

Abstract
The hepatotoxicity of acetaminophen (APAP) occurs by initial metabolism to N-acetyl-p-benzoquinone imine which depletes GSH and forms APAP-protein adducts. Subsequently, the reactive nitrogen species peroxynitrite is formed from nitric oxide (NO) and superoxide leading to 3-nitrotyrosine in proteins. Toxicity occurs with inhibited mitochondrial function. We previously reported that in hepatocytes the nNOS (NOS1) inhibitor NANT inhibited APAP toxicity, reactive nitrogen and oxygen species formation, and mitochondrial dysfunction. In this work we examined the effect of trifluoperazine (TFP), a calmodulin antagonist that inhibits calcium induced nNOS activation, on APAP hepatotoxicity and reactive nitrogen formation in murine hepatocytes and in vivo. In freshly isolated hepatocytes TFP inhibited APAP induced toxicity, reactive nitrogen formation (NO, GSNO, and 3-nitrotyrosine in protein), reactive oxygen formation (superoxide), loss of mitochondrial membrane potential, decreased ATP production, decreased oxygen consumption rate, and increased NADH accumulation. TFP did not alter APAP induced GSH depletion in the hepatocytes or the formation of APAP protein adducts which indicated that reactive metabolite formation was not inhibited. Since we previously reported that TFP inhibits the hepatotoxicity of APAP in mice without altering hepatic APAP-protein adduct formation, we examined the APAP treated mouse livers for evidence of reactive nitrogen formation. 3-Nitrotyrosine in hepatic proteins and GSNO were significantly increased in APAP treated mouse livers and decreased in the livers of mice treated with APAP plus TFP. These data are consistent with a hypothesis that APAP hepatotoxicity occurs with altered calcium metabolism, activation of nNOS leading to increased reactive nitrogen formation, and mitochondrial dysfunction.
Keywords: Acetaminophen, neuronal nitric oxide, oxidative stress, mitochondria
Graphical Abstract
Introduction
One of the most widely used analgesic/antipyretic drug in the world is acetaminophen (paracetamol, APAP; N-acetyl-p-aminophenol). At therapeutic doses APAP is believed to be safe but when an overdose it may cause hepatic centrilobular necrosis (1, 2). Annually approximately 500 deaths occur in the United States due to overdose of APAP (3). Because of their susceptibility to acute doses of APAP and the similarity to the human toxicity, mice have been the most frequently studied experimental model for APAP toxicity studies. The deduced mechanisms have proven to be applicable to the human toxicity. These studies have revealed that, APAP is metabolized in hepatocytes by cytochrome P450 (CYP450 to the reactive metabolite N-acetyl-p-benzoquinone imine (NAPQI) (4). Following a therapeutic dose, glutathione (GSH) efficiently detoxifies NAPQI but in overdose total hepatic GSH is depleted (2, 5). Under conditions of GSH depletion NAPQI reacts with available sulfhydryl on proteins to form 3-(cystein-S-yl)-acetaminophen (APAP-Cys) protein adducts (6). This so called covalent binding correlates with hepatic toxicity. The type of toxicity is hepatocyte necrosis.
Whereas the exact mechanism by which necrosis occurs is unclear, available data suggest that oxidant stress mediated by reactive nitrogen may play a significant role. Previously our laboratory reported the presence of nitrated tyrosine (3-nitrotyrosine) in hepatic proteins of APAP-treated mice (7). The nitrated proteins occurred in the same hepatocytes that contained APAP-protein adducts and were undergoing necrotic changes. Nitration of tyrosine was postulated to be mediated by peroxynitrite, a highly reactive species generated from superoxide and nitric oxide (NO) (8–10). Peroxynitrite is both a nitrating as well as an oxidizing agent and can be detoxified by GSH (11). GSH is depleted in APAP toxicity (2). Our laboratory reported nitration of MnSOD with loss of activity (12). In addition, mitochondrial aldehyde dehydrogenase, glutathione peroxidase, ATP synthase, and 3-ketoacyl-CoA thiolase (13) have been reported to be nitrated in murine APAP toxicity. N-Acetylcysteine (NAC) treatment has been reported to decrease hepatic protein nitration in APAP toxicity (13).
A major focus of our research has been to understand the role of reactive nitrogen in APAP hepatotoxicity. We and others have previously shown that iNOS (inducible nitric oxide synthase) knockout mice are equally sensitive to APAP hepatotoxicity as wild type mice (14, 15) and we reported that pharmacological inhibitors of iNOS do not decrease hepatotoxicity in mice (16). Subsequently we examined APAP in freshly isolated hepatocytes. These hepatocytes still have high levels of CYP enzymes necessary for metabolism of APAP to the reactive metabolite NAPQI leading to GSH depletion and covalent binding. Two pharmacological inhibitors of iNOS (L-NIL and SAIT) did not decrease toxicity; however, the neuronal nitric oxide synthase (nNOS) inhibitor 7-nitroindazole inhibited toxicity (17). We reported that hepatotoxicity of APAP in mice was delayed in nNOSα knockout mice compared to the wild type mice (18). Moreover, we recently reported that the specific nNOS inhibitor NANT inhibited APAP toxicity in freshly isolated hepatocytes and blocked production of reactive nitrogen and oxygen species (19). These data led to the hypothesis that nNOS is the source of the NO leading to oxidant stress in APAP hepatotoxicity. nNOS is known to occur in hepatocytes (20).
Trifluoperazine (TFP) has been previously reported to block APAP toxicity in mice (21–23). Our laboratory previously examined APAP toxicity in freshly isolated hepatocytes, a model of toxicity where GSH levels and CYP levels are still high compared to cultured hepatocytes and time to toxicity is similar to that occurring in vivo in mice. We found that TFP inhibited APAP toxicity in freshly isolated hepatocytes (17). Since TFP is a mitochondrial permeability transition (MPT) inhibitor and another MPT inhibitor, cyclosporine A, blocked APAP toxicity (24, 25), MPT was postulated to be important in toxicity. However, the molecular events causing the APAP-induced MPT are poorly defined. To further understand the mechanism of how TFP blocked APAP toxicity, Chaudhuri et al (21), examined the effect of APAP on hepatic phospholipase A2 (PLA2) activity. Mehendale and coworkers (26) previously postulated a role of phospholipase A2 in APAP induced liver injury. Histological evidence of the APAP induced necrosis and APAP-induced increases of serum ALT were significantly decreased by co-administration with TFP: however, APAP induced GSH depletion and covalent binding were not significantly altered by co-administration with TFP. In livers of APAP treated mice there was a significant early increase in both cytosolic and secretory PLA2 activities, both were partially decreased by co-administration with TFP but the role of PLA2 in APAP toxicity was unclear (21). However, TFP inhibits the calcium-calmodulin activation of nNOS activity by inhibiting electron flow (27–29). Therefore, in this manuscript we have examined the effect of TFP on APAP toxicity and reactive nitrogen and oxygen formation in freshly isolated hepatocytes, and hepatic reactive nitrogen formation in APAP treated mice.
