
Right ventricular enlargement is grossly evident in treated dogs. Gross appearance of the heart of an untreated affected dog euthanized at 10 months of age (A) and in a treated affected dog euthanized at 21 months (B). The right ventricle (arrows) has a flabby appearance in the older dog, and the left ventricle is enlarged in this dog. Small pale foci are evident on the epicardial surface (circled) that are more prominent in the older treated dog.
Microscopically, there were small, apparently randomly distributed foci of fibrosis with clusters of histiocytic inflammatory cells replacing myocytes in the myocardium of all affected treated dogs (Figure 8A). Many of the remaining viable myocytes were clearly necrotic (Figure 8A). The numbers of fibrotic foci and necrotic muscle fibers increased with increasing age of the dogs. With increasing age there was also a dramatic increase in foci in which interstitial collagen replaced muscle fibers within the myocardium (Figure 8B and C).
Figure 8.
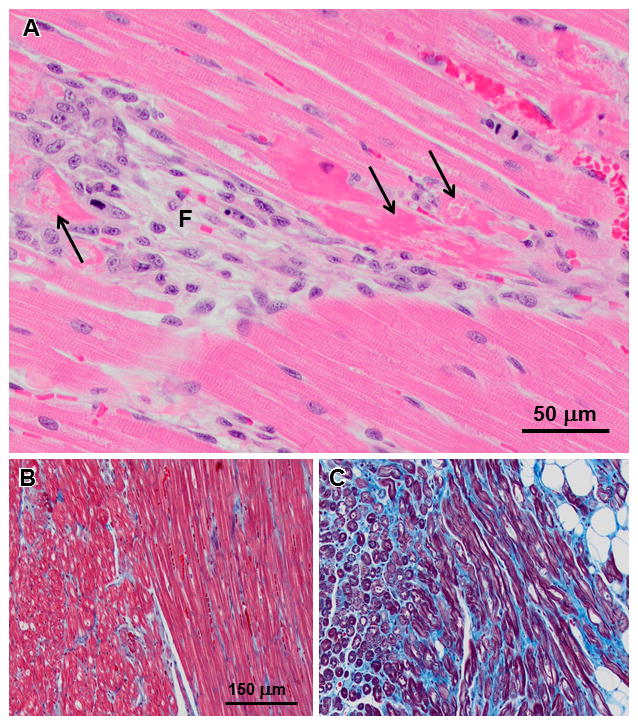
(A) H&E stained right ventricular myocardium from a 16 month old affected treated dog contains fibrotic foci with loss of myofibers (F) as well as necrotic myofibers in the adjacent tissue (arrows in A). (B and C) Masson Trichrome stained sections of right ventricular myocardium from a 10 month old untreated affected dog (B) and a 21 month old affected treated dog (C). Collagen stains blue and myofibers stain red. In the older dog a substantial amount of interstitial collagen has replaced muscle fibers. Bar in (B) indicates magnification of images in (B) and (C).
Due to the functional cardiac deficits observed antemortem, the lungs were examined for pathological abnormalities associated with heart failure. Dogs necropsied between 18 and 21 months of age had pulmonary edema, as shown in Figure 9. Occasional alveolar macrophages contained iron pigment, consistent with hemosiderin staining commonly seen in congestive failure.
Figure 9.
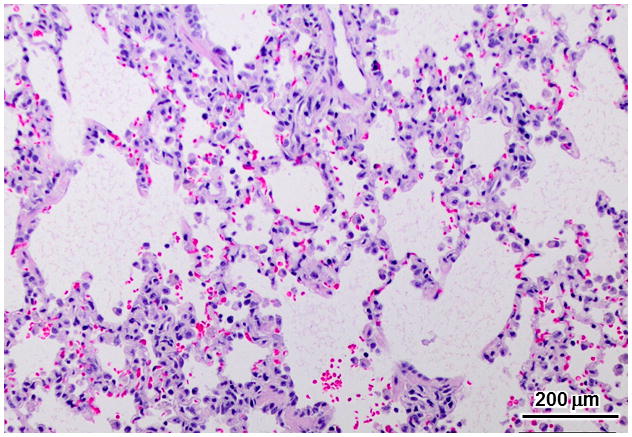
Alveolar histiocytosis in the lung. Mild hemorrhage and hemosiderosis, consistent with left congestive failure, was found in the lungs of the 3 oldest affected treated dogs euthanized at 17 to 21 months of age.
Autofluorescent storage body accumulation persists in cardiac muscle and liver
We previously demonstrated that administration of a TPP1 gene therapy vector to the CSF resulted in a substantial reduction of autofluorescent storage body accumulation in the central nervous system of CLN2-affected dogs, including the spinal cord 10. A similar reduction in autofluorescent storage body storage accumulation in the central nervous system was observed after repeated administration of recombinant TPP1 protein to the CSF 25. Since the affected treated dogs exhibited elevated blood levels of enzymes indicative of heart and liver pathology over time, these tissues were examined for the presence of the disease-specific autofluorescent inclusions typical of the NCLs. In both tissues there was substantial storage body accumulation present at the time of euthanasia in all affected treated dogs that was not present in dogs that were not homozygous for the mutant TPP1 allele (Figure 10).
Figure 10.
