

Abstract
Aggregation of amyloid-β (Aβ) peptide in the brain in the form of neuritic plaques and cerebral amyloid angiopathy (CAA) is a key feature of Alzheimer’s disease (AD). Microglial cells surround aggregated Aβ and are believed to play a role in AD pathogenesis. A therapy for AD that has entered clinical trials is the administration of anti-Aβ antibodies. One mechanism by which certain anti-Aβ antibodies have been proposed to exert their effects is via antibody-mediated microglial activation. Whether, when, or to what extent microglial activation occurs following systemic administration of anti-Aβ antibodies has not been fully assessed. We administered an anti-Aβ antibody (m3D6) which binds to aggregated Aβ to PDAPP mice, an AD mouse model that was bred to contain fluorescent microglia. Three days following systemic administration of m3D6, there was a marked increase in both the number of microglial cells and processes per cell visualized in vivo by multiphoton microscopy. These changes required the Fc domain of m3D6 and were not observed with an antibody specific to soluble Aβ. These findings demonstrate that some effects of antibodies that recognize aggregated Aβ are rapid, involve microglia, and provide insight into the mechanism of action of a specific passive immunotherapy for AD.
Keywords: microglia, beta-amyloid, passive immunization, Alzheimer’s disease, neuritic plaques, cerebral amyloid angiopathy
Introduction
Alzheimer’s disease (AD) is characterized by the presence of two pathological hallmarks, amyloid plaques and neurofibrillary tangles. Plaques consist primarily of extracellular deposits of amyloid-β (Aβ) in the brain parenchyma and in arterioles in the form of cerebral amyloid angiopathy (CAA) (Mandybur, 1975; Glenner et al., 1981; Vinters, 1987); tangles are composed primarily of aggregated, hyperphosphorylated forms of tau (Brion et al., 1985; Selkoe, 2001).
Another important feature of AD pathology are the inflammatory changes that occur, particularly involving microglia. In the AD brain as well as in AD mouse models, microglia cluster around plaques and CAA. It was recently demonstrated that microglial cells move towards newly formed plaques within 24 hours of plaque formation (Meyer-Luehmann et al., 2008) as well as towards existing plaques over the course of 24 hours (Bolmont et al., 2008). Plaque-associated microglia display an activated phenotype and are associated with an enhanced expression of immune cell surface markers and the production of pro-inflammatory cytokines and chemokines (Akiyama et al., 2000).
A promising therapy for AD that has entered human clinical trials is the peripheral administration of anti-Aβ antibodies or passive immunization (Brody and Holtzman, 2008). Peripheral administration of certain anti-Aβ antibodies has been shown to have potentially beneficial effects such as plaque clearance and cognitive improvement as well as toxic effects such as CAA-associated hemorrhage in animal models (Bard et al., 2000; Wilcock et al., 2004b; Wilcock et al., 2004a; Wilcock et al., 2006; Vasilevko et al., 2007). One mechanism by which certain anti-Aβ antibodies have been hypothesized to exert their beneficial as well as their toxic effects is via a small amount of the peripherally administered antibodies crossing the blood-brain-barrier and binding to aggregated Aβ, leading to antibody Fc domain-mediated microglial activation and Aβ phagocytosis (Bard et al., 2000; Wilcock et al., 2004b). Several studies have assessed the effects of anti-Aβ antibodies administered directly into the CNS over days on microglial activation and Aβ clearance (Bacskai et al., 2001; Bacskai et al., 2002; Pfeifer et al., 2002; Wilcock et al., 2003; Wilcock et al., 2004b; Wilcock et al., 2004a; Racke et al., 2005; Wilcock et al., 2006; Burbach et al., 2007; Garcia-Alloza et al., 2007). These studies suggest that 1) antibodies to aggregated forms of Aβ can clear parenchymal plaques by both Fc receptor dependent and independent mechanisms, 2) a marked increase in the number of microglia is observed with antibodies that bind aggregated Aβ with an intact Fc domain, 3) CAA is very difficult to clear, and 4) neuritic dystrophy can rapidly resolve. However, anti-Aβ antibodies being administered to humans are being delivered outside the blood-brain-barrier. Whether, when, or to what extent microglial activation in the CNS occurs following systemic administration of anti-Aβ antibodies, especially soon after administration, has not been assessed. Herein, we examined the effects of peripherally administered anti-Aβ antibodies in an AD mouse model that contains amyloid plaques and fluorescent microglia. We assessed baseline microglial behavior and whether the antibodies rapidly influenced microglial morphology in the brain and the properties of the antibodies required for the effects observed.
Methods
Animals
PDAPP+/−;CX3CR1/GFP+/− double transgenic mice were generated by crossing PDAPP+/+ (Games et al., 1995) with CX3CR1/GFP+/+ (Jung et al., 2000) mice. Double-transgenic mice used for these experiments were 4 months, 14 months, 18 months, or 22 months of age, as denoted for each experiment. The Institutional Review Board at Washington University approved all of the animal procedures used in this study.
