

Abstract
Immunotherapies emerged as an alternative for cancer treatment, yet their clinical efficacies are still limited, especially in case of solid tumors. Myeloid immune cells, such as macrophages and myeloid-derived suppressor cells (MDSCs), are often hijacked by tumors and become pivotal inhibitors of antitumor immunity. Immunosuppressive functions of tumor-associated myeloid cells result from the activity of Signal Transducer and Activator of Transcription 3 (STAT3), a transcription factor with well-defined tumorigenic and tolerogenic roles in human cancers. To overcome challenges in the development of pharmacological STAT3 inhibitors, we recently developed oligonucleotide-based strategies for cell-selective, in vivo STAT3 targeting. Conjugation of a STAT3siRNA or decoy STAT3 inhibitors to synthetic Toll-like Receptor 9 (TLR9) agonists, CpG oligonucleotides, allowed for selective delivery into TLR9-positive cells. Cellular targets for CpG-STAT3 inhibitors include non-malignant, tumor-associated myeloid cells, such as polymorphonuclear MDSCs, as well as cancer cells in acute myeloid leukemia, B cell lymphoma and in certain solid tumors. The chemically modified CpG-STAT3 inhibitors resist blood nucleases and thus can be administered intravenously. Their potency relies on the intracellular gain-of-function effect: release of the central immune checkpoint regulator (STAT3) to unleash proinflammatory signaling (CpG/TLR9) in the same antigen-presenting cell. At the cellular level, CpG-STAT3 inhibitors exert two-pronged effect by rescuing T cells from the immune checkpoint control while decreasing survival of cancer cells. In this article, we review the preclinical data on CpG-STAT3 inhibitors and discuss perspectives of using TLR9-targeted delivery of oligonucleotide therapeutics for the generation of novel, more effective and safer cancer immunotherapies.
Keywords: MDSC, myeloid-derived suppressor cells, targeted delivery, TLR9, STAT3, cancer immunotherapy, Regulatory Myeloid Suppressor Cells, CpG, oligonucleotides
Immunosuppressive signaling networks in the tumor microenvironment
Recent clinical advances in immunotherapy of advanced cancers highlighted therapeutic potential of targeting immune checkpoints[1] while underscoring the complexity of signaling networks within the tumor microenvironment.[2] There is ample evidence that human cancers, and especially solid tumors, employ a wide array of immune checkpoint mechanisms extending beyond PD-1 or CTLA4 triggering.[3] It is also widely recognized that cancer resistance to various treatments, including immunotherapy, results from combination of intrinsic properties of cancer cells and extrinsic influences of tumor stroma.[4, 5] Tumor microenvironment hosts a variety of immune cell subsets that modulate therapeutic efficacy. While T lymphocytes are effector cells essential for durable antitumor immune responses, diverse types of myeloid cells may positively or negatively affect the outcome of therapy.[6, 7] This is an obvious consequence of the physiological role of myeloid cells in the control of tissue homeostasis and wound healing. By modulating intracellular signaling, tumors hijack myeloid cells to promote cancer survival, neovascularization and immune evasion. Signal transducer and activator of transcription 3 (STAT3) was originally described as an oncogenic and pro-survival molecule in cancer cells.[8] Later studies revealed that in vivo STAT3 is activated in both cancer cells and in the tumor-associated myeloid cells such as immature dendritic cells (DCs), tumor-associated macrophages (TAMs) and myeloid-derived suppressor cells (MDSCs), thereby promoting tumors by a variety of mechanisms.[8, 6] Activation of STAT3 inhibits maturation of antigen-presenting cells such as DCs, resulting in decreased expression of MHC class II complexes, costimulatory molecules (CD40, CD80, CD86) and lower IL-12 production.[9, 10] With functionally impaired DCs, STAT3 redirects differentiation of immature myeloid cells into TAMs and MDSCs, that actively support tumor progression, neovascularization and immune evasion.[6, 8, 11, 12] Multitasking in the tumor microenvironment reflects essential function of STAT3 in wound healing and resolution of inflammation, at least partly though shifting transcriptional activity of NF-κB from pro-inflammatory to tumorigenic target genes.[13, 14] Therefore, STAT3 can be considered the central immune checkpoint regulator and the nodal point for immunosuppressive signaling in tumor-associated myeloid cells.[8] This unique role and the contribution of STAT3 to survival of cancer cells, provide a strong rationale for therapeutic interventions targeting this molecule.[8, 13] Importantly, genetic loss of STAT3 activity in humans is not lethal although it leads to complex immunodeficiency (autosomal-dominant hyper-immunoglobulin E syndrome; AD-HIES) associated with skin and lung infections, eosinophilia and high levels of IgE.[15] These manifestations are likely caused by impaired development of Th17 cells, and follicular helper T cells that in turn results in abnormal B cell functions. In addition, risks of inhibiting STAT3 in immune cells include impaired generation of central memory T cells, which are essential for control of chronic viral infections and long term antitumor immunity.[16, 17] As demonstrated in earlier genetic studies, blocking STAT3 in tumor-associated myeloid cells alone, without affecting STAT3 signaling in cancer cells, was sufficient to induce antitumor immunity and inhibit growth of various solid tumor models.[10] When combined with local immunostimulation or tumor irradiation, STAT3 deletion resulted in complete regression of large established tumors and protected mice from tumor recurrence.[12, 18] These proof-of-principle experiments defined the two key elements for generation of effective antitumor immunity: release of the STAT3 checkpoint and immune receptor-triggering to jump-start a cascade of innate and adaptive antitumor responses.