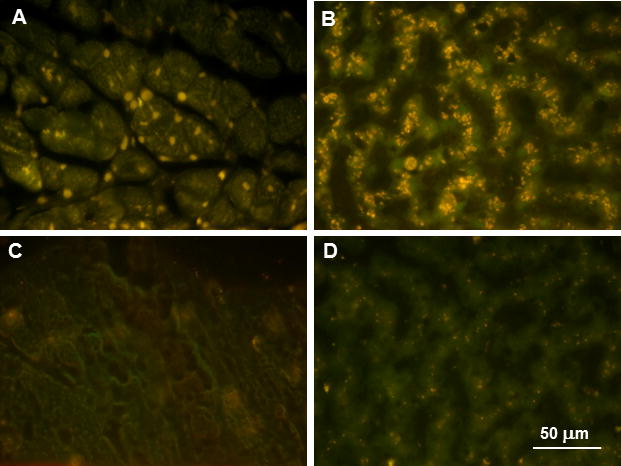
Fluorescence micrographs of unstained cryostat sections of heart ventricular muscle (A) and liver (B) from an affected dog that had been treated with ICV TPP1 gene therapy and was euthanized at 18 months of age and of heart ventricular muscle (C) and liver (D) from a healthy 2 year old long haired Dachshund that was heterozygous for the TPP1 mutant allele. The yellow-emitting autofluorescent inclusions are characteristic of CLN2 disease. A small number of punctate inclusions were present in the livers of the normal dogs and probably represent the normal age pigment lipofuscin.
Discussion
Our data show clear evidence of progressive pathology in tissues outside of the central nervous system of CLN2-diseased dogs despite cerberoventricular administration of a TPP1 gene therapy vector that significantly delayed the onset and progression of neurological disease signs and prolonged lifespan 10. The evidence for cardiac pathology was particularly compelling, but increasing blood levels of ALT and CK activity in addition to the elevated plasma concentrations of cTn1 indicate that disease-related pathology outside of the CNS becomes widespread when inhibiting the progression of neurological signs prolongs lifespan. In every affected treated dog there was a consistent progressive increase in blood cTn1 concentration and ALT activity level with increasing age, reflecting increasing heart and liver pathology.
Incremental increases in the severity of gross morphological cardiac lesions and histolopathologic changes in cardiac muscle and signs of cardiac functional impairment were consistent with the plasma troponin data indicating progressive cardiac pathology with increasing survival time. The elevated heart rates and reduced ventricular systolic function (i.e. decreased ejection fractions and fractional shortening and increased ESV, ESVI, and LVIDs) in affected treated dogs whose lifespans were extended by TPP1 gene therapy administered to the CSF reflect impaired heart function in the affected dogs that was not prevented by the gene therapy treatment. This indicates that despite the fact that some TPP1 in the CSF does reach the systemic circulation and peripheral tissues, including the heart 10, 26, it was not sufficient to prevent the disease-related changes in heart function. No elevation of TPP1 enzyme activity above background was observed in the liver of the gene therapy treated dogs 10, consistent with the observed evidence of liver pathology in the treated dogs. Immunohistochemical labeling of cardiac muscle from TPP1−/− dogs that received the CSF TPP1 gene therapy indicated that a fraction of the cardiomyocytes contained TPP1 protein at the time of euthanasia 10. The distribution of TPP1-postive cells in cardiac muscle was patchy, so the majority of cardiac cells did not receive detectable amounts of TPP1. The same patchy distribution of TPP1-positive cells was observed in the spleens of these dogs 10. It is not apparent how in these dogs some cells of peripheral tissues took up TPP1 while immediately adjacent cells of the same tissue did not, but the fact that the majority of cardiac muscle cells did not contain detectable TPP1 protein is consistent with the fact that the treated dogs exhibited cardiac pathology.
Although the data indicated significant cardiac functional impairment when the data from 12 to 17 months were pooled, we did not evaluate a sufficient number of dogs to assess whether the functional impairment progressed with advancing age. This was due in part to the fact that some of the affected treated dogs reached end-stage neurological disease and had to be euthanized at various ages before 17 months.
The blood CK activities, on the other hand were within the normal reference interval for most of the affected treated dogs until at or near end-stage neurological disease, at which time they became elevated. Elevated CK concentrations are usually associated with muscle pathology related to primary muscle disease or trauma. CK can also be elevated due to the myoclonic fasiculations and seizure activity which are both indicators of end stage disease. Due to the short half-life of CK, the concentrations can vary greatly from day to day if muscle damage is episodic. Sequential monitoring is important in diseases like CLN2 in which episodic seizure activity occurs. Although all of the affected treated dogs exhibited impaired mobility at end stage disease 6, 10, it is not known whether this was due to secondary injury from falling or reflected direct functional impairment in the skeletal and cardiac muscles. To our knowledge blood levels of these enzymes have not been monitored in children affected with CLN2 disease nor is cardiac function typically evaluated. Based on our data affected children should routinely be monitored for signs of extra-neural pathology, particularly in heart, liver and skeletal muscle. This will be especially important for children currently enrolled in clinical trials in which only the CNS is being targeted for treatment, particularly since cardiac failure has been documented as the proximate cause of death in at least one CLN2 disease patient 20. Elevated blood levels of cTn1, ALT activity and CK activity in these children will indicate a need to complement the CNS treatment with systemic treatment that would prevent pathology outside of the CNS. Similarly, serial echocardiography revealed progressively increased left ventricular volumes and diameters along with reductions in ejection fraction and fractional shortening indicating progressive left ventricular systolic dysfunction. The presence of arrhythmias and conduction disturbances noted in the affected treated dogs during their final evaluations may be due to the pathological changes found in the myocardium at necropsy and in the cardiac conduction system. Clinical cardiac failure did not occur in the affected treated dogs, all of which were euthanized due to the progression of the neurological signs of CLN2 disease.
