

Abstract
Antibodies (Abs) produced by immunoglobulin (IG) genes are the most diverse proteins expressed in humans. While part of this diversity is generated by recombination during B cell development and mutations during affinity maturation, the germline IG loci are also diverse across human populations and ethnicities. Recently, proof-of-concept studies have demonstrated genotype-phenotype correlations between specific IG germline variants and the quality of Ab responses during vaccination and disease. However, the functional consequences of IG genetic variation in Ab function and immunological outcomes remain underexplored. Here we outline interconnections between IG genomic diversity and Ab expressed repertoires and structure. We further propose a strategy for integrating IG genotyping with functional Ab profiling data as a means to better predict and optimize humoral responses in genetically diverse human populations, with immediate implications for personalized medicine.
The molecular basis for antibody diversity
Antibodies (Abs) have long been appreciated as key constituents of the adaptive immune response. Their function is to enable selective recognition and mediate immune responses to novel foreign antigens. This is accomplished through the somatic generation of vast repertoires of hundreds of millions of unique Ab receptors that can be selected, matured and ultimately participate in the formation of long-term memory during B cell development and activation. As a consequence of this diversity, even after nearly a century of research, the complexity of the Ab response within and between individuals is only beginning to be delineated at the molecular and genetic level.
Hundreds of variable (V) and dozens of diversity (D) and joining (J) immunoglobulin (IG) germline gene segments across three primary loci in the human genome comprise the necessary building blocks of the expressed Ab heavy and light chain repertoires [1]. Whereas the heavy chain is encoded by genes at the IG heavy chain locus (IGH), the light chain can be encoded by genes at either the IG kappa or lambda chain loci (IGK; IGL) [1]. The naive Ab repertoire is formed by assembling variants of these building blocks using a specialized V(D)J recombination process that somatically joins various V, D, and J segments (or V and J at IGK and IGL), with the introduction and deletion of P-and N-nucleotides at V(D)J junctions, dramatically increasing diversity, and ultimately by the pairing of different heavy and light chains (Figure 1) [2]. Considering these processes alone, a given baseline or primary naive repertoire can theoretically sample from 1015 different Abs [3]. The extraordinary diversity of the naive repertoire ensures that it will likely contain a naive Ab with at least weak initial binding against a vast array of antigens.
Figure 1. A basic overview of key elements that contribute to the diversity of naïve and memory repertoires.
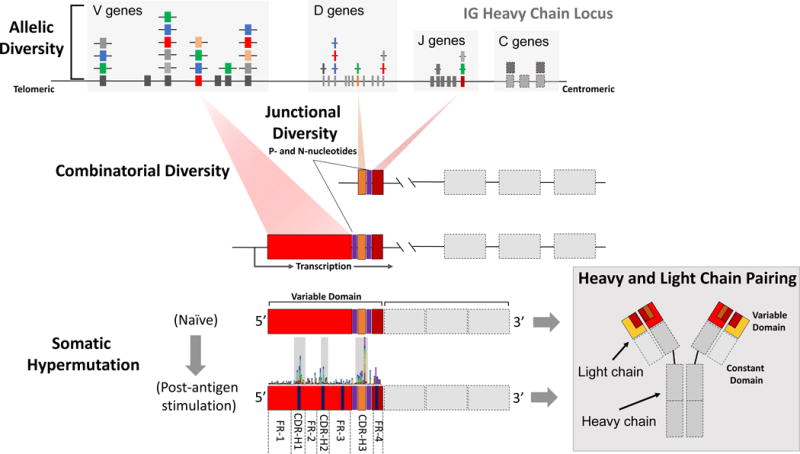
A basic schematic of the germline IGH locus is shown (not to scale), consisting of clusters of tandemly arrayed IGH V, D, J, and C gene segments. For a subset of these segments, multiple alleles are shown, representing population-level “Allelic Diversity” (see Table 1). During the initial formation of the naïve repertoire a single IGH D and J gene segments on one of two chromosomes in a given B cell are somatically recombined, followed by an additional round of recombination joining the paired D and J segment with an accompanying V segment; at each of these steps, P-and N-nucleotides are added at the D-J and V-D junctions (“Junctional Diversity”). This process, known as V(D)J rearrangement, is the basis for “Combinatorial Diversity”. The recombined V (red), D (orange), and J (maroon) segments will then be transcribed, and following splicing, will be paired with a C gene (gray). The somatic recombination process also occurs at one of two loci encoding the Ab light chain gene segments (IGK and IGL; except it involves only V (yellow), J (maroon) and C (light gray) gene segments). Two identical heavy chains and two identical light chains are ultimately paired to form a functional Ab; thus, additional diversity in the expressed Ab repertoire comes from “Heavy and Light Chain