MATERIALS AND METHODS
Reagents
Acetaminophen (APAP; 4-acetamidophenol), Percoll, Hepes, (heparin sodium salt grade I-A from porcine intestinal mucosa), penicillin G (sodium salt), RPMI-1640 modified media (with L-glutamine and without sodium bicarbonate and phenol red), 0.4% trypan blue solution, GSH, GSSG, S-nitrosoglutathione (GSNO), 3NT, NADH, NAD, and trifluoperazine (TFP; 10-[3-(4-methylpiperazin-1-yl)propyl]-2-(trifluoromethyl)-10H-phenothiazine), ATP Bioluminescent Assay kit, were products of Sigma Chemical Company (St Louis, MO). Collagenase A from Clostridium histolyticum was purchased from Roche Diagnostics (Indianapolis, IN). MitoSOX Red, 4-Amino-5-methylamino-2′,7′-difluorofluorescein diacetate (DAF-FM), and JC1 were obtained from Life technologies (Eugene, OR). LDH (lactate dehydrogenase) cytotoxicity detection kit was a product of from Roche Diagnostic Corporation (Indianapolis).
Animals
Male 6-week old mice (B6C3F1) were bred and pruchase from Harlan Laboratories (Indianapolis, IN). All animal experimentation and protocols was approved by the UAMS Institutional Animal Care and Use Committee (IACUC). Experiments were carried out in accordance with the Guide for the Care and Use of Laboratory Animals as published by the U.S. National Institutes of Health. Mice were fed ad libitum and were acclimated one week prior to sacrifice.
Hepatocyte Isolation and Incubation Mixtures
As previously described freshly isolated hepatocytes were obtained from mice by collagenase perfusion (17, 19) Hepatocytes >40 million cell viability >90% was determined by Trypan blue exclusion. The hepatocytes (1 million cells/ml) in RPMI 1640 media supplemented with 25 mM HEPES, 10 IU heparin/ml, and 500 IU penicillin G/ml were incubated in 125 ml Erlenmeyer flasks at 37°C under an atmosphere of 95% O2-5% CO2. APAP (1 mM) was added to experimental hepatocytes (17). Control flasks did not contain APAP. Some incubation contained 10 μM TFP. Incubations were performed in three or four separate experiments performed on different days.
Spectrophotometric Assays
Toxicity was determined by LDH release from hepatocytes as previously reported (19). Briefly, the hepatocytes were isolated from the media by centrifugation. Following kit directions the supernatants (100 μl) was mixed with the reaction mixture from the detection kit (100 μl) and subsequently heated at 37°C for 30 min in the dark. The absorbance of the samples was determined spectrophotometrically in a Bio-rad 550 plate reader at a 490 nm. Cytotoxicity was evaluated as previously described (19). reactive oxygen (superoxide) was evaluated by increased fluorescence of MitoSOX Red as previously described (30, 31). 4-Amino-5-methylamino-2′,7′-difluorofluorescein diacetate (DAF- FM) was utilized for assay of reactive nitrogen (NO) (32). Briefly, hepatocytes (1 ml) were centrifuged at 140 × g for 2 min and supernatant discarded. The hepatocytes were resuspended with in 2ml of phosphate-buffered saline containing DAF-FM (10μM) or MitoSOX (5μM) and incubated at 37°C for 20 min in an atmosphere of 95% O2/5% CO2. The cells were subsequently centrifuged to remove excess dye and resuspended in 2 ml of phosphate-buffered saline. Fluometrical analysis for Mitosox for DAF-FM (excited/emitted at 495/515 nm) and for MitoSOX (510/580 nm) was performed using a SpectraMax M2e fluorescence spectrophotometer. The relative mitochondrial membrane potential was determined using JC1, a mitochondrial membrane specific cationic as previously described (19). Briefly, hepatocytes (2ml) were centrifuged at 140 g for 2 min and the supernatant discarded. Cells were resuspended in 3 ml JC1 buffer (6.5 μM JC1) and incubated for 25 min at 37°C in atmosphere of 95% O2/5% CO2. Subsequently, cells were centrifuged and washed to remove excess dye and resuspended in JC1 buffer (2 ml). Fluometric analysis was performed by excitation at 490nm and emission at 530 and 590 nm. The ratio of absorbance at 590 nm to 530 nm (590:530 ratio) was evaluated as as the relative mitochondrial membrane potential (25, 33). Adenosine 5′-triphosphate (ATP) production in isolated hepatocytes was determined utilizing manufacturer’s protocol, using a TD20/20 luminometer (Turner Design, Sunnyvale, CA, USA). Quantification of ATP was obtained from an ATP standard curve. These assays described were performed as we previously reported (19).
HPLC Assays
High-performance liquid chromatography (HPLC) was used to quantify GSH, GSSG, GSNO and 3-nitrotyrosine (3-NT). Briefly, approximately 2 million hepatocytes were homogenized in ice-cold phosphate-buffered saline (PBS) buffer and protein s were precipitated by treatment with 10% metaphosphoric acid for 30 min on ice. The samples were centrifuged at 18,000 g at 4°C for 15 min. Twenty μl of the resulting supernatants were injected into the HPLC column for metabolite quantification. The pellet was used for protein analysis using BCA protein assay. The methodological details for HPLC analysis of GSH, GSSG, GSNO and 3-NT have been previously described (34, 35). NAD+ and NADH levels were determined utilizing a Dionex Ultimate 3000 HPLC-UV system as previously described (36). APAP covalently bound to proteins (APAP-cysteine) in hepatocytes was determined following protease treatment of hepatocyte homogenates followed by high performance liquid chromatography-electrochemical analysis for as previously reported (37).