Although some TPP1 from the CSF does reach the blood circulation and peripheral organs, in studies in which recombinant TPP1 was administered by infusion into the CSF, peak blood levels of TPP1 were at least 1,000-fold lower than CSF levels 26. In the intracerebroventricular TPP1 gene therapy studies, detectable levels of TPP1 enzyme activity were found in the heart 10. Our current data indicates that these amounts of TPP1 are insufficient to prevent progressive effects of TPP1 deficiency outside of the central nervous system. We previously demonstrated that the intracerebroventricular administration of the TPP1 gene therapy vector was ineffective in preventing progressive retinal degeneration and decline in retinal function in the canine CLN2 disease model 27. Therefore, it seems likely that current efforts to treat the disease by exclusively targeting delivery of TPP1 to the CNS will not only fail to prevent disease-related blindness, but will also likely result in the appearance of clinically evident functional impairment of non-neuronal organs, particularly the heart, when lifespan is prolonged due to the delay in neurological sign progression. Indeed, even in a human CLN2 disease patient that had not received any therapeutic intervention, cardiac functional impairment severe enough to be deemed the proximate cause of death has been reported 20. Cardiac functional impairment and histopathology have also been reported in other forms of NCL in both humans and dogs 19, 21, 28, 29.
Summary
Based on our findings, it appears that therapeutic interventions targeted exclusively to the CNS will be insufficient for treating CLN2 disease and likely other forms of NCL and other similar lysosomal storage diseases as well. A variety of approaches could be tested to overcome this problem. In lysosomal storage diseases without significant CNS involvement that result from deficiencies in soluble lysosomal enzymes, intravenous infusion of a functional form of the defective enzyme has shown considerable therapeutic efficacy 30–33. Therefore, for CLN2 disease, a combination of intracerebroventricular and intravenous administration of either recombinant TPP1 or a TPP1 gene therapy vector may be effective in inhibiting the progression of both neuronal and non-neuronal signs. Alternatively, systemic administration of a gene therapy vector that crosses the blood-brain barrier may be effective in treating all tissues affected by TPP1 deficiency. Intravenous administration of AAV9 vectors have been shown to be very effective in producing trans gene expression throughout the body, including the CNS because these vectors can cross the blood-brain barrier 34–38. Therefore, systemic administration of AAV9-TPP1 vectors should be considered as a possible improved means of treating CLN2 disease and other similar lysosomal storage diseases.
Materials and methods
Animals
A colony of miniature longhaired Dachshunds was developed from a foundation established by breeding two dogs that were heterozygous for a naturally occurring one nucleotide deletion in TPP1 (c.325delC). The mutation causes a frame shift and a premature stop codon 4. Tissues from affected dogs have no detectable TPP1 enzyme activity 4. The colony has been maintained for a number of years by performing a combination of carrier to carrier, normal to carrier and affected to carrier breedings. Untreated affected dogs seldom live to sexual maturity, so most of the affected dogs used for breeding were those whose life spans were extended via TPP1 enzyme delivery to the CSF 6, 10. To avoid potential health problems unrelated to the TPP1 mutation that could arise from close inbreeding, dogs were only bred if they did not have a common ancestor for at least the two most recent previous generations. Unrelated healthy miniature Dachshunds from various sources are periodically bred to our research dogs and resulting puppies heterozygous for the TPP1 mutation are incorporated into the breeding colony to minimize excessive inbreeding. Puppies were genotyped at the TPP1 locus containing the mutation using an allelic discrimination assay 4. Dogs that were homozygous for the mutant and normal alleles or heterozygous for the TPP1 mutation were employed in this study. The latter served as normal controls for cardiac function analyses. Dogs were housed and cared for in an Association for Assessment and Accreditation of Laboratory Animal Care accredited facility, were maintained on a 12:12 daily light cycle and were socialized daily by trained staff. All studies were performed in compliance with the United States National Research Council Guide for the Care and Use of Laboratory Animals and were approved by the University of Missouri Animal Care and Use Committee.
CNS Gene Therapy Treatment
Dogs that were homozygous for the TPP1 null mutation were treated with infusion into the CSF of a rAAV2 vector directing expression of wildtype canine TPP1 (AAV2.caTPP1) and were then evaluated for the efficacy of this treatment in ameliorating disease progression. These studies were described in detail previously 10. At approximately 10 to 11 weeks of age, dogs homozygous for the mutant TPP1 null allele received unilateral, intraventricular injection of rAAV2.caTPP1, which resulted in nearly complete transduction of the ependymal lining of the lateral cerebral ventricles, and additional transduction of the ependyma in the third and fourth ventricles 10. This resulted in sustained levels of TPP1 enzyme in the CSF that were substantially above levels in untreated normal dogs and widespread uptake of the protein in cells throughout the brain. This treatment significantly delayed the onset and slowed the progression of neurological disease signs and substantially prolonged lifespan 10. All of the treated dogs exhibited periodic elevations in CSF levels of nucleated cells indicative of CNS inflammation at various times in the many months after gene therapy vector administration. These dogs were treated with oral prednisone until the levels of cells in the CNS returned to normal. After discontinuation of the prednisone treatment, most of the dogs eventually again exhibited elevations of CSF nucleated cells counts, and each time these were brought down by systemic prednisone treatment. Untreated affected dogs typically reach end-stage disease based on the severity of neurologic signs requiring euthanasia at 10.5 to 11 months of age. Treated dogs lived to be as old as 21 months of age before reaching neurological end-stage disease. The treated dogs were evaluated for evidence of disease-related extra-neuronal pathology.
