

Abstract
Purpose
Current guidelines recommend first-tier chromosome microarray analysis (CMA) and Fragile X syndrome (FX) testing for males with isolated intellectual disabilities/learning delay (ID/LD) and autism spectrum disorders (ASD).
Methods
Males in our clinic with ID/LD or autism (310) were analyzed for positive results from CMA and/or FX testing.
Results
Males with ID/LD were tested by CMA and 29% were found to have abnormalities. Males with ASD were tested by CMA with a detection rate of up to 9% (including VUS and AOH). Males with ID/LD were tested for Fragile X, with a detection rate of 2.5% (2/80) and these two had dysmorphic features and maternal family history. No males with ASD had positive FX testing.
Conclusions
This study’s CMA detection rate in males with isolated ID/LD is higher than the literature (10–20%). CMA results for males with ASD (9%) and FX testing for males with ID/LD (2.5%) overlap with the literature (7–10% and 2%, respectively). The yield of FX testing for patients with ASD was zero, which is close to the literature (0.5–2%). These results suggest that FX testing as a first-tier test may not be necessary, unless other criteria suggest Fragile X syndrome.
Keywords: autism, chromosomal microarray analysis, Fragile X syndrome, learning delay, intellectual impairment
INTRODUCTION
Intellectual disability (ID) and autism spectrum disorders (ASD) are relatively common and have significant impact on the patient and their family. For this reason, identifying possible genetic and environmental causes is important. ID has a prevalence of 3%1,2 in the United States with multiple etiologies, ranging from trauma to genetic disorders. A genetic basis has been demonstrated in multiple studies, and has been shown to be due to chromosome abnormalities, single genes, copy number variants (CNV) and multifactorial inheritance. The prevalence of ASD was 1 in 68 children or approximately 1.47% in 2012 per the CDC3. The heterogeneous phenotype of ASD includes autistic disorder, Asperger syndrome, and pervasive developmental disorder not otherwise specified (PDD-NOS) and occurs in all racial, ethnic, and social groups.4 It is characterized by impairments in social interaction, communication and language development, and rigid and repetitive behaviors4 with an onset prior to 3 years of age.5 The etiology for ASD in most cases is unknown, but in approximately 20–25% it is found to be related to genetic changes or risks.6 The reported incidence of ID has increased significantly while that of ASD has roughly tripled over the last 12 years,3 leading to an increased number of referrals to genetic clinics to attempt to find an etiology.5
Fragile X syndrome (FX) is considered the most common inherited cause of ID worldwide.7 In addition, FX is identified in 0.5–2% of ASD cases4,8,9 and 20% of boys with FX have a diagnosis of ASD.10,11 FX is an X-linked disorder that in 99% of cases is due to a CGG-repeat expansion (> 200 repeats) in the FMR1 gene resulting in a penetrance of 100% in males. The main and most consistent feature of FX is ID, while physical and behavioral features vary with age and gender.12 The prevalence of males with a full mutation is ~1/4,000 – 1/7,000 in the total population13,14 and all ethnic groups and races appear to be susceptible to the expansion of the FMR1 CGG region. Fragile X (FX) testing has a diagnostic yield of ~2% in males and females with ID15 and ~0.46–5% for ASD. The FX test is unlikely to miscategorize an individual with a sensitivity of >99%, with 100% specificity.
Another common cause of developmental delay/ID (DD/ID) is chromosomal abnormalties. Chromosomal microarray (CMA) provides higher diagnostic yield4,15 for genetic testing of patients with unexplained nondysmorphic DD/ID (10–20%) and ASD (~7–10%) than a G-banded karyotype (3%).5,16
Therefore, CMA and FX test are recommended the American College of Medical Genetics and Genomics (ACMGG) and American Academy of Pediatrics (AAP) as the first-tier tests for individuals with ID/LD and ASD.4,5,17,18
In our experience, FX is not as common in either ID/LD or ASD male populations as reported in the literature. Positive results were only noted in male patients with FX facial features and/or family history of FX in siblings or pre-mutation mother (CGG repeats of 55–200). We hypothesized that the yield of FX testing is low and CMA is a more sensitive test to provide a molecular diagnosis for such male patients, unless other criteria including physical and psychological features and/or family history are strongly suggestive of Fragile X syndrome.