Oxygen Consumption Rate
Utilizing previously described methods the Oxygen consumption rate (OCR) was determined at 37°C using an XF96 extracellular flux analyzer (Seahorse Bioscience, Billerica, MA) (19). Briefly, freshly isolated hepatoyctes (8,000 per well) were plated in CellTak coated plates, using unbuffered DMEM containing 4 mM glutamate and incubated in a non-CO2 incubator at 37°C for 1 h. Baseline measurements (three) were acquired before sequential injection of TFP (10 μM) followed by APAP (1 mM). Oxygen consumption rates were calculated using the Seahorse XF-96 software. The data represent an average of 20–32 measurements on two different days (10–16 wells per mouse per day).
Statistical Analyses
Analysis of variance was performed with a Bonferroni post hoc test using the Prism GraphPad 6.0 (San Diego, CA). Statistical significance was defined as the experimental being p < 0.05 compared to control.
APAP toxicity, GSH depletion, and protein adduct formation in hepatocytes
The effect of TFP on APAP mediated toxicity in freshly isolated hepatocytes (hepatocyte incubations) was investigated to determine the role of nNOS. TFP has been reported to be an inhibitor of nNOS (29). Toxicity (LDH release into media) was significantly increased in APAP treated hepatocytes (Figure 1A) at 1.5–3 h compared to control hepatocytes. Addition of 10 μM TFP eliminated toxicity produced by APAP (Figure 1A).
Figure 1.

Effect of TFP on APAP-induced toxicity, glutathione, and APAP-CYS in freshly isolated hepatocytes. Hepatocytes were incubated with APAP (1 mM), APAP plus TFP (10 uM), TFP alone or with media alone (Con) for 0–3h. (A) relative toxicity (LDH release), (B) GSH, (C) APAP-CYS, and (D) GSSG in hepatocytes. Significant increase was indicated by * (p≤0.05) from control group. Samples were n=3 from 3 separate mice and hepatocytes were isolated on 3 different days. The data are presented as mean ± SE.
APAP metabolism by CYP enzymes to the reactive metabolite NAPQI and its role in GSH depletion and covalent binding have been described (1). Since inhibition of CYP mediated APAP metabolic activation is known to decrease toxicity, the effect of TFP on GSH depletion and covalent binding by APAP was evaluated. In the APAP treated hepatocytes GSH levels were significantly depleted by 70% at 0.5 h compared to the control hepatocytes (Figure 1B). APAP mediated GSH depletion was not altered by TFP. Interestingly, oxidized GSH (GSSG) levels were significantly increased to APAP alone compared to the APAP/TFP treated hepatocytes; however, the amount is low compared to total GSH (Figure 1D). These data are consistent with some oxidative stress occurring in the APAP only treated hepatocytes (38). As shown in Figure 1C, TFP did not alter covalent binding of APAP to protein (APAP cysteine protein adducts). These data and the GSH depletion data, indicate that the mechanism by which TFP decreased APAP toxicity in hepatocytes was not by inhibition of CYP metabolism of APAP to form NAPQI leading to GSH depletion and APAP covalent binding (Cys adduct formation).
Formation of reactive oxygen and nitrogen species in hepatocytes
Since TFP has been reported to be a nNOS inhibitor its effect on reactive nitrogen formation in APAP toxicity was determined. 3-Nitrotyrosine in hepatocyte proteins of the APAP treated mice was quantified. 3-Nitrotyrosine is believed to be formed by nitration of tyrosine residues in proteins by peroxynitrite, a reactive nitrogen species formed from NO and superoxide. 3-Nitrotyrosine levels in proteins were significantly increased at 0.5 h in APAP treated hepatocytes compared to control hepatocytes and remained significantly increased to 3.0 h (Figure 2A). 3-Nitrotyrosine levels remained at control levels for the duration of the experiment (0–3 h) (Figure 2A).
Figure 2.

Effect of TFP on APAP-induced reactive nitrogen and oxygen formation in freshly isolated hepatocytes. Hepatocytes were incubated with APAP (1mM), APAP plus TFP (10uM), TFP alone or with media alone (Con) for 0–3 h. (A) 3-NT in proteins, (B) Nitric Oxide (C) Superoxide production, and (D) GSNO in hepatocytes. Significant increase was indicated by * (p≤0.05) from control group. Samples were n=3 from 3 separate mice and hepatocytes were isolated on 3 different days. The data are presented as mean ±SE
Since peroxynitrite is formed from nitric oxide (NO) and superoxide the effect of TFP on these reactive species was determined. NO levels in APAP treated hepatocytes significantly increased at 0.5 h and subsequently decreased. In hepatocytes containing APAP plus TFP, NO levels were not significantly different from control levels at 0–3 h (Figure 2B). In APAP treated hepatocytes superoxide levels were significantly increased at 1 h and subsequently decreased to control levels. TFP blocked the increase in superoxide at 1 h. Interestingly, in the presence of TFP with or without APAP, background levels of superoxide were significantly inhibited (1.5–3 h) compared to non-TFP treated hepatocytes.
The formation of GSNO was also quantified in the hepatocytes. Nitrosylation of proteins has been postulated to important in cell signaling (39). GSNO was significantly increased in the APAP treated hepatocytes at 1.5–3 h (Figure 2D). TFP significantly blocked the APAP induced increase in GSNO formation.
Alteration in NAD+ and NADH levels in hepatocytes
Hepatocyte treated with APAP were found to have a dramatic increase in NADH levels starting at 0.5 h and continuing the duration of the incubation time compared to control hepatocytes. TFP blocked the APAP induced increase in NADH levels in the hepatocytes. NAD levels were significantly lower in APAP, APAP plus TFP, and TFP hepatocytes compared to control hepatocytes at 3 h. (Figure 3B).
Figure 3.

Effect of APAP and TFP on NAD and NADH levels in freshly isolated hepatocytes. Hepatocytes were incubated with APAP (1 mM), APAP plus TFP (10 uM), TFP alone and with media alone (Con) for 0–3h. (A) NADH and (B) NAD + in the hepatocytes. Significant increase was indicated by * (p≤0.05) from control group. Samples were n=3 from 3 separate mice and hepatocytes were isolated on 3 different days. The data are presented as mean ± SE.
Oxygen Consumption Rate (OCR), mitochondrial membrane potential and ATP production in hepatocytes
Using an extracellular flux analyzer the effect of APAP on OCR was determined. A decrease in OCR was observed between 0 and 0.5 h in both APAP treated hepatocytes and in control hepatocytes. Subsequently, there was a gradual decrease in OCR between 1 and 3 h in the APAP treated hepatocytes (Figure 4A). TFP blocked this decrease in the decreased OCR in the APAP treated hepatocytes.
Figure 4.