Assessment of biochemical markers of peripheral tissue pathology
Serial blood samples were collected for complete blood counts (CBC) and biochemical and electrolyte profiles, and cardiac troponin-1 (cTn1) determinations. Among the analytes included in the biochemical profiles were activity levels of alanine amino transferase (ALT), gamma-glutamyl transpeptidase (GGT) and alkaline phosphatase (ALP), indexes of hepatic function (albumin, glucose, cholesterol, bilirubin, blood urea nitrogen [BUN]), creatinine. Cardiac troponin-1 is a highly specific and sensitive marker for myocardial cellular damage in many mammalian species. Its structure is highly conserved across species, and assays used for humans have been validated in dogs. Cardiac troponin-1 concentration is rapidly elevated after cardiomyocyte damage 39 and the protein has a half-life in plasma of 7 hours 40. ALT is an enzyme present in the cytoplasm of hepatocytes, and an elevation in its activity level in plasma or serum is indicative of sublethal hepatocyte injury or hepatocyte necrosis. The serum half-life of ALT is 60 hours in dogs. Plasma or serum ALT activity levels also may be increased with severe muscle damage, but elevated blood ALT activity levels are considered relatively liver specific in dogs 41, 42. Creatine kinase (CK) is a cytosolic enzyme with high levels in muscle, heart and brain, but the vast majority of CK activity in plasma is derived from muscle. Because the half-life of CK in the circulation is 2 hours its activity levels in plasma or serum are a measure of recent muscle damage 43. Red and white blood cell counts, hemoglobin, hematocrit, platelet count, mean corpuscular volume, mean cell hemoglobin, mean corpuscular hemoglobin concentration and reticulocyte count were performed on EDTSA-anticoagulated blood using a Sysmex XT 2000 iV hematology analyzer (Sysmex Corporation, Kobe, Japan). For the CBC plasma total protein was estimated via refractometry. The white blood cell differential counts and blood cell morphologic evaluations were done manually by blood smear examination, using wedge smears stained with a modified Wright-Giemsa stain. The biochemistry panels were done using a Beckman AU400 chemistry analyzer(Beckman Coulter, Brea, CA). Cardiac troponin-1 was determined on heparinized plasma using an i-Stat analyzer (Abbott Laboratories, Abbott Park, IL). An example of a blood analysis report is included in the Supplemental Materials section (Supplemental Figure 3).
Assessment of cardiac function
Serial echocardiographic and electrocardiographic (ECG) evaluations of TPP1 gene therapy treated CLN2-affected dogs were performed starting at 8.5 months of age and continued until time of death. Healthy related Dachshunds heterozygous for the TPP1 mutation were used as age-matched controls for each time point. Evaluations were performed at 8.5 months, 10.5 months, 12 months, 13 months, 14 months, 15 months, 16 months, and 17 months of age. All dogs were sedated with acepromazine (0.03 mg/kg) and butorphanol (0.2 mg/kg) given intramuscularly 10 minutes prior to evaluation. To minimize inter-examiner variances all ECGs and echocardiograms were performed by a single investigator (SBL) board certified in veterinary cardiology. The investigator was blinded to the dog’s genotype.
Standard 6-lead ECGs (leads I, II, III, aVR, aVL, and aVF) were recorded using a Philips Pagewriter TC30 ECG machine (Royal Philips Electronics, Eindhoven Area, Netherlands) with dogs placed in right lateral recumbency. All ECGs were recorded with a calibration of 1 cm/mV and paper speed of 25 mm/s and 50 mm/s. Evaluation of heart rate and rhythm was made using recordings obtained at 25 mm/s. Standard measurements of amplitudes and intervals were performed using lead II with a paper speed of 50 mm/s. Variables measured included P-wave amplitude and duration, P-R interval, R-wave amplitude, QRS duration, and Q-T interval. When a wandering atrial pacemaker was present, the highest amplitude P-wave was measured.
