

Abstract
Background
Orthostatic hypotension (OH) is a sustained fall in blood pressure on standing which can cause symptoms of organ hypoperfusion. OH is associated with increased morbidity and mortality and leads to a significant number of hospital admissions particularly in the elderly (233 per 100,000 patients over 75 years of age in the US). OH can be due to volume depletion, blood loss, large varicose veins, medications, or due to defective activation of sympathetic nerves and reduced norepinephrine release upon standing (i.e., neurogenic OH).
Methods and Findings
Literature review. Neurogenic OH is a frequent and disabling problem in patients with synucleinopathies such as Parkinson disease, multiple system atrophy, and pure autonomic failure, and is commonly associated with supine hypertension. Several pharmacological and non-pharmacological therapeutic options are available.
Conclusions
Here we review the epidemiology, diagnosis, and management of neurogenic OH, and provide an algorithm for its treatment emphasizing the importance of removing aggravating factors, implementing non-pharmacologic measures, and selecting appropriate pharmacological treatments.
Keywords: Autonomic failure, Droxidopa, Multiple system atrophy, Parkinson disease, Supine hypertension, Synucleinopathies
INTRODUCTION
Orthostatic hypotension (OH) is a sustained fall in blood pressure (BP) on standing. The current definition of OH, based on expert consensus,1 is a fall of at least 20 mmHg in systolic BP or 10 mmHg in diastolic BP within 3 minutes of standing or upright tilt.
OH can impair perfusion to organs above the heart, most notably the brain, resulting in symptoms of tissue hypoperfusion. Symptoms can be very disabling, have a profound impact on a patient’s quality of life, and increase morbidity and mortality.2, 3
OH is a frequent problem in the general population, particularly in the frail elderly, and can be due to a variety of medical conditions, such as intravascular volume depletion, blood pooling (i.e., varicose veins4), severe anemia, antihypertensive therapies, and physical deconditioning. In theses cases, OH usually improves dramatically or resolves after the underlying cause is treated.
In a minority of patients, OH is due to reduced norepinephrine release from postganglionic sympathetic nerves, leading to defective vasoconstriction when in the upright posture.1 This is referred to as neurogenic OH (nOH)5 and occurs frequently in patients with neurodegenerative disorders caused by abnormal accumulation of α-synuclein (i.e., synucleinopathies) in the nervous system such as Parkinson disease (PD), dementia with Lewy bodies, multiple system atrophy (MSA), and pure autonomic failure (PAF).5, 6
Complicating nOH management is arterial hypertension when supine (SH), which occurs in up to 70% of patients with nOH.7, 8
Here we review the epidemiology, evaluation, and management of nOH, with emphasis on patients with PD and other synucleinopathies, summarize the non-pharmacological and pharmacological treatment strategies, and provide practical advice on the management of patients with this debilitating condition.
EPIDEMIOLOGY AND PATHOPHYSIOLOGY
The prevalence of OH in the general population increases with age and varies according to the clinical setting.1, 3, 9 The overall prevalence of OH in patients over age 65 is ~20%.10 In the Cardiovascular Health study the prevalence of OH was 18% in subjects age 65 years or older, although only 2% were symptomatic.10 OH is more common in the elderly due, at least in part, to the frequent use of antihypertensive medications.11 Drugs frequently associated with OH are vasodilators (e.g., nitrates, calcium channel blockers, alpha adrenergic blockers), tricyclic antidepressants, opioids, and alcohol. OH is associated with increased morbidity, particularly syncope and falls,12, 13 and with incident coronary artery disease, stroke, and heart failure.14, 15 OH is a common reason for, or contributor to, hospitalization in elderly patients. OH is present in 25% of patients evaluated in the emergency department for syncope.16 The estimate of OH-related hospitalization is 36 per 100,000 adults, and the OH hospitalization rate can be as high as 233 per 100,000 patients over 75 years of age, with a median length of stay of three days and an overall in-hospital mortality rate of 0.9 percent.9 In inpatient series, the prevalence of OH in elderly patients is as high as 60%.17, 18 OH is an independent predictor of mortality.19-21
When OH occurs in middle aged-patients in the absence of volume depletion or drug effects it is usually neurogenic, i.e., due to impaired norepinephrine release from sympathetic postganglionic neurons. This occurs in approximately one third of patients with OH22 and is a common problem in patients with PD, DLB, and MSA, as well as in diabetic autonomic neuropathy, autoimmune autonomic neuropathy and rare genetic disorders (e.g., dopamine beta-hydroxylase deficiency). Neurogenic OH (nOH) is best understood as a neurotransmitter disorder. In patients with PD, dopamine deficiency in the nigrostriatal pathway causes the motor abnormalities, while impaired release of the neurotransmitter norepinephrine from sympathetic postganglionic neurons causes OH (Figure 1). In contrast to regular OH, nOH is an orphan condition as it affects less than 200,000 people in the US, although the true prevalence of nOH may be underestimated, as BP is not always measured in the upright posture.
Figure 1. Neurotransmitter disorders in Parkinson disease.

Motor dysfunction is mainly due to a dopaminergic deficit in basal ganglia neurons. Dopamine deficit can be treated with oral administration of the dopamine precursor L-dopa or with direct dopaminergic agonists. Defective vasoconstriction leading to neurogenic orthostatic hypotension is due to impaired release of norepinephrine from post-ganglionic sympathetic terminals. Norepinephrine deficit can be treated with oral administration of the norepinephrine precursor droxidopa or with direct adrenergic agonists like midodrine.
An estimated 30 to 50% of patients with PD have nOH.23 The prevalence of nOH in PD increases with age and disease duration.8 Although the prevalence of nOH in PD is relatively high, not all patients have symptoms of organ hypoperfusion. In a recent study of 210 patients with PD, only 16% of patients had symptomatic nOH.8 In that study, symptomatic nOH was associated with an upright mean BP below 75 mmHg. This value (a standing mean BP <75 mmHg) had a sensitivity of 97% and a specificity of 98% for detecting symptomatic nOH, and seems to be the lower limit of cerebrovascular autoregulation in patients with PD and nOH, below which patients develop symptoms of cerebral hypoperfusion. Patients fulfilling criteria for nOH that also had SH, were less likely to develop symptomatic nOH after 3-min standing (Supplementary Figure 1).
For MSA, the criteria for nOH are more stringent (a sustained fall of 30 mmHg in systolic or 15 mmHg in diastolic blood pressure).1 Even according to these criteria, nOH is extremely frequent, although not universal, in MSA, with an estimated prevalence of 70-80%.24, 25
Interestingly, nOH can be a feature of the pre-motor stage of patients with both PD and DLB as well as MSA.6, 25, 26 Therefore, patients with nOH without significant motor or cognitive deficits, and therefore labeled as pure autonomic failure (PAF), need to be followed up closely to detect early signs of phenoconversion to a manifest CNS synucleinopathy.6
Despite similarities in their clinical presentation, the physiopathology of nOH in PD/DLB and PAF is different to that in MSA. In patients with MSA, alpha-synuclein-driven neurodegeneration occurs in central autonomic neurons, while peripheral autonomic nerves are mostly spared. As a result, patients with MSA usually have intact post-ganglionic sympathetic fibers and preserved residual sympathetic tone. In contrast, post-ganglionic sympathetic fibers are severely affected in patients with PD, DLB and PAF. These differences are reflected biochemically with near normal plasma norepinephrine levels in patients with MSA but lower levels in those with PD with nOH or PAF.27 These differences also explain the different pressor responses to norepinephrine enhancing medications (e.g., droxidopa, atomoxetine), as will be later discussed.
APPROACH TO THE PATIENT WITH ORTHOSTATIC HYPOTENSION
OH can be symptomatic or asymptomatic. Typical symptoms of OH are lightheadedness, dizziness, blurry vision, and, when the fall in BP is pronounced, loss of consciousness and postural tone (syncope). Symptoms occur only when standing, less frequently when sitting, and abate when lying down. Patients with OH may also complain of generalized weakness, fatigue, leg buckling, occipital headache, neck and shoulder (“coat hanger”) discomfort, and shortness of breath due to ventilation/perfusion mismatch in the apical lung areas.