Effect of TFP on APAP-induced alterations of oxygen consumption, mitochondrial membrane potential, and ATP levels in freshly isolated hepatocytes. Hepatocytes were incubated with APAP (1 mM), APAP plusTFP (10 uM), TFP alone and with media alone (Con) for 0–3 h. Oxygen Consumption Rate (OCR) (A) in the above mention groups were measured using Seahorse XF96 analyzer 1 in the graph represent the time at which APAP is injected in the groups and TFP in the groups. (B) Relative mitochondrial membrane potential and (C) ATP production in the hepatocytes. Significant increase was indicated by * (p≤0.05) from control group. Samples were n=3 from 3 separate mice and hepatocytes were isolated on 3 different days. The data are presented as mean ± SE.
In previous research we reported that APAP caused a loss of mitochondrial membrane potential (19, 25). The effect of TFP on APAP induced alterations in mitochondrial membrane potential in the hepatocytes was thus examined. Mitochondrial membrane potential was significantly reduced in hepatocytes treated with APAP between 1.5–3 h (Figure 4B) compared to control hepatocytes. TFP inhibited the APAP induced loss of mitochondrial membrane potential. The effect of APAP on ATP levels in the hepatocytes was also examined. ATP levels in APAP treated hepatocytes were significantly decreased from 0.5 to 3 h (Figure 4C), compared to control hepatocytes. Inclusion of TFP in the APAP treated hepatocytes blocked the decrease in ATP production.
APAP hepatotoxicity and reactive nitrogen formation in livers of mice
Previously we reported that APAP (200 mg/kg) increased hepatic necrosis and ALT levels at 4, 8, 24 and 48 h (21). TFP (10 mg/kg) administered 1 h prior to APAP significantly decreased the resulting histological necrosis and ALT levels at 4, 8, 24, and 48 h, without altering APAP hepatic covalent binding. Data from our previous experiment have been replotted in a line graph and are provided in Figure 5A. The livers have now been analyzed for evidence of reactive nitrogen formation (3-nitrotyrosine in hepatic proteins and hepatic GS-NO). 3-Nitrotyrosine in hepatic protein (Figure 5B) and hepatic GSNO levels (Figure 5C) were significantly increased in the APAP treated mice and were significantly decreased in the livers of the APAP plus TFP treated mice.
Figure 5.

Effect of TFP on APAP hepatotoxicity and reactive nitrogen formation in mice. Mice were administered APAP (200 mg/kg) or APAP plus TFP (10 mg/kg). TFP was administered 1 hour before APAP. Serum and livers samples were collected at the indicated times. (A) Serum ALT data were previously published as a bar graph (Chaudhuri et al., 2012) and the data have been replotted as a line graph. The livers were frozen at −80° C until analysis. (B) 3-Nitrotyrosine protein adducts. (C) Nitrosoglutathione (GSNO) in hepatocytes.
DISCUSSION
In previous research we reported TFP decreased APAP toxicity in hepatocytes (25) nd in mice without altering metabolic activation (21). TFP has been previously reported to be a MPT inhibitor and MPT had been implicated in APAP toxicity (25). Since Mehendale and coworkers (26) postulated a role for phospholipase A2 (PLA2) in progression of liver injury induced by APAP toxicity we previously examined its role in APAP hepatotoxicity and the effect of TFP. In mice we found that cytoplasmic and secretary phospholipase A2 (sPLA2) were increased in murine APAP hepatotoxicity. The levels of these enzymes were significantly decreased in mice treated with TFP plus APAP.
Since TFP has been previously reported to be a nNOS inhibitor, in this manuscript we examined its effect on APAP induced reactive nitrogen and oxygen species formation. We previously reported that two nNOS inhibitors (7-nitroindazole and NANT) blocked APAP toxicity in freshly isolated hepatocytes. Freshly isolated hepatocytes are superior to cultured hepatocytes because CYP levels and GSH levels are similar to those in vivo whereas in cultured hepatocytes CYP levels are very low. The nNOS inhibitor 7-nitroindazole was found to inhibit APAP mediated toxicity, 3-nitrotyrosine formation, and loss of mitochondrial membrane potential when added in the late phase of toxicity (17). More recently, we examined the effect of the nNOS inhibitor NANT. Importantly, NANT did not inhibit metabolic activation of APAP and was used to examine the effect of nNOS on early events in APAP toxicity in hepatocytes. It was found that NANT when added at time 0 inhibited APAP mediated toxicity, and APAP mediated formation of reactive nitrogen and oxygen species (NO, 3-nitrotyrosine in protein, superoxide, and GSNO). APAP caused a decrease in the hepatocyte oxygen consumption rate, loss of mitochondrial membrane potential, and a decrease in ATP production. All of the events were blocked by NANT (19). In addition, APAP also caused a dramatic increase in NADH which was blocked by NANT. This latter finding suggested that APAP toxicity occurred with inhibition of mitochondrial complex I. Three possibilities were suggested whereby reactive nitrogen species could produce mitochondrial dysfunction leading to toxicity: 1) peroxynitrite mediated nitration of critical mitochondrial proteins in Complex I, 2) peroxynitrite mediated oxidation of ubiquinol, or 3) GSNO mediated nitrosylation of critical mitochondrial proteins in Complex I (19). In this manuscript we have extended our investigation on the role of nNOS and reactive nitrogen in APAP hepatotoxicity by examining the effect of TFP on APAP mediated toxicity and reactive nitrogen and oxygen formation both in freshly isolated hepatocytes and in mice. Previously we reported that TFP blocked APAP toxicity in freshly isolated hepatocytes when added in the late phase of toxicity (25). In this manuscript we found that addition of TFP at time 0 to the APAP containing hepatocyte incubations inhibited APAP toxicity (Figure 1), protein nitration (Figure 2A), NO formation (Figure 2B), reactive oxygen formation (superoxide) (Figure 2C), and GSNO formation (Figure 2D). Toxicity and reactive nitrogen and oxygen species formation were accompanied by decreased oxygen consumption rate (Figure 4A), decreased ATP production (Figure 4B), and decreased mitochondrial membrane potential (Figure 4C). All of these decreases were significantly blocked in the incubations containing APAP plus TFP. In addition, we report that APAP induced accumulation of NADH, loss of mitochondrial membrane potential, and inhibition of ATP biosynthesis are reversed when TFP is included in the APAP containing incubations. Thus, TFP is an effective inhibitor of APAP toxicity and formation of reactive nitrogen and oxygen species. These data are similar to what we previously reported for the nNOS inhibitor NANT and clearly indicate a role for reactive nitrogen and oxygen species in APAP hepatotoxicity.