Antibodies
m3D6 was a kind gift from Eli Lilly and Company (Indianapolis, IN). IgG2b was purchased from Zymed Laboratories/Invitrogen (Carlasbad, CA). Antibody mHJ5.1 was produced by using Aβ peptide fragment 17–28. This peptide was then used as an immunogen for production of a monoclonal antibody according to protocols established by the Washington University Hybridoma Center (http://pathology.wustl.edu/research/hybridoma.php). Fab fragments were generated from m3D6 using a Pierce kit. Briefly, purified IgG was dialyzed against a buffer containing 20mM sodium phosphate and 10mM EDTA, pH 7.0. It was then concentrated using a Pierce Slide-A-Lyzer dialysis cassette and Pierce Slide-A-Lyzer Concentrating Solution to ~20mg/ml. Immobilized papain was washed twice in a glass test tube with a digestion buffer of 42mg cysteine·HCL in 12ml Phosphate Buffer (pH 10) prior to resuspension in 0.5ml of digestion buffer and addition of concentrated IgG. The mixture was shaken for 45 minutes at 37°C. The digest was separated from the immobilized papain and added to Protein A sepharose and rotated for 45 minutes at room temperature. The digest was removed and contained the Fab portion of the IgG. An elution buffer was added to the Protein A sepharose to remove any undigested IgG and bound Fc. All samples were concentrated and run on a 4–12% Bis-tris gel (Invitrogen) for confirmation by size. For immunohistochemical analysis, rabbit-anti-Iba-1 was from Wako (Richmond, VA), goat-anti-rabbit-Cy3 was from Jackson Immunoresarch (West Grover, PA), anti-CD45 was from Serotec (Raleigh, North Carolina), and the goat-anti-rat was from Vector Laboratories (Burlingame, California).
Antibody injection
Three days prior to cranial window surgery and imaging, mice were injected intrperitoneally with 500 μg of m3D6, mHJ5.1, IgG2b, or equimolar amounts of m3D6 Fab fragments.
Surgical procedure
The cranial window procedure was performed as described before (D’Amore et al., 2003; Brendza et al., 2005). Briefly, the mice were anesthetized with avertin prior to being placed in a custom made stereotaxic device. After removing the hair and cleaning the skin on top of the head with betadine and 70% isopropanol, the skin was cut away from just posterior of the eyes to the base of the skull. A hole was carved in the skull, approximately 7.5 mm in diameter, slightly anterior to bregma and slightly anterior to lambda. The carved portion of the skull was then removed to expose a fraction of the brain. The exposed brain region was then irrigated with PBS and packed with gel foam. Gel foam was removed and PBS was then added prior to the placement of an 8 mm glass cover slip. The glass cover slip was held into place with dental cement and superglue. A ring of paraffin was placed over the dental cement mixture in order to create a wall to hold immersion fluid for imaging.
In vivo brain imaging using 2-photon microscopy
Approximately 18 hours prior to imaging, mice were injected intraperitoneally with 10 mg/kg methoxy-XO4, which binds and labels amyloid in the brain. In cases where angiograms were used to label vessels, the mice were injected with Texas-red dextran in a lateral tail vein minutes before imaging. Anesthetized mice with cranial windows were placed on the stage of a 2-photon microscope (Zeiss LSM 510 Meta NLO system with a Coherent Chameleon Ti:Sa laser) while in a custom made stereotaxic device. Mice were imaged immediately after placement of the cranial windows. For simultaneous imaging of methoxy-X04, GFP, and Texas red an excitation wavelength of 800nm was used. Fluorescence emission from amyloid bound methoxy-X04, GFP, and Texas Red is collected in the ranges of 435–485 nm, 500–550 nm, and 560 to 650 nm, respectively. Images were acquired as 512 × 512 arrays of 8 bit pixels in z-series stacks. The fields of view were approximately 230 μm × 230 μm. Near amyloid pathology refers to microglial cells that were analyzed within the 230 × 230 μm field of view that contained amyloid pathology. Not near amyloid pathology refers to microglial cells that were analyzed in fields of view that did not contain amyloid deposition within the 230 × 230 μm field of view. Microglial cells that were completely in the field of view were studied for analysis.
Data Analysis
Image stacks from each imaging session were processed and analyzed using both Zeiss and Image J software. Three-dimensional reconstruction of microglia from z-series stacks were made using maximum intensity projections. The number of processes extending from cell bodies as well as the number of cell bodies were counted in each field of view. All statistics were performed with Graphpad Prism (v5.0).
Histology
Coronal sections (50 μm) were cut on a freezing microtome. For immunohistochemistry, sections were blocked with 3% goat serum prior to overnight incubation with rabbit-anti-Iba-1 antibody. Sections were then washed 3 times and incubated with a goat-anti-rabbit-Cy3 antibody. For immunohistochemistry of CD45, sections were incubated with an anti-CD45 antibody and detected with a goat-anti-rat antibody.
Protein analysis
Three days prior to sacrifice PDAPP+/−;CX3CR1/GFP+/− mice were intraperitoneally injected with m3D6 (500 μg) or PBS. The cortices were removed, weighed and homogenized in 9x volume of 50mM Tris-HCl buffer with 2mM EDTA and protease inhibitors. The tissue homogenates were then sent to Rules Based Medicine (Austin, TX) and analyzed in a multiplex assay.