This study compares the molecular diagnostic yields of CMA and FX testing in two pediatric populations: males with ID/LD without autism, and males with ASD.
MATERIALS AND METHODS
All patient visits to the Children’s National Health System (CNHS) Genetics Department from 07/01/13 to 02/19/15 were collected, based on data from the Billing Department. This amounted to 10,757 charges. Of these, visits for patients with identifiable genetic syndromes, gross chromosomal differences (trisomies), and metabolic disorders were eliminated from analysis, as were all charges from female patients.
These studies focused on two populations of male pediatric patients : 1. patients with isolated ID/LD (without autism) and 2. patients with ASD, ages ranging from 14 months to 19 years. Males were chosen to increase the yield of possible positive testing results due to the X-linked nature of FX.
Selection of the groups was done according to whether they had any of the visit ICD9 codes as discussed below.
Patients with isolated intellectual disability/learning delay (ID/LD)
To identify male patients with isolated ID who had a CMA and/or FX testing, the following selection was done. From the total of 10,757 charges, only those billed with at least one of the ICD9 codes listed were retained (ICD9 codes under 315, except 315.4 (coordination disorder), for delays in development, 317 (mild ID), 318 (moderate ID), 318.1 (severe ID), 318.2 (profound ID) and 319 (mental retardation)), leaving 1760 charges in this pool. Among these 1760 charges, visits which were also charged with ICD9 codes corresponding to metabolic disorders, trisomies and neurofibromatosis (listed below) were removed, leaving 1476 charges of interest. Of these 1476 charges, those billed with the ICD9 code 299.0 (autism disorder and PDD-NOS) were removed, leaving 1317 charges of interest. Some of these charges corresponded to several visits of the same patient. Duplicate charges were removed, leaving 1115 patients of interest. Female patients were removed from that group, leaving 675 male patients. 202 of the 675 male patients were tested with CMA and/or FX testing. (Figure 1 illustrates this selection process.)