Importantly, TFP blocks APAP induced hepatic necrosis and increased serum ALT levels in mice Chaudhuri et al. (21). These ALT data, which are replotted as a line graph in Figure 5A, shows that APAP alone increased serum ALT to approximately 4500 IU/L by 8h. Co-administration of TFP (10 mg/kg) with the APAP decreased ALT at this time to approximately 1000 IU/L. Higher doses of TFP were not examined. Thus, it is unclear if higher doses would have completely eliminated toxicity (21). However, the major point of this experiment is that TFP decreases APAP toxicity. It is unclear if TFP has any clinical application to APAP toxicity in humans.
The finding that TFP blocks APAP hepatotoxicity suggests altered calcium metabolism is a key event leading to toxicity. In response to elevated calcium nNOS binds to calmodulin leading to synthesis of NO (27, 28). TFP blocks activation of nNOS by calcium/calmodulin and thus inhibits NO formation. The mechanism appears to be a distortion of the calmodulin structure necessary for activation (29). Interestingly, PLA2 activity is also mediated by a calcium/calmodulin activation and thus may explain our previous finding that PLA2 activity is increased in APAP hepatotoxicity (21).
Altered calcium metabolism has been previously postulated to be important in APAP hepatotoxicity (40–43). Burcham and Harmon (41) and Timerstein and Nelson (43) reported increases in hepatic calcium levels following toxic doses of APAP to mice. Tsokos-Kuhn and coworkers reported that toxic doses of APAP significantly decreased hepatic plasma membrane calcium-ATPase activity (43, 44). This enzyme is important in the removal of calcium from the cell because extracellular calcium enters the cell due to a large electrochemical gradient driving this ion into the cell (45). Inhibition of the enzyme results in calcium accumulation in the cell. The authors suggested that covalent binding of the reactive metabolite of APAP to the calcium-ATPase was responsible for the loss of its activity (43, 44). This enzyme has not been reported to be adducted by the reactive metabolite NAPQI; however, high levels of protein adducts were observed in the hepatic plasma membrane fraction of APAP treated mice (46).
The effect of the calcium specific chelators on APAP induced toxicity in freshly isolated hamster hepatocytes was examined by Boobis et al.(47). The calcium chelator Quin 2-AM prevented the loss of viability (47). Moreover, they found that an increase in cytosolic calcium correlated with development of toxicity (48). Corcoran et al. found a toxic dose APAP to mice caused an increase in nuclear calcium levels using cultured hepatocytes (49) and that EGTA, a calcium ion chelator, inhibited hepatocyte death; a finding that suggested toxicity was mediated by extracellular calcium since EGTA is ionized and does not enter the cells. In freshly isolated hepatocytes we previously found that EGTA, as well as Quin-2, inhibited APAP hepatotoxicity (50).
It has been recently reported that the transient receptor potential melanostatine 2 (TRPM2) channels are increased in APAP toxicity (51). TRPM2 channels are calcium-permeable cation channels that are regulated by free intracellular ADP-ribose and are activated in response to oxidative stress induced by hydrogen peroxide. It was shown that preincubation of hepatocytes with APAP or hydrogen peroxide and subsequent addition of calcium to the media resulted in increased levels of calcium in the cells. The increases were blocked by addition of TRPM2 siRNA but not by control siRNA. TRPM2 knockout mice were shown to be significantly less sensitive to APAP hepatotoxicity (increases in ALT, AST and cellular necrosis) compared to wildtype mice. However, GSH depletion and covalent binding were not analyzed to be certain that differences in APAP metabolic activation did not occur.
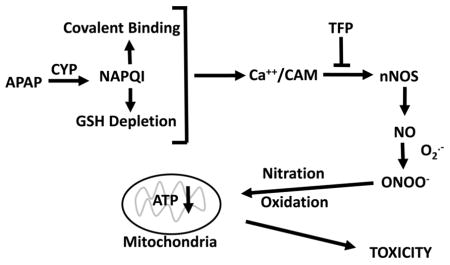
A role for TRPM2 channels is particularly intriguing because previous data have suggested a role for Fenton mediated oxidative stress in APAP hepatotoxicity. Hydrogen peroxide has been reported to accumulate in livers of APAP treated mice before development of toxicity (52). (52). This increase correlates with the depletion of GSH which is important in its detoxification. Hydrogen peroxide plus ferrous ions catalyze a Fenton mediated oxidative stress reaction and the iron chelator deferoxamine (DFO) has been reported to decrease the toxicity of acetaminophen in both rat and mouse hepatocytes (50, 53, 54). Moreover, administration of DFO to either rats (55) or mice (56) significantly delayed development of toxicity. Lemasters and coworkers reported that in APAP hepatocyte toxicity there is an early increase in cytosolic ferrous ions (57). Thus in APAP toxicity a Fenton mediated oxidative stress (ferrous ions plus hydrogen peroxide) may lead to activation of TRPM2 channels resulting in increased levels of calcium in the hepatocyte. Activation of TMR2 channels results in extracellular calcium entering the hepatocyte with activation of nNOS via a calmodulin mediated mechanism (Figure 6). Thus, APAP hepatotoxicity may involve not only a Fenton mediated oxidative stress but as shown in this manuscript a reactive nitrogen mediated oxidative stress.
Figure 6.

Postulated mechanisms important in APAP mediated toxicity.
In this manuscript we show that APAP hepatotoxicity both in mice and in hepatocyte can be decreased by the calmodulin inhibitor trifluoperazine with a decrease in reactive nitrogen formation. The data strong suggest that critical steps in APAP hepatotoxicity are: alteration of intracellular calcium, activation of nNOS leading to formation of reactive nitrogen, and mitochondrial dysfunction with decreased ATP production. These steps are summarized in Figure 6.
Highlights.
Increased reactive nitrogen and oxygen species formation leads to APAP hepatoxicity
TFP is known to block nNOS both in vivo as well as in vitro
The nNOS inhibitor TFP blocks toxicity and the increased RNS/ROS formation
Toxicity occurs with increased 3- nitro tyrosine both in vivo as well as in vitro
nNOS inhibition by TFP leads to decreasing 3-nitro tyrosine in vivo as well as in vitro.