Challenges in targeting STAT3 in tumor-associated myeloid cells
Despite numerous attempts, STAT3 targeting using pharmacologic approaches remains challenging.[19] Until today, there are no FDA-approved small molecule STAT3 inhibitors. Inhibitors of Janus kinases (Jak), upstream from STAT3 and multiple other signaling pathways, have been intensely studied for therapy of cancer and autoimmune diseases.[20] However, in late clinical studies some of the most promising Jak inhibitors caused unexpected adverse effects, likely not related to STAT3 inhibition.[21] Beyond such toxicities, broad inhibition of Jak/STAT signaling may impede IFN-mediated antitumor immunity and/or STAT3-mediated generation of memory T cells.[13] These observations emphasize the need for both molecular and cellular selectivity in targeting STAT3 in order to maximize immunotherapeutic efficacy while reducing potential toxicities. Oligonucleotide-based therapeutics (ONTs), such as siRNA, antisense oligonucleotides (ASO) or decoy oligodeoxynucleotides (dODN), emerged as potential alternatives to small molecule STAT3 inhibitors. Both STAT3 antisense and decoy oligonucleotides as well as the first small molecule inhibitor have reached clinical testing (Table 1). Active trials focus now on the antisense strategy (AZD9150) in combination with immune checkpoint blocking antibodies to PD-L1 to improve therapeutic efficacy.
Table 1.
Clinical Trials of STAT3 Oligonucleotide Inhibitors.
| Oligonucleotide | Formulation | Company or sponsor | Indication | Stage of development | Status |
|---|---|---|---|---|---|
| STAT3 DECOY | DNA competitive inhibitor | University of Pittsburgh | Head and neck cancer | Phase 0 | Completed |
| AZD9150 | Antisense Oligonucleotide inhibitor | AstraZeneca, Ionis Pharmaceuticals | Diffuse large B-cell lymphoma, advanced lymphoma | Phase I/II | Active, not recruiting |
| AZD9150 | Antisense Oligonucleotide inhibitor | National Cancer Institute (NCI) | Ovarian cancer/neoplasms, Gastrointestinal cancer/neoplasms | Phase II | Terminated |
| AZD9150 | Antisense Oligonucleotide inhibitor | AstraZeneca, Ionis Pharmaceuticals | Advanced adult hepatocellular carcinoma, Metastatic hepatocellular carcinoma | Phase I | Completed |
| AZD9150 in combination with MEDI4736 | Antisense Oligonucleotide inhibitor Monoclonal anti- PD-L1 monoclonal antibody | MedImmune | Diffuse large B-cell lymphoma | Phase I | Recruiting |
| AZD9150 in combination with MEDI4736 | Antisense Oligonucleotide inhibitor Monoclonal anti- PD-L1 monoclonal antibody | AstraZeneca, MedImmune | Metastatic head and neck squamous cell carcinoma | Phase I/II | Recruiting |
| AZD9150 in combination with MEDI4736 | Antisense Oligonucleotide inhibitor Monoclonal anti- PD-L1 monoclonal antibody | M. D. Anderson Cancer Center, AstraZeneca | Advanced pancreatic, non- small cell lung and colorectal cancers | Phase II | Not yet recruiting |
Information collected from www.clinicaltrials.gov.
While these strategies offer high specificity in targeting STAT3 at the molecular level, their clinical development continues to be hindered by the lack of methods for efficient and cell-selective oligonucleotide delivery. In addition, the immunogenicity of ONTs has long been considered an unwanted and potentially dangerous side effect alleviated only by extensive chemical modifications. However, immune cells may themselves be essential therapeutic targets in cancer therapy.[22] Synthetic oligodeoxynucleotides comprising an unmethylated CpG motif, CpG ODNs, have been used in dozens of clinical trials against a variety of human cancers, albeit with limited efficacy.[23] CpG ODNs are recognized by the innate immune receptor, Toll-like Receptor 9 (TLR9), which activates immune cells and stimulates the release of proinflammatory cytokines. TLR9 agonists are potent immunoadjuvants for vaccines, however in the tumor microenvironment their potency is strongly limited or even tumorigenic, regardless whether it is triggered by synthetic ligands or release of natural agonists, such as mitochondrial DNA from dying cells.[12] The functional dichotomy of TLR9 outcome seems to be defined by a multilayered negative feedback regulation through STAT3. As shown by our previous studies, TLR9 signaling induced by CpG ODN or endogenous ligands from irradiated cancer cells, triggers Jak/STAT3 activation in myeloid cells mediated by delayed release of IL-6.[12, 18] During infections such negative feedback regulation can limit potential collateral damage caused by the unrestricted proinflammatory TLR9 signaling, however in the tumor microenvironment, it rather serves to sustain STAT3 activity and to promote tumor immune evasions as well as neovascularization.[12, 18] These observations stimulated development of a platform technology for cell-selective delivery of oligonucleotide-based STAT3 inhibitors into TLR9-expressing immune cells.
It takes two to deliver: TLR9 and scavenger receptors
In contrast to many ONTs, single-stranded CpG ODNs are quickly and efficiently internalized by specialized target cells thereby stimulating immune responses.[24] Since the mature and fully functional TLR9 is not present on cell surface, it is not directly involved in ligand internalization. Instead CpG ODNs are recognized by the dextran sulfate-sensitive receptors of the scavenger family.[25, 26–28] The scavenger receptors (SRs) are structurally heterogeneous group recognizing a diverse set of ligands ranging from endogenous proteins and lipoproteins to conserved microbial structures.[25] Due to partial functional redundancy of SRs, several different receptors have been implicated in the uptake of CpG ODNs, including SR-A1/MARCO,[29] SR-BI,[27] CXCL16,[26] CD14,[30] CD205, [31] or RAGE.[32] Correspondingly, our CpG-conjugate internalization studies did not find dominant role of any single tested SR so far. We confirmed partial contribution of SR-A1, CXCL16 and likely other SRs in the uptake by myeloid cells.(Moreira and Kortylewski, unpublished data) SRs are known to be expressed mostly by immune cells specialized in uptake of macromolecules, such as DCs, macrophages and B cells, and therefore coupled to the panel of endosomal TLRs to enable recognition of potential danger signals.[24] The intracellular localization of TLR9, similar to TLR3, TLR7 and TLR8, creates an opportunity for utilizing TLR9 ligands for delivery of therapeutics into the cytosol. In contrast to ubiquitous expression in myeloid cells in mice, in humans TLR9 is restricted to plasmacytoid DCs and B lymphocytes under normal physiological conditions.[24] However, TLR9 levels can be quickly upregulated under inflammatory conditions or in the tumor microenvironment.[33, 34] Our own analyses of cancer patients’ specimens identified TLR9 expression in two therapeutically important immune cell populations: in tumor-associated macrophages and in a subset of myeloid derived suppressor cells as discussed later in this article.[12, 35] TLR9 is also commonly elevated in cancer cells in many hematologic malignancies, including acute myeloid leukemia (AML), multiple myeloma and B cell lymphoma as well as in certain solid tumors, such as prostate cancers or malignant glioma.[23, 36–38] The efficacy of CpG-conjugates is determined not only by TLR9, as TLR9 partners with SRs on the cell surface for conjugate uptake. Noteworthy, SRs expression has been reported in hematologic malignancies and also in various solid tumors, e.g. SR-BI was found in breast, prostate and pancreatic cancer cells.[39] Therefore, the ONT delivery via the SR/TLR9 tandem provides a possibility for simultaneous targeting of tumor-associated myeloid cells as well as cancer cells in several types of human tumors.