Figure 1.
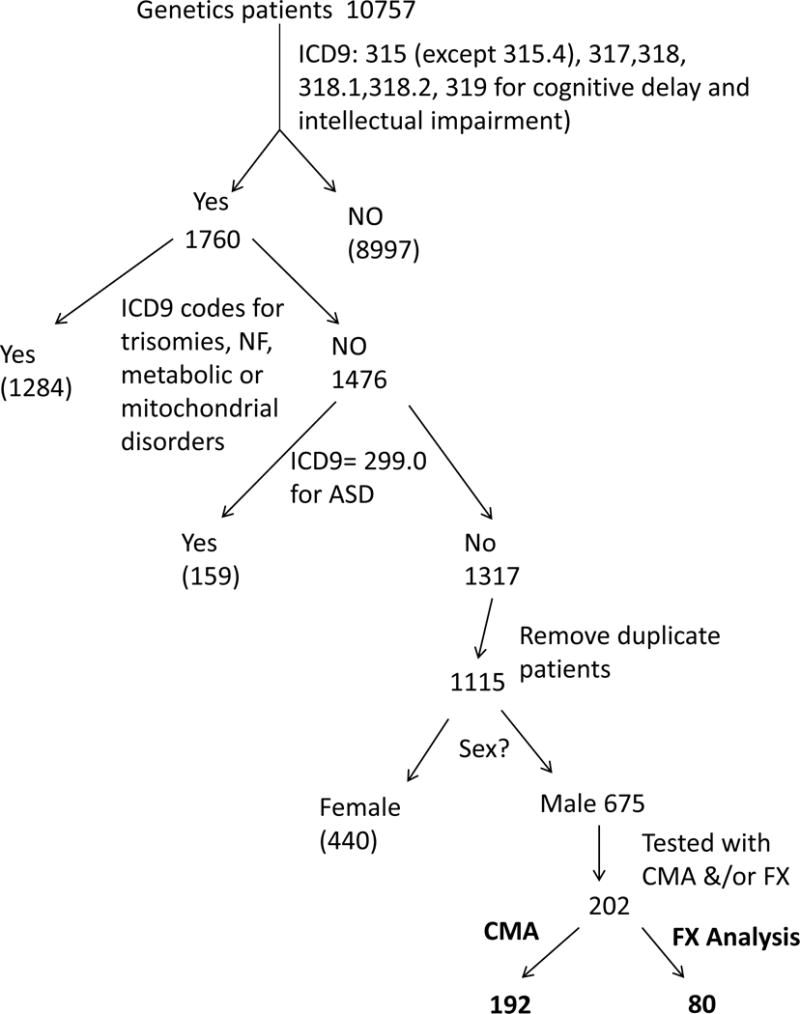
Selection of pediatric male patients with ID/LD (no diagnosis of autism spectrum disorder (ASD)) tested by CMA and/or Fragile X analysis
Patients with autism spectrum disorder (ASD)
To identify male patients with autism who had a CMA and/or FX testing, the following approach was taken (Figure 2). From the 10,757 charges obtained from the billing department, only those billed for males were retained, leaving 5928 charges. Among these 5928 charges, those with the ICD9 code 299.00 (autism disorder and PDD-NOS) were retained. Visits which were also charged with ICD9 codes corresponding to metabolic disorders, trisomies and neurofibromatosis were removed, as explained previously for the ID/LD population, leaving 317 charges. Some of the remaining charges corresponded to several visits of the same patient. Duplicate charges were removed from the 317, leaving 270 patients of interest. 108 of the 270 male patients were tested with CMA and/or FX testing. (Figure 2 illustrates this selection process.)
Figure 2.
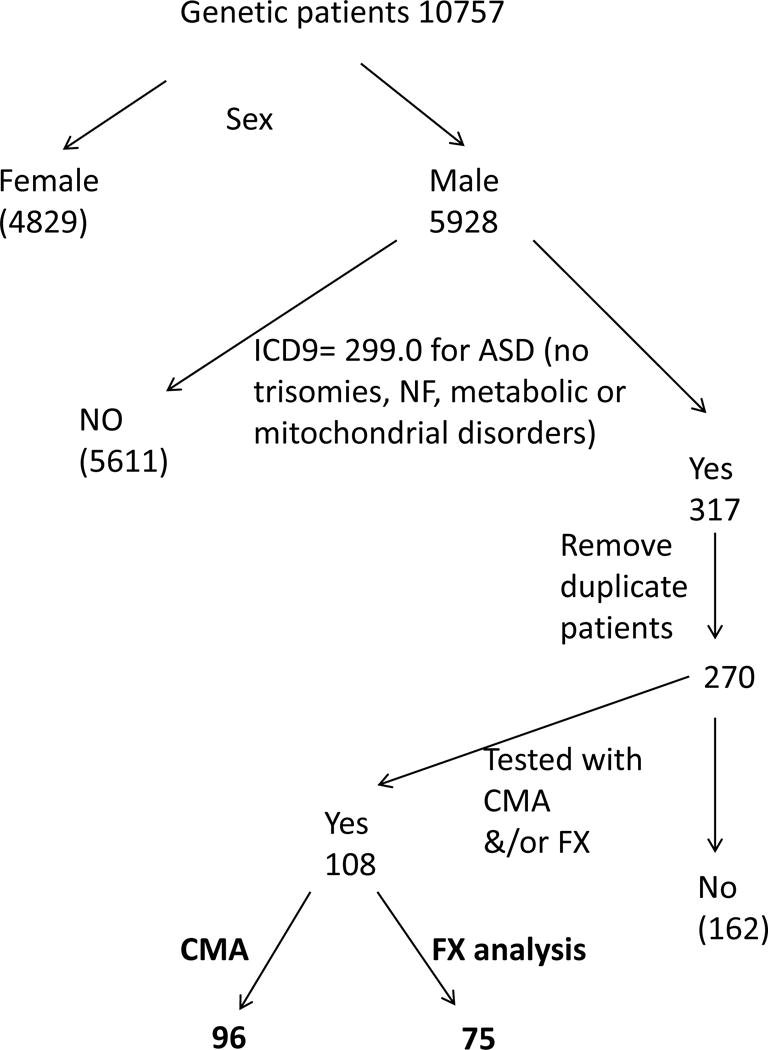
Selection of pediatric male patients with autism spectrum disorder (ASD) tested by CMA and/or Fragile X analysis
Clinical testing (CMA and FX testing)
Test results were collected and analyzed. The clinical notes from the geneticists who had evaluated patients and ordered testing were reviewed for patients whose CMA results were of uncertain significance or included areas of homozygosity. The pictures of the two patients diagnosed with FX were observed for clinical features.
CMA and FX testing were performed by Children’s National Molecular Diagnostics Laboratory. Testing method for CMA: total genomic DNA isolated from peripheral leukocytes was analyzed for CNV and areas of homozygosity by Affymetrix CytoScan Dx SNP-based chromosomal microarray using the GeneChip System 3000Dx and analyzed by Chromosome Analysis Suite Dx Software (ChAS DX Software), approved by the FDA.
