

Abstract
Glycoprotein gp120 is a surface antigen and virulence factor of the human immunodeficiency virus-1 (HIV-1). Broadly neutralizing antibodies (bNAbs) that are reactive to gp120 from a variety of HIV isolates offer hope for the development of broadly effective immunogens for vaccination purposes, if the interactions between gp120 and bNAbs can be understood. From a structural perspective, gp120 is a particularly difficult system due to its size, the presence of multiple flexible regions, and the large amount of glycosylation, all of which are important in gp120-bNAb interactions. Here, the interaction of full-length, glycosylated gp120 with the bNAb b12 is probed using high resolution hydroxyl radical protein footprinting (HR-HRPF) by fast photochemical oxidation of proteins (FPOP). HR-HRPF allows for the measurement in changes of average solvent accessible surface area of multiple amino acids without the need for measures that might alter the protein conformation, such as mutagenesis. HR-HRPF of the gp120-b12 complex coupled with computational modeling shows a novel extensive interaction of the V1/V2 domain, probably with the light chain of b12. Our data also reveal HR-HRPF protection in the C3 domain due to interaction of the N330 glycan with the b12 light chain. In addition to providing information on the interactions of full-length, glycosylated gp120 with b12, this work serves as a template for the structural interrogation of full-length glycosylated gp120 with other bNAbs to better characterize the interactions that drive the broad specificity of the bNAb.
TOC image
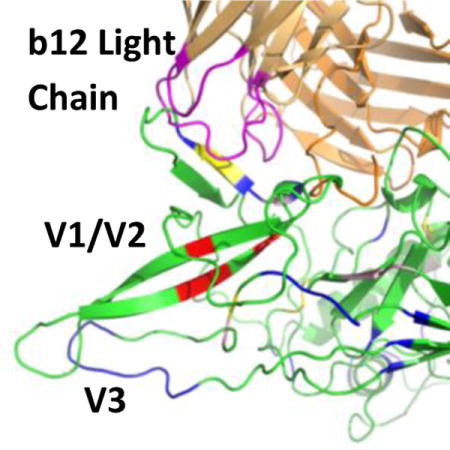
The human immunodeficiency virus-1 (HIV-1) gp120 envelope glycoprotein is the major target for neutralizing antibodies1, 2. The gp120 molecule consists of a polypeptide core of roughly 60 kDa. Extensive modification by N-linked glycosylation increases the molecular weight of the molecule to ~120 kDa3. The amino acid sequence of gp120 is composed of five conserved regions (C1–C5) and five variable regions (V1–V5), many of which are highly flexible. The majority of antibodies raised against gp120 have very narrow ranges of effectiveness and are eventually evaded by the virus. However, a subset of raised antibodies have been found to be effective against a broader array of isolates. The development of a vaccine immunogen which elicits these broadly neutralizing antibodies (bNAbs) and confers protective immunity still remains a challenge. Improved knowledge of the Env structure and what constitutes a full neutralization epitope will help rational immunogen design in order to elicit potent bNAbs. However, gp120 is a very challenging molecule for structural biology. The extensive glycosylation, diversity of isoforms and broad conformational flexibility of gp120 pose formidable barriers for crystallization. To surmount these difficulties and construct a crystal structure for gp120, sources of likely conformational heterogeneity such as N-linked carbohydrates, flexible or mobile N and C termini, and variable internal loops (like V1/V2, and/or V3) are often reduced or eliminated, and ligands such as CD4 are used to restrict conformational mobility as well as to alter the crystallization surface4–12. These stabilized structures give valuable information at high resolution, but at the cost of eliminating regions which have been shown to be important for many gp120-antibody interactions13.
The first broadly neutralizing human monoclonal antibody (mAb) b12 was isolated from clade B infected patients and binds to gp120 at and near its CD4 binding site (CD4bs)7, 10, 14. Binding of b12 to the surface of gp120 blocks attachment of CD4 and thus prevents the entry of HIV-1 into a target cell7, 10, Therefore, gp120 appears to present the b12 epitope in conjunction with several other weakly neutralizing and overlapping epitopes. However, while several other CD4bs antibodies have been discovered since with greater potency and breadth than b12, b12 remains a valuable model for anti-CD4bs bNAbs due to its history of experimental study15–17. A crystal structure of b12 in complex with a truncated, deglycosylated, and mutationally stabilized gp120 core (PDBID 2NY7) has revealed that the contacts between b12 and gp120 are centered around the CD4-binding loop spanning residues 364–373, but involves many other residues10. The truncated, deglycosylated, and mutationally stabilized gp120 core has important differences from its mature counterpart, including a fully truncated V1/V2 domain. It was found that the removal of V1/V2 loops significantly decreases the binding of b12 to gp12017. The removal of a single N-linked glycosylation site at the V3 loop increased the neutralization sensitivity by CD4bs-antibodies18. Because of their absence in the crystal structure of gp120 core in complex with b12, it remains unclear how the V1/V2 and V3 loops interact with b12. The characterization of the contact sites between mature gp120 and b12 will provide a better understanding of the specific broadly neutralization activity of b12 against gp120. As there is no crystal structure available of an intact, glycosylated gp120 in complex with b12, molecular modeling has been used to predict the interface between b12 and gp120 core at the truncated area by using available crystal structures as the basis for homology modeling5, 7, 19. However, such molecular modeling has limitations in accuracy and reliability, and should ideally be coupled with experimental testing and constraints20.
In this study, we used high resolution hydroxyl radical protein footprinting (HR-HRPF) by fast photochemical oxidation of proteins (FPOP) coupled with mass spectrometry (MS) to characterize the binding interface between mature HIV-1 JR-FL gp120 and b12. Hydroxyl radical protein footprinting is a relatively new method for probing changes in the topography of a protein. The measured rate of the reaction of hydroxyl radicals at any particular amino acid side chain depends primarily upon two factors: the chemical reactivity of the side chain (which is invariant between conformations) and the accessibility of the side chain to the radical. Therefore, by monitoring changes in the HR-HRPF reactivity, we can determine changes in average solvent accessibility due to changes in conformation and/or dynamics or direct shielding by protein-protein or protein-ligand binding. MS-based hydroxyl radical protein footprinting has shown great promise in the measurement of protein conformational changes, protein-protein interactions and protein-ligand binding events21–27. HR-HRPF by certain techniques, including FPOP28 and electron accelerator radiolysis29, stably modifies solvent accessible amino acid side chains without deforming the protein structure during the timescale of modification, allowing for heavy surface labeling of the native structure.
In order to measure changes in HR-HRPF reactivity, the stable modifications to the protein side chains are analyzed by liquid chromatography-mass spectrometry (LC-MS), relatively quantifying modified and unmodified peptides, and the MS signal of the oxidized version(s) of each peptide is compared to the MS signal of the unoxidized version of the same peptide to quantify oxidation at the peptide level. By measuring the rate of reaction of each amino acid side chain of a protein under two different structural conditions (in this case, b12-bound vs. free gp120), relative changes in the accessibility of each amino acid can be measured25. Previous work from our group has shown that, using ETD-based methods for quantifying multiple adjacent sites of isomeric oxidation products, we can accurately quantify changes in the hydroxyl radical footprint down to single amino acid spatial resolution30, 31, providing structural information with higher spatial resolution and more accuracy. This method offers an important alternative method for characterizing protein-protein and protein-ligand interactions in cases in which it is not possible to determine high resolution three-dimensional structures of complexes due to heterogeneity, flexibility, and/or size of the target(s).
A glycosylated, full-length homology model of JR-FL gp120 was constructed and a 500 ns molecular dynamics simulation was performed. The resulting simulation was aligned to the available structure of gp120 HXBc2 in complex with b12 allowing an estimation of the contact area between b12 and glycosylated JR-FL. This provided a structure-based rationalization of the experimentally determined protection sites as well as detailed insights into the regions of JR-FL affected by complexation with b12.
Materials and Methods
Materials and Reagents
Hydrogen peroxide (30%) was purchased from J.T. Baker (Phillipsburg, NJ). Dithiothreitol (DTT), HPLC-grade acetonitrile were purchased from Fisher Scientific (Fair Lawn, NJ). Methionine amide was purchased from Bachem (Torrace, CA, USA). Catalase, adenine, formic acid, L-glutamine and phosphate buffer saline (PBS) were obtained from the Sigma-Aldrich Corporation (St. Louis, MO). Sequencing-grade modified trypsin and Glu-C were purchased from Promega Corporation (Madison, WI, USA). PNGase F (500,000 U/mL) was purchased from New England Biolabs (Ipswich, MA). All reagents were used without further purification. Purified water (18 MΩ) was obtained from an in-house Milli-Q Synthesis system (Millipore, Billerica, MA).
Construction, Expression and Purification of Gp120 and Human mAbs IgG1 b12
Recombinant HIV-1 gp120 protein vaccine was produced from gp120-JR-FL DNA vaccine transfected 293F cells. Codon optimized gp120-JR-FL DNA vaccine was constructed into the pJW4303 DNA vaccine vector as previously reported32. At 72 hours after the transient transfection of 293F cells, the culture supernatant was harvested and the secreted gp120 protein from were purified using a lectin column. The purified gp120 protein were verified and analyzed by SDS-PAGE and Western blot analysis (data not shown). The monoclonal antibody (mAb) IgG1 b12, specific for gp120 CD4 binding site (CD4bs), was purchased from Polymun Scientific (Austria).
HR-HRPF by FPOP
6 μL gp120 JR-FL (25 μM) was incubated with 6 μL PBS either with or without 25 μM b12 at 37°C for 1 h. HR-HRPF by FPOP was then performed in triplicate as previously described. Briefly, 4 μL glutamine (100 mM) and 2 μL adenine (200 μg/mL) were mixed with the 12 μL gp120 alone or gp120-b12 reaction solution. Adenine was added as a radical dosimeter to monitor the available radical dose in each sample. 2 μL of 3% hydrogen peroxide (1 M, freshly prepared) was added into the mixture immediately prior to irradiation. The FPOP labeling of the total volume of 20 μL mixture was immediately performed The KrF excimer laser power (GAM Laser Inc., Orlando, FL) was adjusted to 75 mJ/pulse and its pulse frequency set to 5 Hz. The flow rate was set to 12.19 μL/min to ensure a 10% exclusion volume to avoid repeat hydroxyl radical exposure28. Each replicate was collected in a microcentrifuge tube containing a quench mixture 10 μL of 140 mM H-Met-NH2 and 10 μL of 2 μM catalase to eliminate secondary oxidants such as remaining hydrogen peroxide, protein peroxides, superoxide, etc. The samples were incubated in the quench solution for 30 min at room temperature with pipette mixing. Control samples were handled in the same manner as those submitted to FPOP, but they were not laser irradiated; instead, they were incubated for 2 min with H2O2, after which the solution was added to the collection solution containing catalase and H-Met-NH2. All experiments were performed in triplicate for statistical analysis.
