

Abstract
Purpose
Nail-Patella syndrome is a dominantly inherited genetic disorder characterized by abnormalities of the nails, knees, elbows and pelvis. Nail abnormalities are the most constant feature of Nail-Patella syndrome. Pathogenic mutations in a single gene, LMX1B, a mesenchymal determinant of dorsal-ventral patterning, explain approximately 95% of Nail-Patella syndrome cases. 5% of cases remain unexplained.
Methods
Here we present the exome sequencing and analysis of a four generation family with a dominantly inherited Nail-Patella-like disorder, nail dysplasia with some features of Nail-Patella syndrome, who tested negative for LMX1B mutation.
Results
We identify a loss of function mutation in WIF1 (NM_007191 p.W15*), involved in mesoderm segmentation, as the suspected cause of the Nail-Patella-like disorder observed in this family.
Conclusions
Mutation of WIF1 is a potential novel cause of a Nail-Patella-like disorder. Testing in additional patients negative for LMX1B mutation is needed to confirm this finding and further clarify the phenotype.
Keywords: Nail-Patella syndrome, HOOD syndrome, Fong disease, Turner-Kieser syndrome, nail dysplasia
INTRODUCTION
Nail-Patella syndrome is an autosomal dominant multi-system disorder primarily affecting skeletal and connective tissues. Primary features of the syndrome include dysplasia of the nails, knees and elbows. Nails are typically hypoplastic, absent, or dystrophic. Patellae are often small or shaped irregularly. Elbows abnormalities include dysplasia of the radial head and hypoplasia of the lateral epicondyle and capitellum. Joints may also swell, dislocate, display hypermobility, or display early degenerative arthritis. Iliac horns are observed radiologically in ~70% of cases. Other features include renal dysfunction and, less frequently, glaucoma. Renal dysfunction is observed in ~40% of cases, with end-stage renal diseases occurring in ~5% of cases. Glaucoma is observed more frequently and at a younger age in this population. The overall presentation can vary widely from one affected individual to another individual, with various manifestations ranging from severe to totally absent on a case by case basis. Nail changes are the most constant feature of Nail-Patella syndrome, and are observed in ~98% of cases. The incidence of Nail-Patella syndrome is thought to be 1 in 50,000 1,2.
Loss of function mutations in LMX1B are the only known genetic cause of Nail-Patella syndrome. LMX1B is a WNT signaling responsive transcription factor crucial for mesenchymal dorsoventral patterning in developing limbs 3. WNT signaling plays a major role in a wide variety of embryonic development processes, and it is thought that the timing and restricted expression of LMX1B, as well as other cooperating and compensating signaling pathways, underlie both the constraints on abnormalities typically observed in Nail-Patella syndrome, as well as the heterogeneity of clinical presentation observed from patient to patient 6. LMX1B mutations account for 95% of all Nail-Patella syndrome cases 2, leaving a some cases unexplained even after comprehensive LMX1B mutation profiling.
Here we present the exome sequencing findings of a four generation family with a nail dysplasia with some features of Nail-Patella syndrome. Prior to referral for exome sequencing, the proband of this family was worked up for Nail-Patella syndrome, however her pelvic radiographs were negative for iliac horns, her patellas are not small on X-ray, and she tested negative for LMX1B mutation. Although many of the classical findings for Nail-Patella syndrome were not observed, given the similarity of the nail dysplasia observed in this family to that observed in Nail-Patella syndrome and the co-occurrence of joint laxity, the case was referred for exome sequencing for a suspected Nail-Patella-like disorder or other connective tissue disorder. Exome sequencing, bioinformatic analysis, and application of standard inheritance-based, population-based, and coding impact filters resulted in the conclusion that a loss of function mutation in WIF1, a WNT signaling regulator involved in mesoderm segmentation 5, is the suspected novel cause of the Nail-Patella-like disorder observed in this family.
MATERIALS AND METHODS
Study Consent
Study participants provided written informed consent under a protocol approved by the institutional review board of Scripps.