SR/TLR9-targeted delivery and intracellular processing of CpG-STAT3 inhibitors
Our group successfully utilized the SR/TLR9 receptors for ONT delivery by generating two types of targeted STAT3 inhibitors. In the first design, a CpG ODN was tethered to a STAT3 siRNA in a cleavable design to allow for Dicer endonuclease-mediated uncoupling of the processed 21/21mer siRNA after uptake.[40, 41] To overcome the limited serum-stability of CpG-siRNA, we recently generated a clinically relevant second-generation CpG-STAT3 inhibitor suitable for intravenous delivery.[42] In this alternative design, the CpG moiety is linked to a STAT3 decoy ODN (CpG-STAT3dODN), a high-affinity STAT3 DNA binding sequence that acts as a competitive inhibitor and prevents the transcriptional activity of STAT3 (Figure 1).[19] Despite chemical and structural differences, both types of CpG-STAT3 inhibitors are rapidly internalized (within 30–60 min) by a variety of TLR9-positive immune cells both in vitro and in vivo. In mice, CpG-STAT3 inhibitors penetrate DCs, macrophages and B cells but not TLR9-negative NK cells or T lymphocytes.[36, 40, 42, 43] To allow for further optimization of these strategies, our group unraveled the intracellular mechanisms of CpG-siRNA or CpG-dODN processing and function.[28, 41, 42] Similar to unconjugated CpG ODNs, both CpG-siRNA and CpG-dODN conjugates undergo SR-mediated endocytosis and localize within minutes to early endosomes (EE) in target cells. While in the EE compartment, CpG-siRNA conjugates become substrates for Dicer endonuclease, which transiently interacts with the target molecule to cleave off a diced siRNA. To ensure efficient target gene silencing, the siRNA molecules need to escape EE before endosomal acidification and the eventual lysosomal degradation of the content. Unexpectedly, in contrast to many other receptor-mediated ONT delivery strategies (e.g. antibody-mediated delivery),[44] TLR9-targeted delivery strategies are not limited by the endosomal retention. As demonstrated for CpG-siRNA, TLR9 activation permitted rapid endosomal escape of siRNA within the first 2–3 h after internalization.[28] In Tlr9-deficient macrophages, lack of TLR9 activity caused retention of siRNA molecules in EEs and thus impaired target gene silencing. Thus, TLR9 triggering has beneficial role for the CpG-STAT3 inhibitor efficacy despite not being directly involved in the uptake of CpG-conjugates. While a detailed mechanism of TLR9-dependent endosomal release of ONTs remains to be defined, it likely corresponds to the known role of TLR9 in cross-presentation of antigens by APCs. TLR9 and other endosomal TLRs seem to control the release of internalized microbial products using specialized peptide transporters in the EE membrane to enable pathogen recognition by cytosolic sensors.[45]
Figure 1. The intracellular mode of action of bi-functional CpG-STAT3 decoy oligodeoxynucleotides.
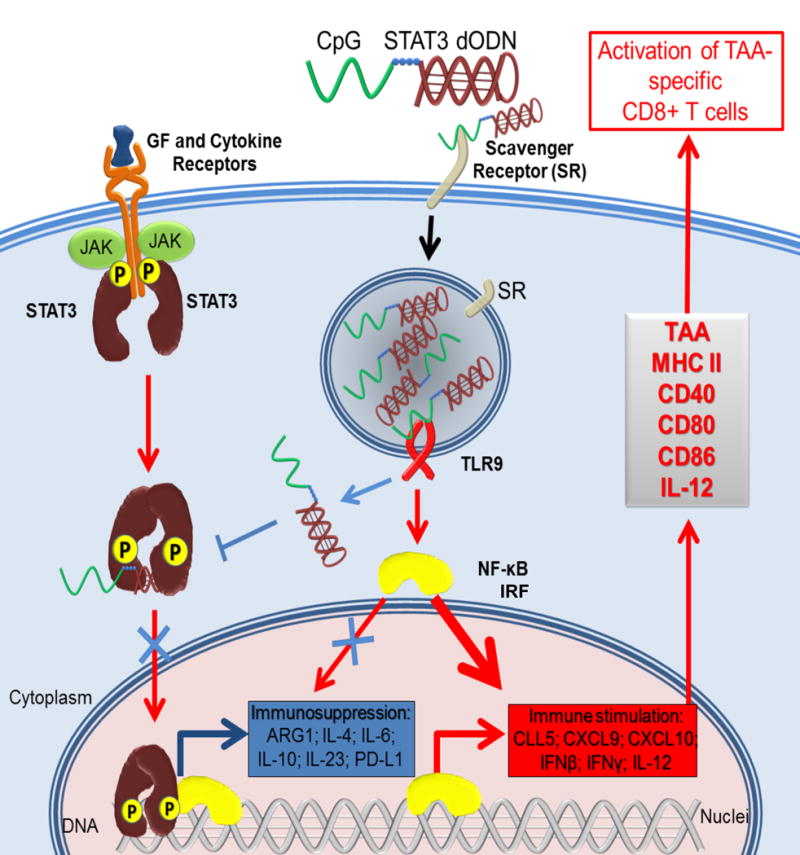
CpG-STAT3dODN conjugates are recognized by scavenger receptors and quickly internalized through endocytosis into target cells. While in early endosomes, some of the internalized conjugates bind to TLR9 triggering downstream immunostimulatory signaling. In addition, TLR9 activation facilitates the release of an excess of unbound CpG-STAT3dODNs from endosomes into cytoplasm. The CpG-STAT3dODN binds to the dimers of STAT3 activated by upstream growth factor/cytokine signaling. The sequestration of the decoy-bound STAT3 in the cytoplasm prevents its transcriptional activity, shifting the balance from immunosuppression to the production of pro-inflammatory cytokines/chemokines and towards the processing of tumor-associated antigens (TAA). Altogether, these effects generate systemic CD8+ T cell-mediated immune responses against specific tumor antigens.