Testing method for Fragile X: A three primer PCR amplification strategy (triplet primed PCR) was performed to amplify the CGG repeat region in the 5′untranslated region of FMR1. Fragment analysis was then performed on an ABI3130XL to determine the size of each allele’s repeat region. The fragment size was converted to number of repeats, which were reported using the ACMGG Standards and Guidelines for Clinical Genetics Laboratories.
RESULTS
CMA results of males with isolated ID/LD
Of the 202 male patients considered, 192 were tested by CMA. Fifty-six of the 192 had an abnormal CMA result, which had the potential to cause the patients’ findings (28 of which had yet to undergo confirmatory testing). Twenty-eight of the patients tested harbored pathogenic or likely pathogenic copy number variations (CNVs) associated with their phenotype (sensitivity: 28/192 = 15%).
Of the 28 cases (28/192) that had abnormalities which were not pathogenic yet may provide an answer with further evaluation, two patients harbored a likely pathogenic CNV (but it is unknown whether the CNV correlates with the patient’s phenotype (2/192)), and eight other patients (8/192) harbored areas of homozygosity (AOH) (the autosomal recessive genes located in these areas had not yet been sequenced). Sixteen patients (16/192) harbored CNVs categorized as variants of unknown clinical significance (VUS) (and neither parent has been tested for their child’s VUS, limiting our ability to further interpret these variants), and two patients (2/192) harbored CNVs categorized as VUS and had AOH with yet to be completed parental testing making the interpretation of these VUSes not possible and sequencing performed on the autosomal recessive genes of the AOH was uninformative. (Data are summarized in Table 1.)
TABLE 1.
Summary of CMA results in males with intellectual disability (no autism)
| Description | Number |
|---|---|
| Total male patients with ID/LD | 192 |
| Pathologic or likely pathologic CNV | 28 (15%) |
| Likely pathological CNV but unknown in this phenotype | 2 (1%) |
| AOH, no sequencing completed on the autosomal recessive genes in region | 8 (4%) |
| VUS, no parental testing | 16 (8%) |
| VUS and AOH, no parental testing and sequencing not informative | 2 (1%) |
Table 1 highlights that the CMA sensitivity for 192 males diagnosed with ID/LD ranges from 15% (28/192) to 29% (56/192).