Following irradiation, 2 μL of the sample (40 μL) was analyzed to determine the oxidation of adenine after HR-HRPF by UV at 260 nm (Thermo Scientific NanoDrop 2000c UV-Vis spectrophotometer) to ensure comparable amounts of available hydroxyl radical for oxidizing the analyte, as previously reported33. The adenine oxidation amounts are similar among all tested samples with an average of around 53% oxidized adenine and CV lower than 5% (data not shown), which indicates the same level of hydroxyl radical available among all samples.
The lyophilized powder was resuspended in 50 μL of 100 mM ammonium bicarbonate buffer. DTT (10 μL, 50 mM) was added and incubated at 65°C for 30 min to denature and reduce the protein. After cooling to room temperature, a 1:20 weight ratio of Glu-C:protein was added to the protein sample and incubated at room temperature for 2 hrs. Glu-C was deactivated by heating the sample to 95°C for 10 min, then after cooling the samples to room temperature a 1:20 weight ratio of trypsin was added to the samples for overnight digestion at 37 °C while rotating. Digestion was terminated by adding 12 μL DTT (50 mM) and heating the samples to 95°C for 10 min. Finally, when the samples were cooled to room temperature, 150 units of PNGase F was added into the digested samples and incubated at 37 °C for 16 hrs. The reaction was terminated by adding 12 μL DTT (50 mM) and heating the sample to 95°C for 10 min. Samples were stored at −20°C until nanoLC-MS/MS analysis.
HR-HRPF samples were analyzed on a LTQ Orbitrap XL (Thermo Fisher Scientific) controlled by Xcalibur 2.0.7 software (Thermo Fisher, San Jose, CA). Samples were loaded on a 10 cm column with a PicoFrit tip, bomb-packed with C18 reverse phase material (Magic, 0.075 mm × 100 mm, 5 μm, 300 Å, Michrom, Auburn, CA). Chromatography was performed using mobile phase A (0.1% formic acid in water) and B (0.1% formic acid in 80% acetonitrile) with a 160-min gradient consisting of 0–60% solvent B over 60 min at 135 μL/min, ramped to 100% B over 20 min, hold for 9 min, then returned to 0% B over 1 min and hold for 70 min. Peptides were eluted directly into the nanospray source of an LTQ Orbitrap XL using the bomb-packed column as the nanospray emitter. All data were acquired in positive ion mode. CID (collision induced dissociation) and/or ETD (electron transfer dissociation) were used to fragment peptides. The spray voltage was set to 2.0 kV and the temperature of the heated capillary was set to 200°C. In CID mode, full MS scans were acquired from m/z 400 to 1500 followed by eight subsequent MS2 scans on the top eight most abundant peptide ions. In ETD mode, the parent ions of all identified peptides under CID mode were listed in the parent ion mass list. ETD-based precursor activation was carried out for 100 ms, including charge-state-dependent with supplemental activation enabled. Precursor ions were isolated with width of 3 m/z units.
HR-HRPF Data Analysis
Unoxidized gp120 controls, oxidized gp120 and oxidized gp120-b12 complex peptide sequences were initially identified using ByOnic V1.2-250 software (Protein Metrics, CA, USA). The enzyme was defined to cleave the protein after lysine, arginine, asparatic acid and glutamine. Deamination on both asparagine and glutamine and all possible major oxidation modifications25 were included as variable modification for database searches. All tandem mass spectra assignments and sites of oxidation were verified manually due to peptide modification complexity. The LC-MS peak intensity of the digested peptides and corresponding oxidation products were used to calculate the average oxidation events per peptide in the sample. Peptide level quantitation is calculated by summing the ion intensities of all the oxidized peptide multiplied by the number of oxidation events required for the mass shift (e.g., one event for +16, two events for +32), then divided by the sum of the ion intensities of all unoxidized and oxidized peptide masses (eq 1):
| (1) |
where P denotes the oxidation events at peptide level and I donates the peak intensities of oxidized and unoxidized peptides.
Gp120 control samples unilluminated by the UV laser, but otherwise identical to the experimental samples, were analyzed to ensure that background oxidation would not be intense enough to interfere with HR-HRPF data. Illumination-free controls showed less than 10% of the oxidation extent of gp120 sample with UV laser for peptides 19–31, 37–63, 64–69, 178–197, 261–274, 386–396 and 397–404 (data not shown). The small amount of oxidation happening occurred at M or C due to sample preparation process (especially for residue M), protein expression and purification. The interference of this background is not significant enough to affect the comparison of the oxidation extent between gp120 and gp120-b12 samples, and was subtracted from experimental samples.
In cases where oxidation at specific sites can be identified either based on the mass differences between non-isomeric oxidation products or the presence of only a single oxidation site within a peptide, residue level quantitation is calculated from the LC-MS signal intensities of each peptide containing a specific oxidized amino acid (Ioxidized), relative to the total of all intensities associated with that peptide sequence (Ioxidized + Iunoxidized) using the following straightforward relationship24:
| (2) |
In cases where the peptide isomers with oxidation at multiple adjacent sites in a single sequence that results in an identical mass shift, residue level quantitation is calculated from the fragment ion intensities from ETD to determine oxidation extent at specific residue site based on our previous studies30, 31. Briefly, oxidized peptide with multiple sites of oxidation can generate both oxidized and unoxidized sequence ions in its tandem mass spectrum. The fractional oxidation of a given sequence ion is defined as the ratio between the oxidized sequence ion intensity to the sum of the intensity of the corresponding oxidized and unoxidized sequence ion (eq 3).
| (3) |
Where f(Ci) denotes the fractional oxidization of sequence ion i (e.g., oxidized C3 ions generated by ETD), and I(Ci) denotes the intensity of the sequence ion i, whether the oxidized and unoxidized form. The absolute level of oxidation for a given amino acid residue i is based on both the average oxidation event of peptide and the fractional oxidation of the corresponding sequence ions, as shown in eq 4:
| (4) |
Where P is the average oxidation events per peptide as derived from eq 1, and the term in square brackets is the fractional oxidation difference of two adjacent sequence ions i and i −1. Multiplying the average number of oxidations per peptide by the fraction of that oxidation that occurs on a given amino acid residue yields the average oxidation events per residue. In cases where ETD did not yield sufficient product ions for quantification, CID was used to provide semi-quantitative information on changes in footprint at a residue level.
The protection by b12 binding is defined as the ratio of the difference of oxidation extent between gp120 sample alone and gp120-b12 binding sample to the oxidation extent of gp120 sample alone, as shown in eq 5:
| (5) |
Glycosylation Analysis
An aliquot of each sample was denatured by incubating with 10 mM of dithiothreitol at 56 °C for an hour and alkylated by 55 mM of iodoacetamide for 45 minutes in dark prior to digestion with proteases optimized based on amino acid sequence of each target protein. Following digestion, the samples were deglycosylated by PNGaseF in the presence of O18-water which covert Asn residues carrying N-linked glycans to O18-Asp residues. The resulting peptides were separated on a 75 μm (I.D.) × 15 cm C18 capillary column (packed in house, YMC GEL ODS-AQ120ǺS-5, Waters) and eluted into the nano-electrospray ion source of an Orbitrap Fusion™ Tribrid™ mass spectrometer (Thermo Fisher Scientific) with a 180-min linear gradient consisting of 0.5–100% solvent B over 150 min at a flow rate of 200 nL/min. The spray voltage was set to 2.2 kV and the temperature of the heated capillary was set to 280 °C. Full MS scans were acquired from m/z 300 to 2000 at 120k resolution, and MS2 scans following collision-induced fragmentation were collected in the ion trap for the most intense ions in the Top-Speed mode within a 3-sec cycle using Fusion instrument software (v1.1, Thermo Fisher Scientific). The resulting spectra were analyzed using SEQUEST (Proteome Discoverer 1.4, Thermo Fisher Scientific) with full MS peptide tolerance of 20 ppm and MS2 peptide fragment tolerance of 0.5 Da, and filtered using ProteoIQ (v2.7, Premier Biosoft) at the protein level to generate a 1% false discovery rate for protein assignments. Site occupancy was calculated using spectral counts of O18-Asp peptides to the identical peptide containing Asn. The sole exception is N88, which was wholly unobserved as glycosylated or non-glycosylated by this method. Glycosylation of this site was assigned based on the high abundance of the N→D substitution of this site after PNGase F treatment during HR-HRPF data acquisition and analysis, as detailed above.
A site was assigned as occupied for the purposes of modeling if the site was predominantly glycosylated across all peptides in which the site was observed. Notably, sites N241 and N262, which are co-resident on a single peptide and were detected as individually glycosylated 56% of the time, but simultaneous glycosylation was detected for only 6% of observed peptides. Thus, the predominant state was for only one site to be glycosylated. Given their equal rate of occupancy, we arbitrarily chose the N241 for glycosylation in the model.
Model Building
Blastp34 was used to find 3D structures from the protein data bank (PDB) with sequences similar to JR-FL. A modified JR-FL has been crystallized previously (PDB 2B4C), however, the N- and C-termini were truncated and the V1/V2 region was substituted with a GAG tripeptide. The model used here is based on 2B4C and employs additional crystal structures (PDBID 4NCO, 3JWD, 4HPO, 4R4H, 4TVP, and 9B4C) to model a complete JR-FL. Modeller35 incorporated each of these structures (see alignments in Supplementary Material, Figure S1) to generate five models of the complete gp120.