Pairing”. Together, the V, D, and J segment depicted comprise the variable domain of the heavy chain of a functional antibody, and together with the variable domain of the light chain, encoded by V and J segments, are responsible for Ag-binding. The C genes of both heavy and light chains comprise the constant domain, providing structural and/or effector functions of the Ab. As shown here for the heavy chain, the variable domain is partitioned into four framework regions (FRs) and three complementarity-determining regions (CDRs). Following Ag stimulation, “Somatic Hypermutation” introduces additional variation in the variable domain of the Ab (vertical purple bars), with the aim of improving binding affinity. While mutations that arise via SHM can occur across the FR and CDR regions, these are most prevalent in CDR regions, as illustrated by the hypothetical frequency histogram shown between the unmutated and mutated IG heavy chain RNA. While the general molecular mechanisms outlined here have long been realized as the primary determinants of diversity within a given expressed Ab repertoire, there is a growing appreciation for the contribution of “Allelic Diversity” as well, particularly as this pertains to repertoire differences observed between unrelated individuals.
Even so, this impressive baseline diversity can be subsequently augmented when a B cell encounters and is stimulated by antigen to undergo somatic hypermutation (SHM; Figure 1), resulting in lineages of tens of thousands of clonally derived affinity maturation variants of the initial Ab. Specifically, SHM introduces somatic mutations throughout the variable portion of the Ab, including targeted hotspots residing within the antigen-contacting hypervariable complementarity-determining regions (CDRs). This process ultimately increases the affinity and specificity of the Ab for binding the target epitope, facilitating a highly focused antigen-specific response.
While the prevailing paradigm for investigating B cell and Ab-mediated responses has placed emphasis on the importance of the unique molecular mechanisms cited above in the generation of key functional antibodies, there is a growing appreciation for the fact that IG genes are highly variable at the germline level, exhibiting extreme allelic polymorphism and gene copy number variation (CNV) between individuals and across populations [4–9]. Recent studies have begun to highlight that, in addition to diversity introduced during V(D)J recombination, heavy and light chain pairing, and SHM, IG germline variation (e.g., allelic variation; Figure 1) play a part in determining the development of the naive repertoire, with downstream impacts on signatures observed in the memory compartment, and the capacity of an individual to mount an Ab response to specific epitopes [10–16].
IG loci haplotype diversity in the human population
Recent genomic sequencing indicates that IG loci, specifically IGH, may be among the most polymorphic in the human genome [17]. Across IGH, IGK, and IGL, there are currently >420 alleles catalogued in the ImMunoGeneTics information system database (IMGT) [18–21] that have been described from germline DNA in the human population, with an enrichment of non-synonymous variants (Table 1). Although the validity of some alleles in IMGT have been called into question [22], the number of polymorphic alleles continues to grow [11,23,24], especially as IG gene sequencing is conducted in increasing numbers of non-Caucasian samples [7,9,25]. A recent study conducted in 28 indigenous South Africans identified 122 non-IMGT IGHV alleles [9]. In addition to IG allelic variation and single nucleotide polymorphisms (SNPs), CNVs, including large deletions, insertions and duplications (~8–75 Kb in length), are also prevalent in IG regions (Table 1). Using IGH as an example, up to 29 of the 58 functional/ORF IGHV genes may vary in genomic copy number [4,6,7,11,26–28]; CNVs of IGH D (“diversity”) and constant (C) region genes are also known [11,12,29]. Until recently, primarily due to technical difficulties caused by the complex genomic architecture of the IG loci, none of the known CNVs in IGHV had been sequenced at nucleotide resolution [7]; many likely remained undescribed at the genomic level.
Table 1.