Echocardiography was performed using a Toshiba Aplio Artida ultrasound machine (Toshiba Medical Systems Corporation, Otawa, Tochigi, Japan) and a 5 mHz phased-array probe. Two-dimensional (2D), M-mode, and Doppler echocardiographic studies were performed using standard views with continuous ECG monitoring 44, 45. Two-dimensional measurement of the left atrial long axis diameter (LAlax) was performed using the right parasternal long-axis 4 chamber view 46. Left ventricular (LV) volume measurements (end-diastolic volume [EDV] and end-systolic volume [ESV]) for calculations of ejection fraction (EF) and volume indices were obtained from a right-parasternal long-axis 4-chamber view using the disk summation method and optimizing the image to include the LV apex 47. The EDV and ESV were indexed to body surface area (BSA) to determine the end-diastolic volume index (EDVI) and end-systolic volume index (ESVI), respectively. Body surface area was calculated as [10.1 × (weight in kg)2/3/100 = M2]. LV parameters including the internal diameters in diastole and systole (LVIDd and LVIDs), and LV fractional shortening were calculated using 2D guided M-mode images at the level of the papillary muscles using the right parasternal short-axis view. Left ventricular diastolic function was evaluated by measuring transmitral flow patterns, and mitral annular motion velocities 48. Mitral inflow pulsed-wave Doppler measurements of peak early (E) wave, peak late (A) wave, and E/A ratio were obtained by placing the sample gate between the open mitral valve leaflets tips. Pulsed-waved tissue Doppler imaging (TDI) was performed to evaluate LV longitudinal motion by placing the sample gate on the lateral wall of the mitral valve annulus using the left apical four-chamber view. The nyquist limit, gain, and filter settings were optimized to detect low-velocity, high-amplitude signals of the myocardium. The early diastolic (Ea) and late diastolic (Aa) motion of the mitral annulus were measured. For all measured echocardiographic variables, 3–5 consecutive cardiac cycles were evaluated.
Tissue histopathology
Affected treated dogs were humanely euthanized at various ages. In addition, for autofluorescence analyses a liver sample obtained from a healthy carrier Dachshund euthanized at approximately 1.5 years of age and cardiac muscle from a 3 year old Dachshund mix that was euthanized for non-neurological disease were evaluated. Within 10 to 15 minutes of euthanasia, the heart was removed and any grossly visible abnormalities in the affected dogs were photographically documented. To evaluate the relationship between cTn1 release and morphological changes, the hearts of 5 treated, end-stage dogs were dissected and weighed immediately thereafter, according to standard procedures 49. Proportional weights of the ventricles to total heart weight, to each other, and to body weight were recorded. The comparison group was made up of normal dogs of varied breeds and ages that were evaluated from dogs euthanized over a period of years. Values were considered abnormal if the ratios exceeded 2 standard deviations above or below the mean of the normal dogs. Portions of the heart muscle were fixed by immersion in neutral buffered formalin (pH 7), and prepared for microscopic examination. Sections of the paraffin embedded tissues were cut a thickness of 4 μm and mounted on glass slides. Sections were stained with hematoxylin and eosin (H&E) for general examination, Masson trichrome for fibrous tissue and picrosirius red. Portions of the lungs were also dissected at necropsy, fixed in neutral buffered formalin and prepared in the same manner as the heart tissue for microscopic examination.
The sections were stained as indicated in the figure captions. After heart weight data were obtained, a small portion of the right ventricular wall was removed and fixed for fluorescence microscopy in immuno fixative (3.5% paraformaldehyde, 0.05% glutaraldehyde, 120 mM sodium cacodylate, 1 mM CaCl2, pH 7.4). Likewise, a slice of liver approximately 5 mm by 5 mm by 2 mm was fixed in immuno fixative. For fluorescence microscopy, slices of the immuno-fixed heart and liver were embedded in TissueTek O.C.T. compound (Sakura Finetek, Torrance, CA) and frozen on a block of dry ice. Sections of the frozen samples were cut at a thickness of 8 μm and mounted on Fisher Superfrost slides in 0.17 M sodium cocylate, pH 7.4. The sections were examined for NCL storage body-specific autofluorescence using a Zeiss Axiophot microscope as described previously 50. Images of the sections were obtained with an Olympus DP72 digital camera.
Statistical Analyses
Electrocardiographic and echocardiographic data obtained from five normal and three treated affected dogs at monthly intervals from 12 to 17 months of age were evaluated for statistically significant differences. Using SigmaPlot statistical software for analysis, the composite data failed both the normality test, so the typical measures of variance used for normally distributed data (standard deviation and standard error) could not be calculated for these parameters. Because the data were not normally distributed the nonparametric Mann-Whitney rank sum test was used to evaluate all of the heart function data. For this test, the median value of each parameter was determined for each group of dogs (affected-treated and normal control) at each time point and is plotted in the summary graphs in the results section. The Mann-Whitney procedure was used to calculate the probabilities that the median values of the affected-treated and control groups were significantly different over the range of ages at which the data were acquired. The latter statistical test was performed using the SigmaPlot software.
Supplementary Material
Acknowledgments
This work was supported in part by grants EY018815 and EY023968 from the U.S. National Institutes of Health, the Children’s Hospital of Philadelphia and by a grant from the Knights Templar Eye Foundation.
Our thanks to Lani Castaner with her assistance in many aspects of the research including managing the care and socialization of the dogs and assisting with most procedures, including sample collections and cardiac function assessments. We also thank Drs. Dawna Voelkl and Dietrich Volkmann for managing the breeding of the dogs used in this study and Dr. Beverly Davidson for her previous collaboration on assessing the efficacy of TPP1 gene therapy on the nervous system in the dogs used in this study. This work was supported in part by grants EY018815 and EY023968 from the U.S. National Institutes of Health, the Children’s Hospital of Philadelphia and by a grant from the Knights Templar Eye Foundation.
Supplementary information is available at Gene Therapy’s website.
Footnotes
Conflict of interest: The authors declare no conflict of interest.