Patients with chronic nOH due to neurological disorders usually tolerate very low BPs with only mild or no symptoms at all but syncope can occur with added orthostatic stressors (e.g., large carbohydrate-rich meals, alcohol intake, very warm weather, dehydration, and antihypertensive treatment).
Not all patients with nOH are symptomatic. Symptoms of cerebral hypoperfusion emerge when BP standing falls below the lower limit of the cerebral autoregulatory range. As mentioned above, in patients with PD, this usually occurs when mean BP standing is below 75 mmHg, which corresponds to ~90/60 mmHg (systolic/diastolic) at the heart level.8
Symptoms of OH typically disappear after the patient resumes the sitting or lying position because cerebral blood flow is restored to levels above the lower limit of autoregulatory capacity (Figure 2). The chronic nature of nOH allows remarkable adaptive changes in cerebral autoregulatory mechanisms.28 Indeed, patients with nOH are frequently able to tolerate wide swings in BPs and often remain conscious at pressures that would otherwise induce syncope in healthy subjects.29
Figure 2. Blood pressure and cerebral blood flow in a patient with neurogenic orthostatic hypotension.

The upper tracing displays blood flow velocity as measured by transcranial Doppler ultrasound of the middle cerebral artery (MCA), which is proportional to cerebral blood flow. The lower tracing shows continuous blood pressure recorded with plethysmography. When the patient is in the supine position, BP is normal (120/85 mmHg) and MCA velocity (Vm) is 55 cm/s indicating normal cerebral blood flow. When the patient stands up, BP drops rapidly to 68/55 mmHg and cerebral blood flow falls by nearly 50% as shown by Vm down to 28 cm/s. The patient becomes symptomatic, feels faint and is unable to remain standing (indicated by a swirl). The patient then sits down and his BP increases to 95/62 mmHg. Although this BP value is still low, the patient is not symptomatic because Vm increased to 46 cm/s indicating almost normal cerebral blood flow. This tracing shows that for a patient to become asymptomatic BP does not have to return to normal values but only to increase above the lower limit of cerebral autoregulation.
Symptoms of nOH can be non-specific, including fatigue and difficultly concentrating and may sometimes mimic a levodopa “off” motor state in PD patients. In these cases, the diagnosis of nOH may be missed unless BP is measured in the standing position. Conversely, it is important to realize that in patients with PD postural lightheadedness mimicking nOH may be caused by abnormal postural reflexes, vestibular deficits or orthostatic tremor.30
In contrast to vasovagal (neurally-mediated) syncope, syncope in nOH occurs without signs of autonomic activation such as diaphoresis, tachycardia, nausea or abdominal discomfort. Following syncope, as soon as they resume the supine position, patients with nOH usually recover quickly and may be unaware of the event. Patients report that symptom severity varies from day-to-day and fluctuates throughout the day. The morning hours tend to be most difficult as OH symptoms are aggravated by intravascular volume loss overnight.31 Meals, particularly carbohydrate-rich, lead to splanchnic vasodilatation and post-prandial hypotension (i.e., fall in BP within 2 hours of eating). The severity of postprandial hypotension is directly related to insulin release.32 This has therapeutic implications as will be later discussed. Physical inactivity and prolonged bed rest are common in patients with nOH. This leads to cardiovascular deconditioning further worsening the fall in BP and increasing symptoms leading to a vicious cycle.
Patients at high risk should be routinely screened for OH, even in the absence of symptoms. These include patients with a synucleinopathy (PD/DLB, MSA or PAF), elderly subjects (>70 years old), or on multiple medications.
DIAGNOSIS OF NEUROGENIC ORTHOSTATIC HYPOTENSION
The diagnosis of OH requires BP readings while supine and upright, either during active standing or during a tilt-table test, to determine the presence of a sustained orthostatic fall of at least 20 mmHg systolic or 10 mmHg diastolic BP. BP and heart rate should be measured after the patient has been supine for several minutes and after standing still (or passively tilted) for 1-3 minutes. The magnitude of the BP fall and symptom severity vary at different times of the day; thus it may be necessary to re-test the patient in the morning when the orthostatic fall in pressure is more pronounced or after a meal if the history suggest post-prandial hypotension.
The changes in heart rate on standing help to determine whether the OH is neurogenic in origin. In patients with nOH, reduced sympathetic innervation causes heart rate to increase much less than expected considering the magnitude of the BP fall. 2, 33 Marked increases in heart rate suggest that the OH is non-neurogenic. To ascertain the diagnosis of nOH may require autonomic testing including the BP response to the Valsalva maneuver and plasma norepinephrine levels while supine and standing. During the Valsalva maneuver, patients with nOH fail to show the classical BP “overshoot” after release of the strain (phase 4) (Supplementary Figure 2). An increase in plasma norepinephrine after 5-10 minutes of standing of less than 100% suggest defective baroreflex-mediated sympathetic activation and a diagnosis of nOH (Table 1).
Table 1.
Features of neurogenic and non-neurogenic orthostatic hypotension
| Non-neurogenic
orthostatic hypotension |
Neurogenic orthostatic hypotension |
|
|---|---|---|
| Frequency | Frequent (particularly in
the elderly) |
Rare (< 200,000 in the U.S.) |
| Onset | Variable | Usually chronic (Acute or subacute in immune- mediated neuropathies and ganglionopathies) |
| Causes | Intravascular volume loss
(e.g., dehydration, anemia) Blood pooling (e.g., large varicose veins, skeletal muscle atrophy) Advanced heart failure Adrenal insufficiency Physical deconditioning Antihypertensive medications |
Defective norepinephrine release from sympathetic post- ganglionic nerves upon standing up |
| Prognosis | Resolves when underlying cause is corrected |
Chronic disorder |
| Sympathetic tone | Increased | Low or absent |
|
Increase in heart rate upon
standing |
Pronounced | Mild or absent |
|
Blood pressure overshoot
(phase 4) in Valsalva maneuver |
Present | Absent |
|
Increase in plasma
norepinephrine levels upon standing |
Normal or enhanced (at least x2) |
Reduced or absent (less than x2) |
|
Other symptoms of autonomic
failure |
No | Constipation Erectile dysfunction (men) Urinary abnormalities Sweating abnormalities |
|
Concomitant neurological
deficits |
None (or if present, they are not related to OH) |
None Parkinsonism Cerebellar signs Cognitive impairment Sensory neuropathy |
Ambulatory BP monitoring (ABPM) can assist in the diagnosis and management of nOH 34. Affected patients typically have a reversal of the normal circadian blood pressure pattern with higher BP during the night when the patient is supine in bed than during the day. Nocturnal SH causes pressure natriuresis with exaggerated sodium and water loss causing overnight depletion of intravascular volume, worsening OH in the morning. ABPM and a detailed diary of activities is also useful to specifically tailor the use of short acting pressor agents only at times when OH is severe in patients that may remain seated for long periods of the day or are wheelchair-bound.
Some patients with PD can experience symptoms suggesting OH but neither a significant fall in BP nor reduced cerebral perfusion on standing can be documented during office visits. In these cases, ABPM, a high-calorie challenge, and autonomic testing are all normal. This has been recently described as “inebriation-like syndrome”, and its pathophysiology is uncertain.30 A misdiagnosis of nOH can lead to unnecessary treatment with pressor agents and increase the risk of side effects.
MANAGEMENT OF NEUROGENIC ORTHOSTATIC HYPOTENSION
The goal of treatment is not to normalize standing BP, but to reduce symptom burden, to improve quality of life. Consensus guidelines for the treatment of nOH are currently lacking and there are no long-term studies analyzing the impact of treatment on survival, falls, or quality of life.
A noteworthy percentage of patients with nOH also have SH, which poses a difficult therapeutic challenge. In a multicenter study including 210 patients with PD from the US and Europe, 44% had a supine BP >140/90 mmHg.8 Similar results (45% prevalence of SH) were found in a sample of 72 patients with PD from Japan.35 Another study found that 71% of patients with PD had absent or reversed nocturnal BP dipping, as measured by ABPM, which is another way of quantifying SH.7
Drugs that can increase BP while in the upright position can worsen SH. Therefore, pharmacological treatment of nOH requires careful consideration of the potential risks and actual benefits.