Whole Exome Sequencing, Variant Calling, and Filtration
DNA was extracted from freshly drawn blood and WES was pursued utilizing Agilent SureSelect exome hybridization followed by barcoding and sequencing of paired 100bp reads on an Illumina HiSeq2500 instrument. Read mapping and variant calling and quality filtration was performed using a BWA-GATK best practices variant quality score recalibration approach. A mean coverage of 86X per individual was achieved with 98.1% of the target exome covered by >10 reads. Variant annotation was performed using the SG-ADVISER system 6. A series of filters was applied to derive a set of candidate disease causative variants (Table 2): (1) population-based filtration, given the rarity of this condition, removed variants present at >0.2% allele frequency in the Exome Aggregation Consortium 7, 1,000 Genomes 8, NHLBI Exome Sequencing Project 9, or Scripps Wellderly populations 10; (3) functional impact-based filtration to remove variants that are not nonsynonymous, frameshift, inframe, nonsense, or do not affect canonical splice-site donor/acceptor sites, and (4) inheritance-based filters to remove variants that do not segregate in the family in a manner consistent with autosomal dominant inheritance.
Table 2.
Inheritance of Candidate Variants
| Gene | Coordinates (hg19) | Protein Impact | Allele Frequency | ID41P | ID41M | ID41F | ID41K4 | ID41K5 | ID41K6 | ID41K7 | ID41K8 | ID41K9 | ID41K10 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DYNC2LI1 | chr2:44023025 snp G>A | c.*360-1G>A | 0.19% | 01 | 01 | 00 | 01 | 01 | 00 | 01 | 00 | 01 | 01 |
| FOXN2 | chr2:48602076 snp C>T | p.L264F | <0.001% | 01 | 01 | 00 | 01 | 01 | 00 | 01 | 00 | 01 | 01 |
| FAT4 | chr4:126370186 snp A>T | p.D2672V | 0.20% | 01 | 01 | 00 | 01 | 01 | 00 | 00 | 01 | 01 | 01 |
| RDH16 | chr12:57348805 snp T>C | p.R153G | 0.006% | 01 | 01 | 00 | 01 | 01 | 01 | 01 | 01 | 01 | 01 |
| KRT74 | chr12:52962172 snp C>A | p.R379L | 0.047% | 01 | 01 | 00 | 01 | 01 | 01 | 01 | 01 | 01 | 01 |
| WIF1 | chr12:65514927 snp C>T | p.W15X | <0.001% | 01 | 01 | 00 | 01 | 01 | 01 | 01 | 01 | 01 | 01 |
| DACT1 | chr14:59113231 snp G>T | p.K630N | 0.31% | 01 | 01 | 00 | 01 | 01 | 01 | 01 | 00 | 00 | 01 |
| EIF2AK4 | chr15:40268999 ins->GACGAC | p.D736_D737dup | 0.005% | 01 | 01 | 00 | 01 | 01 | 01 | 01 | 01 | 01 | 00 |
| PAK6 | chr15:40558610 snp C>G | p.R258G | 0.054% | 01 | 01 | 00 | 01 | 01 | 01 | 01 | 00 | 01 | 00 |
| VPS18 | chr15:41191521 snp A>G | p.I169V | 0.08% | 01 | 01 | 00 | 01 | 01 | 01 | 01 | 00 | 01 | 00 |
| ATP6V0A1 | chr17:40652829 snp C>T | p.T602M | 0.058% | 01 | 01 | 00 | 01 | 01 | 00 | 00 | 00 | 01 | 01 |
| SLC4A1 | chr17:42331864 snp G>A | p.T686M | 0.014% | 01 | 01 | 00 | 01 | 01 | 00 | 00 | 00 | 01 | 01 |
Genotypes: 0 – reference allele, 1 – alternate allele. Variants observed in all affected family members are bolded.
RESULTS
Phenotype
The proband is a 45 year old female presenting with dysplastic ridged nails, joint laxity, and a history of multiple fractures with minimal trauma, a torn anterior cruciate ligament, subluxations of the left shoulder, knee and temporomandibular joints, spondylolisthesis, and arthritis. Prior to referral for exome sequencing, she was evaluated for Nail-Patella syndrome with radiographs of the knees and pelvis which documented normal sized patellas and no iliac horns. Genetic testing for pathogenic deletions, duplications, and mutations in LMX1B was negative (Fulgent Diagnostics).