Model Glycosylation
Previous work revealed that expression of gp120 from Ba-L (a Clade B isolate) in 293F cells resulted in a mixture of complex and high mannose glycans, and that interaction of the B-clade gp120 with the b12 bNAb was increased by expression in cells that increased high mannose glycosylation36. Since the full diversity of possible glycan structures are impractical to simulate even for individual compositions of complex glycans, Man9GlcNAc2 structures were modeled into all sites assigned as glycosylated, as this glycan is a single, biologically relevant structure that explores the extended reach of glycans in examining both glycan-antibody interactions and glycan shielding of the antibody binding surface. GLYCAM-Web (www.glycam.org) was used to generate 3D structures of the common rotamers of Man9GlcNAc2. These structures were superimposed onto the Asn side chain at each assigned glycosylation site. The glycan structure was adapted to the local protein environment and any previous glycans in the following manner: the four most populated conformations of N-linked Asn side chains found in the PDB were assessed in series by altering the chi, phi and psi angles of the N-linked glycan. Any vdW overlaps between the glycan and the glycoprotein were minimized by altering the torsion angles of the Asn-GlcNAc and interglycosidic linkages by ±20° in 5° increments. The first glycan rotamer and Asn side chain conformer which allowed the glycan to fit without large vdW overlaps was added to the glycoprotein structure.
Model Selection
A single model of JR-FL was chosen based on the number of experimentally determined glycosylation sites that were accessible for glycosylation and the DOPE score provided by Modeller. A recent crystal structure of a complete JR-FL (PDBID 5FYK)37, released during manuscript preparation, allowed validation of the model generated here. A Needleman-Wunsch alignment using a BLOSUM-62 matrix38 gave a relatively large RMSD of 5.2 Å over 450 atom pairs. However, most of the deviation is in the flexible N and C-terminal domains that are ordered by the presence of the gp41 in the crystal structure. When these regions are discounted the RMSD drops to 1.0 Å over 303 atom pairs indicating that the homology model successfully predicted the remaining 3D structure.
Molecular Dynamics Simulation
All simulations were performed with the CUDA implementation of PMEMD39, 40 in the Amber14 software suite41. The carbohydrate was modeled using the GLYCAM06j-1 force field42, while the Amber ff14SB force field43 was employed for the protein. A Berendsen barostat with a time constant of 1 ps was employed for pressure regulation, while a Langevin thermostat with a collision frequency of 2 ps−1 was employed for temperature regulation. A nonbonded interaction cutoff of 8 Å was employed. Long-range electrostatics were treated with the particle-mesh Ewald (PME) method44. Covalent bonds involving hydrogen were constrained with the SHAKE algorithm allowing a time step of 2 fs45.
Each system was placed in a cubic box of TIP5P water46 with an 8 Å buffer between solute and the periodic boundary. Five sodium ions were added to neutralize the overall charge. Energy minimization was performed for 20,000 steps (10,000 steepest decent, followed by 10,000 conjugant gradient) with Cartesian restraints (5 kcal/mol-Å2) on solute heavy atoms. This was followed by a 400 ps solvent equilibration phase at 300 K under nPT conditions. Cartesian restraints (5 kcal/mol-Å2) were employed on the Cα atoms of the first and last three residues (N- and C-terminal) during a 1 ns structural equilibration phase and 500 ns production phase (nPT).
Structure of gp120 (JR-FL) in Complex with the b12 Antibody
A structure-based sequence alignment of gp120 (HXBc2) in complex with the b12 Ab (PDB 2NY7) to 100 snapshots from the MD simulation of JR-FL was performed using Sequoia47, with an RMSD of 4.14 Å for 245 equivalent matches based on aligning to the original model, with a Sequoia alignment score of 0.08 Based on each alignment, an estimation of the contact area between b12 and JR-FL was obtained by averaging the percentage change in the respective solvent accessibilities48 for each snapshot in the presence and absence of the b12 structure.
RESULTS
Glycosylation analysis of gp120 and the gp120-b12 complex
The recombinant glycoprotein gp120 used in this study was expressed as a 477-amino acid mature protein containing intact N and C termini, five variable regions (V1/V2, V3, V4, and V5) and five conserved regions (C1 to C5). Following Glu-C and trypsin digests of gp120, thirty-two unique peptides were identified by CID based MS/MS data, covering 72% of the overall sequence. Eighteen highly occupied N-linked glycosylation sites were identified. The details of the oxidation sites and glycosylation sites are listed in Figure 1.
Figure 1. Sequence of HIV-1 gp120 (JR-FL).

The sequence covered by HR-HRPF experiments are overlined with green. The identified oxidation sites are shown in red text with protected sites underlined and the sole exposed site in italics. The heavily occupied N-linked glycosylation sites are shown in green text. The variable domains are labeled above the sequence.
HR-HRPF of gp120-b12 Complex
Twenty-nine peptides were seen to be modified by FPOP footprinting. Sixty-one oxidized residues were identified in these modified peptides based on Byonic database search and verified manually from the CID and/or ETD MS/MS spectra. Almost one in five residues in the peptides detected was modified, indicating a high yield of protein labeling though the short FPOP reaction window. The oxidation extent of the modified twenty-nine peptides was determined at the peptide level by measuring the intensity of the oxidized peptide ion compared to the sum of oxidized and unoxidized peptide ions as calculated by Eq 1. The side-by-side comparison of the oxidation extent of sequenced peptides in gp120 and gp120-b12 samples was shown in Figure 2. The overall number of average oxidations per peptide for different peptides varies significantly from 0.003 for peptide 179–192 to 3 for peptide 198–207. Most peptides show comparable amounts of oxidation between binding and non-binding samples. Eleven peptides including 92–97, 98–106, 156–168, 173–178, 179–192, 316–327, 336–343, 358–370, 410–419, 433–440, and 467–476 showed a significant decrease in oxidation extent upon b12 binding, indicating a structural event that shielded the sites from the radical.
Figure 2. Peptide-level HRPF of gp120 footprinting for gp120 alone (white) and gp120-b12 complex (grey) (mean +/− SD, n=3).
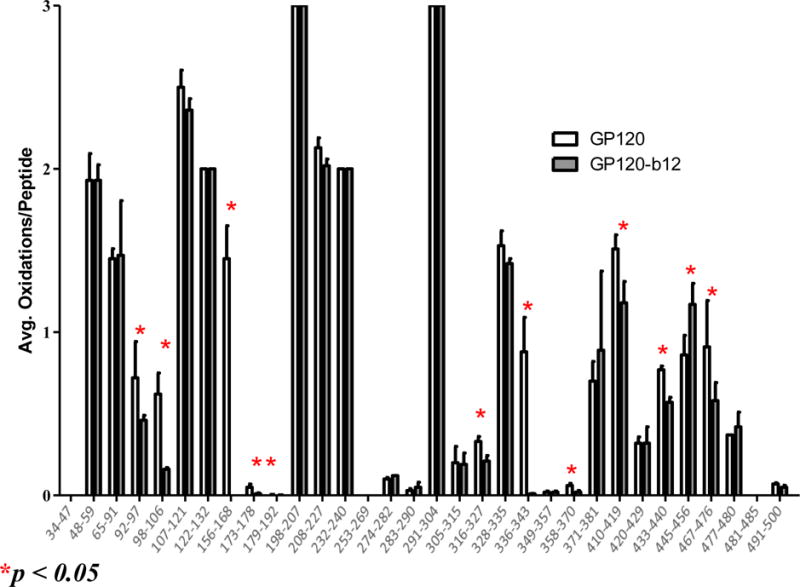
Peptides highlighted with a red star showed a statistically significant change of oxidation extent upon gp120-b12 binding (α≤0.05).
To investigate structural changes upon binding between gp120 and b12 at a high spatial resolution, the CID- and ETD-based tandem mass spectrum of these eleven peptides were analyzed. A representative ETD spectrum is shown in Figure 3. Based on the CID/ETD fragmentation ions, most of the eleven peptides were found to have multiple oxidation sites except peptides 156–168 and 336–343. For peptides with only one oxidation site, the oxidation extent at residue level was calculated by Eq 2. For oxidation isomers having multiple oxidation sites on single sequence (peptides 143–148, 149–162, 284–296, 326–338, 375–383, and 397–404), the oxidation extent at residue level was calculated by Eq. 3 and Eq. 4. Oxidized residues that exhibit HR-HRPF protection upon b12 binding are shown in Table 1. Due to the semi-quantitative nature of CID-based oxidation quantification30, CID-based data were selected only when ETD data wasn’t available (residues M72, M76, C127).
Figure 3. The ETD spectrum of peptide 143-YALFYK-148 and its oxidation products.
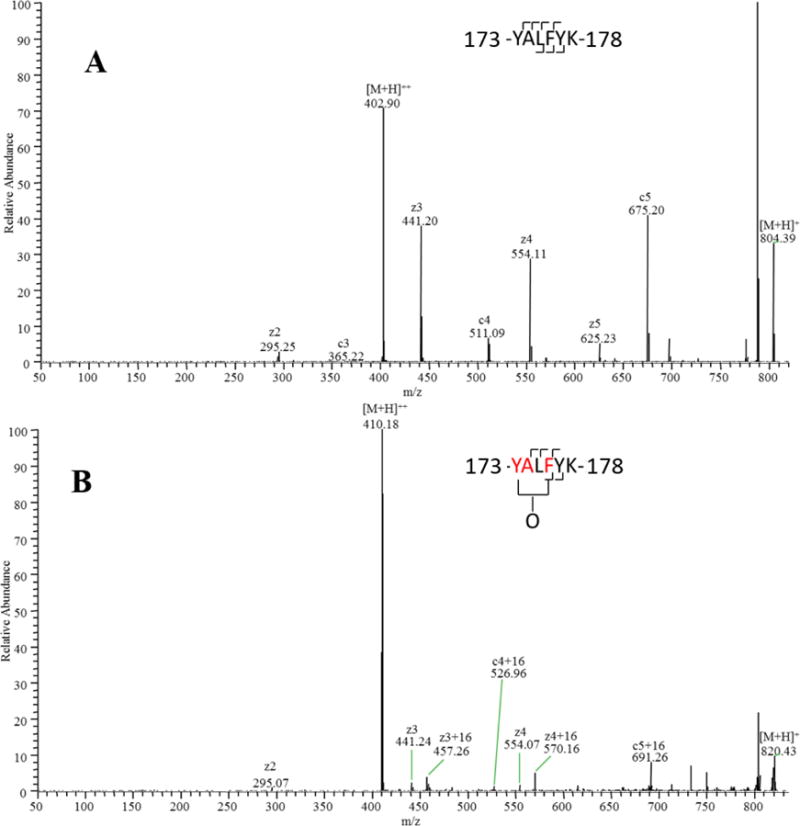
(A) Unoxidized peptide 173-YALFYK-178. (B) Mixture of singly oxidized isomers. By measuring the ratio of [oxidized product ions]:[total product ions] for each fragment, oxidation can be quantified to occur on F176 and the Y173A174 fragment (based on CID data and the relative reactivities of alanine and tyrosine, almost no oxidation occurs on A174).