The steps in management include: a) correcting aggravating factors, b) implementing non-pharmacological measures and c) drug therapies (Figure 3).
Figure 3. Treatment algorithm in patients with neurogenic orthostatic hypotension.
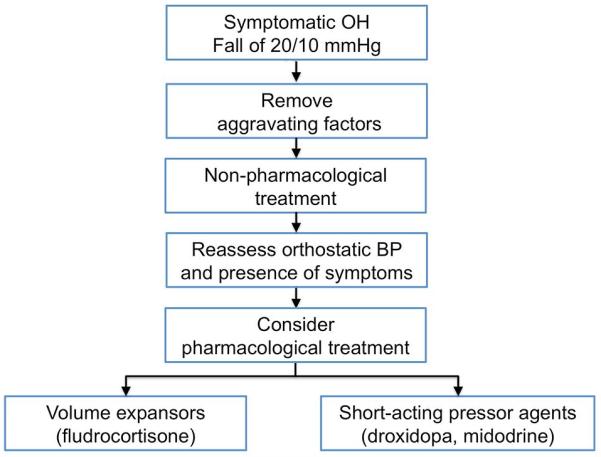
Removal of aggravating factors and initiation of non-pharmacological measures must always predate pharmacological agents.
CORRECTION OF AGGRAVATING FACTORS
Discontinuation of hypotensive drugs
Drugs that reduce intravascular volume, induce vasodilatation or block the release or activity of norepinephrine at the neurovascular junction exacerbate OH and worsen symptoms. Common offenders include diuretics, α-blockers (prescribed for benign prostatic hypertrophy), phosphodiesterase-5 inhibitors like sildenafil (for erectile dysfunction), nitrates, centrally acting α2-agonists like clonidine, calcium-channel blockers, beta-blockers, and tricyclic antidepressants. Levodopa and dopamine agonists may also lower BP and a dose adjustment may be considered based on an individual risk-benefit assessment. Conversely, angiotensin-converting enzyme (ACE) inhibitors are infrequently associated with OH.11, 15, 20, 36
Correction of anemia and vitamin deficiencies
Patients with nOH have an increased incidence of anemia of chronic disease.37 Anemia worsens OH by reducing blood viscosity and oxygen-carrying capacity. Interestingly, because hemoglobin scavenges nitric oxide, a powerful vasodilator38, it is possible that nitric oxide-mediated mechanisms worsen vasodilation in anemic patients with nOH, as it does in other disorders39, although this remains to be proven. Anemia should be investigated and treated appropriately. Small case series show that raising red cell mass with recombinant erythropoietin therapy lessens the fall in BP on standing.40 Vitamin B12 deficiency (<250 pg/ml with elevated methylmalonic acid levels) can also cause or aggravate OH and should be corrected.41
NON-PHARMACOLOGICAL MANAGEMENT
Non-pharmacological measures and patient education are key to the successful management of nOH. Patients should understand the effects of posture on BP and learn physical maneuvers to raise BP.
Volume expansion
Intravascular volume depletion reduces circulating blood volume and worsens the fall in BP on standing. Many elderly patients are chronically volume depleted.42 Patients should be aware of the diuretic effects of caffeine and alcohol, which is also a vasodilator. Similarly, patients should avoid sugary beverages (e.g., bottled juices, sodas) due to the hypotensive effects of high-glycemic index carbohydrates.32
Volume expansion with salt and water is necessary, and fluid intake should be 2-2.5 L of water per day. Patients should be encouraged to increase salt intake by adding 1–2 teaspoon of salt to a healthy diet. Other patients prefer using salt tablets (e.g., 0.5 g or 1.0 g tablets) but they may cause abdominal discomfort.
In patients with nOH, drinking half a liter of water (16 oz. or 1 pint) produces a marked increase in BP. 43 This can be used as a rescue measure because the pressor effect is quick; the increase in BP occurs within 5–10 min and peaks in around 30 min, but it is short-lived. The increase in BP is thought to be due to induction of hypo-osmolality in the portal vein43 triggering a spinal sympathetic reflex,44 although other mechanisms, such as aquaporin-1 regulation,45 might be involved.
Life-style, physical activity, and meals
Hot and humid environments exacerbate symptoms of nOH. A hot shower or sitting in a sauna cause skin vasodilatation and should be avoided because the normal compensatory splanchnic vasoconstriction that maintains BP is lacking in these patients.
Even short periods of bed rest worsen nOH. Symptomatic burden can quickly lead to a reluctance to stand up and avoidance of physical activity. Physical immobility and skeletal muscle loss worsens the severity of OH, which leads to a “vicious cycle” of deconditioning (Supplementary Figure 3).2 Exercise is therefore important and should be performed in the recumbent or seated position, e.g., using a stationary bicycle or a rowing machine, if exercising upright is not tolerated. Exercise in a pool is ideal, as the hydrostatic pressure of water counteracts the gravity-induced fall in BP and increases orthostatic tolerance markedly. Importantly, patients should be extremely careful when exiting the pool, as the sudden decrease of hydrostatic pressure can contribute to venous pooling and a symptomatic drop in BP.
Eating results in blood pooling within the splanchnic circulation.46 Normally, this is compensated for by increases in sympathetic activity producing vasoconstriction. In patients with nOH, however, vasoconstrictor nerve activity is deficient and many patients become severely hypotensive within 2 hours of eating.1, 47 Postprandial hypotension is more pronounced after a carbohydrate-rich meal as insulin release is a contributing factor. Alcohol is also a powerful vasodilator and should be reserved for the evening, prior to laying supine.
Physical countermaneuvers
Physical counter-maneuvers are helpful for managing nOH. Time should be spent making certain patients understand the effect of gravitational fluid shifts on BP and orthostatic symptoms. They should be instructed to change positions gradually, and briefly sit before standing. Patients should be made aware that Valsalva-like maneuvers (straining with closed glottis) produce an abrupt and frequently severe fall in BP and should be avoided. Straining during bowel movements is a common cause of syncope, hence constipation must be treated aggressively. Maneuvers like leg crossing, standing on tiptoes and buttock clenching effectively increase venous return, raise BP and lessen orthostatic symptoms.48
Compression stockings and abdominal binders
Splanchnic venous pooling is a major hemodynamic determinant of OH. Compression stockings apply counter pressure to the lower limbs and abdomen, reducing venous pooling and capillary filtration 49. High-waist stocking that produce at least 15–20 mmHg compression are an effective way to increase venous return. Patients with movement disorders struggle to put the stockings on, which limits their usefulness in everyday life. Elastic abdominal binders are a good alternative. 50, 51 A recently developed abdominal binder that inflates automatically only on standing and provides sustained splanchnic venous compression (40 mmHg) showed promising results in patients with nOH.52
Sleep with the head of the bed raised
Supine hypertension (SH) is frequent in patients with nOH. It is a side effect of anti-hypotensive treatment but it also occurs in untreated patients. The pathophysiology of SH in patients with autonomic failure is not well understood. In patients with MSA, residual sympathetic neurovascular tone may contribute to SH. In contrast, SH in patients with PAF and PD has a mechanism other than increased sympathetic outflows, yet to be fully identified.53, 54
Managing SH in patients with nOH can be challenging. During daytime, avoiding the supine position is the best treatment. If patients need to rest, they should sit in a reclining chair with the feet on the floor. During the night, tilting the bed at least 6 to 9 inches to achieve a 30-degree angle (so that the patient sleeps with his/her head and torso above his/her legs) lowers BP.55 This is best accomplished with an electric bed or electric mattress, rather than just minimally raising the head of the patient with extra pillows. Lowering BP during the night reduces the exaggerated nocturnal diuresis and natriuresis characteristic of these patients, therefore reducing the overnight fluid loss and improving OH in the morning. The use of pressor agents should be avoided before bedtime. Other non-pharmacological measures such as eating carbohydrates snacks or drinking a glass of wine before bedtime also contribute to decrease nocturnal SH.