Of note, there were two patients (2/192) harboring AOH but who were eventually diagnosed by another genetic test, unrelated to CMA or FX testing. Two other patients (2/192), who are not listed in the table, harbored AOH in which sequencing of the autosomal recessive genes in these areas was negative.
CMA results of patients with autism
Of the 108 male patients considered, 96 were tested by CMA. Five (5/96) harbored pathogenic or likely pathogenic CNV(s) associated with their phenotype (sensitivity: 5% (5/96)). Two (2/96) harbored AOH. The autosomal recessive genes located in these areas were not sequenced. Two patients (2/96) harbored CNVs categorized as VUSes. Since no parental testing was completed, further interpretation of these VUSes was not possible. It is possible that further investigation of these 4 cases (4/96) would lead to a molecular diagnosis. (Results are summarized in Table 2.)
TABLE 2.
Summary of CMA results in males with ASD
| Description | Number |
|---|---|
| Total male ASD patients | 96 |
| Pathogenic or likely pathogenic CNVs | 5 (5%) |
| AOH, but autosomal recessive genes in region not sequenced | 2 (2%) |
| CNVs that VUS, no parental testing done | 2 (2%) |
Thus, the CMA yield for 96 males diagnosed with autism ranges from 5% (5/96) to 9% (9/96).
Of note, an additional patient harbored a pathogenic CNV, however it is not known to be associated with autism or autistic features. Two other patients harbored AOHs and sequencing of the autosomal recessive genes located in these regions did not provide a diagnosis. Finally, two other patients harbored CNVs categorized as VUSes but inherited from unaffected parents (one maternally and the other paternally inherited), thereby decreasing the chance that these CNVs were causative of the patients’ autism.
Fragile X results of males with isolated ID/LD
Of the 202 male patients considered, 80 were tested for FX. Two were positive, identified with fully expanded allele (Yield: 2/80 = 2.5%). Both our patients had physical findings and family histories concerning for FX.
The clinical characteristics of these two patients as indicated in their genetic clinic notes are reviewed below.
Patient 1 was first evaluated at 40 months when FX testing was recommended. At that time, he had frontal bossing, up-slanting palpebral fissures, long eyelashes of the upper and lower eyelids, bilateral epicanthal folds, large ears, developmental delay, and autistic features (including repetitive behaviors, delayed nonverbal/problem solving skill and history of social impairment: lack of interest to play with other children but during the developmental assessment he showed moderate social interest) without a formal diagnosis of ASD. His family history was consistent with a maternal seven-year-old half-sister and a maternal uncle with a diagnosis of autism. His testing identified one fully expanded allele with more than 200 CGG repeats.
Patient 2 was first evaluated at 14 months of age when FX testing was recommended. He had slight frontal bossing with a recessed hairline, very large and fleshy ears with slight overfolded and squared off helices, axial and peripheral hypotonia and global developmental delay. He was nonverbal and he had a tendency to propel himself out of caregivers arms and throw his body around the room including onto the floor when unhappy. Family history was consistent with mild learning differences in the boys and men on the maternal side of the family. He was found to have size mosaicism for FX with with more than 200 CGG repeats and a premutation size of 146 CGG repeats.
Fragile X test results of patients with autism
Of the 108 male patients considered, 75 were tested for Fragile X. All had a negative result.
DISCUSSION
In summation, the yield of CMA measured by this study in pediatric males with ID/LD of 15–29% is slightly higher than that reported in the literature of 10–20%. The yield of CMA measured by this study in pediatric males with ASD of 5–9% overlaps that reported in the literature of 7–10%. The yield of FX testing measured by this study in pediatric males with LD/ID of 2.5% is close to that in the literature of 2%. The yield of FX testing measured by this study in pediatric males with ASD of 0% is lower than that reported in the literature of 0.5–2%. Here we demonstrate that CMA provides more diagnoses than FX testing in males with ID/DD or autism. Moreover, in the two patients with positive FX testing, there was a strong suspicion of Fragile X syndrome prior to molecular testing, based on their facial features and family history of DD/LD/autism in several individuals on the maternal side of the family. This brings to question whether FX testing should be done as a first-tier test in DD/ID and in the absence of physical findings or family history or rather should be second-tier test.