Acknowledgments
This work was supported by the National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases to JAH [Grant R01-DK0709008]. A part of the work was supported by National Institute of Environmental Health Sciences [Grant R15 ES022781], National Institute of General Medical Sciences [Grant P20 GM109005] and Arkansas Science and Technology Authority [ASTA 15-B-19] to NAB and KJK. A part of the work was supported by Arkansas Biosciences Institute (ABI) Grant to SBM.
Conflict of Interest: Drs. Hinson and James are part-owners of Acetaminophen Toxicity Diagnostics, LLC and have a patent pending for the development of a rapid assay for the measurement of 3-(cystein-S-yl)-acetaminophen (APAP-Cys) adducts in human samples.
Abbreviations
- APAP
Acetaminophen
- NAPQI
N-Acetyl-p-benzoquinone imine
- GSH
reduced glutathione
- APAP-cys
3-(cystein-S-yl)-acetaminophen
- NO
Nitric Oxide
- iNOS
Inducible nitric oxide synthase (NOS2)
- nNOS
Neuronal nitric oxide synthase (NOS1)
- GSNO
S-Nitrosoglutathione
- GSSG
glutathione disulfide
- NANT
N-[(4S)-4-amino-5-[(2-aminoethyl) amino] pentyl]-N′-nitroguanidinetris (trifluoroacetate)
- MPT
Mitochondrial Permeability Transition
- LDH
Lactate Dehydrogenase
- OCR
Oxygen Consumption Rate
- 3-NT
3-Nitrotyrosine
- TFP
Trifluoperazine
Footnotes
This work was has not been previously presented.
Authorship Contributions:
Participated in research design: Banerjee, Melnyk, Aykin-Burn, James, Hinson
Conducted experiments: Banerjee, Melnyk, Krager, Aykin-Burn, McCullough
Contributed new reagents or analytic tools: Melnyk, Krager, Aykin-Burn, James
Performed data analysis: Banerjee, Melnyk, Aykin-Burn, James, Hinson
Wrote or contributed to the writing of the manuscript: Banerjee, Melnyk, Aykin-burn, James, Hinson
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.James LP, Mayeux PR, Hinson JA. Acetaminophen-induced hepatotoxicity. Drug Metab Dispos. 2003;31:1499–1506. doi: 10.1124/dmd.31.12.1499. [DOI] [PubMed] [Google Scholar]
- 2.Mitchell JR, et al. Acetaminophen-induced hepatic necrosis. I. Role of drug metabolism. J Pharmcol Exp Ther. 1973a;187:185–194. [PubMed] [Google Scholar]
- 3.Lee WM, Squires RH, Jr, Nyberg SL, Doo E, Hoofnagle JH. Acute liver failure: Summary of a workshop. Hepatology. 2008;47:1401–1415. doi: 10.1002/hep.22177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dahlin DC, Miwa GT, Lu AY, Nelson SD. N-acetyl-p-benzoquinone imine: a cytochrome P-450-mediated oxidation product of acetaminophen. Proc Natl Acad Sci U S A. 1984;81:1327–1331. doi: 10.1073/pnas.81.5.1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jollow DJ, Thorgeirsson SS, Potter WZ, Hashimoto M, Mitchell JR. Acetaminophen-induced hepatic necrosis. VI. Metabolic disposition of toxic and nontoxic doses of acetaminophen. Pharmacology. 1974;12:251–271. doi: 10.1159/000136547. [DOI] [PubMed] [Google Scholar]
- 6.Hoffmann KJ, Streeter AJ, Axworthy DB, Baillie TA. Identification of the major covalent adduct formed in vitro and in vivo between acetaminophen and mouse liver proteins. Mol Pharmacol. 1985;27:566–573. [PubMed] [Google Scholar]
- 7.Hinson JA, Pike SL, Pumford NR, Mayeux PR. Nitrotyrosine-protein adducts in hepatic centrilobular areas following toxic doses of acetaminophen in mice. Chem Res Toxicol. 1998;11:604–607. doi: 10.1021/tx9800349. [DOI] [PubMed] [Google Scholar]
- 8.Beckman JS. Oxidative damage and tyrosine nitration from peroxynitrite. Chem Res Toxicol. 1996;9:836–844. doi: 10.1021/tx9501445. [DOI] [PubMed] [Google Scholar]
- 9.Pryor WA, Squadrito GL. The chemistry of peroxynitrite: a product from the reaction of nitric oxide with superoxide [see comments] Am J Physiol. 1995;268:L699–722. doi: 10.1152/ajplung.1995.268.5.L699. [DOI] [PubMed] [Google Scholar]
- 10.Reiter CD, Teng RJ, Beckman JS. Superoxide reacts with nitric oxide to nitrate tyrosine at physiological pH via peroxynitrite. J Biol Chem. 2000;275:32460–32466. doi: 10.1074/jbc.M910433199. [DOI] [PubMed] [Google Scholar]
- 11.Sies H, Sharov VS, Klotz LO, Briviba K. Glutathione peroxidase protects against peroxynitrite-mediated oxidations. A new function for selenoproteins as peroxynitrite reductase. J Biol Chem. 1997;272:27812–27817. doi: 10.1074/jbc.272.44.27812. [DOI] [PubMed] [Google Scholar]
- 12.Agarwal R, et al. Acetaminophen-induced hepatotoxicity in mice occurs with inhibition of activity and nitration of mitochondrial manganese superoxide dismutase. The Journal of pharmacology and experimental therapeutics. 2011;337:110–118. doi: 10.1124/jpet.110.176321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abdelmegeed MA, Jang S, Banerjee A, Hardwick JP, Song BJ. Robust protein nitration contributes to acetaminophen-induced mitochondrial dysfunction and acute liver injury. Free Radic Biol Med. 2013;60:211–222. doi: 10.1016/j.freeradbiomed.2013.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Michael SL, et al. Acetaminophen-induced hepatotoxicity in mice lacking inducible nitric oxide synthase activity. Nitric Oxide. 2001;5:432–441. doi: 10.1006/niox.2001.0385. [DOI] [PubMed] [Google Scholar]
- 15.Bourdi M, et al. Protection against acetaminophen-induced liver injury and lethality by interleukin 10: role of inducible nitric oxide synthase. Hepatology. 2002;35:289–298. doi: 10.1053/jhep.2002.30956. [DOI] [PubMed] [Google Scholar]
- 16.Hinson JA, Bucci TJ, Irwin LK, Michael SL, Mayeux PR. Effect of inhibitors of nitric oxide synthase on acetaminophen-induced hepatotoxicity in mice. Nitric Oxide. 2002;6:160–167. doi: 10.1006/niox.2001.0404. [DOI] [PubMed] [Google Scholar]