Antihypertensive drugs may be necessary in patients with severe nocturnal SH that persists after non-pharmacological measures, particularly if they already have end-organ target damage. (e.g., renal failure)31 When antihypertensive drugs are prescribed, patients should be warned about the increased risk of hypotension and falls when they get up at night to urinate. In these cases, the use of a urinal or bedside commode should be encouraged.
PHARMACOLOGICAL MANAGEMENT
While non-pharmacological methods are very effective when performed properly, many patients with nOH still require pharmacological treatment to improve symptoms. Two complementary strategies are used: a) Expanding intravascular volume with the synthetic mineralocorticoid fludrocortisone and b) Increasing peripheral vascular resistance with the pressor agents midodrine or droxidopa. Selection of one or the others or both depends on the specific features and needs of each patient. Fludrocortisone can be combined with midodrine or droxidopa. No studies have directly compared midodrine and droxidopa, so whether one exerts more symptomatic relief than the other is unknown.
All available drugs that raise BP in the standing position also raise BP in the supine position, therefore increasing the risk or worsening SH. Although there are no specific data on cardio- and cerebrovascular events induced by SH in patients with nOH, treating physicians should be aware of this potential side effect. Before beginning treatment with fludrocortisone, midodrine or droxidopa, the patient’s medication should be carefully reviewed.
Combination therapy of agents that increase BP (e.g., fludrocortisone, ephedrine, midodrine, droxidopa and triptans) increases the risk of SH.
Patients should be instructed to avoid the supine position during the day, to sleep with the head of the bed raised 30-degrees, and to ensure that they take their final dose of droxidopa or midodrine at least 4-h before bedtime. Droxidopa or midodrine should be reduced and, if necessary, discontinued if severe SH persists. BP should be rechecked supine at a 30-degree angle if increased doses are required. Safety in patients with BP higher than 180 mmHg at a 30-degree angle has not been established as these patients were excluded from the clinical trials that led to drug approval.
Fludrocortisone
Fludrocortisone (9α-fluorocortisol) is a synthetic mineralocorticoid that increases renal sodium and water re-absorption, therefore expanding intravascular volume and increasing supine, sitting, and standing BP.
Although not approved by the U.S. Food and Drug Administration (FDA) for this indication, fludrocortisone is often prescribed for the treatment of nOH. Because mineralocorticoid receptor activation in the kidney triggers inflammation and fibrosis and may have a direct nephrotoxic effect leading to a faster decline in renal function and hypertension,56 fludrocortisone should be used with extreme caution in the treatment of OH, and dosage should not be higher than 0.1 or 0.2 mg/day. Higher dosages are rarely more effective but amplify side effects.
Clinical effects of fludrocortisone usually require 1-2 weeks of treatment. Frequent side effects include SH, hypokalemia and ankle edema.57 Experimental data suggest that fludrocortisone enhance the pressor effect of norepinephrine and angiotensin II. Long-term use exacerbates SH and end-organ target damage 56, including left ventricular hypertrophy58 and renal failure.56 To reduce the risk of hypokalemia, patients taking fludrocortisone should be instructed to eat potassium-rich foods or to take potassium supplements (potassium chloride 20 mEq a day).
Midodrine
Midodrine is an oral prodrug converted peripherally into the active metabolite desglymidodrine, an α-1-adrenoceptor agonist that constricts arteriolar and venous vasculature producing an increase in BP. Midodrine was approved by the US FDA in 1996 for the treatment of symptomatic OH based on clinical trials showing an increase in systolic BP after standing for 1 minute.59-61 Trials were randomized, double blind and parallel-design and enrolled patients with OH of different etiologies with supine-to-standing fall of systolic BP of at least 15 mmHg accompanied by at least moderate dizziness/lightheadedness. A recent phase IV randomized, double blind, placebo-controlled trial confirmed that midodrine also improves symptoms of OH.62
Administration of midodrine raises standing, sitting, and supine systolic and diastolic BP in patients with nOH. Standing systolic BP increases by 10 mmHg to 30 mmHg 1 hour after a 10 mg dose of midodrine, with some effect persisting for 2-3 hours. Treatment should begin with a 2.5 or 5 mg dose, which can then be increased up to 10 mg to be taken up to 3 times a day.
As with other pressor agents SH is common with midodrine therapy, hence patients should not take midodrine less than 3-4 hours before bedtime. Other reported side effects are pilo-erection (“goosebumps”), itching of the scalp and urinary retention. Midodrine has no effect on heart rate as it does not stimulate cardiac beta-adrenergic receptors and, owing to its poor diffusion across the blood–brain barrier, has no central nervous system side effects.63
Droxidopa
Droxidopa (L-threo-3,4-dihydroxyphenyl-serine or L-DOPS) is a synthetic amino acid that after oral administration is converted to the naturally occurring sympathetic neurotransmitter norepinephrine.64 Droxidopa is decarboxylated to norepinephrine by the enzyme aromatic amino-acid decarboxylase (AAAD) the same enzyme the converts L-dopa to dopamine. AAAD is ubiquitously expressed throughout the body.
The mechanism of action of droxidopa is not completely understood. Norepinephrine production after droxidopa administration occurs in neuronal and non-neuronal tissues. Thus, patients with nOH and extensive degeneration of sympathetic neurons (such as those with PAF, PD or DLB) retain the ability to convert droxidopa into norepinephrine and increase their BP.65 Droxidopa may also convert into norepinephrine within sympathetic terminals and the newly synthesized norepinephrine is then released as a neurotransmitter upon activation of sympathetic neurons.
Droxidopa has been approved in Japan since 1989 for the treatment of nOH in PD, MSA, and familial amyloid polyneuropathy.66-69 The US FDA approved droxidopa in 2014 for the treatment of orthostatic dizziness, lightheadedness or “feeling about to faint” in adult patients with symptomatic nOH associated with PD, MSA, PAF, dopamine β-hydroxylase deficiency and non-diabetic autonomic neuropathy, all disorders characterized by defective norepinephrine release from sympathetic nerves upon standing.5 The FDA approved droxidopa, for short-term use, based on the results of 3 phase-III clinical trials.70-73 Clinical experience with droxidopa shows that it is safe and well tolerated. The most commonly observed adverse reactions were headache, dizziness, nausea, and hypertension.
Droxidopa dosage selection
Peak plasma concentrations of droxidopa are reached by 1-4 h (mean of approximately 3 h) after oral administration. In clinical trials, droxidopa was taken three times daily: upon arising in the morning, at noon and in the late afternoon at least 3 hours prior to bedtime to reduce the risk of SH during sleep. Dosages ranged from 100 mg to 600 mg three times/day.
Because the pressor effect of droxidopa varies among patients, a titration procedure supervised by a clinician is recommended. BP is measured in the 30-degree supine position and standing, and the patient is asked to grade his symptoms. Then, droxidopa 100 mg is administered and 1.5-2 hours later, BP measurements are repeated and the patient is asked again about symptoms. If the patient does not experience full symptomatic relief, and SH is not pronounced, he/she may receive 200 mg. The procedure is repeated until the optimal dose is identified or until the maximum recommended dose (600 mg three times/day) is reached.
Although in the pivotal phase III clinical trials patients took droxidopa three times daily, clinical experience indicates that the dosage should be tailored to each patient’s needs considering the periods of time when he/she is going to be active or inactive. For instance, it is reasonable to administer a single morning dose of droxidopa and skip the afternoon and evening dosages in a patient with MSA and reduced mobility that remains active for only a few hours in the morning when taking a shower, cooking breakfast, or going to physical therapy. Other patients may take droxidopa twice daily only, or take a higher dose in the morning and then lower doses in the afternoon and evening, depending on their daily activities.
Some patients with nOH do not respond to droxidopa. A recent study showed that, in patients with nOH, lower resting plasma norepinephrine levels are associated with a greater pressor response to droxidopa.74 This response is probably related to the degree of denervation supersensitivity. Therefore, supine norepinephrine levels in plasma may be useful to predict appropriate dosing of droxidopa. AMBM is useful to evaluate the BP profile after initiating treatment with droxidopa.75
Does carbidopa block the effect of droxidopa?