Nail dysplasia and joint laxity co-occur in a variety of disorders, all of which are associated with additional more severe findings. A search for disorders with combined nail dysplasia (human phenotype ontology identifiers - HP:0002164) and joint laxity (HP:0001388) via the Phenomizer tool 11 returns possible diagnoses that are either severe syndromic disorders (Meier-Gorlin syndrome, Costello syndrome, chromosome 2q32-q33 deletion syndrome, Weaver syndrome, Coffin-Lowry syndrome, craniofrontonasal syndrome, and focal dermal hypoplasia), or severe bone development disorders (metaphyseal chondrodysplasia, and cranioectodermal dysplasia), as well as Nail-Patella syndrome. Thus, although many of the classical findings for Nail-Patella syndrome were not observed, we concluded the phenotype observed in the proband is a nail dysplasia with some features of Nail-Patella Syndrome, what we refer to as a Nail-Patella-like disorder.
Family history includes four generations of 14 total individuals with nail dysplasia and a variety of connective tissue concerns (Table 1, Figure 1, Figure S2–S3). All affected family members belong to the maternal lineage of the proband, including the mother, grandmother, and maternal aunts, uncles, cousins, brother and niece of the proband (see pedigree in Figure 2) – two instances of male to male transmission support autosomal dominant inheritance in the maternal lineage. To identify candidate genetic variants underlying the Nail-Patella-like disorder observed in this four-generation family, whole exome sequencing was pursued in 5 individuals of this family, the mother-father-proband trio and two cousins (the most distantly related family members available for sequencing). Follow-up genotyping of candidate variants was performed in these individuals plus an additional 5 members of this family.
Table 1.
Phenotype Description
| Subject | Age | Nail Dysplasia | Iliac Horns | Joint Problems | Renal Disease | Glaucoma | Other |
|---|---|---|---|---|---|---|---|
| ID41P (proband) | 45 | Present | Absent | lax joints, multiple subluxation, torn ACL, multiple fractures, arthritis | Irritable bowel | ||
| ID41M (mother) | 73 | Present | Unknown | patellar tendon rupture, 3 knee replacements, scoliosis | chronic kidney disease | Present | breast cancer, chronic cough |
| ID41K4 (cousin) | 48 | Present | Unknown | ||||
| ID41K5 (cousin) | NA | Present | Unknown | Crohn’s disease | |||
| ID41K6 (daughter) | 15 | Present | Absent | hip dysplasia, torn ulnar collateral ligament, chronic back pain | |||
| ID41K7 (daughter) | 13 | Present | Unknown | recurrent ankle sprain, microinstability at fibular fracture | |||
| ID41K8 (son) | 13 | Present | Unknown | torn ankle tendon | cranial nerve palsy | ||
| ID41K9 (uncle) | NA | Present | Unknown | chronic cough | |||
| ID41K10 (aunt) | NA | Present | Unknown | multiple joint complaints, hip replacement | chronic cough | ||
| Grandmother | 78 | Present | Unknown | Arthritis | renal failure | Present |
All familial relations are maternal in respect to the proband.
Figure 1. Images of the Proband and Children’s Nail Dysplasia.