Results
Assessment of microglia in living PDAPP mice
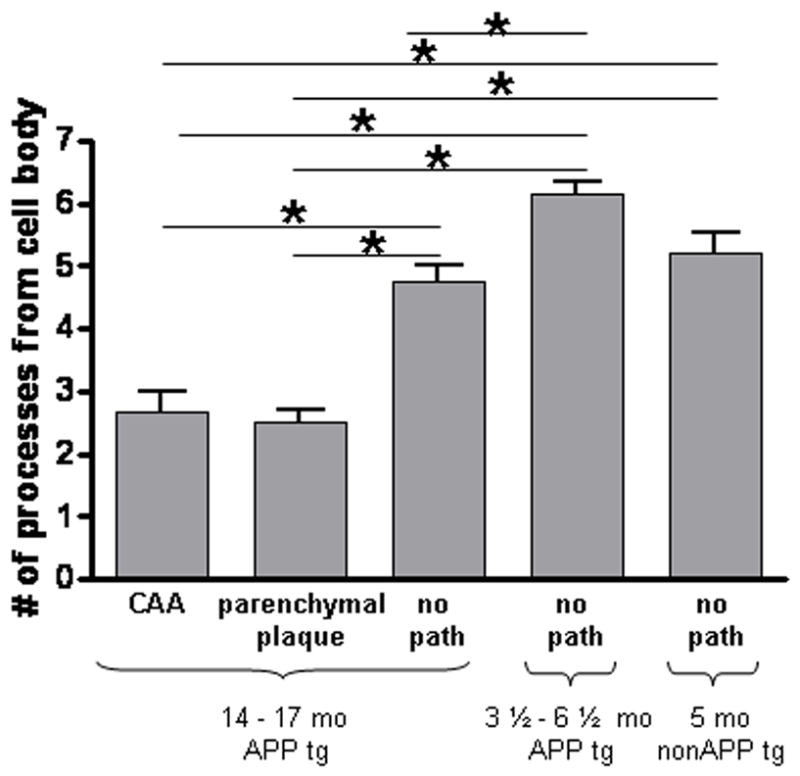
Recently, mice were generated in which green fluorescent protein (GFP) is inserted into the CX3CR1 gene (Jung et al., 2000). CX3CR1 is specifically expressed in microglia in the CNS which can be imaged in vivo (Nimmerjahn et al., 2005). We crossed PDAPP mice to CX3CR1 mice to generate mice that developed amyloid plaques in the presence of fluorescent microglia. Real time in vivo images were taken to assess the morphology and behavior of microglial cells in the brains of PDAPP+/−;CX3CR1/GFP+/− mice. These double transgenic animals begin to develop amyloid pathology around 9 months of age while their microglial cells express GFP from birth onward. Through cranial windows, we observed vessels labeled with dextran, amyloid deposition labeled with Methoxy-XO4, and GFP-positive microglial cells (Figure 1). Microglial cells in 14–17 month old animals were compared between areas in the vicinity of CAA or parenchymal plaques and in fields of view that did not contain amyloid deposition. These animals were also compared to young PDAPP animals, 3.5 months old, prior to the deposition of amyloid as well as 5 month old non-APP transgenic mice. The microglial cells in young APP-transgenic mice had smaller cell bodies than those observed in older APP-transgenic animals. Additionally, microglia in the young mice had longer processes than the microglia around plaques in the older mice. Microglial cells around amyloid pathology (within the 230 μm × 230 μm field of view) were noticeably different in appearance than those in areas of the brain distant from amyloid pathology (in a 230 μm × 230 μm field of view that did not contain pathology). These cells had fewer processes and larger cell bodies than cells distant from areas containing amyloid (Figure 2).
Figure 1.
Microglia have altered morphology and cluster around amyloid plaques in PDAPP+/−;CX3CR1/GFP+/− mice. 3-D reconstructed z-series stack images taken of cortical microglia in (A) PDAPP+/−;CX3CR1/GFP+/− mice at 3 ½ months of age (in the absence of plaques), in (B) 14 month old PDAPP+/−;CX3CR1/GFP+/− mice around amyloid plaques (blue), in (C) 14 month old PDAPP+/−;CX3CR1/GFP+/− mice in areas lacking plaques, and in (D) 5 month old PDAPP−/−;CX3CR1/GFP+/− mice. GFP labeled microglia are green. Vessels are labeled with Texas-Red dextran. Amyloid fluoresces blue after injection of Methoxy-X04. Scale bar is 20 μm.
Figure 2.

Decrease in the number of microglial processes around amyloid deposition. The number of processes extending from the cell body were counted in areas containing CAA, parenchymal amyloid plaques, and no amyloid pathology (no path). Microglia were compared in 14–17 month old mice around CAA, parenchymal amyloid plaques, and in areas with no amyloid pathology as well as in 3 ½ – 6 ½ month old PDAPP+/−;CX3CR1/GFP+/− mice lacking amyloid pathology and 5 month old PDAPP−/−;CX3CR1/GFP+/− mice. Five mice were studied per age group in APP transgenic mice and 6 fields of view were imaged in each animal. Six fields of view were studied in 2 non-APP transgenic mice. Data is presented as mean +/− SEM. * p<0.001.
Next, we assessed microglial dynamics around amyloid pathology. Images were taken of the same area 45–60 minutes apart. These images were compared to each other to detect changes in microglial process extensions and retractions that occurred over 45–60 minutes in the presence and absence of amyloid pathology and in mice of different ages. Microglial cells in young mice extended and retracted their processes significantly more than microglial cells in older mice (Figure 3). The microglial cells immediately around amyloid pathology in older mice as well as areas distant from pathology appeared to be relatively stable and exhibited significantly less process movement as compared to young mice.
Figure 3.

Decrease in microglial process movement in older PDAPP mice. The number of microglial process (A) extensions and (B) retractions were counted in young (3 ½ – 6 ½ month) and old (14–17 month) PDAPP+/−;CX3CR1/GFP+/− mice near and distant from amyloid pathology. Five mice were studied per age group and 6 fields of view were imaged in each animal. Data is presented as mean +/− SEM. *p<0.001.