References
- 1.Mole SE, Williams RE, Goebel HH. The Neuronal Ceroid Lipofuscinoses (Batten Disease) 2. Oxford University Press; Oxford: 2011. [Google Scholar]
- 2.Mole SE, SLC Gentics of the neuronal ceroid lipofuscinoses (Batten disease) Biochimica Bipphysica Acta. 2015;1852:2237–2241. doi: 10.1016/j.bbadis.2015.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sleat DE, Gin RM, Sohar I, Wisniewski K, Sklower-Brooks S, Pullarkat RK, et al. Mutational analysis of the defective protease in classic late-infantile neuronal ceroid lipofuscinosis, a neurodegenerative lysosomal storage disorder.[Erratum appears in Am J Hum Genet. 2004 Dec;75(6):1158] Am J Hum Genet. 1999;64(6):1511–23. doi: 10.1086/302427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Awano T, Katz ML, O’Brien DP, Sohar I, Lobel P, Coates JR, et al. A frame shift mutation in canine TPP1 (the ortholog of human CLN2) in a juvenile Dachshund with neuronal ceroid lipofuscinosis. Mol Genet Metab. 2006;89(3):254–60. doi: 10.1016/j.ymgme.2006.02.016. [DOI] [PubMed] [Google Scholar]
- 5.Katz ML, Coates JR, Cooper JJ, O’Brien DP, Jeong M, Narfstrom K. Retinal pathology in a canine model of late infantile neuronal ceroid lipofuscinosis. Invest Ophthalmol Vis Sci. 2008;49(6):2686–95. doi: 10.1167/iovs.08-1712. [DOI] [PubMed] [Google Scholar]
- 6.Katz ML, Coates JR, Sibigtroth CM, Taylor JD, Carpentier M, Young WM, et al. Enzyme replacement therapy attenuates disease progression in a canine model of late-infantile neuronal ceroid lipofuscinosis (CLN2 disease) J Neurosci Res. 2014;92(11):1591–8. doi: 10.1002/jnr.23423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sanders DN, Kanazono S, Wininger FA, Whiting RE, Flournoy CA, Coates JR, et al. A reversal learning task detects cognitive deficits in a Dachshund model of late-infantile neuronal ceroid lipofuscinosis. Genes Brain Behav. 2011;10(7):798–804. doi: 10.1111/j.1601-183X.2011.00718.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Whiting RE, Narfstrom K, Yao G, Pearce JW, Coates JR, Castaner LJ, et al. Pupillary light reflex deficits in a canine model of late infantile neuronal ceroid lipofuscinosis. Exp Eye Res. 2013;116:402–10. doi: 10.1016/j.exer.2013.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Whiting RE, Pearce JW, Castaner LJ, Jensen CA, Katz RJ, Gilliam DH, et al. Multifocal retinopathy in Dachshunds with CLN2 neuronal ceroid lipofuscinosis. Exp Eye Res. 2015;134:123–32. doi: 10.1016/j.exer.2015.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Katz ML, Tecedor L, Chen Y, Williamson BG, Lysenko E, Wininger FA, et al. AAV gene transfer delays disease onset in a TPP1-deficient canine model of the late infantile form of Batten disease. Science Translational Medicine. 2015;7:313ra180. doi: 10.1126/scitranslmed.aac6191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Anderson GW, Goebel HH, Simonati A. Human pathology in NCL. Biochim Biophys Acta. 2013;1832(11):1807–26. doi: 10.1016/j.bbadis.2012.11.014. [DOI] [PubMed] [Google Scholar]
- 12.Goebel HH, Schochet SS, Jaynes M, Bruck W, Kohlschutter A, Hentati F. Progress in neuropathology of the neuronal ceroid lipofuscinoses. Mol Genet Metab. 1999;66(4):367–72. doi: 10.1006/mgme.1999.2808. [DOI] [PubMed] [Google Scholar]
- 13.Goebel HH, Zeman W, Pilz H. Significance of muscle biopsies in neuronal ceroid-lipofuscinoses. J Neurol Neurosurg Psychiatry. 1975;38(10):985–93. doi: 10.1136/jnnp.38.10.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kieseier BC, Goebel HH. Immunelectronmicroscopic characterization of T4 and T8 lymphocytes and natural killer cells in neuronal ceroid-lipofuscinosis. Am J Med Genet. 1995;57(2):222–4. doi: 10.1002/ajmg.1320570221. [DOI] [PubMed] [Google Scholar]
- 15.Gilliam D, Kolicheski A, Johnson GS, Mhlanga-Mutangadura T, Taylor JF, Schnabel RD, et al. Golden Retriever dogs with neuronal ceroid lipofuscinosis have a two-base-pair deletion and frameshift in CLN5. Mol Genet Metab. 2015;115(2–3):101–9. doi: 10.1016/j.ymgme.2015.04.001. [DOI] [PubMed] [Google Scholar]
- 16.Kolicheski A, Johnson GS, O’Brien DP, Mhlanga-Mutangadura T, Gilliam D, Guo J, et al. Australian Cattle Dogs with neuronal ceroid lipofuscinosis are homozygous for a CLN5 nonsense mutation previously identified in Border Collies. Journal of Internal Veterinary Medicine. 2016 May; doi: 10.1111/jvim.13971. 2016: online. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ashwini A, D’Angelo A, Yamato O, Giordano C, Cagnotti G, Harcourt-Brown T, et al. Neuronal ceroid lipofuscinosis associated with an MFSD8 mutation in Chihuahuas. Mol Genet Metab. 2016 May; doi: 10.1016/j.ymgme.2016.05.008. online. [DOI] [PubMed] [Google Scholar]