Table 1.
Protection of Residues from HR-HRPF Upon b12 Binding
| Residuea | % HR-HRPF Protection upon b12 Bindingb | Direct Protection in MD upon b12 Binding (Δ%)c |
|---|---|---|
| M95 | 31.9 | 0% |
| W96 | 81.8 | 0% |
| M100 | 84.6d | 0% |
| M104 | 66.0d | 0% |
| C157 | 100d | 64% |
| Y173 A174 | 100 | 53% 76% |
| F176 | 100 | 70% |
| D180 | 64.5 | 35% |
| V182 P183 | 21.3 | 32% 54% |
| I322a | 43.6 | 0% |
| I323 | 28.2 | 14% |
| W338 | 100 | 0% |
| V360 | 72.4 | 0% |
| F361 | 73.3 | 0% |
| H363 | 70.4 | 0% |
| P417 C418 R419 | 45 | 0% 0% 0% |
| M434 | 28.5 | 0% |
| Y435 | 28.2 | 0% |
| C445 | −35.7 | 0% |
| M475 | 11.3 | 0% |
The residue numbering used is based on the HXB2 numbering system.
The protection upon binding for each residue was calculated by the oxidation extent changes between gp120 and gp120-b12 divided by the oxidation extent of gp120.
The direct protection is calculated by measuring the change in solvent accessibility after alignment of b12. The values are averages from the MD simulation.
CID-based data were used in HR-HRPF results and should be considered semi-quantitative.
It should be noted that the HR-HRPF technique cannot differentiate changes in solvent accessibility as a result of direct binding from those due to conformational or dynamic perturbations. Therefore even a preliminary 3D model is valuable in interpreting the HR-HRPF data, especially when dealing with a protein with as many dynamic domains as full-length, glycosylated gp120. With the exception of the small V5 domain, we were able to probe at least one amino acid in each domain by HR-HRPF. Residues that show significant differences in oxidation due to altered average solvent accessibility after complex formation can either be directly involved in b12 binding or experiencing conformational/dynamic changes that occur upon b12 binding. Residues that were oxidized, yet showed no protection upon b12 binding, experienced no net change in the average solvent accessible surface area of the side chains probed.
Alignment of b12
The 3D structure of gp120 (HXBc2) in complex with b12 (PDB ID 2NY7) was aligned to the gp120 (JR-FL) model generated here. Residues that experienced protection from HR-HRPF oxidation upon b12 binding are plotted against a full-length model of glycosylated gp120 aligned with the stabilized gp120-b12 complex structure (RMSD 2.3 Å) and shown in Figure 4. The alignment provided an estimation of the direct contact area between b12 and gp120 (JR-FL), and changes in solvent accessibility due to direct interaction between b12 and gp120 were calculated (Table 1). During the MD simulation the V1/V2 region samples conformations that overlap with the area occupied by a bound b12, indicating that b12 binding would impact the ensemble of conformations adopted by the V1/V2 region, either by inducing new conformations or just limiting the available conformational space.
Figure 4. Glycosylation and HRPF of gp120.

A model of full-length glycosylated gp120 (ribbon protein in light grey, licorice Man9GlcNAc2 glycans in dark grey) was generated and relaxed by MD simulation, then aligned with the crystal structure of stabilized gp120 bound to the b12 Fab (aligned gp120 not shown, ribbon b12 in dark grey) (PDB ID 2NY7). Residues that showed protection from HRPF upon b12 binding are colored red. Residues that showed no protection from HRPF are colored blue. Residue C445 (also blue) showed an exposure to HRPF upon b12.
The combination of HR-HRPF data and modeling allows us to predict which of those residues are directly protected from oxidation upon complexation (residues D180, V182, P183, P417, R419 and M426) and which become protected via induced conformational/dynamics changes in the gp120 (JR-FL) structure (residues M95, W96, M100, M104, F176, C157, Y173, I322a, I323, W338, M434, Y435, V360, F361, H363). The combination also allows us to determine residues where the model disagrees with the HR-HRPF data, indicating regions of either insufficient modeling or unreliable experimental results to be probed by further experimentation (L179 and I184).
DISCUSSION
The X-ray crystal structure of monomeric gp120 in complex with b12-Fab includes only 62% of our intact gp120 polypeptide sequence and does not include glycosylation10. The structure lacks most of the residues in the V1/V2 loops (residues 121–203), V3 loop (residues 298–329), V4 loop (residues 393–404), and portions of the amino and carboxy termini (residues 31–82, residues 493–507). In this reported crystal structure, a total of 33 residues were defined as contact sites. In the current HR-HRPF study, 22 residues were identified as protected from hydroxyl radicals upon b12 binding. It is clear that the V1/V2 loops and V3 loop can affect antibody binding to the CD4bs either by direct contact or by transmitted conformational effects49. The deletion of the V1/V2 and V3 loops also has an adverse effect on the binding of b12 to gp12050. The application of a full-length, glycosylated model of gp120 allows us to probe and try to explain the observed roles of these and other gp120 domains in b12 binding.
V1/V2 Domain
The V1/V2 domain is highly dynamic, and is excised from the published X-ray crystal structure of gp120 bound to b12 in order to promote crystallization10. However, alanine scanning mutagenesis data has shown that several amino acids in the V2 region are important for b12 binding50. Similarly, the crystal structure shows no direct interaction between the light chain of the b12 antibody and gp120; however, mutagenesis found that several amino acids in the L1 and L3 CDR loops were important for b12 binding to gp12051. HR-HRPF results of the V1/V2 domain within full-length, glycosylated gp120 is shown in Figure 5, with glycans labeled and shown as 3D-SNFG symbols52 positioned at each residue’s ring center. These results revealed several amino acids that were protected from HR-HRPF upon b12 binding. C157 and Y173 are two residues on adjacent strands of an anti-parallel β-sheet that both experience 100% protection from HR-HRPF upon b12 binding. F176 resides on the C-terminal end of the second strand of that same β-sheet, and also exhibits 100% protection from HR-HRPF upon b12 binding. F176 and C157 present their side chains to the opposite face of the β-sheet from Y173 (Figure 5, inset), suggesting that the protection observed by these residues is probably not all from direct interaction with the b12 antibody, but rather is from stabilization of this β-sheet upon antibody binding. We also observe modest protection of D180 and V182/P183 upon b12 binding, but neighboring residues L179, V181 and I184 are not protected upon b12 binding. This targeted protection is more in line with the effects of direct interaction, as a change in dynamics or gross conformation is likely to affect all amino acids in a region.
Figure 5. HR-HRPF and glycosylation of the V1/V2 and V3 domains.
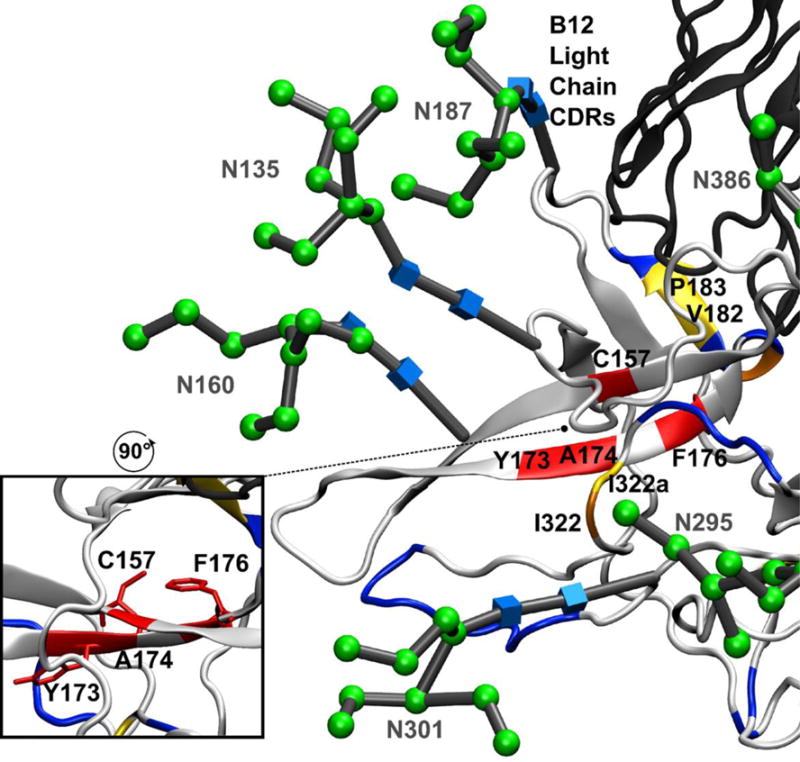
The aligned model of full-length glycosylated gp120 after MD simulation (light grey ribbon) bound to the b12 Fab (dark grey ribbon) (PDB ID 2NY7). Residues that showed >80% protection from HR-HRPF upon b12 binding are colored red; residues that showed between 40% and 80% protection from HR-HRPF upon b12 binding are colored orange; residues that showed statistically significant protection from HR-HRPF <40% are colored yellow. Residues showing no protection from HR-HRPF upon b12 binding are colored blue. The CDR loops of the light chain modeled to potentially interact with the V1/V2 domain are shown. (inset) The orientation of C157, F176 and Y173 side chains relative to the β-sheet.
Modeling of the glycosylated V1/V2 domain into the gp120 structure revealed a potential interaction between the V1/V2 domain and the CDR loops of the b12 light chain, as well as a loop of the b12 heavy chain. However, both L179 and I184 are outliers as their oxidation was not affected by b12 binding, but were predicted to be in direct contact with b12. These outliers are within the flexible V1/V2 region and the inconsistency with the footprinting data may be the result of the dynamics of this region. Previously, alanine scanning mutagenesis was performed on the monomeric gp120 (JR-CSF) to define in more detail which residues on gp120 influence or modulate b12 reactivity. The outcome indicated that in V2 loop, the mutations F176A, D180A, I184A, and D185A significantly reduced the binding affinity of b12, which supports our finding in regard to the contact sites identified in the V2 loop50.