The increased number of ID/LD and ASD diagnoses has led to higher demand for CMA and FX testing, both of which are recommended by the CDC, American Academy of Pediatrics (AAP) and ACMGG as first-tier evaluation18. There is no recent publication regarding the diagnostic yield of FX testing for patients with ID and/or ASD. Since the yield of FX testing is low, we suggest screening potential candidates for FX prior to ordering the test as opposed to systematically ordering FX testing as a first-tier test. Future research could include establishing a pre-test checklist to predict the possibility of a positive FX result. As previously reported in the literature, the use of physical and psychological checklists by a geneticist/dysmorphologist to select patients with a high probability of FX may reduce the number of individuals to be submitted to molecular evaluation by 60–80%.19 Age-appropriate (pre- vs. post-pubertal) checklists could include family history of ID or ASD, elongated face, large ears, hyperextensible finger joints, large testes, plantar crease, hand biting, hand flapping, tactile defensiveness, poor eye contact, brain anomalies and others.15 Since this study was done in our genetics clinic, clinicians are comfortable with identifying physical findings consistent with FX which may influence testing selection. Check lists may assist the non-geneticist in determining whether this testing is indicated.
Since genetic testing is expensive and from our experience insurance may cover only one test, CMA should have precedence over FX testing, unless further clues point to FX. As always, good history taking including family history and physical exam are the cornerstone of making the diagnosis in these patients.
Our study demonstrated the diagnostic yield of CMA for male patients with isolated ID/LD was satisfactory. Compared to the diagnostic yield reported in previous articles, the number from our study was slightly higher (10–20% vs. 15–29%). The higher yield for CMA in our study may be related to the fact that our population was all males and there is a higher prevalence of ID/LD in males. According to CDC, males have twice the prevalence of any DD than females and more specifically have higher prevalence of ADHD, autism, learning disabilities, stuttering/stammering and other DDs. This discrepancy is likely also related to the nature of our practice (non-reference testing with a large cadre of geneticists), and to the fact that variants of uncertain clinical significance were not excluded.
FX testing is relatively inexpensive and there are no currently clinically available therapies compared to some disorders diagnosed by chromosomal microarray and whole exome. Whole exome has a growing role in the genetic evaluation, but currently CMA is also important.20,21 No matter the ultimate diagnosis and testing regimen, inappropriate selection of testing can lead to a diagnostic odessey which can be expensive and frustrating. Currently there are no clinically available interventions for individuals with FX, however, there are several ongoing trials for therapies and so since FX testing is relatively inexpensive, even with a lower yield compared to CMA and whole exome, there continues to be a role for it in appropriately selected patients, especially since diagnosis is also important for reproductive counselling, neurological and endocrine follow up, and treatment for female carriers due to risk for primary ovarian insufficiency.
We grant that there are several limitations to this study, For example, we relied on ICD9 codes for the selection of our patient population. The ICD9 codes were assigned by a number of geneticists at CNHS. The consistency of selection of the ICD9 codes between providers was not checked. At least one patient who did not have autism, but did have delay, was assigned the 299.0 ICD9 code for autism. This error was noticed while exploring the results. That patient was removed from the autism group and counted as a patient with cognitive impairment. It is possible that other errors passed unnoticed. We aimed to limit our studies to males with isolated ID/LD or ASD by using selected ICD9 codes. In addition, we could not guarantee that those within the isolated ID/LD groups did not have syndromic features due to our approach. Therefore, it is possible that males with additional phenotype findings were included in the study.