- 18.Guo J, Johnson GS, Brown HA, Provencher ML, da Costa RC, Mhlanga-Mutangadura T, et al. A CLN8 nonsense mutation in the whole genome sequence of a mixed breed dog with neuronal ceroid lipofuscinosis and Australian Shepherd ancestry. Mol Genet Metab. 2014;112(4):302–9. doi: 10.1016/j.ymgme.2014.05.014. [DOI] [PubMed] [Google Scholar]
- 19.Ostergaard JR, Rasmussen TB, Molgaard H. Cardiac involvement in juvenile neuronal ceroid lipofuscinosis (Batten disease) Neurology. 2011;76(14):1245–51. doi: 10.1212/WNL.0b013e31821435bd. [DOI] [PubMed] [Google Scholar]
- 20.Fukumura S, Saito Y, Saito T, Komaki H, Nakagawa E, Sugai K, et al. Progressive conduction defects and cardiac death in late infantile neuronal ceroid lipofuscinosis. Dev Med Child Neurol. 2012;54(7):663–6. doi: 10.1111/j.1469-8749.2011.04170.x. [DOI] [PubMed] [Google Scholar]
- 21.Hofman IL, van der Wal AC, Dingemans KP, Becker AE. Cardiac pathology in neuronal ceroid lipofuscinoses--a clinicopathologic correlation in three patients. Europ J Paediatr Neurol. 2001;5(Suppl A):213–7. doi: 10.1053/ejpn.2000.0465. [DOI] [PubMed] [Google Scholar]
- 22.Sakajiri K, Matsubara N, Nakajima T, Fukuhara N, Makifuchi T, Wakabayashi M, et al. A family with adult type ceroid lipofuscinosis (Kufs’ disease) and heart muscle disease: report of two autopsy cases. Intern Med. 1995;34(12):1158–63. doi: 10.2169/internalmedicine.34.1158. [DOI] [PubMed] [Google Scholar]
- 23.Adin DB, Oyama MA, Sleeper MM, Milner RJ. Comparison of canine cardiac troponin I concentrations as determined by 3 analyzers. J Vet Intern Med. 2006;20(5):1136–42. doi: 10.1892/0891-6640(2006)20[1136:coccti]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 24.Oyama MA, Sisson DD. Cardiac troponin-I concentration in dogs with cardiac disease. J Vet Intern Med. 2004;18(6):831–9. doi: 10.1892/0891-6640(2004)18<831:ctcidw>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 25.Vuillemenot BR, Katz ML, Coates JR, Kennedy D, Tiger P, Kanazono S, et al. Intrathecal tripeptidyl-peptidase 1 reduces lysosomal storage in a canine model of late infantile neuronal ceroid lipofuscinosis. Mol Genet Metab. 2011;104(3):325–37. doi: 10.1016/j.ymgme.2011.06.018. [DOI] [PubMed] [Google Scholar]
- 26.Vuillemenot BR, Kennedy D, Cooper JD, Wong AM, Sri S, Doeleman T, et al. Nonclinical evaluation of CNS-administered TPP1 enzyme replacement in canine CLN2 neuronal ceroid lipofuscinosis. Mol Genet Metab. 2015;114(2):281–93. doi: 10.1016/j.ymgme.2014.09.004. [DOI] [PubMed] [Google Scholar]
- 27.Whiting REH, Jensen CA, Pearce JW, Castaner LJ, Gillespie LE, Bristow DE, et al. Intracerebroventricular gene therapy that delays neurological disease progression is associated with selective preservation of retinal ganglion cells in a canine model of CLN2 disease. Exp Eye Res. 2016;146:276–282. doi: 10.1016/j.exer.2016.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Armstrong D, Lombard C, Ellis A. Electrocardiographic and histologic abnormalities in canine ceroid-lipofuscinosis (CCL) J Mol Cell Cardiol. 1986;18(1):91–7. doi: 10.1016/s0022-2828(86)80986-5. [DOI] [PubMed] [Google Scholar]
- 29.Michielsen P, Martin JJ, Vanagt E, Vrints C, Gillebert T, Snoeck J. Cardiac involvement in juvenile ceroid lipofuscinosis of the Spielmeyer-Vogt-Sjogren type: prospective noninvasive findings in two siblings. Eur Neurol. 1984;23(3):166–72. doi: 10.1159/000115699. [DOI] [PubMed] [Google Scholar]
- 30.Hollak CE, Wijburg FA. Treatment of lysosomal storage disorders: successes and challenges. J Inherit Metab Dis. 2014;37(4):587–98. doi: 10.1007/s10545-014-9718-3. [DOI] [PubMed] [Google Scholar]
- 31.Kaminsky P, Lidove O. Current therapeutic strategies in lysosomal disorders. Presse Med. 2014;43(11):1174–84. doi: 10.1016/j.lpm.2013.12.022. [DOI] [PubMed] [Google Scholar]
- 32.Noh H, Lee JI. Current and potential therapeutic strategies for mucopolysaccharidoses. J Clin Pharm Ther. 2014;39(3):215–24. doi: 10.1111/jcpt.12136. [DOI] [PubMed] [Google Scholar]
- 33.Ortolano S, Vieitez I, Navarro C, Spuch C. Treatment of lysosomal storage diseases: recent patents and future strategies. Recent Pat Endocr Metab Immune Drug Discov. 2014;8(1):9–25. doi: 10.2174/1872214808666140115111350. [DOI] [PubMed] [Google Scholar]