The oxidation extents of residues M95 and W96 were decreased by 41.9% and 71.3%, respectively upon b12 binding and are likely involved in the binding interface with b12 based on the docking model of the crystal structure of b12 and the CD4-complexed gp120 core structure of HXB2. However, the previous observation that the M95A mutation has a very modest reduction of b12 binding affinity for JR-CSF gp120 (75% of WT binding) suggests that this residue, while protected by the gp120-b12 interface, is probably not energetically involved in stabilizing the binding to any significant extent7, 50. Hydrogen-deuterium exchange (HDX) studies were previously reported for the KNH1144 SOSIP.664 trimer structure bound to b12. These studies largely monitor changes in secondary structure dynamics that alter backbone hydrogen bonding stability, serving as complementary information for the side chain-oriented HR-HRPF data presented here. HDX protection profiles of the V1/V2 domain upon b12 binding were complex, with some peptides showing stabilized secondary structure and others showing destabilized secondary structure, and with changes in stability differing over time. The fact that HDX data are usually limited in spatial resolution to the peptide level further increases the difficulty in interpreting the data. The V1/V2 region is clearly structurally altered by b12 binding in complex ways, with the region of ~100–175 destabilized by b12 binding, and 176–179 slightly stabilized53. Combined with our HR-HRPF data, this suggests that HDX protection of this region is largely limited to the area of direct contact with the b12 antibody, with regions affected conformationally by b12 binding in this region exhibiting increased HDX rates.
V3 Domain
The V3 domain is a highly flexible loop that is missing from the stabilized gp120-b12 crystal structure. The V3 loop is heavily modified by HR-HRPF, suggesting a high degree of solvent accessibility. The vast majority of residues in the V3 domain (C296, K305-R313, D324, I325, R326 and Q327) show no protection upon b12 binding, indicating that the domain remains highly flexible in the b12-bound form. However, slight to moderate protection is observed for I322a and I323. Examining the location of these residues in the MD simulation of full-length glycosylated gp120 (Figure 5), I322a and I323 are adjacent to Y173, an amino acid in the V1/V2 domain that shows complete protection upon b12 binding. We suggest that the b12-induced conformational change in the V1/V2 domain allows for interaction between I322a and I323 and the V1/V2 domain, perhaps through Y173. While the modest extent of protection of I322a and I323 suggests that this interaction is not completely protecting, the protection is both significant and unique in the V3 domain. This suggests that interactions between V3 and other molecules (e.g. heparan sulfate,54) could modulate the affinity of the b12 antibody. This observation is supported by the fact that the V3 domain of KNH1144 SOSIP.664 shows marked increase in HDX rates upon b12 binding, and the V3 peptides most markedly affected are those containing residues 322 and 32353, as well as by mutational and recombinant analysis yielding evidence that the V3 region interacts directly with V1/V2 in DH102 gp12055. Though the deletion of V3 loop (Residues N302-D324) was reported to reduce the binding affinity of b12, it is currently unknown which particular residues mediate the interaction50.
C1 and C5 Domains
Ten residues were observed as oxidized in the C1 domain, four of which experience protection upon b12 binding: M95, W96, M100 and M104. These residues cluster at a loop and the N-terminal region of the large central helix of the C1 domain. Alanine-scanning mutagenesis of b12 binding to JR-CSF gp120 did not cover this end of the helix thoroughly, and did not directly test any of the residues protected by HRPF; K97A caused a modest increase in apparent affinity, while E102A caused a modest decrease. Mutagenesis at the C-terminal end of this helix showed an almost complete loss of binding affinity, suggesting a role for the helix in the interaction50. Hydrogen-deuterium exchange of the KNH1144 SOSIP.664 trimer with b12 indicated this region as exhibiting increased exchange rates upon antibody binding, suggesting that antibody binding destabilizes the secondary structure of the N-terminal end of the C1 helix53. The HDX data (which largely probes backbone secondary structure stability) combined with our HR-HRPF data (which probes side chain solvent accessibility) suggests that antibody binding increases the amount of side chain packing not by stabilizing the helical structure, but by destabilizing the helical structure and granting the side chains additional flexibility to maximize interactions. In our model, the protected N-terminal end of the C1 helix packs against a helix in the C5 domain that contains the only protected residue in C5: M475 (Figure 6). Residues G471 through M475 show substantial contact with the heavy chain of b12, with M475 showing 29.5 Å2 interaction surface with the b12 heavy chain10. Mutagenesis data revealed that mutation of M475 to alanine increased the apparent affinity of b12 for JR-CSF gp120 by a very modest amount, while mutation of the nearby G473 to alanine increased affinity almost ten-fold. However, quadruple mutation of G473, D474, M475 and R476 to alanine increased the apparent relative affinity for b12 only 2.5-fold, and GM and DMR multiple mutations increased the apparent affinity similarly to the M475A mutant alone, suggesting that the large effect of G473A may be due to stabilization of the helix, with the interactions mediated by the amino acids in the helix50. Based on our data and the glycosylated homology model, the binding of the b12 heavy chain fixes the C5 domain helix by direct contact, shielding M475. The binding simultaneously destabilizes the backbone of the N-terminus of the C1 helix as shown by HDX,53 with the destabilized helix packing against the fixed C5 helix, providing the observed shielding of M95, W96, M100 and M104. Residues in domains C1 and C5 that are not protected (F53, C54, H66, H72, and C74 in domain C1; L494 and K500 in the C-terminal tail of domain C5) are distal to this interaction interface, and do not experience any direct contact or significant stabilization by b12 binding, showed no change in HDX experiments,53 and were not in the covered regions of the previously reported alanine mutagenesis work50. C4 Domain. The C4 domain contains three sites that exhibited modest protection upon b12 binding (P417/C418/R419, M434 and Y435) as well as one highly reactive cysteine that showed modest exposure upon b12 binding (C445). The P417/C418/R419 region interacts directly with W100 from CDR H3 of the b12 antibody (Figure 7a). While P417/C418/R419 was not analyzed by alanine mutagenesis, W100 of b12 was shown to be essential for b12 binding51, as was K421 of JR-CSF gp12050; this interaction explains the protection observed here.
Figure 6. HR-HRPF Protection Suggests Stabilization of the α1-helix:α5-helix Interaction Upon b12 Binding to the α5-helix.
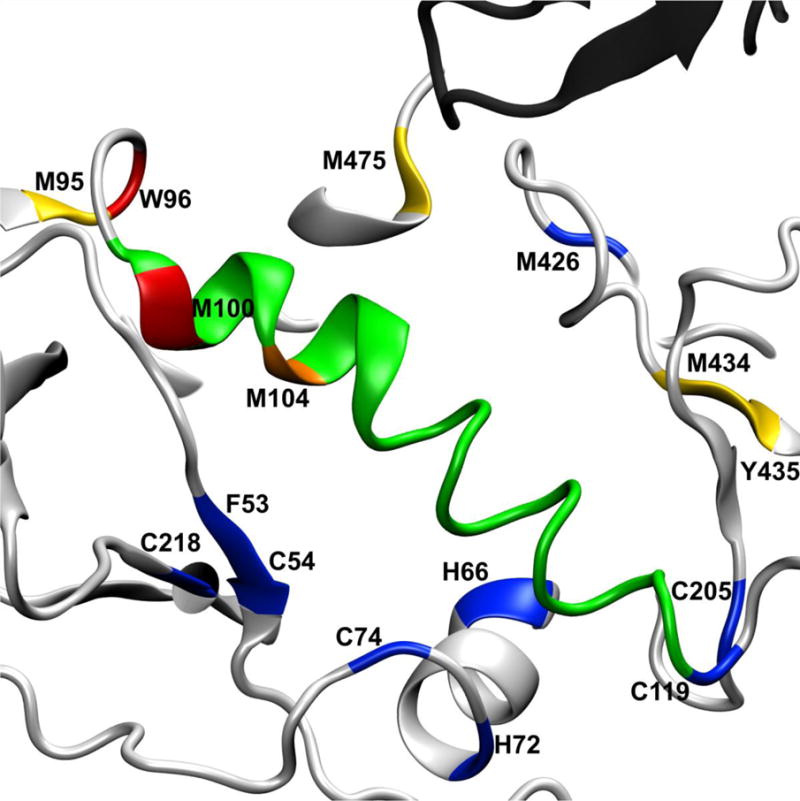
The α1-helix of the C1 domain (green) shows protection of four residues: M95 and W96 in the loop at the N-terminus of the α1-helix, and M99 and M104 within the α1-helix. The sole residue probed in the α5-helix, M475, interacts directly with a CDR loop of the b12 heavy chain (dark grey), with the α5-helix (light grey) packing against the N-terminus of the α1-helix. Residues that showed >80% protection from HR-HRPF upon b12 binding are colored red; residues that showed between 40% and 80% protection from HR-HRPF upon b12 binding are colored orange; residues that showed statistically significant protection from HR-HRPF <40% are colored yellow. Residues showing no protection from HR-HRPF upon b12 binding are colored blue.
Figure 7. b12 Both Interacts Directly with Domain C4 and Alters Its Dynamics.