Effects of antibody to aggregated Aβ on microglial cells
To determine the effects of a peripherally administered anti-Aβ antibody on microglial cells, we injected 500 μg of m3D6 intraperitoneally into 22 month old PDAPP+/−;CX3CR1/GFP+/− mice 3 days prior to imaging. m3D6 recognizes amino acids 1–5 of the Aβ peptide and can bind both aggregated and soluble Aβ (Bacskai et al., 2002; Cirrito et al., 2003). In vivo images were then taken of areas that contained CAA, parenchymal plaques, and areas with little to no amyloid pathology present immediately after placement of the cranial window. Microglial cells in m3D6-injected mice were compared to non-injected mice (Figure 4). The m3D6-injected animals contained significantly more detectable GFP-positive cells and these cells also had almost twice as many processes protruding from their cell bodies (Figure 4, Table 1, and Table 2). These changes were very notable around parenchymal plaques, CAA, and even in areas that did not contain methoxy-X04 staining in the cortex. The changes seen in the GFP-positive cells were also seen with the microglial marker Iba-1, in sections stained at the end of the imaging sessions (Figure 5). Cells in brain slices assessed after in vivo imaging stained with the microglial marker CD45 showed similar morphological changes seen with GFP and Iba-1 staining. However, with CD45, the colocalization was mostly with GFP-positive cells that were immediately around amyloid deposits (Figure 6). This suggests that CD45 staining is relatively localized to “activated” CX3CR1-positive cells. There were no significant differences in dynamic microglial process movements between m3D6-injected and non-injected animals (data not shown). We also compared the levels of several growth factors, cytokines, and other proteins in the cortices of m3D6 vs. vehicle-treated mice 3 days after injection and found that epidermal growth factor, haptoglobin, and MIP1α were modestly and significantly lower in m3D6 treated mice (data not shown). This suggests that specific biochemical changes are occurring in conjunction with the cellular morphological changes observed.
Figure 4.

Peripheral m3D6 administration results in marked morphological changes in microglia. 3-D reconstructed z-series stack images taken of 22 month old PDAPP+/−; CX3CR1/GFP+/− mice injected with 500 μg of m3D6 (A–C), an anti-Aβ antibody, or not injected (D–F). GFP labeled microglia are green. Fibrillar amyloid was labeled with Methoxy-X04 (blue). Scale bar is 20 μm.
Table 1.
Peripheral m3D6 injection increases the number of microglial processes per cell in 22 month old PDAPP+/−;CX3CR1+/− mice. The number of microglial processes were counted in 3-D reconstructed z-series stack images in 22 month old PDAPP+/−; CX3CR1/GFP+/− mice injected with 500 μg of m3D6 or untreated mice. All cortical area samples were quantified and the images were also divided into groups based on the presence of visualized pathology: in the vicinity of CAA, in the vicinity of parenchymal amyloid plaques, and in areas with little to no amyloid pathology. Four mice were studied per treatment group and 6–10 fields of view were imaged in each animal. Data is presented as mean +/− SEM.
| treatment | all cortical areas | CAA | plaques | little/no pathology |
|---|---|---|---|---|
| m3D6 | 4.5 +/− 0.09 | 4.4+/− 0.15 | 5.1 +/− 0.19 | 4.7+/− 0.18 |
| uninjected | 2.1 +/−0.06 | 1.9 +/− 0.10 | 2.4+/− 0.12 | 2.0+/− 0.13 |
Table 2.
Peripheral m3D6 injection increases the number of microglial cells in 22 month old PDAPP+/−;CX3CR1+/− mice. The number of microglial cells were counted in 3-D reconstructed z-series stack images in 22 month old PDAPP+/−;CX3CR1/GFP+/− mice injected with 500 μg of m3D6 or untreated mice. All cortical area samples were quantified and the images were also divided into groups based on the presence of visualized pathology: in the vicinity of CAA, in the vicinity of parenchymal amyloid plaques, and in areas with little to no amyloid pathology. Four mice were studied per treatment group and 6–10 fields of view were imaged in each animal. Data is presented as mean +/− SEM.
| treatment | all cortical areas | CAA | plaques | little/no pathology |
|---|---|---|---|---|
| m3D6 | 31.9+/− 1.87 | 30.2 +/− 2.54 | 36.3 +/− 6.42 | 30.2 +/− 3.04 |
| uninjected | 19.3 +/− 0.77 | 19.7 +/− 1.09 | 21.7+/−1.70 | 16.9 +/− 1.11 |
Figure 5.

Iba-1 staining colocalizes with GFP-labeled microglia and there is increased staining around plaques following m3D6 administration. Brain sections from 18 month old PDAPP+/−;CX3CR1/GFP+/− mice were stained with Iba-1 and images were taken at low power (A–H) and high power (I–P). Iba-1 positive cells are in red (A, E, I, M), GFP labels CX3CR1 positive microglia (B, F, J, N), and Methoxy-X04 labels aggregated amyloid in blue (C, G, K, O). Merged images are also shown (D, G, L, P). Scale bars are 100 μm and 50 μm for low and high power images, respectively.
Figure 6.

CD45 colocalizes with GFP-labeled microglia around amyloid pathology and there is increased staining around plaques following m3D6 administration. Brain sections from 18 month old PDAPP+/−;CX3CR1/GFP+/− mice were stained with CD45 and images were taken at low power (A–H) and high power (I–P). CD45 positive cells are in red (A, E, I, M), GFP labels CX3CR1 positive microglia (B, F, J, N), and Methoxy-X04 labels aggregated amyloid in blue (C, G, K, O). Merged images are also shown (D, G, L, P). Scale bars are 100 μm and 50 μm for low and high power images, respectively.
Microglial activation requires recognition of aggregated Aβ and the Fc domain
We also injected 4 month old mice, an age prior to amyloid deposition, with m3D6, to determine if the observed effects were due to recognition of aggregated vs. soluble forms of Aβ by the antibody. There was no significant difference between GFP-positive, microglial cell number or number of microglial processes when m3D6-injected and non-injected 4 month old mice were compared (Table 3) suggesting the effects of m3D6 are due to the antibody binding aggregated forms of Aβ. Furthermore, we tested other antibodies to see if they also affected microglial number or morphology. We injected 500 μg of m3D6 into 18 month old PDAPP+/−;CX3CR1/GFP+/− mice and compared the effects to mice injected with 500 μg mHJ5.1, IgG2b, or equimolar amounts of m3D6 Fab fragments 3 days after intraperitoneal administration. mHJ5.1 is an antibody that recognizes amino acids 17–24 of soluble Aβ, but does not bind aggregated Aβ in the form of plaques. IgG2b served as an isotype control to m3D6. As opposed to m3D6, treatment of mice with mHJ5.1 or IgG2b did not affect the number of GFP-positive microglia or alter their morphology (Figure 7 A, B). Fab fragments of m3D6 were also injected into 18 month old mice to determine if the full length m3D6 antibody was needed to elicit the change in microglial cell number and morphology. Injection of m3D6 Fab fragments had no effect on GFP-positive microglial cell number or morphology suggesting that the presence of the Fc domain was required to elicit the microglial changes observed in m3D6 treated mice (Figure 7 A, B).