Inhibition of AAAD with high dose carbidopa can abolish the pressor effect of droxidopa by preventing its peripheral conversion to norepinephrine. This was demonstrated in early studies 76 using a 200 mg single dose of carbidopa administered 30 minutes before droxidopa. Because in clinical practice the dose of carbidopa in patients with PD treated with L-dopa is lower than 200 mg, carbidopa appears not to block the pressor effect of droxidopa significantly. Dopamine agonists, amantadine derivatives, and MAO-B inhibitors did not affect the pressor effect of droxidopa. Although no dedicated studies have been performed, norepinephrine reuptake inhibitors (e.g., venlafaxine) and adrenergic agonists (e.g., midodrine) may potentiate the pressor effect of droxidopa and should be used with caution.
Other medications
Pyridostigmine, an inhibitor of cholinesterase, the enzyme that catalyzes the hydrolysis of acetylcholine and terminates its action, potentiates cholinergic neurotransmission in autonomic ganglia, both sympathetic and parasympathetic. A double blind study showed that pyridostimine can increase, on average, 4 mmgHg in systolic BP.77 The combination of 5 mg midodrine with 60 mg pyridostigmine was slightly more effective than pyridostigmine alone.
Erythropoietin (25-50 units/kg, subcutaneous, 3 times a week) in conjunction with iron supplements may be beneficial, especially in patients with nOH that have anemia.40
Atomoxetine (10 or 18 mg) blocks the reuptake of norepinephrine by inhibiting the norepinephrine transporter (NET) and can be helpful to increase BP in some patients with nOH 78. In normal subjects, atomoxetine has little effect on BP because the peripheral effects of NET inhibition that result in noradrenergic vasoconstriction are counteracted by the increase in brain norepinephrine, which reduces sympathetic outflow. In patients with autonomic failure and intact post-ganglionic sympathetic noradrenergic fibers (i.e., MSA), only the peripheral vasoconstriction is apparent. This translates to a significant pressor effect of atomoxetine in MSA, but not in PD, DLB or PAF.27 A recent work including 101 patients with nOH treated with atomoxetine showed that higher supine baseline norepinephrine levels predicted a greater symptomatic improvement of orthostatic symptoms.79 The long-term safety and efficacy of atomoxetine in nOH is being currently assessed in a US FDA-sponsored clinical trial.80
The long term safety and efficacy of these approaches have not been determined. Figure 4 summarizes the mechanism of action of drugs used in patients with nOH.
Figure 4. Mechanism of action of drugs for neurogenic orthostatic hypotension.

Commonly used short-acting vasoconstrictor agents include midodrine, an alpha-1-adrenergic receptor agonist; and droxidopa, an artificial aminoacid, which is converted to norepinephrine (NE) by the enzyme aromatic aminoacid decarboxylase (AAAD) in the postganglionic sympathetic terminal, but also extra-neurally in other organs (e.g., kidney). Less commonly used, pyridostigmine, an acetylcholinesterase inhibitor, enhances cholinergic (ACho) transmission in the pre to postganglionic synapse of the sympathetic pathways, but its pressor effect is less consistent. Atomoxetine is a norepinephrine transporter (NET) inhibitor that increases norepinephrine levels; clinical trials are underway to ascertain if chronic treatment with atomoxetine exerts a significant pressor effect in patients with nOH. Fludrocortisone (not shown) is a mineralocorticoid that retains sodium and water therefore increasing intravascular volume. CNS: Central nervous system.
CONCLUSIONS
nOH is a disabling disorder that occurs frequently in patients with PD and other synucleinopathies. Mildly-moderately affected patients need a combination of non-pharmacological and pharmacological therapies, e.g., the synthetic mineralocorticoid fludrocortisone and the pressor agents midodrine or droxidopa. Severely affected patients are unable to stand but for a few seconds, making it impossible to perform even simple activities of daily living. The risk of falls and injuries is increased and patients can become socially isolated due to the burden of symptoms. In these severe cases of nOH success with available agents is only partial, and many patients continue to suffer severe symptoms. Exercise becomes intolerable, which inevitably leads to physical deconditioning and muscle atrophy, which, in turn, worsen the fall in BP. Despite its importance, there is a paucity of treatment options for this condition, the most recently available being droxidopa. New treatment options are needed.
Supplementary Material
Supplementary Figure 1. Blood pressure supine and after 3-min standing in patients with Parkinson disease and orthostatic hypotension (OH according to the 20/10 mmHg criteria). Patients with asymptomatic OH (red) were more likely to have hypertensive BP values in the supine position.
Supplementary Figure 2. Valsalva maneuver in a patient with nOH (A) and a healthy subject (B). The phase IV overshoot of the Valsalva maneuver is absent in nOH (A) and present in healthy subjects (B).
Supplementary Figure 3. The vicious circle of physical deconditioning. Because patients with nOH have intolerable symptoms when standing they avoid moving; lack of physical activity results in striated and cardiac muscle atrophy, which further impairs cardiovascular control. This, in turn, further increases the orthostatic fall in blood pressure, worsening symptoms in a perpetual cycle. Improving symptoms can break this cycle.
Acknowledgments
FUNDING SOURCES AND CONFLICT OF INTERESTS
This work was funded by grants from the National Institutes of Health (U54-NS065736-01), and the Dysautonomia Foundation.
Dr. Palma receives research support from The Dysautonomia Foundation, Inc.; the MSA Coalition; the National Institutes of Health (U54-NS065736-01); the Michael J. Fox Foundation, the Food and Drug Administration, and has received compensation as a consultant/advisory board member for Lundbeck.
Dr. Kaufmann research support from the National Institutes of Health (U54NS065736 [PI]) 1U01NS078025-01, the Food and Drug Administration (FD-R-3731-01 [PI]), the Michael J. Fox Foundation, and the Dysautonomia Foundation, Inc.; has received compensation as a consultant/advisory board member for Lundbeck, Eli Lilly, Pfizer, and Astra Zeneca.
FINANCIAL DISCLOSURES IN THE PREVIOUS 12 MONTHS
Dr. Palma receives research support from The Dysautonomia Foundation, Inc.; the MSA Coalition; the National Institutes of Health (U54-NS065736-01); the Michael J. Fox Foundation, the Food and Drug Administration, and has received compensation as a consultant/advisory board member for Lundbeck.
Dr. Kaufmann research support from the National Institutes of Health (U54NS065736 [PI]) 1U01NS078025-01, the Food and Drug Administration (FD-R-3731-01 [PI]), the Michael J. Fox Foundation, and the Dysautonomia Foundation, Inc.; has received compensation as a consultant/advisory board member for Lundbeck, Eli Lilly, Pfizer, and Astra Zeneca.
Footnotes
AUTHORS’ ROLES
Dr. Palma: conception, design, drafting the article and editing the manuscript for important intellectual content.
Dr. Kaufmann: conception, design, drafting the article and editing the manuscript for important intellectual content.
ETHICAL COMPLIANCE STATEMENT
We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this work is consistent with those guidelines.