CpG-STAT3 inhibitors: targeting solid tumor-associated myeloid cells
In parallel to studies on the intracellular mode of action, we tested the feasibility of using CpG-STAT3 inhibitors against a variety of human and mouse tumors.[12, 40, 46] CpG-STAT3siRNA conjugates were injected locally or systemically into melanoma, colon, glioma, bladder and prostate tumors to demonstrate internalization and efficient STAT3 silencing in tumor-associated dendritic cells, macrophages and B cells. These studies provided the first evidence that targeted STAT3 inhibition combined with TLR9 triggering by CpG-STAT3siRNA oligonucleotides can break immune tolerance and induce potent T-cell mediated anti-tumor immunity (Figure 2).[40, 46] The immune activation following STAT3-inhibition/TLR9-activation was two-step: starting with rapid tumor infiltration by innate immune cells (neutrophils, macrophages and in some tumor models NK cells) and leading later to the development of adaptive immune responses, as manifested by CD8+ T cell infiltration into tumors and tumor-draining lymph nodes. At the same time, CpG-STAT3 inhibitors would reduce tolerogenic populations of MDSCs and regulatory T cells in both locations. These effects were associated with elevated CD8+ T cell responses against specific tumor antigens, and with protection from rechallenge with the same cancer type. Successful generation of Th1-type antitumor immunity resulted from improved functions of APCs, such as DCs in tumor-draining lymph nodes, after CpG-STAT3siRNA treatment.[40, 46] At the same time, the temporary inhibition of STAT3, usually limited to 2–4 weeks, did not affect generation of DCs which is a known effect of Stat3 ablation.[13, 40] In fact, local intratumoral injections of CpG-STAT3siRNA drastically improved effector functions of adoptively transferred CD8+ T cells, increasing their killing activity and tumor infiltration. Improved tumor antigen-presentation also contributed to the synergistic effect of CpG-STAT3siRNA combined with localized tumor RT.[12] However, in this case STAT3 inhibition interfered mainly with the proangiogenic activity of tumor-infiltrating macrophages, thereby disrupting initial step of the tumor revascularization and recurrence. Altogether, these preclinical studies on CpG-STAT3siRNA validated the concept of bi-functional immunotherapeutic strategy, based on the proof-of-principle experiments in the genetic model of Stat3-deletion discussed earlier.[18] Jump-starting potent and multilayered antitumor immunity depended on a combination of TLR9 triggering (“push”) and STAT3 inhibition (“release”) within the same target antigen-presenting cell, best accomplished by incorporating both functions into a single therapeutic agent.
Figure 2. Two-pronged therapeutic effect of CpG-STAT3 inhibitors against TLR9-positive human cancers.
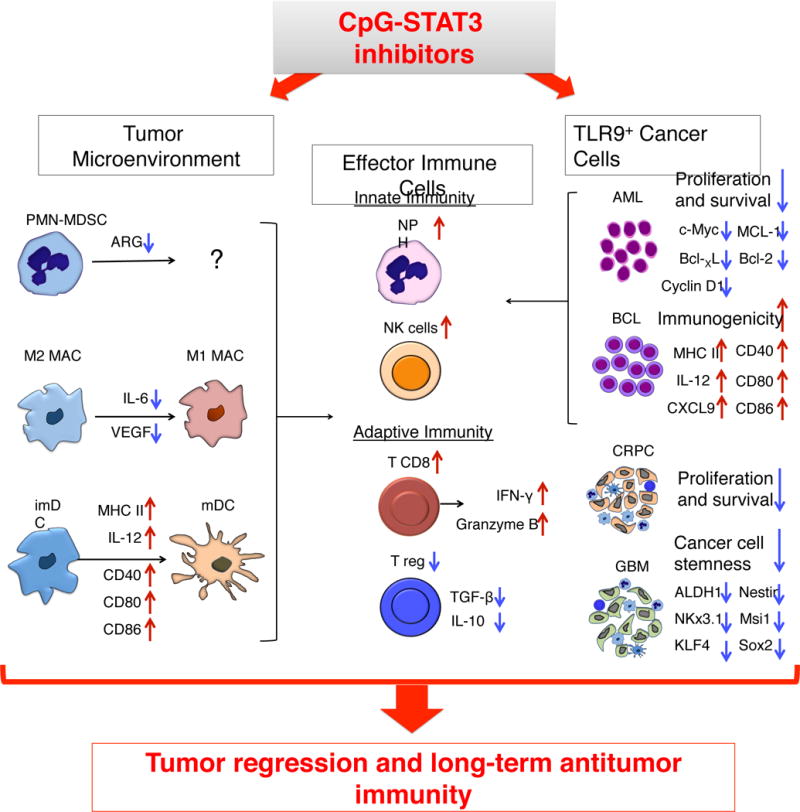
TLR9-targeted delivery of STAT3 inhibitors allows for targeting both TLR9-positive tumor-associated myeloid cells (imDC = immature DC; MAC = macrophages; PMN-MDSC = polymorphonuclear myeloid-derived suppressor cells) as well as certain human cancers, such as castration-resistant prostate cancer (CRPC) and glioma (GBM). TLR9-activation/STAT3-inhibition in the tumor microenvironment reduces production of immunosuppressive mediators by myeloid cells while stimulating DC and MAC maturation and promoting presentation of tumor-specific antigens and release proinflammatory mediators. While CpG-STAT3 inhibitors alleviate immunosuppressive functions of PMN-MDSCs, it is yet unclear whether these effects are accompanied by loss of viability and/or differentiation of MDSCs. At the same time, CpG-STAT3 inhibitors can increase immunogenicity of cancer cells in AML and B cell lymphoma, while decreasing survival and tumorigenic potential of cancer stem-like cells in CRPC and GBM. The combination of breaking immune suppression in the tumor microenvironment and decreasing cancer cell survival is likely to augment the overall therapeutic efficacy against human TLR9-positive cancers.