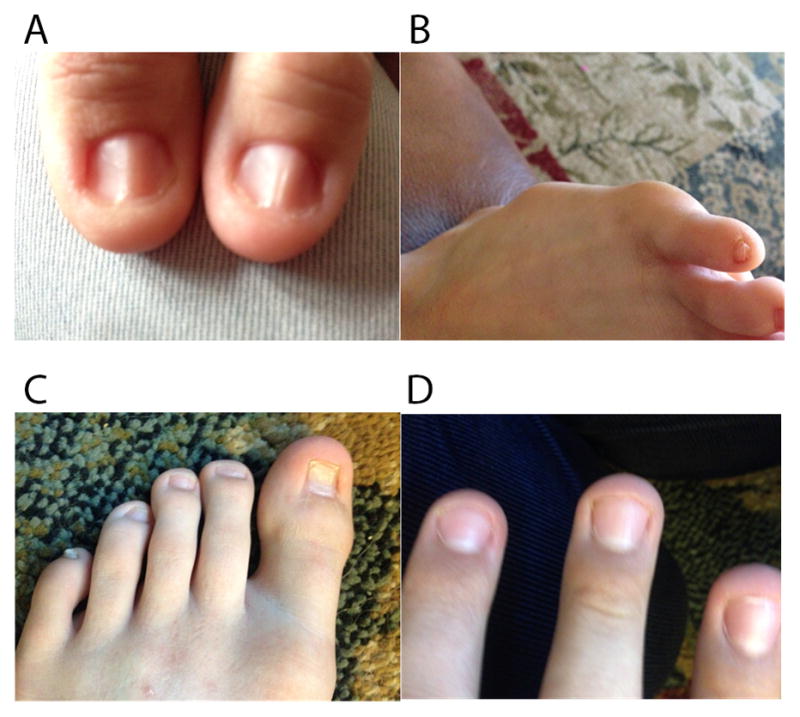
A. Both thumbs of the proband (ID41P). B. 5th toe nail of the proband’s daughter (ID41K7). C–D. Toe and finger nails of the proband’s son (ID41K8). Note variable ridging, dysplasia and thin texture.
Figure 2. Family Pedigree.
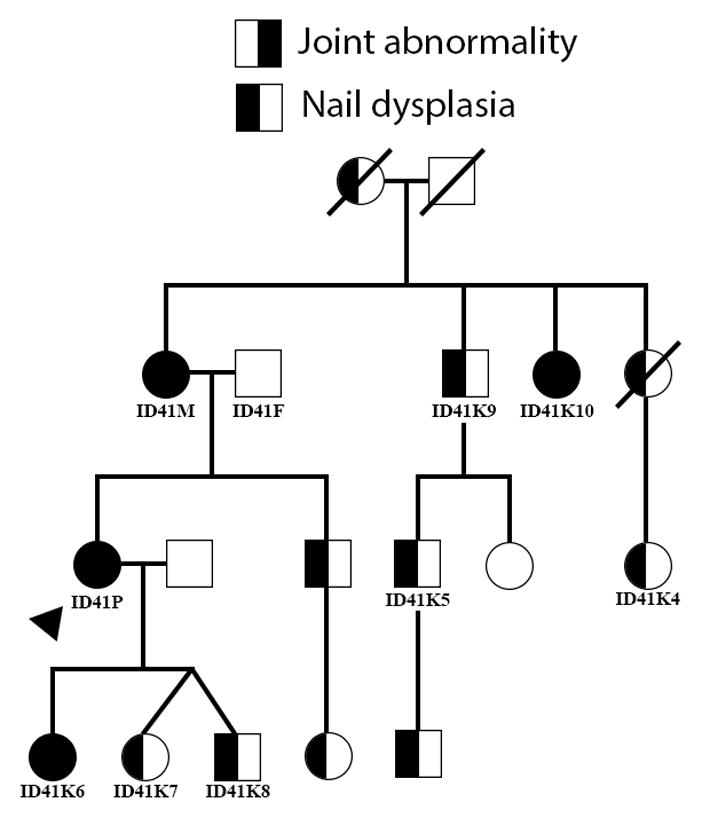
Pedigree of the four generation family with a dominantly inherited Nail-Patella-like disorder. The proband is indicated by the arrow. Identifiers are provided for family members that underwent genetic analysis. Corresponding identifiers can be found in tables. Not all unaffected family members are depicted. Unaffected family members not pictured include three unaffected children of the pictured brother of the proband, four unaffected children of ID41K10, and an unaffected brother of ID41K4.