REFERENCES
- 1.Freeman R, Wieling W, Axelrod FB, et al. Consensus statement on the definition of orthostatic hypotension, neurally mediated syncope and the postural tachycardia syndrome. Clinical autonomic research : official journal of the Clinical Autonomic Research Society. 2011;21(2):69–72. doi: 10.1007/s10286-011-0119-5. [DOI] [PubMed] [Google Scholar]
- 2.Freeman R. Clinical practice. Neurogenic orthostatic hypotension. The New England journal of medicine. 2008;358(6):615–624. doi: 10.1056/NEJMcp074189. [DOI] [PubMed] [Google Scholar]
- 3.Masaki KH, Schatz IJ, Burchfiel CM, et al. Orthostatic hypotension predicts mortality in elderly men: the Honolulu Heart Program. Circulation. 1998;98(21):2290–2295. doi: 10.1161/01.cir.98.21.2290. [DOI] [PubMed] [Google Scholar]
- 4.Arenander E. Hemodynamic effects of varicose veins and results of radical surgery. Acta Chir Scand Suppl. 1960;(Suppl 260):1–76. [PubMed] [Google Scholar]
- 5.Kaufmann H, Biaggioni I. Autonomic failure in neurodegenerative disorders. Semin Neurol. 2003;23(4):351–363. doi: 10.1055/s-2004-817719. [DOI] [PubMed] [Google Scholar]
- 6.Kaufmann H, Norcliffe-Kaufmann L, Palma JA, et al. The Natural History of Pure Autonomic Failure: a U.S. Prospective Cohort. Annals of neurology. 2017 doi: 10.1002/ana.24877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berganzo K, Diez-Arrola B, Tijero B, et al. Nocturnal hypertension and dysautonomia in patients with Parkinson's disease: are they related? Journal of neurology. 2013;260(7):1752–1756. doi: 10.1007/s00415-013-6859-5. [DOI] [PubMed] [Google Scholar]
- 8.Palma JA, Gomez-Esteban JC, Norcliffe-Kaufmann L, et al. Orthostatic hypotension in Parkinson disease: how much you fall or how low you go? Movement disorders : official journal of the Movement Disorder Society. 2015;30(5):639–645. doi: 10.1002/mds.26079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shibao C, Grijalva CG, Raj SR, Biaggioni I, Griffin MR. Orthostatic hypotension-related hospitalizations in the United States. Am J Med. 2007;120(11):975–980. doi: 10.1016/j.amjmed.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 10.Rutan GH, Hermanson B, Bild DE, Kittner SJ, LaBaw F, Tell GS. Orthostatic hypotension in older adults. The Cardiovascular Health Study. CHS Collaborative Research Group. Hypertension. 1992;19(6 Pt 1):508–519. doi: 10.1161/01.hyp.19.6.508. [DOI] [PubMed] [Google Scholar]
- 11.Fotherby MD, Potter JF. Orthostatic hypotension and anti-hypertensive therapy in the elderly. Postgrad Med J. 1994;70(830):878–881. doi: 10.1136/pgmj.70.830.878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ooi WL, Hossain M, Lipsitz LA. The association between orthostatic hypotension and recurrent falls in nursing home residents. Am J Med. 2000;108(2):106–111. doi: 10.1016/s0002-9343(99)00425-8. [DOI] [PubMed] [Google Scholar]
- 13.Jonsson PV, Lipsitz LA, Kelley M, Koestner J. Hypotensive responses to common daily activities in institutionalized elderly. A potential risk for recurrent falls. Arch Intern Med. 1990;150(7):1518–1524. [PubMed] [Google Scholar]
- 14.Luukinen H, Koski K, Laippala P, Airaksinen KE. Orthostatic hypotension and the risk of myocardial infarction in the home-dwelling elderly. J Intern Med. 2004;255(4):486–493. doi: 10.1111/j.1365-2796.2004.01313.x. [DOI] [PubMed] [Google Scholar]
- 15.Rose KM, Tyroler HA, Nardo CJ, et al. Orthostatic hypotension and the incidence of coronary heart disease: the Atherosclerosis Risk in Communities study. Am J Hypertens. 2000;13(6 Pt 1):571–578. doi: 10.1016/s0895-7061(99)00257-5. [DOI] [PubMed] [Google Scholar]
- 16.Sarasin FP, Louis-Simonet M, Carballo D, Slama S, Junod AF, Unger PF. Prevalence of orthostatic hypotension among patients presenting with syncope in the ED. Am J Emerg Med. 2002;20(6):497–501. doi: 10.1053/ajem.2002.34964. [DOI] [PubMed] [Google Scholar]
- 17.Feldstein C, Weder AB. Orthostatic hypotension: a common, serious and underrecognized problem in hospitalized patients. Journal of the American Society of Hypertension : JASH. 2012;6(1):27–39. doi: 10.1016/j.jash.2011.08.008. [DOI] [PubMed] [Google Scholar]
- 18.Aung AK, Corcoran SJ, Nagalingam V, Paul E, Newnham HH. Prevalence, associations, and risk factors for orthostatic hypotension in medical, surgical, and trauma inpatients: an observational cohort study. The Ochsner journal. 2012;12(1):35–41. [PMC free article] [PubMed] [Google Scholar]
- 19.Angelousi A, Girerd N, Benetos A, et al. Association between orthostatic hypotension and cardiovascular risk, cerebrovascular risk, cognitive decline and falls as well as overall mortality: a systematic review and meta-analysis. J Hypertens. 2014;32(8):1562–1571. doi: 10.1097/HJH.0000000000000235. discussion 1571. [DOI] [PubMed] [Google Scholar]
- 20.Rose KM, Eigenbrodt ML, Biga RL, et al. Orthostatic hypotension predicts mortality in middle-aged adults: the Atherosclerosis Risk In Communities (ARIC) Study. Circulation. 2006;114(7):630–636. doi: 10.1161/CIRCULATIONAHA.105.598722. [DOI] [PubMed] [Google Scholar]
- 21.Xin W, Lin Z, Mi S. Orthostatic hypotension and mortality risk: a meta-analysis of cohort studies. Heart. 2014;100(5):406–413. doi: 10.1136/heartjnl-2013-304121. [DOI] [PubMed] [Google Scholar]
- 22.Goldstein DS, Sharabi Y. Neurogenic orthostatic hypotension: a pathophysiological approach. Circulation. 2009;119(1):139–146. doi: 10.1161/CIRCULATIONAHA.108.805887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Velseboer DC, de Haan RJ, Wieling W, Goldstein DS, de Bie RM. Prevalence of orthostatic hypotension in Parkinson's disease: a systematic review and meta-analysis. Parkinsonism & related disorders. 2011;17(10):724–729. doi: 10.1016/j.parkreldis.2011.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Low PA, Reich SG, Jankovic J, et al. Natural history of multiple system atrophy in the USA: a prospective cohort study. Lancet neurology. 2015;14(7):710–719. doi: 10.1016/S1474-4422(15)00058-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roncevic D, Palma JA, Martinez J, Goulding N, Norcliffe-Kaufmann L, Kaufmann H. Cerebellar and parkinsonian phenotypes in multiple system atrophy: similarities, differences and survival. J Neural Transm (Vienna) 2014;121(5):507–512. doi: 10.1007/s00702-013-1133-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Palma JA, Kaufmann H. Autonomic disorders predicting Parkinson's disease. Parkinsonism & related disorders. 2014;20(Suppl 1):S94–98. doi: 10.1016/S1353-8020(13)70024-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jordan J, Shibao C, Biaggioni I. Multiple system atrophy: using clinical pharmacology to reveal pathophysiology. Clinical autonomic research : official journal of the Clinical Autonomic Research Society. 2015;25(1):53–59. doi: 10.1007/s10286-015-0271-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fuente Mora C, Palma JA, Kaufmann H, Norcliffe-Kaufmann L. Cerebral autoregulation and symptoms of orthostatic hypotension in familial dysautonomia. J Cereb Blood Flow Metab. 2016 doi: 10.1177/0271678X16667524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Horowitz DR, Kaufmann H. Autoregulatory cerebral vasodilation occurs during orthostatic hypotension in patients with primary autonomic failure. Clinical autonomic research : official journal of the Clinical Autonomic Research Society. 2001;11(6):363–367. doi: 10.1007/BF02292768. [DOI] [PubMed] [Google Scholar]