The strong efficacy in mouse tumor models in vivo prompted an optimization of the CpG-STAT3siRNA strategy for human system. For broader range of targeted human immune cells, we decided on two types of CpG ODN conjugates based on A and B types of CpG ODNs: CpG(A) – known for preferentially stimulating pDCs, and CpG(B) – mainly activating B cells.[23] As expected, these specificities were retained by CpG(A)-conjugates, which effectively target human TLR9+ pDCs, mDCs and myeloid leukemia cells, and by CpG(B)-conjugates, which show enhanced uptake by human B cells and B cell lymphoma cells.[36] As mentioned earlier, inflammation and genotoxic stress cause upregulation of TLR9 in additional types of myeloid cells or in cancer cells at least partly through p53-dependent mechanism.[34] The heterogeneous population of MDSCs plays a pivotal role in cancer progression and poor patients’ survival.[47] The MDSCs are immature myeloid cells deprived of the majority of immune cell surface markers, which makes them elusive targets for antibody-based therapies.[48] Recently, we identified for the first time that TLR9 is expressed specifically by the subset of polymorphonuclear MDSCs (PMN-MDSC: Lin−HLA-DR−CD14−CD15HICD33LO) accumulating in the circulation of prostate cancer patients with progression of the disease.[35] We found a gradual increase of the percentage of TLR9+ PMN-MDSCs, but not of TLR9− M-MDSCs, between healthy subjects, patients with localized or metastatic prostate cancers. Similar as reported in tumor-associated myeloid cells in mice,[10, 11] STAT3 activity was elevated in human PMN-MDSCs and correlated with their potent immunosuppressive effects on T cells. Importantly, ex vivo CpG(A)-STAT3siRNA effectively silenced STAT3 in primary PMN-MDSCs. CpG(A)-STAT3siRNA also reduced expression of the arginase-1 (ARG1), a known downstream target of STAT3,[49] which is and a potent immunosuppressive factor secreted by PMN-MDSCs to blunt T-cell responses. Overall, CpG(A)-STAT3siRNA alleviated tolerogenic effects of PMN-MDSCs on T-cell proliferation and effector functions, such as IFNγ and granzyme B production. Ongoing studies should determine whether loss of PMN-MDSC activity after TLR9-triggering/STAT3-inhibition is accompanied by their differentiation and whether downstream TLR9 signaling contributes in any way to tolerogenic functions. The population of PMN-MDSCs is commonly elevated in peripheral blood from patients with many other human cancers, including hematologic malignancies, head and neck, glioma, lung or gastrointestinal cancers.[50, 51] Importantly, TLR9 expression does not seem to be a unique property of prostate cancer-associated PMN-MDSCs and it is observed also in PMN-MDSCs from head and neck cancer patients as verified in our laboratory.(Kortylewski, unpublished data) Therefore, TLR9-targeted delivery can provide a platform technology for immunotherapeutic application in a relatively broad spectrum of human cancers. As shown recently in several solid tumors, such as castration-resistant prostate cancers or in malignant glioma, cancer stem-like cells can acquire ability to express innate immune receptors such as TLR9.[37, 38] While beyond scope of this article, in such setting CpG-STAT3 inhibitors can exert a two-pronged therapeutic effect against the tumor and its microenvironment (Figure 2). TLR9-targeted STAT3 inhibition would not only eliminate tumor immune evasion but also reduce the tumorigenic potential of cancer cells and therapeutic resistance.