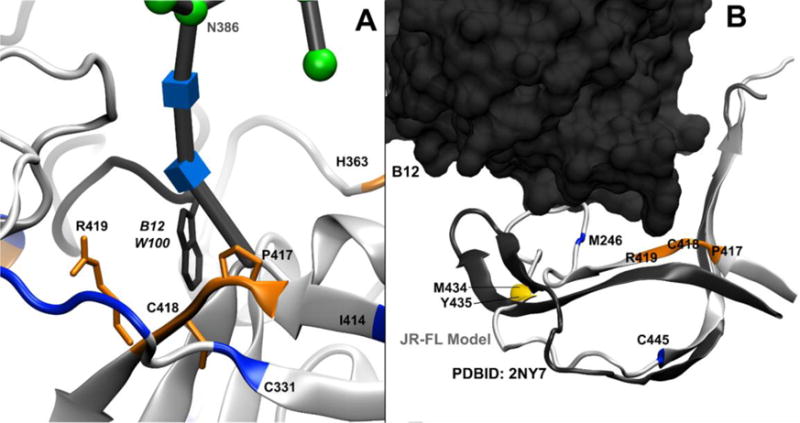
Residues that showed >80% protection from HR-HRPF upon b12 binding are colored red; residues that showed between 40% and 80% protection from HR-HRPF upon b12 binding are colored orange; residues that showed statistically significant protection from HR-HRPF <40% are colored yellow. Residues showing no protection from HR-HRPF upon b12 binding are colored blue. Residue C445 (also blue) showed an increase in oxidation upon b12. A.) P417/C418/R419 of the β19-strand (orange ribbon and licorice) interact directly with W100 (dark grey ribbon and licorice) of the CDR H3 loop of the b12 heavy chain, which extends out from the bulk of the b12 IgG fold. B.) A model of full-length, glycosylated gp120 (light grey) aligned with the crystal structure of stabilized, b12-bound gp120 (dark grey), with only the C4 domain backbone shown for clarity and the b12 antibody shown as a spacefill model. The β22/β23 strand remains largely unperturbed; however, the β20 and β21strands are much more flexible in the full-length, glycosylated, unbound gp120 MD simulation. HR-HRPF data of M426 shows no protection upon b12 binding, suggesting that there is no appreciable change in the conformation or dynamics of this residue in the β20 strand. However, the protection of M434 and Y435 suggest that the β21 strand does experience a stabilization upon b12 binding.
The slight protection observed of M434 and Y435 and the modest exposure observed by C445 are more difficult to understand. This region does not appear to directly interact with b12 in our MD model, or in the crystal structure. This region shows considerable flexibility in the MD simulations of free gp120 as the C-terminal end of the 413–426 β-strand, as well as the entire 432–436 β-strand, completely unravels, deviating significantly from the crystal structure of the stabilized gp120-b12 complex (Figure 7b). Upon b12 binding, M434 and Y435 in the small β-strand shows protection, suggesting this β-strand may be stabilized by b12 binding. This hypothesis is supported by HDX analysis of KNH1144 SOSIP.664 bound to b12, where binding shows slight protection of peptide 427–433 and very little change of peptide 435–445,53 indicating that the protection is largely occurring in residues associated with this strand. However, M426 at the extreme C-terminal end of the 413–426 β-strand shows no protection, yet shows considerable flexibility in the MD simulation of free gp120 as the C-terminal of its β-strand unravels. C445 in the C4 domain exhibits modest exposure upon b12 binding, but shows little deviation between the MD simulation of free gp120 and the X-ray crystal structure of the stabilized gp120-b12 complex, remaining in a β-strand configuration. While HDX analysis suggests no significant change in the secondary structure stability at C44553 and our MD simulation indicates that the displacement of this residue in the strand is small, b12 binding does result in statistically significant exposure of C445. Of note, previous HR-HRPF results have indicated that cysteine oxidation is sensitive to relatively small changes in solvent accessibility31; it is probable that the modest exposure observed in C445 indicates a small change in the average solvent accessibility of this residue.
Previous alanine scanning mutagenesis data support the importance of this region to b12 binding: K421A and M426A resulted in strong loss of binding affinity of b12 for JR-CSF gp120, while N425A resulted in very slight loss of affinity. Similarly, K432A (the only mutated residue in the 432–436 strand) resulted in a strong loss of affinity. Interestingly, the V430A mutant in the turn between these two strands showed over a 5-fold increase in apparent affinity50, suggesting the reduced bulk of the alanine residue in this turn improves the interaction efficiency, perhaps by improving stabilization of these two β-strands. Based on these data and the MD simulation, we hypothesize that b12 binding stabilizes the C-terminus of the 413–426 strand and the paired 432–436 strand, but the extreme C-terminal end of the 413–426 strand in the stabilized crystal structure remains sufficiently disordered to not register topographical protection in HR-HRPF. The β-strand starting with C445 appears to undergo a slight displacement upon b12 binding by MD simulation, which we can detect only due to the sensitivity of cysteine residues to SASA changes in HR-HRPF.
C3 Domain
Seven residues in the C3 domain were oxidized. Three residues at the edges of the folded domain (C331, F353 and C377) experienced no protection upon b12 binding. These residues mostly exist in loops between secondary structure elements, and these loops are distal to the b12 interaction elements; a lack of protection of these residues upon b12 binding would be anticipated.
Four residues experience strong protection upon b12 binding. Three of these residues cluster around the glysocylation site N362: V360, F361 and H363. The Man9GlcNAc2 glycan modeled on the N362 site is poised to interact with the b12 light chain (Figure 8). Based on the glycosylated gp120 model and our HR-HRPF data, it appears that the interaction of the N362 glycan with the light chain of b12 protects the region around N362. Examination of the CATNAP database56 suggests that an intact glycosylation sequon at N362 is not correlated with b12 binding across various gp120 sequences, suggesting that while interaction with b12 may stabilize topography around this glycosylation site, the interaction is not energetically important for b12 binding.
Figure 8. Interaction of b12 with the C3 domain of gp120 is mediated by N-linked glycosylation.
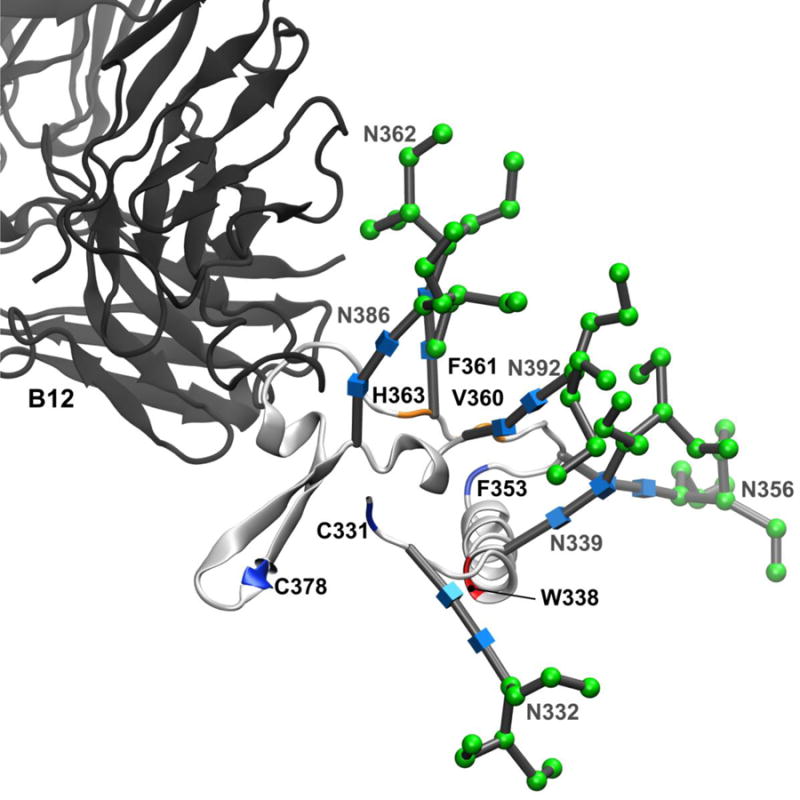
The backbone and N-linked glycans of the C3 domain from the full-length, glycosylated gp120 MD simulation (light grey ribbon) aligned with the b12 Fab fragment (dark grey ribbon). All other gp120 domains have been excluded from the figure for clarity. Residues that showed >80% protection from HR-HRPF upon b12 binding are colored red; residues that showed between 40% and 80% protection from HR-HRPF upon b12 binding are colored orange. Residues showing no protection from HR-HRPF upon b12 binding are colored blue. Glycans are labeled and shown as 3D-SNFG symbols, which are positioned at each residue’s ring center.
W338 in the C3 domain shows complete protection from HR-HRPF upon b12 binding. W338 exists at the N-terminal end of the large α2 helix of the C3 domain. This helix does not interact with b12 in either the X-ray crystal structure or the full-length glycosylated model. W338 is directly adjacent to the N339 glycosylation site. However, in our model, the N339 Man9GlcNAc2 glycan does not directly interact with the b12 antibody. It is possible that b12 binding triggers conformational changes in the core of the C3 domain or in the C3-C4 domain-domain interactions (namely, between the α2 helix of the C3 domain and the β22/β23 strand of the C4 domain) that are observed in the strong HR-HRPF protection of W338 observed. Mutagenesis of the K343 residue in the α2 helix, as well as T450 in the center of the β22/β23 strand both showed a greatly decreased affinity for the b12 antibody50, supporting a potential impact of b12 binding on this region.
It is also possible that the N339 glycan is involved in interactions that are not properly captured by our model. Interestingly, the CATNAP database56 shows a strong positive correlation between an intact N339 glycosylation sequon and b12 binding, suggesting that glycosylation of N339 is involved, directly or indirectly, in the energetics of b12 binding. Previous deglycosylation studies by Koch and co-workers did not examine elimination of the N339 or the N362 glycosylation sites18. While it is not apparent how the N339 glycan could interact directly with the b12 antibody, perhaps the N339 glycan contributes indirectly via another glycan, which interacts directly with b12.
Unprotected Domains
The C2 and V4 domains both had at least one residue probed by HR-HRPF, but no residues that showed protection upon b12 binding. For the C2 domain, eight residues were probed by HR-HRPF (C205, C218, C239, S274, D275, F277, L288, and K289), including three cysteines that are highly sensitive to changes in solvent accessibility. This domain is distal to the b12 antibody binding interface, and is largely made up of flexible loops and small anti-parallel β-sheets. All of the HR-HRPF probed residues are found on regions lacking secondary structure in both the MD model and the X-ray crystal structure of the stabilized gp120-b12 complex, indicating that the binding of b12 does not appreciably change the structure and dynamics of the loops. The residues in the β-sheets were silent in this HR-HRPF experiment. Interestingly, several residues in the C2 domain were found by mutagenesis to affect b12 binding affinity, mostly among residues that are conserved among most gp120 sequences50. Unfortunately, in no case did we receive HR-HRPF data directly on a residue mutated by Pantophlet and co-workers, and the pattern of mutations that changed binding affinity is by no means clear; several of the mutations that decreased b12 binding affinity occurred on the 223–239 stretch of the C2 domain that was unoxidized by HR-HRPF (Figure 2).