- 30.Palma JA, Norcliffe-Kaufmann L, Kaufmann H. An orthostatic hypotension mimic: The inebriation-like syndrome in Parkinson disease. Movement disorders : official journal of the Movement Disorder Society. 2016;31(4):598–600. doi: 10.1002/mds.26516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arnold AC, Biaggioni I. Management approaches to hypertension in autonomic failure. Curr Opin Nephrol Hypertens. 2012;21(5):481–485. doi: 10.1097/MNH.0b013e328356c52f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shibao C, Gamboa A, Diedrich A, et al. Acarbose, an alpha-glucosidase inhibitor, attenuates postprandial hypotension in autonomic failure. Hypertension. 2007;50(1):54–61. doi: 10.1161/HYPERTENSIONAHA.107.091355. [DOI] [PubMed] [Google Scholar]
- 33.Palma JA, Carmona-Abellan MM, Barriobero N, et al. Is cardiac function impaired in premotor Parkinson's disease? A retrospective cohort study. Movement disorders : official journal of the Movement Disorder Society. 2013;28(5):591–596. doi: 10.1002/mds.25431. [DOI] [PubMed] [Google Scholar]
- 34.Norcliffe-Kaufmann L, Kaufmann H. Is ambulatory blood pressure monitoring useful in patients with chronic autonomic failure? Clinical autonomic research : official journal of the Clinical Autonomic Research Society. 2014;24(4):189–192. doi: 10.1007/s10286-014-0229-y. [DOI] [PubMed] [Google Scholar]
- 35.Umehara T, Matsuno H, Toyoda C, Oka H. Clinical characteristics of supine hypertension in de novo Parkinson disease. Clinical autonomic research : official journal of the Clinical Autonomic Research Society. 2016;26(1):15–21. doi: 10.1007/s10286-015-0324-8. [DOI] [PubMed] [Google Scholar]
- 36.Kamaruzzaman S, Watt H, Carson C, Ebrahim S. The association between orthostatic hypotension and medication use in the British Women's Heart and Health Study. Age Ageing. 2010;39(1):51–56. doi: 10.1093/ageing/afp192. [DOI] [PubMed] [Google Scholar]
- 37.Biaggioni I, Robertson D, Krantz S, Jones M, Haile V. The anemia of primary autonomic failure and its reversal with recombinant erythropoietin. Annals of internal medicine. 1994;121(3):181–186. doi: 10.7326/0003-4819-121-3-199408010-00004. [DOI] [PubMed] [Google Scholar]
- 38.Azarov I, Huang KT, Basu S, Gladwin MT, Hogg N, Kim-Shapiro DB. Nitric oxide scavenging by red blood cells as a function of hematocrit and oxygenation. J Biol Chem. 2005;280(47):39024–39032. doi: 10.1074/jbc.M509045200. [DOI] [PubMed] [Google Scholar]
- 39.Allen BW, Piantadosi CA. How do red blood cells cause hypoxic vasodilation? The SNO-hemoglobin paradigm. American journal of physiology Heart and circulatory physiology. 2006;291(4):H1507–1512. doi: 10.1152/ajpheart.00310.2006. [DOI] [PubMed] [Google Scholar]
- 40.Perera R, Isola L, Kaufmann H. Effect of recombinant erythropoietin on anemia and orthostatic hypotension in primary autonomic failure. Clinical autonomic research : official journal of the Clinical Autonomic Research Society. 1995;5(4):211–213. doi: 10.1007/BF01824009. [DOI] [PubMed] [Google Scholar]
- 41.Toru S, Yokota T, Inaba A, et al. Autonomic dysfunction and orthostatic hypotention caused by vitamin B12 deficiency. Journal of neurology, neurosurgery, and psychiatry. 1999;66(6):804–805. doi: 10.1136/jnnp.66.6.804a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weinberg AD, Minaker KL. Dehydration. Evaluation and management in older adults. Council on Scientific Affairs, American Medical Association. JAMA : the journal of the American Medical Association. 1995;274(19):1552–1556. doi: 10.1001/jama.274.19.1552. [DOI] [PubMed] [Google Scholar]
- 43.May M, Jordan J. The osmopressor response to water drinking. American journal of physiology Regulatory, integrative and comparative physiology. 2011;300(1):R40–46. doi: 10.1152/ajpregu.00544.2010. [DOI] [PubMed] [Google Scholar]
- 44.McHugh J, Keller NR, Appalsamy M, et al. Portal osmopressor mechanism linked to transient receptor potential vanilloid 4 and blood pressure control. Hypertension. 2010;55(6):1438–1443. doi: 10.1161/HYPERTENSIONAHA.110.151860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chu YH, Hsu YJ, Lee HS, et al. The osmopressor response is linked to upregulation of aquaporin-1 tyrosine phosphorylation on red blood cell membranes. Hypertension. 2013;62(1):197–202. doi: 10.1161/HYPERTENSIONAHA.111.200147. [DOI] [PubMed] [Google Scholar]
- 46.Kooner JS, Raimbach S, Watson L, Bannister R, Peart S, Mathias CJ. Relationship between splanchnic vasodilation and postprandial hypotension in patients with primary autonomic failure. J Hypertens Suppl. 1989;7(6):S40–41. doi: 10.1097/00004872-198900076-00017. [DOI] [PubMed] [Google Scholar]
- 47.Jansen RW, Lipsitz LA. Postprandial hypotension: epidemiology, pathophysiology, and clinical management. Annals of internal medicine. 1995;122(4):286–295. doi: 10.7326/0003-4819-122-4-199502150-00009. [DOI] [PubMed] [Google Scholar]
- 48.Krediet CT, van Lieshout JJ, Bogert LW, Immink RV, Kim YS, Wieling W. Leg crossing improves orthostatic tolerance in healthy subjects: a placebo-controlled crossover study. American journal of physiology Heart and circulatory physiology. 2006;291(4):H1768–1772. doi: 10.1152/ajpheart.00287.2006. [DOI] [PubMed] [Google Scholar]
- 49.Diedrich A, Biaggioni I. Segmental orthostatic fluid shifts. Clinical autonomic research : official journal of the Clinical Autonomic Research Society. 2004;14(3):146–147. doi: 10.1007/s10286-004-0188-9. [DOI] [PubMed] [Google Scholar]
- 50.Smit AA, Wieling W, Fujimura J, et al. Use of lower abdominal compression to combat orthostatic hypotension in patients with autonomic dysfunction. Clinical autonomic research : official journal of the Clinical Autonomic Research Society. 2004;14(3):167–175. doi: 10.1007/s10286-004-0187-x. [DOI] [PubMed] [Google Scholar]
- 51.Fanciulli A, Goebel G, Metzler B, et al. Elastic Abdominal Binders Attenuate Orthostatic Hypotension in Parkinson's Disease. Movement Disorders Clinical Practice. 2016;3(2):156–160. doi: 10.1002/mdc3.12270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Okamoto LE, Diedrich A, Baudenbacher FJ, et al. Efficacy of Servo-Controlled Splanchnic Venous Compression in the Treatment of Orthostatic Hypotension: A Randomized Comparison With Midodrine. Hypertension. 2016;68(2):418–426. doi: 10.1161/HYPERTENSIONAHA.116.07199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shannon JR, Jordan J, Diedrich A, et al. Sympathetically mediated hypertension in autonomic failure. Circulation. 2000;101(23):2710–2715. doi: 10.1161/01.cir.101.23.2710. [DOI] [PubMed] [Google Scholar]
- 54.Goldstein DS, Pechnik S, Holmes C, Eldadah B, Sharabi Y. Association between supine hypertension and orthostatic hypotension in autonomic failure. Hypertension. 2003;42(2):136–142. doi: 10.1161/01.HYP.0000081216.11623.C3. [DOI] [PubMed] [Google Scholar]
- 55.MacLean AR, Allen EV. Orthostatic hypotension and orthostatic tachycardia - Treatment with the "head-up" bed. Journal of the American Medical Association. 1940;115:2162–2167. [Google Scholar]
- 56.Norcliffe-Kaufmann L, Axelrod FB, Kaufmann H. Developmental abnormalities, blood pressure variability and renal disease in Riley Day syndrome. Journal of human hypertension. 2013;27(1):51–55. doi: 10.1038/jhh.2011.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chobanian AV, Volicer L, Tifft CP, Gavras H, Liang CS, Faxon D. Mineralocorticoid-induced hypertension in patients with orthostatic hypotension. The New England journal of medicine. 1979;301(2):68–73. doi: 10.1056/NEJM197907123010202. [DOI] [PubMed] [Google Scholar]