CONCLUSION
Early genetic diagnosis in males with ID/LD and autism is important as it may give them access to earlier interventions, e.g., special educational services, and consequently improves outcome. Additionally, it enables providers to explain recurrence risks to the family beyond those of multifactorial inheritance, while sparing the family the burden of a costly diagnostic odyssey. As genetic test coverage by insurance is limited, it is important to select first-tier tests with the highest diagnostic yield. This study’s results align with those reported in the literature regarding the considerably higher yield of CMA compared to FX testing for males with ID/LD or ASD. Further studies need to be done to verify these results, however, we suggest that unless other criteria are considered in addition to ID/LD or ASD, performing FX testing as a first-tier test in males may not be necessary and rather it is better suited in this situation to be a second-tier test. We also suggest the use of pre-test, age-dependent algorithms to help screen patients who should undergo FX testing to avoid missing patients.
TABLE 3.
Comparison of CMA and Fragile X test results between patients with isolated ID/LD and patients with autism
| Population | CMA sensitivity | Fragile X test sensitivity |
|---|---|---|
| LD/ID (no autism): | 14.5–29% (28/192–56/192) | 2.5% (2/80) |
| Autism: | 5–9.5% (5/96–9/96) | 0% (0/75) |
| Total: | 11.5–22.5% (33/288–65/288) | 1.5% (2/155) |
Acknowledgments
The authors thank Chanti Smith and Bryce Comstock for their administrative help. We would also like to thank the Section of Genetics and Metabolism support staff and clinicians for their assistance in helping take care of these patients.
Funding
Kimberly A. Chapman is funded in part from NIDDK grant DK105233.
Footnotes
Conflict of interest.
The authors have indicated they have no potential conficts of interest to disclose. The authors have indicated they have no financial relationships relevant to this article to disclose.
Veronique Weinstein has no conflicts of interest to disclose.
Pranoot Tanpaiboon has no conflicts of interest to disclose related to this publication.
Kimberly A Chapman is funded by NIH, but otherwise no conflicts of interest to disclose.
Nicholas Ah Mew has no conflicts of interest to disclose related to this publication.
Sean Hofherr has no conflicts of interest to disclose related to this publication.
Patient consent
All patients were consented for their genetic testing per instutional protocol.
References
- 1.Petersen MC, Kube DA, Palmer FB. Classification of developmental delays. Semin Pediatr Neurol. 1998;5(1):2–14. doi: 10.1016/s1071-9091(98)80012-0. [DOI] [PubMed] [Google Scholar]
- 2.Michelson DJ, Shevell MI, Sherr EH, Moeschler JB, Gropman AL, Ashwal S. Evidence report: Genetic and metabolic testing on children with global developmental delay: report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology. 2011;77(17):1629–1635. doi: 10.1212/WNL.0b013e3182345896. [DOI] [PubMed] [Google Scholar]
- 3.Boyle CA, Boulet S, Schieve LA, et al. Trends in the prevalence of developmental disabilities in US children, 1997–2008. Pediatrics. 2011;127(6):1034–1042. doi: 10.1542/peds.2010-2989. [DOI] [PubMed] [Google Scholar]
- 4.Shen Y, Dies KA, Holm IA, et al. Clinical genetic testing for patients with autism spectrum disorders. Pediatrics. 2010;125(4):e727–735. doi: 10.1542/peds.2009-1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schaefer GB, Mendelsohn NJ, Professional P, Guidelines C. Clinical genetics evaluation in identifying the etiology of autism spectrum disorders: 2013 guideline revisions. Genet Med. 2013;15(5):399–407. doi: 10.1038/gim.2013.32. [DOI] [PubMed] [Google Scholar]