Table 3.
Peripheral m3D6 injection does not alter the number of microglial cells or the number of microglial processes per cell in 4 month old PDAPP+/−;CX3CR1+/− mice. The number of microglial processes and the number of microglial cells were counted in 3-D reconstructed z-series stack images in 4 month old PDAPP+/−;CX3CR1/GFP+/− mice injected with 500 μg of m3D6 and in non-injected (untreated) mice. Four mice were studied per treatment group and 6–10 fields of view were imaged in each animal. Data is presented as mean +/− SEM.
| treatment | # of processes all cortical areas | # of cells all cortical areas |
|---|---|---|
| m3D6 | 3.5+/−0.07 | 12.9 +/− 0.43 |
| uninjected | 3.6 +/− 0.12 | 12.0 +/− 0.50 |
Figure 7.

Microglial activation after antibody treatment requires recognition of aggregated Aβ and the Fc domain. (A) The number of microglial processes and (B) the number of microglial cells were counted in 3-D reconstructed z-series stack images in 18 month old PDAPP+/−;CX3CR1/GFP+/− mice not injected (untreated) or injected with 500 μg of m3D6, 500 μg of IgG2b, 500 μg of mHJ5.1, 500 μg of m3D6 Fab fragments. Four mice were studied per treatment group and 6–10 fields of view were imaged in each animal. Data is presented as mean +/− SEM. * p<0.001.
Discussion
The amyloid hypothesis of AD postulates that the aggregation of Aβ results in a myriad of downstream events leading to the neurodegeneration that typifies AD (Selkoe, 2000). Therefore, studying the relationship between the potentially deleterious accumulation of Aβ and microglial cells, which invariably surround plaques, is important in understanding AD pathogenesis. We studied microglial cells before and after amyloid deposition. We found that microglial processes extended and retracted significantly more in younger than in older APP transgenic mice. Furthermore, the presence of amyloid pathology was associated with altered microglial morphology. We also demonstrated that microglia undergo rapid changes after systemic administration of a specific anti-Aβ antibody. These alterations, an increase in the number of processes per cell and in cell number, were observed in older PDAPP mice but not in younger mice in which only soluble Aβ is present. Lastly, we demonstrated that this rapid microglial response required recognition of aggregated Aβ and the Fc domain of the antibody.
Microglial cells consistently congregate around amyloid pathology in humans and animals. Microglia clustered around amyloid plaques, are classically activated, and release pro-inflammatory cytokines into the milieu (Akiyama et al., 2000; Combs et al., 2000; Combs et al., 2001). Inhibition of the complement cascade has been associated with an increase in Aβ pathology and degenerating neurons as well as an alteration in microglial responses (Wyss-Coray et al., 2002; Maier et al., 2008). Despite the fact that under certain conditions microglia have been shown to phagocytose Aβ and have been implicated in Aβ clearance (Bard et al., 2000; Wilcock et al., 2003; Koenigsknecht and Landreth, 2004; Wilcock et al., 2004b), microglial cells do not appear to efficiently or effectively clear amyloid in the AD brain. While it is clear that microglia cells play a role in Aβ-induced brain changes, the extent to which microglia play a positive vs. negative role in the AD brain is not well understood. In this study we demonstrate that microglial morphology is altered in the presence of amyloid pathology in APP transgenic mice in that the cells are less ramified, cell bodies are larger, and processes are shorter. We report a decrease in microglial process dynamics in older mice containing amyloid pathology whereas Bolmont et al. observed similar degrees of microglial process movement compared to non-APP transgenic mice (Bolmont et al., 2008). This difference may be due to several factors. Bolmont et al. utilized a different mouse model (Iba-1-GFP;APPPS1). This APPPS1 model begins to develop amyloid pathology at about 6 weeks of age (Radde et al., 2006), and were imaged at 3 to 4 months. In our study the APP-transgenic mice developed amyloid pathology around 9 months of age and were imaged later. In addition, we imaged our mice immediately after placement of the cranial window, whereas the Bolmont group imaged their mice 1 week after cranial window surgery.