Sequencing and Candidate Variant Identification
Whole exome sequencing (~86X coverage, 98.1% of target exome covered by 10+ reads) was performed on the affected proband (ID41P), unaffected father (ID41F), affected mother (ID41M) and two affected maternal cousins (ID41K4 and ID41K5) (see Methods for details) (Figure 1B). Variants discovered from WES were annotated by SG-ADVISER 6. Nonsynonymous, in-frame, frameshift, nonsense, and consensus splice site variants, at a threshold population allele frequency of <0.2%, were filtered under an autosomal dominant model of inheritance. This allele frequency threshold is 100-fold above the incidence of Nail-Patella syndrome (1 in 50,000). Twelve candidate causative variants were retained after filtration (Table 2).
Follow-up genotyping, via Sanger sequencing, for the twelve candidate variants was performed in the above mentioned individuals plus three affected children (ID41K6, ID41K7, ID41K8), an affected maternal uncle (ID41K9), and an affected maternal aunt (ID41K10) of the proband (Figure 1B). This process ultimately resulted in the retention of 3 candidate variants: KRT74 p.R379L, RDH16 p.R153G, and WIF1 p.W15* (Table 2 – bolded).
DISCUSSION
Relaxed allele frequency based filtration of coding variants inherited in autosomal dominant fashion in the study family resulted in three candidate genes for the observed Nail-Patella-like disorder; KRT74, RDH16, and WIF1. It should be noted that the underlying assumption leading to the identification of these three candidate variants is that the disorder is driven by a coding mutation captured by exome sequencing, and that the Nail-Patella-like disorder observed in this family is rare. No assumptions regarding the specific phenotype were included in this variant prioritization approach – i.e. variants underlying other known causes of nail dysplasia or other connective tissue disorders would have been captured by our analysis approach. Given our initial assumptions, further filtration of the candidate variants by allele frequency under the assumption of complete penetrance, and prioritization based on biological relevance of the candidate genes, results in the conclusion that mutation of WIF1 is the suspected cause of Nail-Patella-like disorder in this family.
The candidate variant in KRT74 can likely be removed from consideration on the basis of phenotype as well as allele frequency. KRT74 is an epithelial keratin known to cause autosomal dominant woolly hair and autosomal recessive ectodermal dysplasia 12. The family in this report has normal hair, teeth, skin, and sweat glands. Moreover, this particular variant in KRT74 (rs143748352) has been observed at 0.047% allele frequency in Europeans 7, which is at approximately 25-fold excess of the incidence of Nail-Patella syndrome. Thus, under the assumption of near complete penetrance, which is appropriate given the pattern of inheritance observed in this family, the variant in KRT74 is unlikely to be a cause of the observed Nail-Patella-like disorder. However, we cannot fully exclude the possibility that the variant contributes to a portion of the phenotype observed in this family.
The candidate variant in RDH16 cannot definitively be eliminated from consideration due to allele frequency but its known function does not appear to fit the observed phenotype. The variant in RDH16 (rs372580365) has been observed at 0.006% allele frequency in Europeans 7, which is at a slight (3X) excess of the incidence of Nail-Patella syndrome – an excess which would be amplified further given 95% of cases are already explained by mutation in LMX1B mutation (60X excess). Moreover, RDH16 is a liver specific retinol dehydrogenase which plays no known role in connective tissue biology. Finally, RDH16 knockout mice have no significant abnormalities 13.
On the other hand, the variant in WIF1 is a strong candidate as the cause of a novel Nail-Patella-like disorder. The nonsense variant (WIF1 p.W15*) observed in this gene has not been previously observed in over 100,000 genomes 7. Thus, the frequency of this variant is consistent with a fully penetrant cause of the observed disorder. Moreover, WIF1 is a secreted antagonist of WNT signaling, predominantly expressed at the superficial layer of epiphyseal and articular cartilage, and is known to promote chondrogenesis 14. In mouse models of arthritis, loss of WIF1 aggravates cartilage damage 15. The observed nonsense mutation in WIF1 is expected to result in the complete loss of the N-terminal signal peptide of WIF1 (amino acids 1 – 28) and thus inhibit its secretion into the extracellular matrix where it inhibits WNT factors. Thus, loss of WIF1 function would be expected to lead to joint cartilage abnormalities. Moreover, WIF1 functions within the same molecular signaling network as LMX1B – a WNT responsive transcription factor 16.