- 58.Vagaonescu TD, Saadia D, Tuhrim S, Phillips RA, Kaufmann H. Hypertensive cardiovascular damage in patients with primary autonomic failure. Lancet. 2000;355(9205):725–726. doi: 10.1016/S0140-6736(99)05320-9. [DOI] [PubMed] [Google Scholar]
- 59.Wright RA, Kaufmann HC, Perera R, et al. A double-blind, dose-response study of midodrine in neurogenic orthostatic hypotension. Neurology. 1998;51(1):120–124. doi: 10.1212/wnl.51.1.120. [DOI] [PubMed] [Google Scholar]
- 60.Jankovic J, Gilden JL, Hiner BC, et al. Neurogenic orthostatic hypotension: a double-blind, placebo-controlled study with midodrine. Am J Med. 1993;95(1):38–48. doi: 10.1016/0002-9343(93)90230-m. [DOI] [PubMed] [Google Scholar]
- 61.Low PA, Gilden JL, Freeman R, Sheng KN, McElligott MA. Efficacy of midodrine vs placebo in neurogenic orthostatic hypotension. A randomized, double-blind multicenter study. Midodrine Study Group. JAMA : the journal of the American Medical Association. 1997;277(13):1046–1051. [PubMed] [Google Scholar]
- 62.Smith W, Wan H, Much D, Robinson AG, Martin P. Clinical benefit of midodrine hydrochloride in symptomatic orthostatic hypotension: a phase 4, double-blind, placebo-controlled, randomized, tilt-table study. Clinical autonomic research : official journal of the Clinical Autonomic Research Society. 2016;26(4):269–277. doi: 10.1007/s10286-016-0363-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McTavish D, Goa KL. Midodrine. A review of its pharmacological properties and therapeutic use in orthostatic hypotension and secondary hypotensive disorders. Drugs. 1989;38(5):757–777. doi: 10.2165/00003495-198938050-00004. [DOI] [PubMed] [Google Scholar]
- 64.Kaufmann H, Norcliffe-Kaufmann L, Palma JA. Droxidopa in neurogenic orthostatic hypotension. Expert review of cardiovascular therapy. 2015;13(8):875–891. doi: 10.1586/14779072.2015.1057504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Goldstein DS, Holmes C, Kaufmann H, Freeman R. Clinical pharmacokinetics of the norepinephrine precursor L-threo-DOPS in primary chronic autonomic failure. Clinical autonomic research : official journal of the Clinical Autonomic Research Society. 2004;14(6):363–368. doi: 10.1007/s10286-004-0221-z. [DOI] [PubMed] [Google Scholar]
- 66.Birkmayer W, Birkmayer G, Lechner H, Riederer P. DL-3,4-threo-DOPS in Parkinson's disease: effects on orthostatic hypotension and dizziness. J Neural Transm. 1983;58(3-4):305–313. doi: 10.1007/BF01252816. [DOI] [PubMed] [Google Scholar]
- 67.Sakoda S, Suzuki T, Higa S, et al. Treatment of orthostatic hypotension in Shy-Drager syndrome with DL-threo-3,4-dihydroxyphenylserine: a case report. European neurology. 1985;24(5):330–334. doi: 10.1159/000115820. [DOI] [PubMed] [Google Scholar]
- 68.Senda Y, Muto T, Matsuoka Y, Takahashi A, Sobue I. [Clinical effects of oral L-threo-3,4-dihydroxyphenylserine on orthostatic hypotension in patients with Shy-Drager syndrome] Rinsho shinkeigaku = Clinical neurology. 1987;27(3):300–304. [PubMed] [Google Scholar]
- 69.Kachi T, Iwase S, Mano T, Saito M, Kunimoto M, Sobue I. Effect of L-threo-3,4-dihydroxyphenylserine on muscle sympathetic nerve activities in Shy-Drager syndrome. Neurology. 1988;38(7):1091–1094. doi: 10.1212/wnl.38.7.1091. [DOI] [PubMed] [Google Scholar]
- 70.Kaufmann H, Freeman R, Biaggioni I, et al. Droxidopa for neurogenic orthostatic hypotension: a randomized, placebo-controlled, phase 3 trial. Neurology. 2014;83(4):328–335. doi: 10.1212/WNL.0000000000000615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kaufmann H, Malamut R, Norcliffe-Kaufmann L, Rosa K, Freeman R. The Orthostatic Hypotension Questionnaire (OHQ): validation of a novel symptom assessment scale. Clinical autonomic research : official journal of the Clinical Autonomic Research Society. 2012;22(2):79–90. doi: 10.1007/s10286-011-0146-2. [DOI] [PubMed] [Google Scholar]
- 72.Hauser RA, Isaacson S, Lisk JP, Hewitt LA, Rowse G. Droxidopa for the short-term treatment of symptomatic neurogenic orthostatic hypotension in Parkinson's disease (nOH306B) Movement disorders : official journal of the Movement Disorder Society. 2015;30(5):646–654. doi: 10.1002/mds.26086. [DOI] [PubMed] [Google Scholar]
- 73.Elgebaly A, Abdelazeim B, Mattar O, Gadelkarim M, Salah R, Negida A. Meta-analysis of the safety and efficacy of droxidopa for neurogenic orthostatic hypotension. Clinical autonomic research : official journal of the Clinical Autonomic Research Society. 2016;26(3):171–180. doi: 10.1007/s10286-016-0349-7. [DOI] [PubMed] [Google Scholar]
- 74.Palma JA, Martinez J, Perez M, Norcliffe-Kaufmann L, Kaufmann H. Predictors of response to droxidopa in patients with neurogenic orthostatic hypotension. Clin Aut Res. 2016;26(5):343. [Google Scholar]
- 75.Kaufmann H, Norcliffe-Kaufmann L, Hewitt LA, Rowse GJ, White WB. Effects of the novel norepinephrine prodrug, droxidopa, on ambulatory blood pressure in patients with neurogenic orthostatic hypotension. Journal of the American Society of Hypertension : JASH. 2016;10(10):819–826. doi: 10.1016/j.jash.2016.07.009. [DOI] [PubMed] [Google Scholar]
- 76.Kaufmann H, Saadia D, Voustianiouk A, et al. Norepinephrine precursor therapy in neurogenic orthostatic hypotension. Circulation. 2003;108(6):724–728. doi: 10.1161/01.CIR.0000083721.49847.D7. [DOI] [PubMed] [Google Scholar]
- 77.Singer W, Sandroni P, Opfer-Gehrking TL, et al. Pyridostigmine treatment trial in neurogenic orthostatic hypotension. Archives of neurology. 2006;63(4):513–518. doi: 10.1001/archneur.63.4.noc50340. [DOI] [PubMed] [Google Scholar]
- 78.Ramirez CE, Okamoto LE, Arnold AC, et al. Efficacy of atomoxetine versus midodrine for the treatment of orthostatic hypotension in autonomic failure. Hypertension. 2014;64(6):1235–1240. doi: 10.1161/HYPERTENSIONAHA.114.04225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shibao C, Norcliffe-Kaufmann L, Kaufmann H, Biaggioni I. Baseline supine norepinephrine levels predict the improvement in orthostatic symptoms after atomoxetine in patients with neurogenic orthostatic hypotension. Clin Aut Res. 2016;26(5):347. [Google Scholar]
- 80. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT02796209.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Blood pressure supine and after 3-min standing in patients with Parkinson disease and orthostatic hypotension (OH according to the 20/10 mmHg criteria). Patients with asymptomatic OH (red) were more likely to have hypertensive BP values in the supine position.
Supplementary Figure 2. Valsalva maneuver in a patient with nOH (A) and a healthy subject (B). The phase IV overshoot of the Valsalva maneuver is absent in nOH (A) and present in healthy subjects (B).
Supplementary Figure 3. The vicious circle of physical deconditioning. Because patients with nOH have intolerable symptoms when standing they avoid moving; lack of physical activity results in striated and cardiac muscle atrophy, which further impairs cardiovascular control. This, in turn, further increases the orthostatic fall in blood pressure, worsening symptoms in a perpetual cycle. Improving symptoms can break this cycle.