Currently, a promising AD therapeutic strategy is immunization. Animal model studies have shown that active immunization and passive immunization with certain anti-Aβ antibodies results in decreased amyloid pathology and improved cognitive performance (Brody and Holtzman, 2008). To determine the effects of short term systemic treatment of a previously studied anti-Aβ antibody (m3D6), a humanized version of which is now in phase III clinical trials in humans called Bapineuzumab (www.clinicaltrials.gov), on microglial cells, we intraperitoneally injected mice with m3D6 and imaged the mice 3 days later. We observed remarkable differences in the microglial cells between m3D6-injected and non-injected mice. The microglial cells in the m3D6-injected mice contained significantly more processes per cell protruding from their cell somas than microglial cells in non-injected mice. There were also more microglia observed in the m3D6-injected mice than in the non-injected mice. These changes were observed in areas that contained amyloid pathology as well as areas that contained little to no pathology. Thus, m3D6 is able to initiate rapid changes in microglial cells which may, in part, explain the results of previous studies which have observed clearance of amyloid following treatment with certain antibodies including m3D6 (Bard et al., 2000; Bacskai et al., 2001; Bacskai et al., 2002; Wilcock et al., 2003; Wilcock et al., 2004b; Wilcock et al., 2004a; Wilcock et al., 2006; Burbach et al., 2007; Prada et al., 2007). Importantly, treatment with mHJ5.1, an antibody that binds soluble Aβ but not fibrillar Aβ did not induce the rapid changes in microglia. m3D6 binds both soluble and aggregated Aβ. Injection of antibodies, including the chronic use of m3D6, has resulted in clearance of plaques as compared to mice treated with various controls (Bard et al., 2000; Wilcock et al., 2003; Wilcock et al., 2004b; Wilcock et al., 2004a; Schroeter et al., 2008). Furthermore, it has been demonstrated that placement of certain anti-Aβ antibodies on the brain surface results in the rapid clearance of amyloid plaques which was also accompanied by an increase in microglial cells around the area of antibody treatment (Bacskai et al., 2001; Prada et al., 2007). Although there is strong evidence for microglial-dependent amyloid clearance following CNS infusion of anti-Aβ antibodies, there is also evidence for microglial-independent mechanisms. A two-phase mechanism for Aβ clearance via Aβ antibodies was introduced following intracranial administration of an N-terminal Aβ antibody (Wilcock et al., 2003). The first, non-microglial dependent, phase took place in the initial 24 hours. The second phase, between 1 and 3 days after antibody injection, revealed the presence of microglial activation and clearance of amyloid deposits. This result argues that multiple mechanisms, both dependent and independent of microglia, likely play a role in Aβ plaque clearance. Wilcock and colleagues have reported clearance of amyloid deposits and behavioral improvement after chronic systemic treatment with anti-Aβ antibodies (Wilcock et al., 2004b). Over months, they found that antibody enters the brain, binds deposits, and likely clears Aβ through Fc receptor-mediated phagocytosis. There are also potential side effects. It has been found that certain anti-Aβ antibodies increase microhemorrhages and macrohemorrhages associated with CAA (Pfeifer et al., 2002; Wilcock et al., 2004a; Racke et al., 2005). It is possible that an increase in activated microglia results in an increase in secretion of inflammatory cytokines which may cause damage to vascular endothelium and smooth muscle cells thereby leading to hemorrhages. It is possible that the rapid changes in microglia after antibody treatment reported in this study may explain some of the positive responses (such as plaque clearance) as well as negative responses (such as vasogenic edema and CAA-associated hemorrhage) that have been observed in other anti-Aβ antibody studies.
The source of the increased number of microglial cells as the result of m3D6 treatment in our study is currently unknown. One possibility is that some of these cells come from an infiltration of cells into the brain from the periphery. There is evidence suggesting that some microglial cells are recruited to the brain parenchyma from the periphery by chemoattraction to surround amyloid plaques (Simard et al., 2006). This infiltration of microglia was demonstrated in a model system that used bone-marrow chimeras that resulted from transplantation of lethally irradiated recipients. In a recent study, parabiosis rather than lethal irradiation was used, to produce chimeric mice (Ajami et al., 2007). This model did not result in the recruitment of peripherally derived microglia. However, in a set of animals that were lethally irradiated, there was recruitment of microglia from the periphery. In addition, it has been demonstrated that irradiation of mice while protecting their heads from irradiation does not result in microglia infiltration into the brain from the periphery after axotomy (Mildner et al., 2007). Thus, it has been argued that microglial recruitment in the lethally irradiated model was the result of a compromised blood brain barrier or due to a non-physiological system. An alternative possibility for the increase in microglial cell number observed after antibody treatment is proliferation of resident cells in the brain parenchyma. For example, it has been shown that microglia proliferate around amyloid plaques (Bornemann et al., 2001). Some of our recent observations in the brains of PDAPP mice that we have examined, suggests that the increased microglial cells following m3D6 treatment is not solely the result of infiltration of cells from the periphery. We have found that cells are not increased exclusively around venules, the location where immune cells enter the brain from the periphery in inflammatory disease models (Lassmann et al., 1981; Saida et al., 1981). It is likely that the increase in microglial cell number seen in mice treated with m3D6 is the result of the proliferation of resident cells as well as potentially infiltration of some cells from the periphery. Further studies to clarify this issue will be required.
In sum, the rapid changes in microglia observed could result in some of the beneficial changes seen in animals and possibly humans. These alterations in microglial cells may also be, in part, responsible for deleterious side effects such as edema and increased hemorrhages associated with CAA seen with some antibodies. It is possible that the side effects may be surmountable and avoided by utilization of antibodies lacking the property of induction of microglial activation.
Acknowledgments
We thank E. Nolley and K. Hyrc for technical assistance. This work was supported by NIH grants F32 AG029044, NS32636, the Neuroscience Blueprint Center Core Grant P30 NS057105 to Washington University, and Eli Lilly and Co.