- 17.Burke AS, MacMillan-Crow LA, Hinson JA. Reactive nitrogen species in acetaminophen-induced mitochondrial damage and toxicity in mouse hepatocytes. Chem Res Toxicol. 2010;23:1286–1292. doi: 10.1021/tx1001755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Agarwal R, et al. Acetaminophen-induced hepatotoxicity and protein nitration in neuronal nitric-oxide synthase knockout mice. The Journal of pharmacology and experimental therapeutics. 2012;340:134–142. doi: 10.1124/jpet.111.184192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Banerjee S, et al. The neuronal nitric oxide synthase inhibitor NANT blocks acetaminophen toxicity and protein nitration in freshly isolated hepatocytes. Free Radic Biol Med. 2015;89:750–757. doi: 10.1016/j.freeradbiomed.2015.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Villanueva C, Giulivi C. Subcellular and cellular locations of nitric oxide synthase isoforms as determinants of health and disease. Free Radic Biol Med. 2010;49:307–316. doi: 10.1016/j.freeradbiomed.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chaudhuri S, et al. Acetaminophen hepatotoxicity and HIF-1alpha induction in acetaminophen toxicity in mice occurs without hypoxia. Toxicol Appl Pharmacol. 2011;252:211–220. doi: 10.1016/j.taap.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dimova S, Koleva M, Rangelova D, Stoythchev T. Effect of nifedipine, verapamil, diltiazem and trifluoperazine on acetaminophen toxicity in mice. Arch Toxicol. 1995;70:112–118. doi: 10.1007/BF02733671. [DOI] [PubMed] [Google Scholar]
- 23.Yamamoto H. Antagonism of acetaminophen-induced hepatocellular destruction by trifluoperazine in mice. Pharmacol Toxicol. 1990;67:115–119. doi: 10.1111/j.1600-0773.1990.tb00795.x. [DOI] [PubMed] [Google Scholar]
- 24.Kon K, Kim JS, Jaeschke H, Lemasters JJ. Mitochondrial permeability transition in acetaminophen-induced necrosis and apoptosis of cultured mouse hepatocytes. Hepatology. 2004;40:1170–1179. doi: 10.1002/hep.20437. [DOI] [PubMed] [Google Scholar]
- 25.Reid AB, Kurten RC, McCullough SS, Brock RW, Hinson JA. Mechanisms of acetaminophen-induced hepatotoxicity: role of oxidative stress and mitochondrial permeability transition in freshly isolated mouse hepatocytes. The Journal of pharmacology and experimental therapeutics. 2005;312:509–516. doi: 10.1124/jpet.104.075945. [DOI] [PubMed] [Google Scholar]
- 26.Bhave VS, Donthamsetty S, Latendresse JR, Cunningham ML, Mehendale HM. Secretory phospholipase A-mediated progression of hepatotoxicity initiated by acetaminophen is exacerbated in the absence of hepatic COX-2. Toxicol Appl Pharmacol. 251:173–180. doi: 10.1016/j.taap.2011.01.013. [DOI] [PubMed] [Google Scholar]
- 27.Abu-Soud HM, Stuehr DJ. Nitric oxide synthases reveal a role for calmodulin in controlling electron transfer. Proc Natl Acad Sci U S A. 1993;90:10769–10772. doi: 10.1073/pnas.90.22.10769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Abu-Soud HM, Yoho LL, Stuehr DJ. Calmodulin controls neuronal nitric-oxide synthase by a dual mechanism. Activation of intra- and interdomain electron transfer. J Biol Chem. 1994;269:32047–32050. [PubMed] [Google Scholar]
- 29.Lee SJ, Stull JT. Calmodulin-dependent regulation of inducible and neuronal nitric-oxide synthase. J Biol Chem. 1998;273:27430–27437. doi: 10.1074/jbc.273.42.27430. [DOI] [PubMed] [Google Scholar]
- 30.Kudin AP, Bimpong-Buta NY, Vielhaber S, Elger CE, Kunz WS. Characterization of superoxide-producing sites in isolated brain mitochondria. J Biol Chem. 2004;279:4127–4135. doi: 10.1074/jbc.M310341200. [DOI] [PubMed] [Google Scholar]
- 31.Robinson KM, et al. Selective fluorescent imaging of superoxide in vivo using ethidium-based probes. Proc Natl Acad Sci U S A. 2006;103:15038–15043. doi: 10.1073/pnas.0601945103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McQuade LE, Lippard SJ. Fluorescent probes to investigate nitric oxide and other reactive nitrogen species in biology (truncated form: fluorescent probes of reactive nitrogen species) Curr Opin Chem Biol. 14:43–49. doi: 10.1016/j.cbpa.2009.10.004. [DOI] [PubMed] [Google Scholar]
- 33.Reers M, et al. Mitochondrial membrane potential monitored by JC-1 dye. Methods Enzymol. 1995;260:406–417. doi: 10.1016/0076-6879(95)60154-6. [DOI] [PubMed] [Google Scholar]
- 34.Melnyk S, Pogribna M, Pogribny I, Hine RJ, James SJ. A new HPLC method for the simultaneous determination of oxidized and reduced plasma aminothiols using coulometric electrochemical detection. J Nutr Biochem. 1999;10:490–497. doi: 10.1016/s0955-2863(99)00033-9. [DOI] [PubMed] [Google Scholar]
- 35.Melnyk S, Pogribna M, Pogribny IP, Yi P, James SJ. Measurement of plasma and intracellular S-adenosylmethionine and S-adenosylhomocysteine utilizing coulometric electrochemical detection: alterations with plasma homocysteine and pyridoxal 5′-phosphate concentrations. Clin Chem. 2000;46:265–272. [PubMed] [Google Scholar]
- 36.Stocchi V, et al. Simultaneous extraction and reverse-phase high-performance liquid chromatographic determination of adenine and pyridine nucleotides in human red blood cells. Analytical biochemistry. 1985;146:118–124. doi: 10.1016/0003-2697(85)90405-1. [DOI] [PubMed] [Google Scholar]