The WNT pathway controls a variety of embryonic development processes. The specificity of WNT signaling is achieved through a variety of WNT ligands, each of which are subject to highly organized temporal and spatially restricted patterns of expression to coordinate embryonic development 17. A complex, and not fully understood, interplay between WNT and other signaling cascades is known to mediate joint development 18. Similarly, WNT signaling is known to play a central role in nail development 19, though the precise molecular mechanisms have not been well described. It is thought that the role of LMX1B as both a necessary and sufficient mediator of WNT signaling to specify dorsal limb patterning, specifically through induction by the WNT ligand WNT7A, underlies the primary abnormalities observed in Nail-Patella syndrome 16. However, LMX1B is also involved in a variety of other complex WNT dependent processes, where LMX1B function may be compensated for by other factors, potentially leading to the phenotypic heterogeneity observed across Nail-Patella syndrome cases 4. Similarly, WIF1 interacts with a variety of WNT ligands, including WNT7A, and thus participates in an overlapping set of WNT signaling functions as LMX1B 20. However, the details of the role of WIF1 in embryonic and other developmental processes are not well described.
Further study of additional WIF1 positive cases would help elucidate the role of WIF1 mutation in Nail-Patella-like disorder, and the role of WNT signaling in nail and joint development. The phenotype in this family evolves over time. The majority of affected individuals have abnormal nails at birth but do not manifest other features until they become competitively active at adolescence. Multiple joint dislocations and arthritis are most troublesome. Two older members of the family have developed renal issues and one has glaucoma. It is unclear if these features are related to the WIF1 mutation or are unrelated. Longitudinal follow up of family members is clearly warranted. The complete phenotypic spectrum due to loss of WIF1 function may be clarified if more families are reported.
Supplementary Material
A. Thumbs of ID41K5. B. 5th toe of ID41K5. C. Toes of ID415K. Note prematurely wrinkled skin. ID41K5 is 48 years old.
A–B. Finger and toes of proband’s mother (ID41M). C. Fingers of proband’s maternal uncle (ID41K9). D. Toes of proband’s maternal aunt (ID41K10). Note variability in severity of nail dysplasia.
Acknowledgments
This work is supported by Scripps Genomic Medicine, an NIH-NCATS Clinical and Translational Science Award (CTSA; 5 UL1 RR025774) to STSI, as well as funding from the Shaffer Family Foundation and the Anne and Henry Zarrow Foundation. Further support is from the Lavin Family Foundation, NIH-U01 HG006476 and U54GM114833 (AT). We would like to thank the members of our review panel for their dedication and support: Kelly Bethel, Joel Diamant, Nelson Hywnn, James Mason, Evan Muse, Brad Patay, Paul Pockros, Ron Simon, and Gary Williams.
Footnotes
Conflict of Interest Statement:
All authors declare no conflict of interest.