- 34.Dufour BD, McBride JL. Intravascular AAV9 Administration for Delivering RNA Silencing Constructs to the CNS and Periphery. Methods Mol Biol. 2016;1364:261–75. doi: 10.1007/978-1-4939-3112-5_21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gong Y, Mu D, Prabhakar S, Moser A, Musolino P, Ren J, et al. Adenoassociated virus serotype 9-mediated gene therapy for x-linked adrenoleukodystrophy. Mol Ther. 2015;23(5):824–34. doi: 10.1038/mt.2015.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mattar CN, Wong AM, Hoefer K, Alonso-Ferrero ME, Buckley SM, Howe SJ, et al. Systemic gene delivery following intravenous administration of AAV9 to fetal and neonatal mice and late-gestation nonhuman primates. Faseb J. 2015;29(9):3876–88. doi: 10.1096/fj.14-269092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Walia JS, Altaleb N, Bello A, Kruck C, LaFave MC, Varshney GK, et al. Long-term correction of Sandhoff disease following intravenous delivery of rAAV9 to mouse neonates. Mol Ther. 2015;23(3):414–22. doi: 10.1038/mt.2014.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Weismann CM, Ferreira J, Keeler AM, Su Q, Qui L, Shaffer SA, et al. Systemic AAV9 gene transfer in adult GM1 gangliosidosis mice reduces lysosomal storage in CNS and extends lifespan. Hum Mol Genet. 2015;24(15):4353–64. doi: 10.1093/hmg/ddv168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sleeper MM, Clifford CA, Laster LL. Cardiac troponin I in the normal dog and cat. J Vet Intern Med. 2001;15(5):501–3. doi: 10.1892/0891-6640(2001)015<0501:ctiitn>2.3.co;2. [DOI] [PubMed] [Google Scholar]
- 40.Ljungvall I, Hoglund K, Tidholm A, Olsen LH, Borgarelli M, Venge P, et al. Cardiac troponin I is associated with severity of myxomatous mitral valve disease, age, and C-reactive protein in dogs. J Vet Intern Med. 2010;24(1):153–9. doi: 10.1111/j.1939-1676.2009.0428.x. [DOI] [PubMed] [Google Scholar]
- 41.Hoffmann WE, Solter PF. Diagnostic enzymology of domestic animals. In: Koneko JJ, Harvey JW, Bruss ML, editors. Clinical Biochemistry of Domestic Animals. 6. Elsevier; Oxford: 2008. pp. 351–378. [Google Scholar]
- 42.Moss DW, Henderson AR. Enzymes. In: Burtis CA, Ashwood ER, editors. Tietz Textbook of Clinical Chemistry. 2. W.B. Saunders; Philidelphia: 1994. pp. 735–896. [Google Scholar]
- 43.Aktas M, Auguste D, Lefebvre HP, Toutain PL, Braun JP. Creatine kinase in the dog: a review. Vet Res Commun. 1993;17(5):353–69. doi: 10.1007/BF01839386. [DOI] [PubMed] [Google Scholar]
- 44.Thomas WP, Gaber CE, Jacobs GJ, Kaplan PM, Lombard CW, Moise NS, et al. Recommendations for standards in transthoracic two-dimensional echocardiography in the dog and cat. Echocardiography Committee of the Specialty of Cardiology, American College of Veterinary Internal Medicine. J Vet Intern Med. 1993;7(4):247–52. doi: 10.1111/j.1939-1676.1993.tb01015.x. [DOI] [PubMed] [Google Scholar]
- 45.Wingfield WE, Boon J, Miller CW. Echocardiographic assessment of mitral valve motion, cardiac structures, and ventricular function in dogs with atrial fibrillation. J Am Vet Med Assoc. 1982;181(1):46–9. [PubMed] [Google Scholar]
- 46.Rishniw M, Erb HN. Evaluation of four 2-dimensional echocardiographic methods of assessing left atrial size in dogs. J Vet Intern Med. 2000;14(4):429–35. doi: 10.1892/0891-6640(2000)014<0429:eofemo>2.3.co;2. [DOI] [PubMed] [Google Scholar]
- 47.Wess G, Maurer J, Simak J, Hartmann K. Use of Simpson’s method of disc to detect early echocardiographic changes in Doberman Pinschers with dilated cardiomyopathy. J Vet Intern Med. 2010;24(5):1069–76. doi: 10.1111/j.1939-1676.2010.0575.x. [DOI] [PubMed] [Google Scholar]
- 48.Boon J. Evaluation of size, function and hemodynamics. In: Boon J, editor. Veterinary Echcardiography. 2. Wiley-Blackwell; Oxford: 2011. pp. 153–166. [Google Scholar]
- 49.Turk JR, Root CR. Necropsy of the canine heart: a simple technique for quantifying ventricular hypertrophy and valvular alterations. Compendium of Continuing Education for the Practicing Veterinarian. 1983;5:905–910. [Google Scholar]
- 50.Katz ML, Gao CL, Rice LM. Long-term variations in cyclic light intensity and dietary vitamin A intake modulate lipofuscin content of the retinal pigment epithelium. J Neurosci Res. 1999;57(1):106–16. doi: 10.1002/(SICI)1097-4547(19990701)57:1<106::AID-JNR11>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.