- 6.Miles JH. Autism spectrum disorders–a genetics review. Genet Med. 2011;13(4):278–294. doi: 10.1097/GIM.0b013e3181ff67ba. [DOI] [PubMed] [Google Scholar]
- 7.Gallagher A, Hallahan B. Fragile X-associated disorders: a clinical overview. J Neurol. 2012;259(3):401–413. doi: 10.1007/s00415-011-6161-3. [DOI] [PubMed] [Google Scholar]
- 8.Roesser J. Diagnostic yield of genetic testing in children diagnosed with autism spectrum disorders at a regional referral center. Clin Pediatr (Phila) 2011;50(9):834–843. doi: 10.1177/0009922811406261. [DOI] [PubMed] [Google Scholar]
- 9.McGrew SG, Peters BR, Crittendon JA, Veenstra-Vanderweele J. Diagnostic yield of chromosomal microarray analysis in an autism primary care practice: which guidelines to implement? J Autism Dev Disord. 2012;42(8):1582–1591. doi: 10.1007/s10803-011-1398-3. [DOI] [PubMed] [Google Scholar]
- 10.Clifford S, Dissanayake C, Bui QM, Huggins R, Taylor AK, Loesch DZ. Autism spectrum phenotype in males and females with fragile X full mutation and premutation. J Autism Dev Disord. 2007;37(4):738–747. doi: 10.1007/s10803-006-0205-z. [DOI] [PubMed] [Google Scholar]
- 11.Hatton DD, Sideris J, Skinner M, et al. Autistic behavior in children with fragile X syndrome: prevalence, stability, and the impact of FMRP. Am J Med Genet A. 2006;140A(17):1804–1813. doi: 10.1002/ajmg.a.31286. [DOI] [PubMed] [Google Scholar]
- 12.Hagerman RJ, Amiri K, Cronister A. Fragile X checklist. Am J Med Genet. 1991;38(2–3):283–287. doi: 10.1002/ajmg.1320380223. [DOI] [PubMed] [Google Scholar]
- 13.Hill MK, Archibald AD, Cohen J, Metcalfe SA. A systematic review of population screening for fragile X syndrome. Genet Med. 2010;12(7):396–410. doi: 10.1097/GIM.0b013e3181e38fb6. [DOI] [PubMed] [Google Scholar]
- 14.Coffee B, Keith K, Albizua I, et al. Incidence of fragile X syndrome by newborn screening for methylated FMR1 DNA. Am J Hum Genet. 2009;85(4):503–514. doi: 10.1016/j.ajhg.2009.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Christofolini DM, Abbud EM, Lipay MV, et al. Evaluation of clinical checklists for fragile X syndrome screening in Brazilian intellectually disabled males: proposal for a new screening tool. J Intellect Disabil. 2009;13(3):239–248. doi: 10.1177/1744629509348429. [DOI] [PubMed] [Google Scholar]
- 16.Miller DT, Adam MP, Aradhya S, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010;86(5):749–764. doi: 10.1016/j.ajhg.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sherman S, Pletcher BA, Driscoll DA. Fragile X syndrome: diagnostic and carrier testing. Genet Med. 2005;7(8):584–587. doi: 10.1097/01.GIM.0000182468.22666.dd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moeschler JB, Shevell M. Comprehensive evaluation of the child with intellectual disability or global developmental delays. Pediatrics. 2014;134(3):e903–918. doi: 10.1542/peds.2014-1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mandel JL, Biancalana V. Fragile X mental retardation syndrome: from pathogenesis to diagnostic issues. Growth Horm IGF Res. 2004;14(Suppl A):S158–165. doi: 10.1016/j.ghir.2004.03.034. [DOI] [PubMed] [Google Scholar]
- 20.Yang Y, Muzny DM, Xia F, et al. Molecular findings among patients referred for clinical whole-exome sequencing. Jama. 2014;312(18):1870–1879. doi: 10.1001/jama.2014.14601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Posey JE, Harel T, Liu P, et al. Resolution of Disease Phenotypes Resulting from Multilocus Genomic Variation. The New England journal of medicine. 2017;376(1):21–31. doi: 10.1056/NEJMoa1516767. [DOI] [PMC free article] [PubMed] [Google Scholar]