- 37.Muldrew KL, et al. Determination of acetaminophen-protein adducts in mouse liver and serum and human serum after hepatotoxic doses of acetaminophen using high-performance liquid chromatography with electrochemical detection. Drug Metab Dispos. 2002;30:446–451. doi: 10.1124/dmd.30.4.446. [DOI] [PubMed] [Google Scholar]
- 38.Jaeschke H. Glutathione disulfide formation and oxidant stress during acetaminophen-induced hepatotoxicity in mice in vivo: the protective effect of allopurinol. The Journal of pharmacology and experimental therapeutics. 1990;255:935–941. [PubMed] [Google Scholar]
- 39.Burwell LS, Nadtochiy SM, Tompkins AJ, Young S, Brookes PS. Direct evidence for S-nitrosation of mitochondrial complex I. Biochem J. 2006;394:627–634. doi: 10.1042/BJ20051435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Corcoran GB, Wong BK, Neese BL. Early sustained rise in total liver calcium during acetaminophen hepatotoxicity in mice. Res Commun Chem Pathol Pharmacol. 1987;58:291–305. [PubMed] [Google Scholar]
- 41.Burcham PC, Harman AW. Effect of acetaminophen hepatotoxicity on hepatic mitochondrial and microsomal calcium contents in mice. Toxicol Lett. 1988;44:91–99. doi: 10.1016/0378-4274(88)90134-8. [DOI] [PubMed] [Google Scholar]
- 42.Tsokos KJ. Evidence in vivo for elevation of intracellular free Ca2+ in the liver after diquat, acetaminophen, and CCl4. Biochem Pharmacol. 1989;38:3061–3065. doi: 10.1016/0006-2952(89)90016-6. [DOI] [PubMed] [Google Scholar]
- 43.Tirmenstein MA, Nelson SD. Subcellular binding and effects on calcium homeostasis produced by acetaminophen and a nonhepatotoxic regioisomer, 3′-hydroxyacetanilide, in mouse liver. J Biol Chem. 1989;264:9814–9819. [PubMed] [Google Scholar]
- 44.Tsokos-Kuhn JO, Hughes H, Smith CV, Mitchell JR. Alkylation of the liver plasma membrane and inhibition of the Ca2+ ATPase by acetaminophen. Biochem Pharmacol. 1988;37:2125–2131. doi: 10.1016/0006-2952(88)90570-9. [DOI] [PubMed] [Google Scholar]
- 45.Strehler EE, Zacharias DA. Role of alternative splicing in generating isoform diversity among plasma membrane calcium pumps. Physiol Rev. 2001;81:21–50. doi: 10.1152/physrev.2001.81.1.21. [DOI] [PubMed] [Google Scholar]
- 46.Pumford NR, Roberts DW, Benson RW, Hinson JA. Immunochemical quantitation of 3-(cystein-S-yl)acetaminophen protein adducts in subcellular liver fractions following a hepatotoxic dose of acetaminophen. Biochem Pharmacol. 1990;40:573–579. doi: 10.1016/0006-2952(90)90558-3. [DOI] [PubMed] [Google Scholar]
- 47.Boobis AR, Seddon CE, Nasseri-Sina P, Davies DS. Evidence for a direct role of intracellular calcium in paracetamol toxicity. Biochem Pharmacol. 1990;39:1277–1281. doi: 10.1016/0006-2952(90)90003-4. [DOI] [PubMed] [Google Scholar]
- 48.Hardwick SJ, Wilson JW, Fawthrop DJ, Boobis AR, Davies DS. Paracetamol toxicity in hamster isolated hepatocytes: the increase in cytosolic calcium accompanies, rather than precedes, loss of viability. Arch Toxicol. 1992;66:408–412. doi: 10.1007/BF02035131. [DOI] [PubMed] [Google Scholar]
- 49.Shen W, Kamendulis LM, Ray SD, Corcoran GB. Acetaminophen-induced cytotoxicity in cultured mouse hepatocytes: effects of Ca(2+)-endonuclease, DNA repair, and glutathione depletion inhibitors on DNA fragmentation and cell death. Toxicol Appl Pharmacol. 1992;112:32–40. doi: 10.1016/0041-008x(92)90276-x. [DOI] [PubMed] [Google Scholar]
- 50.Burke AS, Redeker K, Kurten RC, James LP, Hinson JA. Mechanisms of chloroform-induced hepatotoxicity: oxidative stress and mitochondrial permeability transition in freshly isolated mouse hepatocytes. J Toxicol Environ Health A. 2007;70:1936–1945. doi: 10.1080/15287390701551399. [DOI] [PubMed] [Google Scholar]
- 51.Kheradpezhouh E, Ma L, Morphett A, Barritt GJ, Rychkov GY. TRPM2 channels mediate acetaminophen-induced liver damage. Proc Natl Acad Sci U S A. 2014;111:3176–3181. doi: 10.1073/pnas.1322657111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen C, Krausz KW, Idle JR, Gonzalez FJ. Identification of novel toxicity-associated metabolites by metabolomics and mass isotopomer analysis of acetaminophen metabolism in wild-type and Cyp2e1-null mice. J Biol Chem. 2008;283:4543–4559. doi: 10.1074/jbc.M706299200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kyle ME, Miccadei S, Nakae D, Farber JL. Superoxide dismutase and catalase protect cultured hepatocytes from the cytotoxicity of acetaminophen. Biochem Biophys Res Commun. 1987;149:889–896. doi: 10.1016/0006-291x(87)90491-8. [DOI] [PubMed] [Google Scholar]
- 54.Ito Y, et al. Role of iron and glutathione redox cycle in acetaminophen-induced cytotoxicity to cultured rat hepatocytes. Digestive diseases and sciences. 1994;39:1257–1264. doi: 10.1007/BF02093791. [DOI] [PubMed] [Google Scholar]
- 55.Sakaida I, et al. Protection against acetaminophen-induced liver injury in vivo by an iron chelator, deferoxamine. Scand J Gastroenterol. 1995;30:61–67. doi: 10.3109/00365529509093237. [DOI] [PubMed] [Google Scholar]
- 56.Schnellmann JG, Pumford NR, Kusewitt DF, Bucci TJ, Hinson JA. Deferoxamine delays the development of the hepatotoxicity of acetaminophen in mice. Toxicol Lett. 1999;106:79–88. doi: 10.1016/s0378-4274(99)00021-1. [DOI] [PubMed] [Google Scholar]
- 57.Kon K, Kim JS, Uchiyama A, Jaeschke H, Lemasters JJ. Lysosomal iron mobilization and induction of the mitochondrial permeability transition in acetaminophen-induced toxicity to mouse hepatocytes. Toxicological sciences: an official journal of the Society of Toxicology. 2010;117:101–108. doi: 10.1093/toxsci/kfq175. [DOI] [PMC free article] [PubMed] [Google Scholar]