CpG-STAT3 inhibitors: triggering immunogenicity of cancer cells in hematological malignancies
Extensive prior studies documented the immunostimulatory effect of STAT3 inhibition on tumor-associated myeloid cells, B cells and also malignant B cell lymphoma cells.[10, 18, 40, 52] Since both myeloid leukemia and B cell lymphoma cells commonly express high levels of TLR9 together with activated STAT3, they are ideal targets for CpG-STAT3 inhibitors. Our proof-of-principle studies in xenotransplanted and syngeneic models of acute myeloid leukemia (AML) compared the antitumor efficacy of CpG-STAT3siRNA and CpG-STAT3dODN.[36, 42, 43] As expected, the improved nuclease-resistance of CpG-STAT3dODN proved indispensable for induction of the direct cytotoxicity in disseminated STAT3-dependent human AML. However, both types of CpG-STAT3 inhibitors caused regression of the syngeneic Cbfb/Myh11/Mpl (CMM) model of AML, which does not rely on STAT3 signaling for survival. CpG-STAT3 inhibitors, but not CpG ODN alone or Jak/STAT inhibitors alone, induced potent antitumor responses against established CMM leukemia in immunocompetent mice. Intriguingly, while antitumor effects of CpG-STAT3 inhibitors were primarily immune-mediated, they did not depend on activity of antigen-presenting cells of the host. In TLR9-deficient mice, lacking responsiveness to TLR9 conjugates, CpG-STAT3 inhibitors showed unaltered and potent antitumor effects. Thus, the augmented immunogenicity of CMM cells alone has potential to trigger potent antitumor immunity after CpG-STAT3 inhibitor treatment. Similar to the effect of STAT3-inhibition/TLR9-activation on non-malignant DCs or macrophages, the CMM leukemic cells upregulated surface expression of MHC class II, costimulatory molecules and essential mediators of Th1 type immunity (IFNγ and IL-12), while concomitantly reducing Th2 mediators (IL-4 and IL-6) and arginase expression. In fact, CpG-STAT3 inhibitors induced dramatic changes in cellular morphology of AML cells indicative of differentiation, including asymmetric localization of nuclei and increased number of mitochondria (Kortylewski, unpublished data). Importantly, targeted STAT3-blocking/TLR9-triggering resulted in systemic eradication of leukemia and long-term survival of majority of mice due to successful elimination of quiescent leukemia-initiating cells. We attribute this broad therapeutic effect to the ubiquitous expression of TLR9 in all cellular compartments of human and mouse AML independently from their cytogenetic subtypes and hierarchy, including leukemia-progenitor/-stem cells.[42] Similar to acute myeloid leukemia, B cell lymphoma cells are also known to gain immunogenic phenotype as a result of CpG stimulation or STAT3 inhibition.[52, 53] In fact, our studies provide evidence that systemic administration of CpG-STAT3 inhibitors induces regression of disseminated mouse A20 B cell lymphoma through immune-mediated and not directly cytotoxic effects similar as reported in AML models.[36] These results underscore well-established opposing roles of STAT3 and TLR9/NF-κB signaling in antigen-presentation, cytokine production and cell differentiation. However, there is still much to learn about the pool of tumor-specific epitopes revealed and/or expanded as a result of STAT3-inhibition and TLR9-triggering. This information can have important implications for the design of more effective and safer T cell-based immunotherapies to AML, NHL and potentially other hematologic malignancies.
Conclusions and perspectives
Targeting nodal points of cell signaling networks in the tumor microenvironment, such as TFs,[54] provides an opportunity to maximize efficacy and potency of therapeutic intervention. At the same time, it faces serious challenges related to technical difficulty, specificity and safety, especially when targeting immune cell signaling. Our studies focused on targeting STAT3 underscore the need for specificity at both molecular and cellular level to ensure maximally effective yet safe therapeutic responses. CpG-STAT3 inhibitors represent a new-in-class, cell-selective and dual-function oligonucleotide-based approach to cancer immunotherapy. Their efficiency is a consequence of the intracellular gain-of-function (GOF): the release of the central immune checkpoint regulator (STAT3) to unleash proinflammatory signaling (CpG/TLR9) and antigen-presentation in target cells. The combination of TLR9-triggering/STAT3-inhibition is sufficient to override tolerogenic phenotype of tumor-associated myeloid cells, such as PMN-MDSC or TAMs, and certain malignant myeloid and lymphoid cells. CpG-STAT3 inhibitors have potential to stimulate the presentation/recognition of a broad repertoire of cancer-specific antigens in vivo and provide an opportunity for the design of new combinatorial immunotherapeutic regimens with standard treatments, vaccines or T cell-based therapies. The simplicity and flexibility of the CpG-conjugate design allows for their further adaptation to targeting other tumorigenic and/or immunosuppressive TFs beyond STAT3, such as STAT5 and NF-κB,[37, 54] or even oncogenic micro RNAs as shown recently.[55] Therefore, TLR9-targeted delivery of ONTs can overcome current limitations of small molecule drugs and pave way to novel, cell-selective cancer immunotherapies.
Acknowledgments
This work was supported in part by the National Cancer Institute/National Institutes of Health award number R01CA155367, P50CA107399, P30CA033572 (COH), the Department of Defense grant W81XWH-16-1-0499 and the STOP-CANCER Allison-Tovo-Dwyer Memorial Career-Development Award. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The authors are grateful to Dr. Peter Heinrich for critical reading of the manuscript and Dr. Sumanta Pal for thoughtful comments and suggestions.
Abbreviations
- AML
Acute myeloid leukemia
- ASO
Antisense oligonucleotide
- CD
Cluster of differentiation
- CMM
Cbfb/Myh11/Mpl
- CTLA4
Cytotoxic T-lymphocyte associated protein 4
- DC
Dendritic cell
- DLBCL
Diffuse large B cell lymphoma
- dODN
Decoy oligodeoxynucleotide
- Jak
Janus kinase
- MDSC
Myeloid-derived suppressor cell
- MTD
Maximum tolerated dose
- NHL
Non-Hodgkin lymphoma
- ODN
Oligodeoxynucleotide
- ONT
Oligonucleotide therapeutic
- PD-1
Programmed cell death protein 1
- PMN
Polymorphonuclear
- miRNA
Micro RNA
- NF-κB
Nuclear factor κB
- RAGE
Receptor for advanced glycation endproducts
- RT
Radiation therapy
- siRNA
Short interfering RNA
- STAT3
Signal transducer and activator of transcription 3
- TAM
Tumor-associated macrophage
- TF
Transcription factor
- TLR
Toll-like receptor
Footnotes
Conflict of interest
The authors declare that they have no conflict of interests.