Allelic, copy number, and amino acid variation for IG functional and open reading frame genes catalogued in IMGT*.
| Family | Genes | Alleles | NS Variants | S Variants | CDR-H1 NS Variants | CDR-H2 NS Variants | Genes in CNV |
|---|---|---|---|---|---|---|---|
|
| |||||||
| IGHV1 | 11 | 40 | 19 | 13 | 2 | 3 | 6 |
| IGHV2 | 4 | 23 | 26 | 9 | 3 | 1 | 1 |
| IGHV3 | 27 | 109 | 82 | 57 | 9 | 17 | 12 |
| IGHV4 | 10 | 78 | 92 | 71 | 11 | 8 | 8 |
| IGHV5 | 2 | 9 | 4 | 4 | 0 | 0 | 1 |
| IGHV6 | 1 | 2 | 0 | 1 | 0 | 0 | 0 |
| IGHV7 | 2 | 6 | 4 | 0 | 0 | 0 | 1 |
|
| |||||||
| Sub Total | 58 | 267 | 227 | 155 | 25 | 29 | 29 |
|
| |||||||
| IGKV1 | 20 | 35 | 33 | 17 | 4 | 1 | 1 |
| IGKV2 | 11 | 18 | 14 | 4 | 1 | 1 | 0 |
| IGKV3 | 8 | 18 | 24 | 9 | 2 | 1 | 0 |
| IGKV4 | 1 | 1 | NA | NA | NA | NA | 0 |
| IGKV5 | 1 | 1 | NA | NA | NA | NA | 0 |
| IGKV6 | 3 | 5 | 2 | 0 | 0 | 0 | 0 |
| IGKV7 | 0 | 0 | NA | NA | NA | NA | 0 |
|
| |||||||
| Sub Total | 44 | 78 | 73 | 30 | 7 | 3 | 1 |
|
| |||||||
| IGLV1 | 7 | 12 | 4 | 2 | 0 | 2 | 1 |
| IGLV2 | 6 | 20 | 13 | 8 | 2 | 3 | 0 |
| IGLV3 | 11 | 18 | 14 | 5 | 3 | 3 | 0 |
| IGLV4 | 3 | 6 | 2 | 1 | 0 | 0 | 0 |
| IGLV5 | 5 | 10 | 3 | 2 | 0 | 0 | 1 |
| IGLV6 | 1 | 2 | 2 | 0 | 0 | 0 | 0 |
| IGLV7 | 2 | 3 | 1 | 0 | 0 | 0 | 0 |
| IGLV8 | 1 | 3 | 1 | 1 | 0 | 0 | 1 |
| IGLV9 | 1 | 3 | 0 | 2 | 0 | 0 | 0 |
| IGLV10 | 1 | 3 | 4 | 1 | 1 | 0 | 0 |
| IGLV11 | 1 | 2 | 1 | 1 | 0 | 0 | 0 |
|
| |||||||
| Sub Total | 39 | 82 | 45 | 23 | 6 | 8 | 3 |
|
| |||||||
| Total | 141 | 427 | 345 | 208 | 38 | 40 | 33 |
Abbreviations: NS, Non-synonymous; S, synonymous;
Data accessed from IMGT February, 2017.
The high prevalence of IG allelic and locus structural diversity translates into extreme levels of inter-individual haplotype variation [4–7]. For example, recent comparisons of the two available completed assemblies for the IGHV gene region revealed that two human chromosomes can vary by >100 Kb of sequence, with >2,800 SNPs, and CNVs of 10 IGHV functional/ORF genes [7,17]. In population sequencing experiments, extreme examples of heterozygosity have been noted, with evidence of some individuals carrying >1 allele at every IGHV coding gene [9]. Supporting earlier genetic mapping data [4,5], more recent analysis of inferred haplotypes from Ab repertoire data surveyed in 9 individuals revealed that all 18 haplotypes characterized were unique [6]. Furthermore, at the population level, of the few SNPs and CNVs screened within IGH, allele and genotype frequencies have been shown to vary considerably between ethnic backgrounds [7–9,15], with evidence of selection [7]. Despite the support for elevated germline diversity, genomic resources for IG loci continue to lag behind other regions of the genome [26]. Because of this, the comprehensive and accurate genotyping of IG polymorphisms remains a significant challenge [26,30], and as a result, the full extent of IG polymorphism and implications for human health are yet to be uncovered [26]. However, it is plausible that population level diversity in the IG loci, particularly in IGH, will rival that of other complex immune gene families, such as the human leukocyte antigen (HLA) and killer cell immunoglobulin-like receptor (KIR) genes. These genes are also characterized by extreme haplotype diversity, due to CNV and coding region variation [31,32]; HLA genes, for example, have thousands of known alleles [31]. In contrast to IG genes, HLA and KIR have been studied more extensively across human populations, and have demonstrated critical roles in disease [31,32].