References
- Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FM. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci. 2007;10:1538–1543. doi: 10.1038/nn2014. [DOI] [PubMed] [Google Scholar]
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, O’Banion MK, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyoma I, Van Muiswinkel FL, Veerhuis R, Walker D, Webster S, Wegrzyniak B, Wenk G, Wyss-Coray T. Inflammation and Alzheimer’s disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacskai BJ, Kajdasz ST, McLellan ME, Games D, Seubert P, Schenk D, Hyman BT. Non-Fc-mediated mechanisms are involved in clearance of amyloid-beta in vivo by immunotherapy. J Neurosci. 2002;22:7873–7878. doi: 10.1523/JNEUROSCI.22-18-07873.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacskai BJ, Kajdasz ST, Christie RH, Carter C, Games D, Seubert P, Schenk D, Hyman BT. Imaging of amyloid-beta deposits in brains of living mice permits direct observation of clearance of plaques with immunotherapy. Nat Med. 2001;7:369–372. doi: 10.1038/85525. [DOI] [PubMed] [Google Scholar]
- Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Lieberburg I, Motter R, Nguyen M, Soriano F, Vasquez N, Weiss K, Welch B, Seubert P, Schenk D, Yednock T. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6:916–919. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- Bolmont T, Haiss F, Eicke D, Radde R, Mathis CA, Klunk WE, Kohsaka S, Jucker M, Calhoun ME. Dynamics of the microglial/amyloid interaction indicate a role in plaque maintenance. J Neurosci. 2008;28:4283–4292. doi: 10.1523/JNEUROSCI.4814-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornemann KD, Wiederhold KH, Pauli C, Ermini F, Stalder M, Schnell L, Sommer B, Jucker M, Staufenbiel M. Abeta-induced inflammatory processes in microglia cells of APP23 transgenic mice. Am J Pathol. 2001;158:63–73. doi: 10.1016/s0002-9440(10)63945-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brendza RP, Bacskai BJ, Cirrito JR, Simmons KA, Skoch JM, Klunk WE, Mathis CA, Bales KR, Paul SM, Hyman BT, Holtzman DM. Anti-Abeta antibody treatment promotes the rapid recovery of amyloid-associated neuritic dystrophy in PDAPP transgenic mice. J Clin Invest. 2005;115:428–433. doi: 10.1172/JCI23269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brion JP, Couck AM, Passareiro E, Flament-Durand J. Neurofibrillary tangles of Alzheimer’s disease: an immunohistochemical study. J Submicrosc Cytol. 1985;17:89–96. [PubMed] [Google Scholar]
- Brody DL, Holtzman DM. Active and Passive Immunotherapy for Neurodegenerative Disorders. Annu Rev Neurosci. 2008 doi: 10.1146/annurev.neuro.31.060407.125529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burbach GJ, Vlachos A, Ghebremedhin E, Del Turco D, Coomaraswamy J, Staufenbiel M, Jucker M, Deller T. Vessel ultrastructure in APP23 transgenic mice after passive anti-Abeta immunotherapy and subsequent intracerebral hemorrhage. Neurobiol Aging. 2007;28:202–212. doi: 10.1016/j.neurobiolaging.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Cirrito JR, May PC, O’Dell MA, Taylor JW, Parsadanian M, Cramer JW, Audia JE, Nissen JS, Bales KR, Paul SM, DeMattos RB, Holtzman DM. In vivo assessment of brain interstitial fluid with microdialysis reveals plaque-associated changes in amyloid-beta metabolism and half-life. J Neurosci. 2003;23:8844–8853. doi: 10.1523/JNEUROSCI.23-26-08844.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combs CK, Karlo JC, Kao SC, Landreth GE. beta-Amyloid stimulation of microglia and monocytes results in TNFalpha-dependent expression of inducible nitric oxide synthase and neuronal apoptosis. J Neurosci. 2001;21:1179–1188. doi: 10.1523/JNEUROSCI.21-04-01179.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combs CK, Johnson DE, Karlo JC, Cannady SB, Landreth GE. Inflammatory mechanisms in Alzheimer’s disease: inhibition of beta-amyloid-stimulated proinflammatory responses and neurotoxicity by PPARgamma agonists. J Neurosci. 2000;20:558–567. doi: 10.1523/JNEUROSCI.20-02-00558.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Amore JD, Kajdasz ST, McLellan ME, Bacskai BJ, Stern EA, Hyman BT. In vivo multiphoton imaging of a transgenic mouse model of Alzheimer disease reveals marked thioflavine-S-associated alterations in neurite trajectories. J Neuropathol Exp Neurol. 2003;62:137–145. doi: 10.1093/jnen/62.2.137. [DOI] [PubMed] [Google Scholar]
- Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F, et al. Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature. 1995;373:523–527. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- Garcia-Alloza M, Ferrara BJ, Dodwell SA, Hickey GA, Hyman BT, Bacskai BJ. A limited role for microglia in antibody mediated plaque clearance in APP mice. Neurobiol Dis. 2007;28:286–292. doi: 10.1016/j.nbd.2007.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glenner GG, Henry JH, Fujihara S. Congophilic angiopathy in the pathogenesis of Alzheimer’s degeneration. Ann Pathol. 1981;1:120–129. [PubMed] [Google Scholar]
- Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, Sher A, Littman DR. Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol. 2000;20:4106–4114. doi: 10.1128/mcb.20.11.4106-4114.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenigsknecht J, Landreth G. Microglial phagocytosis of fibrillar beta-amyloid through a beta1 integrin-dependent mechanism. J Neurosci. 2004;24:9838–9846. doi: 10.1523/JNEUROSCI.2557-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassmann H, Kitz K, Wisniewski HM. Histogenesis of demyelinating lesions in the spinal cord of guinea pigs with chronic relapsing experimental allergic encephalomyelitis. J Neurol Sci. 1981;50:109–121. doi: 10.1016/0022-510x(81)90046-0. [DOI] [PubMed] [Google Scholar]