References
- 1.Topalian SL, Weiner GJ, Pardoll DM. Cancer immunotherapy comes of age. J Clin Oncol. 2011;29:4828–4836. doi: 10.1200/JCO.2011.38.0899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Turley SJ, Cremasco V, Astarita JL. Immunological hallmarks of stromal cells in the tumour microenvironment. Nat Rev Immunol. 2015;15:669–682. doi: 10.1038/nri3902. [DOI] [PubMed] [Google Scholar]
- 3.Gajewski TF. The Next Hurdle in Cancer Immunotherapy: Overcoming the Non-T-Cell-Inflamed Tumor Microenvironment. Semin Oncol. 2015;42:663–671. doi: 10.1053/j.seminoncol.2015.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kammertoens T, Schüler T, Blankenstein T. Immunotherapy: target the stroma to hit the tumor. Trends Mol Med. 2005;11:225–231. doi: 10.1016/j.molmed.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 5.Fang H, Declerck YA. Targeting the tumor microenvironment: from understanding pathways to effective clinical trials. Cancer Res. 2013;73:4965–4977. doi: 10.1158/0008-5472.CAN-13-0661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12:253–268. doi: 10.1038/nri3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Engblom C, Pfirschke C, Pittet MJ. The role of myeloid cells in cancer therapies. Nat Rev Cancer. 2016;16:447–462. doi: 10.1038/nrc.2016.54. [DOI] [PubMed] [Google Scholar]
- 8.Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol. 2007;7:41–51. doi: 10.1038/nri1995. [DOI] [PubMed] [Google Scholar]
- 9.Wang T, Niu G, Kortylewski M, et al. Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells. Nat Med. 2004;10:48–54. doi: 10.1038/nm976. [DOI] [PubMed] [Google Scholar]
- 10.Kortylewski M, Kujawski M, Wang T, et al. Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat Med. 2005;11:1314–1321. doi: 10.1038/nm1325. [DOI] [PubMed] [Google Scholar]
- 11.Kujawski M, Kortylewski M, Lee H, et al. Stat3 mediates myeloid cell-dependent tumor angiogenesis in mice. J Clin Invest. 2008;118:3367–3377. doi: 10.1172/JCI35213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gao C, Kozlowska A, Nechaev S, et al. TLR9 signaling in the tumor microenvironment initiates cancer recurrence after radiotherapy. Cancer Res. 2013;73:7211–7221. doi: 10.1158/0008-5472.CAN-13-1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hillmer EJ, Zhang H, Li HS, Watowich SS. STAT3 signaling in immunity. Cytokine Growth Factor Rev. 2016 doi: 10.1016/j.cytogfr.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kortylewski M, Xin H, Kujawski M, et al. Regulation of the IL-23 and IL-12 balance by Stat3 signaling in the tumor microenvironment. Cancer Cell. 2009;15:114–123. doi: 10.1016/j.ccr.2008.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haddad E. STAT3: too much may be worse than not enough! Blood. 2015;125:583–584. doi: 10.1182/blood-2014-11-610592. [DOI] [PubMed] [Google Scholar]
- 16.Cui W, Liu Y, Weinstein JS, et al. An interleukin-21-interleukin-10-STAT3 pathway is critical for functional maturation of memory CD8+ T cells. Immunity. 2011;35:792–805. doi: 10.1016/j.immuni.2011.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Siegel AM, Heimall J, Freeman AF, et al. A critical role for STAT3 transcription factor signaling in the development and maintenance of human T cell memory. Immunity. 2011;35:806–818. doi: 10.1016/j.immuni.2011.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kortylewski M, Kujawski M, Herrmann A, et al. Toll-like receptor 9 activation of signal transducer and activator of transcription 3 constrains its agonist-based immunotherapy. Cancer Res. 2009;69:2497–2505. doi: 10.1158/0008-5472.CAN-08-3031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sen M, Grandis JR. Nucleic acid-based approaches to STAT inhibition. JAKSTAT. 2012;1:285–291. doi: 10.4161/jkst.22312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kontzias A, Kotlyar A, Laurence A, et al. Jakinibs: a new class of kinase inhibitors in cancer and autoimmune disease. Curr Opin Pharmacol. 2012;12:464–470. doi: 10.1016/j.coph.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ratner M. Setback for JAK2 inhibitors. Nat Biotechnol. 2014;32:119–119. doi: 10.1038/nbt0214-119a. [DOI] [PubMed] [Google Scholar]
- 22.Kortylewski M, Nechaev S. Cancer therapy using oligonucleotide-based STAT3 inhibitors: will they deliver? Ther Deliv. 2014;5:239–242. doi: 10.4155/tde.13.152. [DOI] [PubMed] [Google Scholar]
- 23.Krieg AM. CpG still rocks! Update on an accidental drug. Nucleic Acid Ther. 2012;22:77–89. doi: 10.1089/nat.2012.0340. [DOI] [PubMed] [Google Scholar]
- 24.Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5:987–995. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 25.Canton J, Neculai D, Grinstein S. Scavenger receptors in homeostasis and immunity. Nat Rev Immunol. 2013;13:621–634. doi: 10.1038/nri3515. [DOI] [PubMed] [Google Scholar]
- 26.Gursel M, Gursel I, Mostowski HS, Klinman DM. CXCL16 influences the nature and specificity of CpG-induced immune activation. J Immunol. 2006;177:1575–1580. doi: 10.4049/jimmunol.177.3.1575. [DOI] [PubMed] [Google Scholar]
- 27.Zhu P, Liu X, Treml LS, et al. Mechanism and regulatory function of CpG signaling via scavenger receptor B1 in primary B cells. J Biol Chem. 2009;284:22878–22887. doi: 10.1074/jbc.M109.018580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nechaev S, Gao C, Moreira D, et al. Intracellular processing of immunostimulatory CpG-siRNA: Toll-like receptor 9 facilitates siRNA dicing and endosomal escape. J Control Release. 2013;170:307–315. doi: 10.1016/j.jconrel.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Józefowski S, Sulahian TH, Arredouani M, Kobzik L. Role of scavenger receptor MARCO in macrophage responses to CpG oligodeoxynucleotides. J Leukoc Biol. 2006;80:870–879. doi: 10.1189/jlb.0705357. [DOI] [PubMed] [Google Scholar]