The V4 domain had one residue at its extreme C-terminus probed by HR-HRPF: I414, which can be seen near the N-terminal end of the β19 strand in Figure 7a. The interaction with b12 occurs at the C-terminal end of the long β19 strand, and the lack of HR-HRPF protection of I414 upon b12 binding suggests that the N-terminal end of this strand remains stable in the absence of b12 binding. No mutagenesis data for this residue was reported, and CATNAP database analysis showed no correlation between this amino acid identity and b12 affinity.
Conclusion
We demonstrate the ability of the hydroxyl radical-mediated protein footprinting combined with molecular modeling to examine the protein–protein interactions between a highly glycosylated mature gp120 and its bNAb b12. The data presented here identified the highly occupied glycosylation sites and probed the solvent accessibility of sixty-one residues in full-length, glycosylated JR-FL gp120, and identify 23 residues protected upon b12 binding. Analysis of our results in the context of a model of full-length, glycosylated gp120 indicate a prominent role of V1/V2 interactions with the light chain of the b12 antibody, as well as potential roles for the N362 glycan in interacting directly with b12. HR-HRPF was able to detect both direct interactions and conformational changes upon b12 binding without the need to introduce mutations or site-specific chemical labels that could alter the native conformation we seek to probe. The combination of HR-HRPF for providing biophysical data on the full-length glycosylated gp120 protein, combined with molecular modeling to provide a context to interpret changes in HR-HRPF protection, is a powerful combination. Further studies examining the interactions of gp120 with other bNAbs are underway.
Supplementary Material
Acknowledgments
Figures depicting 3D structures were created using Visual Molecular Dynamics (VMD)57. J.S.S. discloses a significant ownership share of Photochem Technologies, LLC, a small company that is active in the area of hydroxyl radical protein footprinting.
Funding Information
This research is supported by the National Institute of General Medical Sciences (1R01GM096049-01A1 and R01GM100058). Instrumentation used for analysis were funded in part by the National Institute of General Medical Sciences-funded “Research Resource for Integrated Glycotechnology” (P41 GM103390) from the National Institutes of Health. Glycosylation occupancy identification by L.W. was supported in part by the National Center for Biomedical Glycomics (P41 GM103490). S.L. acknowledges funding from a Gates Foundation grant (OPP1033112) and the National Institute of Allergy and Infectious Diseases (AI082274 and AI082676). R.J.W was supported by the National Institutes of Health (U01 CA207824 and P41 GM103390).
Footnotes
Supporting Information
Supporting Information available includes a tabulation of glycosylation sequon occupancy for gp120 samples used for this study, and a figure showing the gp120 sequence alignments used to by Modeller to generate the homology model.
References
- 1.Kowalski M, Potz J, Basiripour L, Dorfman T, Goh WC, Terwilliger E, Dayton A, Rosen C, Haseltine W, Sodroski J. Functional regions of the envelope glycoprotein of human immunodeficiency virus type 1. Science. 1987;237:1351–1355. doi: 10.1126/science.3629244. [DOI] [PubMed] [Google Scholar]
- 2.Nara PL, Garrity RR, Goudsmit J. Neutralization of HIV-1: a paradox of humoral proportions. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 1991;5:2437–2455. doi: 10.1096/fasebj.5.10.1712328. [DOI] [PubMed] [Google Scholar]
- 3.Lasky LA, Groopman JE, Fennie CW, Benz PM, Capon DJ, Dowbenko DJ, Nakamura GR, Nunes WM, Renz ME, Berman PW. Neutralization of the AIDS retrovirus by antibodies to a recombinant envelope glycoprotein. Science. 1986;233:209–212. doi: 10.1126/science.3014647. [DOI] [PubMed] [Google Scholar]
- 4.Kwong PD, Wyatt R, Robinson J, Sweet RW, Sodroski J, Hendrickson WA. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature. 1998;393:648–659. doi: 10.1038/31405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wyatt R, Kwong PD, Desjardins E, Sweet RW, Robinson J, Hendrickson WA, Sodroski JG. The antigenic structure of the HIV gp120 envelope glycoprotein. Nature. 1998;393:705–711. doi: 10.1038/31514. [DOI] [PubMed] [Google Scholar]
- 6.Kwong PD, Wyatt R, Desjardins E, Robinson J, Culp JS, Hellmig BD, Sweet RW, Sodroski J, Hendrickson WA. Probability analysis of variational crystallization and its application to gp120, the exterior envelope glycoprotein of type 1 human immunodeficiency virus (HIV-1) The Journal of biological chemistry. 1999;274:4115–4123. doi: 10.1074/jbc.274.7.4115. [DOI] [PubMed] [Google Scholar]
- 7.Saphire EO, Parren PW, Pantophlet R, Zwick MB, Morris GM, Rudd PM, Dwek RA, Stanfield RL, Burton DR, Wilson IA. Crystal structure of a neutralizing human IGG against HIV-1: a template for vaccine design. Science. 2001;293:1155–1159. doi: 10.1126/science.1061692. [DOI] [PubMed] [Google Scholar]
- 8.Chen B, Vogan EM, Gong H, Skehel JJ, Wiley DC, Harrison SC. Structure of an unliganded simian immunodeficiency virus gp120 core. Nature. 2005;433:834–841. doi: 10.1038/nature03327. [DOI] [PubMed] [Google Scholar]
- 9.Huang CC, Tang M, Zhang MY, Majeed S, Montabana E, Stanfield RL, Dimitrov DS, Korber B, Sodroski J, Wilson IA, Wyatt R, Kwong PD. Structure of a V3-containing HIV-1 gp120 core. Science. 2005;310:1025–1028. doi: 10.1126/science.1118398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou T, Xu L, Dey B, Hessell AJ, Van Ryk D, Xiang SH, Yang X, Zhang MY, Zwick MB, Arthos J, Burton DR, Dimitrov DS, Sodroski J, Wyatt R, Nabel GJ, Kwong PD. Structural definition of a conserved neutralization epitope on HIV-1 gp120. Nature. 2007;445:732–737. doi: 10.1038/nature05580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Diskin R, Marcovecchio PM, Bjorkman PJ. Structure of a clade C HIV-1 gp120 bound to CD4 and CD4-induced antibody reveals anti-CD4 polyreactivity. Nature structural & molecular biology. 2010;17:608–613. doi: 10.1038/nsmb.1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McLellan JS, Pancera M, Carrico C, Gorman J, Julien JP, Khayat R, Louder R, Pejchal R, Sastry M, Dai K, O’Dell S, Patel N, Shahzad-ul-Hussan S, Yang Y, Zhang B, Zhou T, Zhu J, Boyington JC, Chuang GY, Diwanji D, Georgiev I, Kwon YD, Lee D, Louder MK, Moquin S, Schmidt SD, Yang ZY, Bonsignori M, Crump JA, Kapiga SH, Sam NE, Haynes BF, Burton DR, Koff WC, Walker LM, Phogat S, Wyatt R, Orwenyo J, Wang LX, Arthos J, Bewley CA, Mascola JR, Nabel GJ, Schief WR, Ward AB, Wilson IA, Kwong PD. Structure of HIV-1 gp120 V1/V2 domain with broadly neutralizing antibody PG9. Nature. 2011;480:336–343. doi: 10.1038/nature10696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kwong PD, Mascola JR, Nabel GJ. Broadly neutralizing antibodies and the search for an HIV-1 vaccine: the end of the beginning. Nat Rev Immunol. 2013;13:693–701. doi: 10.1038/nri3516. [DOI] [PubMed] [Google Scholar]
- 14.Burton DR, Pyati J, Koduri R, Sharp SJ, Thornton GB, Parren PW, Sawyer LS, Hendry RM, Dunlop N, Nara PL, Lamacchia M, Garratty E, Stiehm ER, Bryson YJ, Cao Y, Moore JP, Ho DD, Barbas CF., 3rd Efficient neutralization of primary isolates of HIV-1 by a recombinant human monoclonal antibody. Science. 1994;266:1024–1027. doi: 10.1126/science.7973652. [DOI] [PubMed] [Google Scholar]
- 15.Barbas CF, 3rd, Bjorling E, Chiodi F, Dunlop N, Cababa D, Jones TM, Zebedee SL, Persson MA, Nara PL, Norrby E, Burton DR. Recombinant human Fab fragments neutralize human type 1 immunodeficiency virus in vitro. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:9339–9343. doi: 10.1073/pnas.89.19.9339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barbas CF, 3rd, Collet TA, Amberg W, Roben P, Binley JM, Hoekstra D, Cababa D, Jones TM, Williamson RA, Pilkington GR, Haigwood NL, Cabezas E, Satterthwait AC, Sanz I, Burton DR. Molecular profile of an antibody response to HIV-1 as probed by combinatorial libraries. Journal of molecular biology. 1993;230:812–823. doi: 10.1006/jmbi.1993.1203. [DOI] [PubMed] [Google Scholar]
- 17.Roben P, Moore JP, Thali M, Sodroski J, Barbas CF, 3rd, Burton DR. Recognition properties of a panel of human recombinant Fab fragments to the CD4 binding site of gp120 that show differing abilities to neutralize human immunodeficiency virus type 1. Journal of virology. 1994;68:4821–4828. doi: 10.1128/jvi.68.8.4821-4828.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koch M, Pancera M, Kwong PD, Kolchinsky P, Grundner C, Wang L, Hendrickson WA, Sodroski J, Wyatt R. Structure-based, targeted deglycosylation of HIV-1 gp120 and effects on neutralization sensitivity and antibody recognition. Virology. 2003;313:387–400. doi: 10.1016/s0042-6822(03)00294-0. [DOI] [PubMed] [Google Scholar]
- 19.Liu J, Bartesaghi A, Borgnia MJ, Sapiro G, Subramaniam S. Molecular architecture of native HIV-1 gp120 trimers. Nature. 2008;455:109–113. doi: 10.1038/nature07159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Woods RJ, Tessier MB. Computational glycoscience: characterizing the spatial and temporal properties of glycans and glycan-protein complexes. Curr Opin Struct Biol. 2010;20:575–583. doi: 10.1016/j.sbi.2010.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aye TT, Low TY, Sze SK. Nanosecond laser-induced photochemical oxidation method for protein surface mapping with mass spectrometry. Analytical chemistry. 2005;77:5814–5822. doi: 10.1021/ac050353m. [DOI] [PubMed] [Google Scholar]