The influence of IG germline diversity in the expressed antibody repertoire and Ab function
Our limited knowledge of IG population diversity has hindered our ability to comprehensively test for direct connections between IG germline polymorphisms, variation in the repertoire generated after recombination, amino acid variation in the Ab produced, and ultimately Ab function. However, advances in high-throughput sequencing technology now enable extensive characterization of the expressed Ab repertoire [33–35], creating opportunities for beginning to investigate the heritability of the Ab response at fine scale resolution. Applications of these methods, collectively referred to as repertoire sequencing (“IgSeq” or “RepSeq”), have already led to a wealth of new discoveries in a range of contexts [33,36]. These include general observations that key features of the Ab repertoire show extensive variability between healthy individuals [10,11,13,14,37], and a limited overlap of BCR clones between individuals, even monozygotic (MZ) twins [10,13,14]. However, RepSeq studies have also revealed that these inter-individual differences are not necessarily random, but likely have a strong underlying genetic component, providing initial support for the importance of germline IG polymorphism in determining the naive and Ag-stimulated Ab repertoire. For example, several recent studies have revealed that V, D, and J gene usage in the naive repertoire is much more highly correlated between MZ twins than between unrelated individuals [10,13,14], and that IG gene usage patterns are consistent across time points within a given individual [38]. A role for genetic factors can be seen for other repertoire features in twins as well, including the degree of SHM [13], and the distribution of CDR-H3 length and clone convergence [10,13,14]. Intriguingly, although existing data suggest that features in the memory compartment are more stochastic, likely reflective of random recruitment and transient proliferation, certain genes and repertoire features exhibit predictable patterns even in memory B cells [39,10,13,14].
Studies of repertoire heritability are consistent with a number of examples for which germline IG polymorphisms have been explicitly linked to features in the expressed Ab repertoire [12,15,40–42] (hypothetical examples of IG genotype effects on the repertoire are shown in Box 1, Figure IA). Sasso et al. [40] reported the first direct connection to IG genotype, reporting that CNV of IGHV1-69 was tightly correlated with its relative usage in tonsilar B cells. Our own work has also demonstrated this relationship, but uncovered associations for IGHV1-69 coding and potentially non-coding polymorphism as well as CNV [15]. Inferred deletions of IGHD genes have also been shown to associate with variation in D-J pairing frequencies, demonstrating that germline effects on the repertoire extend beyond V genes [12]. An interesting aspect of IGH CNVs is that, in addition to observed effects of these variants on the genes within the CNV event, they also can impact the usage of genes elsewhere in the locus [12,15]. For example, we recently observed apparent long-range effects of IGHV1-69 CNV in the naive and memory repertoire, in that individuals with fewer IGHV1-69 germline copies and reduced usage showed consistently higher usage of IGHV genes over 200 Kb away [15]. The mechanisms underlying the observed effects of CNVs in human IG loci remain technically difficult to assess experimentally, but it has been speculated that these large changes in locus architecture (i.e., deletions and insertions) could alter regulatory systems related to V(D)J recombination [12,15], for example, by modifying the chromatin landscape, cis-regulatory elements and transcription factor binding, and/or the physical locations of IG V, D, and J genes. All of these factors are known to be key determinants of IG gene accessibility and usage frequencies in the mouse [43,44].
Box 1. The influence of IG germline polymorphism on Ab repertoire variation and functional Ab structural residues.
-
-Although the roles of IG germline variants have not been comprehensively studied there is now convincing evidence that they can influence Ab repertoire variation and function in two main ways (A & B). In addition, known functional variants exhibit allele frequency variation between human populations (C):
-
-(A) Gene copy number changes and coding/non-coding SNPs in IGHV genes have been shown to correlate with gene usage patterns in the naïve repertoire, the memory repertoire, patterns of SHM, class-switch frequency, and circulating Ab titres (Figure IA).
-
-(B) There are now many examples that provide evidence for functional effects of germline variants encoded in CDR-H1 and CDR-H2, many of which are polymorphic and vary between human populations. Based on known IGHV alleles in the IMGT database, residues within CDR-H1/H2 that have a higher probability of making Ag contact, are also more likely to be associated with a polymorphic allele (Figure IB).
-
-(C) Several positions in IGHV genes that encode residues critical for antigen binding are polymorphic and exhibit different genotype frequencies between human populations and ethnicities (Figure IC).