References
- 1.Sweeney E, Hoover-Fong JE, McIntosh I. Nail-Patella Syndrome. GeneReviews. 2014 doi: 10.1111/j.1529-8817.2005.00191.x. [DOI] [PubMed] [Google Scholar]
- 2.Sweeney E, Fryer A, Mountford R, Green A, McIntosh I. Nail patella syndrome: a review of the phenotype aided by developmental biology. J Med Genet. 2003;40(3):153–162. doi: 10.1136/jmg.40.3.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen H, Lun Y, Ovchinnikov D, et al. Limb and kidney defects in Lmx1b mutant mice suggest an involvement of LMX1B in human nail patella syndrome. Nature genetics. 1998;19(1):51–55. doi: 10.1038/ng0598-51. [DOI] [PubMed] [Google Scholar]
- 4.Harita Y, Kitanaka S, Isojima T, Ashida A, Hattori M. Spectrum of LMX1B mutations: from nail-patella syndrome to isolated nephropathy. Pediatr Nephrol. 2016 doi: 10.1007/s00467-016-3462-x. [DOI] [PubMed] [Google Scholar]
- 5.Hsieh JC, Kodjabachian L, Rebbert ML, et al. A new secreted protein that binds to Wnt proteins and inhibits their activities. Nature. 1999;398(6726):431–436. doi: 10.1038/18899. [DOI] [PubMed] [Google Scholar]
- 6.Pham PH, Shipman WJ, Erikson GA, Schork NJ, Torkamani A. Scripps Genome ADVISER: Annotation and Distributed Variant Interpretation SERver. PLoS One. 2015;10(2):e0116815. doi: 10.1371/journal.pone.0116815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Auton A, Brooks LD, et al. Genomes Project C. A global reference for human genetic variation. Nature. 2015;526(7571):68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tennessen JA, Bigham AW, O’Connor TD, et al. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science. 2012;337(6090):64–69. doi: 10.1126/science.1219240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Erikson GA, Bodian DL, Rueda M, et al. Whole-Genome Sequencing of a Healthy Aging Cohort. Cell. 2016;165(4):1002–1011. doi: 10.1016/j.cell.2016.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kohler S, Schulz MH, Krawitz P, et al. Clinical diagnostics in human genetics with semantic similarity searches in ontologies. Am J Hum Genet. 2009;85(4):457–464. doi: 10.1016/j.ajhg.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Raykova D, Klar J, Azhar A, et al. Autosomal recessive transmission of a rare KRT74 variant causes hair and nail ectodermal dysplasia: allelism with dominant woolly hair/hypotrichosis. PLoS One. 2014;9(4):e93607. doi: 10.1371/journal.pone.0093607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van der Weyden L, White JK, Adams DJ, Logan DW. The mouse genetics toolkit: revealing function and mechanism. Genome biology. 2011;12(6):224. doi: 10.1186/gb-2011-12-6-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Surmann-Schmitt C, Widmann N, Dietz U, et al. Wif-1 is expressed at cartilage-mesenchyme interfaces and impedes Wnt3a-mediated inhibition of chondrogenesis. J Cell Sci. 2009;122(Pt 20):3627–3637. doi: 10.1242/jcs.048926. [DOI] [PubMed] [Google Scholar]
- 15.Stock M, Bohm C, Scholtysek C, et al. Wnt inhibitory factor 1 deficiency uncouples cartilage and bone destruction in tumor necrosis factor alpha-mediated experimental arthritis. Arthritis Rheum. 2013;65(9):2310–2322. doi: 10.1002/art.38054. [DOI] [PubMed] [Google Scholar]
- 16.Chen H, Johnson RL. Interactions between dorsal-ventral patterning genes lmx1b, engrailed-1 and wnt-7a in the vertebrate limb. Int J Dev Biol. 2002;46(7):937–941. [PubMed] [Google Scholar]
- 17.MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Developmental cell. 2009;17(1):9–26. doi: 10.1016/j.devcel.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lories RJ, Corr M, Lane NE. To Wnt or not to Wnt: the bone and joint health dilemma. Nat Rev Rheumatol. 2013;9(6):328–339. doi: 10.1038/nrrheum.2013.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Naz G, Pasternack SM, Perrin C, et al. FZD6 encoding the Wnt receptor frizzled 6 is mutated in autosomal-recessive nail dysplasia. Br J Dermatol. 2012;166(5):1088–1094. doi: 10.1111/j.1365-2133.2011.10800.x. [DOI] [PubMed] [Google Scholar]
- 20.Banyai L, Kerekes K, Patthy L. Characterization of a Wnt-binding site of the WIF-domain of Wnt inhibitory factor-1. FEBS Lett. 2012;586(19):3122–3126. doi: 10.1016/j.febslet.2012.07.072. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A. Thumbs of ID41K5. B. 5th toe of ID41K5. C. Toes of ID415K. Note prematurely wrinkled skin. ID41K5 is 48 years old.
A–B. Finger and toes of proband’s mother (ID41M). C. Fingers of proband’s maternal uncle (ID41K9). D. Toes of proband’s maternal aunt (ID41K10). Note variability in severity of nail dysplasia.