- 22.Chance MR. Unfolding of apomyoglobin examined by synchrotron footprinting. Biochemical and biophysical research communications. 2001;287:614–621. doi: 10.1006/bbrc.2001.5628. [DOI] [PubMed] [Google Scholar]
- 23.Goldsmith SC, Guan JQ, Almo S, Chance M. Synchrotron protein footprinting: a technique to investigate protein-protein interactions. Journal of biomolecular structure & dynamics. 2001;19:405–418. doi: 10.1080/07391102.2001.10506750. [DOI] [PubMed] [Google Scholar]
- 24.Charvatova O, Foley BL, Bern MW, Sharp JS, Orlando R, Woods RJ. Quantifying protein interface footprinting by hydroxyl radical oxidation and molecular dynamics simulation: application to galectin-1. Journal of the American Society for Mass Spectrometry. 2008;19:1692–1705. doi: 10.1016/j.jasms.2008.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu G, Chance MR. Hydroxyl radical-mediated modification of proteins as probes for structural proteomics. Chemical reviews. 2007;107:3514–3543. doi: 10.1021/cr0682047. [DOI] [PubMed] [Google Scholar]
- 26.Kiselar JG, Maleknia SD, Sullivan M, Downard KM, Chance MR. Hydroxyl radical probe of protein surfaces using synchrotron X-ray radiolysis and mass spectrometry. International journal of radiation biology. 2002;78:101–114. doi: 10.1080/09553000110094805. [DOI] [PubMed] [Google Scholar]
- 27.Kiselar JG, Mahaffy R, Pollard TD, Almo SC, Chance MR. Visualizing Arp2/3 complex activation mediated by binding of ATP and WASp using structural mass spectrometry. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:1552–1557. doi: 10.1073/pnas.0605380104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gau BC, Sharp JS, Rempel DL, Gross ML. Fast photochemical oxidation of protein footprints faster than protein unfolding. Analytical chemistry. 2009;81:6563–6571. doi: 10.1021/ac901054w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Watson C, Janik I, Zhuang T, Charvatova O, Woods RJ, Sharp JS. Pulsed electron beam water radiolysis for submicrosecond hydroxyl radical protein footprinting. Analytical chemistry. 2009;81:2496–2505. doi: 10.1021/ac802252y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li X, Li Z, Xie B, Sharp JS. Improved identification and relative quantification of sites of peptide and protein oxidation for hydroxyl radical footprinting. Journal of the American Society for Mass Spectrometry. 2013;24:1767–1776. doi: 10.1007/s13361-013-0719-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li Z, Moniz H, Wang S, Ramiah A, Zhang F, Moremen KW, Linhardt RJ, Sharp JS. High structural resolution hydroxyl radical protein footprinting reveals an extended robo1-heparin binding interface. The Journal of biological chemistry. 2015;290:10729–10740. doi: 10.1074/jbc.M115.648410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang S, Arthos J, Lawrence JM, Van Ryk D, Mboudjeka I, Shen S, Chou TH, Montefiori DC, Lu S. Enhanced immunogenicity of gp120 protein when combined with recombinant DNA priming to generate antibodies that neutralize the JR-FL primary isolate of human immunodeficiency virus type 1. Journal of virology. 2005;79:7933–7937. doi: 10.1128/JVI.79.12.7933-7937.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xie B, Sharp JS. Hydroxyl Radical Dosimetry for High Flux Hydroxyl Radical Protein Footprinting Applications Using a Simple Optical Detection Method. Analytical chemistry. 2015 doi: 10.1021/acs.analchem.5b02865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Madden T. In: The BLAST Sequence Analysis Tool. McEntyre J, Ostell J, editors. National Center for Biotechnology Information (US), The NCBI Handbook; 2002. [Google Scholar]
- 35.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. Journal of molecular biology. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 36.Kong L, Sheppard NC, Stewart-Jones GB, Robson CL, Chen H, Xu X, Krashias G, Bonomelli C, Scanlan CN, Kwong PD, Jeffs SA, Jones IM, Sattentau QJ. Expression-system-dependent modulation of HIV-1 envelope glycoprotein antigenicity and immunogenicity. Journal of molecular biology. 2010;403:131–147. doi: 10.1016/j.jmb.2010.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stewart-Jones GB, Soto C, Lemmin T, Chuang GY, Druz A, Kong R, Thomas PV, Wagh K, Zhou T, Behrens AJ, Bylund T, Choi CW, Davison JR, Georgiev IS, Joyce MG, Kwon YD, Pancera M, Taft J, Yang Y, Zhang B, Shivatare SS, Shivatare VS, Lee CC, Wu CY, Bewley CA, Burton DR, Koff WC, Connors M, Crispin M, Baxa U, Korber BT, Wong CH, Mascola JR, Kwong PD. Trimeric HIV-1-Env Structures Define Glycan Shields from Clades A, B, and G. Cell. 2016;165:813–826. doi: 10.1016/j.cell.2016.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 39.Salomon-Ferrer R, Götz AW, Poole D, Le Grand S, Walker RC. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J Chem Theory Comput. 2013;9:3878–3888. doi: 10.1021/ct400314y. [DOI] [PubMed] [Google Scholar]
- 40.Gotz AW, Williamson MJ, Xu D, Poole D, Le Grand S, Walker RC. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born. J Chem Theory Comput. 2012;8:1542–1555. doi: 10.1021/ct200909j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Case DA, Babin V, Berryman JT, Betz RM, Cai Q, Cerutti DS,TE, Cheatham I, Darden TA, Duke RE, Gohlke H, Goetz AW, Gusarov S, Homeyer N, Janowski P, Kaus J, Kolossváry I, Kovalenko A, Lee TS, LeGrand S, Luchko T, Luo R, Madej B, Merz KM, Paesani F, Roe DR, Roitberg A, Sagui C, Salomon-Ferrer R, Seabra G, Simmerling CL, Smith W, Swails J, Walker RC, Wang J, Wolf RM, Wu X, Kollman PA. AMBER 14. University of California; San Francisco: 2014. [Google Scholar]
- 42.Kirschner KN, Yongye AB, Tschampel SM, Gonzalez-Outeirino J, Daniels CR, Foley BL, Woods RJ. GLYCAM06: a generalizable biomolecular force field. Carbohydrates. J Comput Chem. 2008;29:622–655. doi: 10.1002/jcc.20820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Case DA, Babin V, Berryman JT, Betz RM, Cai Q, Cerutti DS, Cheatham TE, Darden TA, III, Duke RE, Gohlke H, Goetz AW, Gusarov S, Homeyer N, Janowski P, Kaus J, Kolossvary I, Kovalenko A, Lee TS, LeGrand S, Luchko T, Luo R, Madej B, Merz KM, Jr, Roe DR,FP, Roitberg A, Sagui C, Salomon-Ferrer R, Seabra G, Simmerling CL, Smith WL, Swails J, Walker RC, Wang J, Wolf RM, Wu X, Kollman PA. The FF14SB force field, AMBER 14 Reference Manual. 2014:29–31. [Google Scholar]
- 44.Darden T, York D, Pedersen L. Particle mesh Ewald: An N⋅ log (N) method for Ewald sums in large systems. The Journal of chemical physics. 1993;98:10089. [Google Scholar]
- 45.Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-Alkanes. J Comput Phys. 1977;23:327–341. [Google Scholar]
- 46.Mahoney MW, Jorgensen WL. A Five-site Model for Liquid Water and the Reproduction of the Density Anomoly by Rigid, Non-polarizible Potential Functions. J Chem Phys. 2000;112:8910–8922. [Google Scholar]
- 47.Bruns CM, Hubatsch I, Ridderstrom M, Mannervik B, Tainer JA. Human glutathione transferase A4-4 crystal structures and mutagenesis reveal the basis of high catalytic efficiency with toxic lipid peroxidation products. Journal of molecular biology. 1999;288:427–439. doi: 10.1006/jmbi.1999.2697. [DOI] [PubMed] [Google Scholar]
- 48.Hubbard SJ, Thornton JM. NACCESS, Department of Biochemistry and Molecular biology. University College of London; 1993. [Google Scholar]
- 49.Zwick MB, Kelleher R, Jensen R, Labrijn AF, Wang M, Quinnan GV, Jr, Parren PW, Burton DR. A novel human antibody against human immunodeficiency virus type 1 gp120 is V1, V2, and V3 loop dependent and helps delimit the epitope of the broadly neutralizing antibody immunoglobulin G1 b12. Journal of virology. 2003;77:6965–6978. doi: 10.1128/JVI.77.12.6965-6978.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pantophlet R, Ollmann Saphire E, Poignard P, Parren PW, Wilson IA, Burton DR. Fine mapping of the interaction of neutralizing and nonneutralizing monoclonal antibodies with the CD4 binding site of human immunodeficiency virus type 1 gp120. Journal of virology. 2003;77:642–658. doi: 10.1128/JVI.77.1.642-658.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zwick MB, Parren PW, Saphire EO, Church S, Wang M, Scott JK, Dawson PE, Wilson IA, Burton DR. Molecular features of the broadly neutralizing immunoglobulin G1 b12 required for recognition of human immunodeficiency virus type 1 gp120. Journal of virology. 2003;77:5863–5876. doi: 10.1128/JVI.77.10.5863-5876.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thieker DF, Hadden JA, Schulten K, Woods RJ. 3D implementation of the symbol nomenclature for graphical representation of glycans. Glycobiology. 2016;26:786–787. doi: 10.1093/glycob/cww076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guttman M, Cupo A, Julien JP, Sanders RW, Wilson IA, Moore JP, Lee KK. Antibody potency relates to the ability to recognize the closed, pre-fusion form of HIV Env. Nat Commun. 2015;6:6144. doi: 10.1038/ncomms7144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Connell BJ, Lortat-Jacob H. Human immunodeficiency virus and heparan sulfate: from attachment to entry inhibition. Front Immunol. 2013;4:385. doi: 10.3389/fimmu.2013.00385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhu CB, Zhu L, Holz-Smith S, Matthews TJ, Chen CH. The role of the third beta strand in gp120 conformation and neutralization sensitivity of the HIV-1 primary isolate DH012. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:15227–15232. doi: 10.1073/pnas.261359098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yoon H, Macke J, West AP, Jr, Foley B, Bjorkman PJ, Korber B, Yusim K. CATNAP: a tool to compile, analyze and tally neutralizing antibody panels. Nucleic Acids Res. 2015;43:W213–219. doi: 10.1093/nar/gkv404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.