- 30.Baumann CL, Aspalter IM, Sharif O, et al. CD14 is a coreceptor of Toll-like receptors 7 and 9. J Exp Med. 2010;207:2689–2701. doi: 10.1084/jem.20101111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lahoud MH, Ahmet F, Zhang J-G, et al. DEC-205 is a cell surface receptor for CpG oligonucleotides. Proc Natl Acad Sci U S A. 2012;109:16270–16275. doi: 10.1073/pnas.1208796109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sirois CM, Jin T, Miller AL, et al. RAGE is a nucleic acid receptor that promotes inflammatory responses to DNA. J Exp Med. 2013;210:2447–2463. doi: 10.1084/jem.20120201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McKelvey KJ, Highton J, Hessian PA. Cell-specific expression of TLR9 isoforms in inflammation. J Autoimmun. 2011;36:76–86. doi: 10.1016/j.jaut.2010.11.001. [DOI] [PubMed] [Google Scholar]
- 34.Shatz M, Menendez D, Resnick MA. The human TLR innate immune gene family is differentially influenced by DNA stress and p53 status in cancer cells. Cancer Res. 2012;72:3948–3957. doi: 10.1158/0008-5472.CAN-11-4134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hossain DMS, Pal SK, Moreira D, et al. TLR9-Targeted STAT3 Silencing Abrogates Immunosuppressive Activity of Myeloid-Derived Suppressor Cells from Prostate Cancer Patients. Clin Cancer Res. 2015;21:3771–3782. doi: 10.1158/1078-0432.CCR-14-3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang Q, Hossain DMS, Nechaev S, et al. TLR9-mediated siRNA delivery for targeting of normal and malignant human hematopoietic cells in vivo. Blood. 2013;121:1304–1315. doi: 10.1182/blood-2012-07-442590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moreira D, Zhang Q, Hossain DMS, et al. TLR9 signaling through NF-κB/RELA and STAT3 promotes tumor-propagating potential of prostate cancer cells. Oncotarget. 2015;6:17302–17313. doi: 10.18632/oncotarget.4029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Herrmann A, Cherryholmes G, Schroeder A, et al. TLR9 is critical for glioma stem cell maintenance and targeting. Cancer Res. 2014;74:5218–5228. doi: 10.1158/0008-5472.CAN-14-1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rajora MA, Zheng G. Targeting SR-BI for Cancer Diagnostics, Imaging and Therapy. Frontiers in pharmacology. 2016;7:326. doi: 10.3389/fphar.2016.00326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kortylewski M, Swiderski P, Herrmann A, et al. In vivo delivery of siRNA to immune cells by conjugation to a TLR9 agonist enhances antitumor immune responses. Nat Biotechnol. 2009;27:925–932. doi: 10.1038/nbt.1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hossain DMS, Moreira D, Zhang Q, et al. TLR9-Targeted SiRNA Delivery In Vivo. Methods Mol Biol. 2016;1364:183–196. doi: 10.1007/978-1-4939-3112-5_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang Q, Hossain DMS, Duttagupta P, et al. Serum-resistant CpG-STAT3 decoy for targeting survival and immune checkpoint signaling in acute myeloid leukemia. Blood. 2016;127:1687–1700. doi: 10.1182/blood-2015-08-665604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hossain DMS, Santos C Dos, Zhang Q, et al. Leukemia cell-targeted STAT3 silencing and TLR9 triggering generate systemic antitumor immunity. Blood. 2014;123:15–25. doi: 10.1182/blood-2013-07-517987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ma Y, Kowolik CM, Swiderski PM, et al. Humanized Lewis-Y specific antibody based delivery of STAT3 siRNA. ACS Chem Biol. 2011;6:962–970. doi: 10.1021/cb200176v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nakamura N, Lill JR, Phung Q, et al. Endosomes are specialized platforms for bacterial sensing and NOD2 signalling. Nature. 2014;509:240–244. doi: 10.1038/nature13133. [DOI] [PubMed] [Google Scholar]
- 46.Herrmann A, Kortylewski M, Kujawski M, et al. Targeting Stat3 in the myeloid compartment drastically improves the in vivo antitumor functions of adoptively transferred T cells. Cancer Res. 2010;70:7455–7464. doi: 10.1158/0008-5472.CAN-10-0736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marvel D, Gabrilovich DI. Myeloid-derived suppressor cells in the tumor microenvironment: expect the unexpected. J Clin Invest. 2015;125:3356–3364. doi: 10.1172/JCI80005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bronte V, Brandau S, Chen S-H, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun. 2016;7:12150. doi: 10.1038/ncomms12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vasquez-Dunddel D, Pan F, Zeng Q, et al. STAT3 regulates arginase-I in myeloid-derived suppressor cells from cancer patients. J Clin Invest. 2013;123:1580–1589. doi: 10.1172/JCI60083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.De Veirman K, Van Valckenborgh E, Lahmar Q, et al. Myeloid-derived suppressor cells as therapeutic target in hematological malignancies. Front Oncol. 2014;4:349. doi: 10.3389/fonc.2014.00349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016;37:208–220. doi: 10.1016/j.it.2016.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cheng F, Wang H, Horna P, et al. Stat3 inhibition augments the immunogenicity of B-cell lymphoma cells, leading to effective antitumor immunity. Cancer Res. 2012;72:4440–4448. doi: 10.1158/0008-5472.CAN-11-3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Decker T, Schneller F, Sparwasser T, et al. Immunostimulatory CpG-oligonucleotides cause proliferation, cytokine production, and an immunogenic phenotype in chronic lymphocytic leukemia B cells. Blood. 2000;95:999–1006. [PubMed] [Google Scholar]
- 54.Darnell JE. Transcription factors as targets for cancer therapy. Nat Rev Cancer. 2002;2:740–749. doi: 10.1038/nrc906. [DOI] [PubMed] [Google Scholar]
- 55.Zhang B, Li L, Chen C, et al. Knockdown (KD) of Mir-126 Expression Enhances Tyrosine Kinase Inhibitor (TKI)-Mediated Targeting of Chronic Myelogenous Leukemia (CML) Stem Cells. 57th Annual Meeting of American Society of Hematology, oral presentation, Orlando, FL. Blood. 2015;126:51. [Google Scholar]