- Maier M, Peng Y, Jiang L, Seabrook TJ, Carroll MC, Lemere CA. Complement C3 deficiency leads to accelerated amyloid beta plaque deposition and neurodegeneration and modulation of the microglia/macrophage phenotype in amyloid precursor protein transgenic mice. J Neurosci. 2008;28:6333–6341. doi: 10.1523/JNEUROSCI.0829-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandybur TI. The incidence of cerebral amyloid angiopathy in Alzheimer’s disease. Neurology. 1975;25:120–126. doi: 10.1212/wnl.25.2.120. [DOI] [PubMed] [Google Scholar]
- Meyer-Luehmann M, Spires-Jones TL, Prada C, Garcia-Alloza M, de Calignon A, Rozkalne A, Koenigsknecht-Talboo J, Holtzman DM, Bacskai BJ, Hyman BT. Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer’s disease. Nature. 2008;451:720–724. doi: 10.1038/nature06616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mildner A, Schmidt H, Nitsche M, Merkler D, Hanisch UK, Mack M, Heikenwalder M, Bruck W, Priller J, Prinz M. Microglia in the adult brain arise from Ly-6ChiCCR2+ monocytes only under defined host conditions. Nat Neurosci. 2007;10:1544–1553. doi: 10.1038/nn2015. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- Pfeifer M, Boncristiano S, Bondolfi L, Stalder A, Deller T, Staufenbiel M, Mathews PM, Jucker M. Cerebral hemorrhage after passive anti-Abeta immunotherapy. Science. 2002;298:1379. doi: 10.1126/science.1078259. [DOI] [PubMed] [Google Scholar]
- Prada CM, Garcia-Alloza M, Betensky RA, Zhang-Nunes SX, Greenberg SM, Bacskai BJ, Frosch MP. Antibody-mediated clearance of amyloid-beta peptide from cerebral amyloid angiopathy revealed by quantitative in vivo imaging. J Neurosci. 2007;27:1973–1980. doi: 10.1523/JNEUROSCI.5426-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racke MM, Boone LI, Hepburn DL, Parsadainian M, Bryan MT, Ness DK, Piroozi KS, Jordan WH, Brown DD, Hoffman WP, Holtzman DM, Bales KR, Gitter BD, May PC, Paul SM, DeMattos RB. Exacerbation of cerebral amyloid angiopathy-associated microhemorrhage in amyloid precursor protein transgenic mice by immunotherapy is dependent on antibody recognition of deposited forms of amyloid beta. J Neurosci. 2005;25:629–636. doi: 10.1523/JNEUROSCI.4337-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radde R, Bolmont T, Kaeser SA, Coomaraswamy J, Lindau D, Stoltze L, Calhoun ME, Jaggi F, Wolburg H, Gengler S, Haass C, Ghetti B, Czech C, Holscher C, Mathews PM, Jucker M. Abeta42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep. 2006;7:940–946. doi: 10.1038/sj.embor.7400784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saida T, Saida K, Silberberg DH, Brown MJ. Experimental allergic neuritis induced by galactocerebroside. Ann Neurol. 1981;(9 Suppl):87–101. doi: 10.1002/ana.410090714. [DOI] [PubMed] [Google Scholar]
- Schroeter S, Khan K, Barbour R, Doan M, Chen M, Guido T, Gill D, Basi G, Schenk D, Seubert P, Games D. Immunotherapy reduces vascular amyloid-beta in PDAPP mice. J Neurosci. 2008;28:6787–6793. doi: 10.1523/JNEUROSCI.2377-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ. Toward a comprehensive theory for Alzheimer’s disease. Hypothesis: Alzheimer’s disease is caused by the cerebral accumulation and cytotoxicity of amyloid beta-protein. Ann N Y Acad Sci. 2000;924:17–25. doi: 10.1111/j.1749-6632.2000.tb05554.x. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- Simard AR, Soulet D, Gowing G, Julien JP, Rivest S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer’s disease. Neuron. 2006;49:489–502. doi: 10.1016/j.neuron.2006.01.022. [DOI] [PubMed] [Google Scholar]
- Vasilevko V, Xu F, Previti ML, Van Nostrand WE, Cribbs DH. Experimental investigation of antibody-mediated clearance mechanisms of amyloid-beta in CNS of Tg-SwDI transgenic mice. J Neurosci. 2007;27:13376–13383. doi: 10.1523/JNEUROSCI.2788-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinters HV. Cerebral amyloid angiopathy. A critical review. Stroke. 1987;18:311–324. doi: 10.1161/01.str.18.2.311. [DOI] [PubMed] [Google Scholar]
- Wilcock DM, Rojiani A, Rosenthal A, Subbarao S, Freeman MJ, Gordon MN, Morgan D. Passive immunotherapy against Abeta in aged APP-transgenic mice reverses cognitive deficits and depletes parenchymal amyloid deposits in spite of increased vascular amyloid and microhemorrhage. J Neuroinflammation. 2004a;1:24. doi: 10.1186/1742-2094-1-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcock DM, DiCarlo G, Henderson D, Jackson J, Clarke K, Ugen KE, Gordon MN, Morgan D. Intracranially administered anti-Abeta antibodies reduce beta-amyloid deposition by mechanisms both independent of and associated with microglial activation. J Neurosci. 2003;23:3745–3751. doi: 10.1523/JNEUROSCI.23-09-03745.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcock DM, Alamed J, Gottschall PE, Grimm J, Rosenthal A, Pons J, Ronan V, Symmonds K, Gordon MN, Morgan D. Deglycosylated anti-amyloid-beta antibodies eliminate cognitive deficits and reduce parenchymal amyloid with minimal vascular consequences in aged amyloid precursor protein transgenic mice. J Neurosci. 2006;26:5340–5346. doi: 10.1523/JNEUROSCI.0695-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcock DM, Rojiani A, Rosenthal A, Levkowitz G, Subbarao S, Alamed J, Wilson D, Wilson N, Freeman MJ, Gordon MN, Morgan D. Passive amyloid immunotherapy clears amyloid and transiently activates microglia in a transgenic mouse model of amyloid deposition. J Neurosci. 2004b;24:6144–6151. doi: 10.1523/JNEUROSCI.1090-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss-Coray T, Yan F, Lin AH, Lambris JD, Alexander JJ, Quigg RJ, Masliah E. Prominent neurodegeneration and increased plaque formation in complement-inhibited Alzheimer’s mice. Proc Natl Acad Sci U S A. 2002;99:10837–10842. doi: 10.1073/pnas.162350199. [DOI] [PMC free article] [PubMed] [Google Scholar]