-
-
Figure I. Impacts of IG germline polymorphism on Ab repertoire/structural diversity.
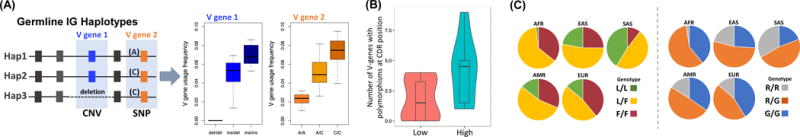
(A) Hypothetical examples of associations between IG gene region CNV (V gene 1 insertion/deletion) and SNP (non-coding regulatory variant, A/C) genotypes and V gene usage frequencies in the expressed Ab repertoire. (B) Violin plot showing non-synonymous polymorphism rates in CDR positions with high (>0.6; “High”, blue) or low (<0.25; “Low”, red) frequency of contact with antigen, as labeled on the X-axis. The Y-axis records, for each CDR-H1 and CDR-H2 position, the number of IMGT IGHV genes that have alleles with nonsynonymous polymorphisms at that position. The positional probability of antigen contact was calculated for each CDR position as the percentage of 150 crystal structures of antibody-antigen complexes from the protein database (PDB) where any atom of that residue is within 5 Angstroms of any antigen atom. Allelic variation is enriched in antigen-contact sites, in that the number of IGHV genes with alleles containing nonsynonymous polymorphisms is greater for high contact probability positions. (C) Genotype frequency differences between five human ethnic groups (Africans, AFR; East Asians, EAS; South Asians, SAS; Central/South American, AMR; and Europeans, EUR), published by the 1000 Genomes Project [80]*, at two SNPs in IGHV1-69 that have been shown to encode functional residues critical for neutralizing Abs against the influenza HA stem (F54 and L54 amino acid associated alleles; SNP rs55891010; left panel), and “NEAT2” domain of Staphlyococcus aureus (R50 and G50 alleles; SNP rs11845244; right panel). In the left panel, the F allele encodes the functional critical phenylalanine residue, and in the right panel, the primary glycine residue is encoded by the G allele. Interestingly, in both cases, the frequency of individuals lacking alleles encoding the critical residues varies among populations, with the L/L and R/R genotypes showing the lowest frequencies in Africans, and the highest frequencies in South Asians. rs55891010 and rs11845244 are in linkage disequilibrium, and thus R50 and L54 amino acids (and likewise, G50 and F54) tend to co-occur in alleles of IGHV1-69. This explains similarities in genotype frequency estimates between the two SNPs in each population. *Although these genotypes may contain error due to confounds of unrepresented CNV information, they can provide insight into potential population differences.
A role for non-coding polymorphisms is also strongly supported by early work conducted in the human IGK region that directly showed that a variant associated with Haemophilus influenza infection susceptibility in the recombination signal sequence (RSS) of IGKV2-29 significantly decreased gene rearrangement frequency [42]. RSSs, which are critical for the recruitment of RAG1/2 genes have also been demonstrated to impact IgHV gene usage in mice [43,44]. Moreover, extensive work in the murine IG gene loci have uncovered important roles for other key cis-regulatory sequences and transcription factors as well [45,46]. Such analyses have not yet been comprehensively conducted in humans, and as a result our knowledge of the IG regulatory elements involved in the formation of the expressed Ab repertoire is restricted to canonical RSS, promoter, enhancer elements, and class switch regions (CSRs). However, even for these well-known non-coding regulatory regions, limited data on human population-level variation exists, and thus the broader consequences of polymorphism in these elements on Ab repertoire variability has not been explored.
Although direct links between repertoire variability and human IG CNVs and non-coding polymorphisms remains limited to the few examples discussed above, additional evidence from expressed Ab repertoire studies in unrelated individuals also highlights the clear potential for these variants to have pervasive impacts on Ab repertoire features, particularly gene usage in the naïve compartment. Most demonstrable is the fact that many of the genes with the most variability in naïve repertoire usage across individuals are also known to be in CNV, including examples of the complete absence of genes in the expressed Ab repertoires of some donors [6,10–12]. In addition, allele-specific usage in the naïve Ab repertoires of individuals heterozygous at a given IGHV gene has been demonstrated, also clearly suggesting a role for non-coding variation and CNV [11]. Moreover, although effects of germline IG polymorphism may be most evident on a per gene basis, it is worth discussing that findings from MZ twins demonstrating that certain CDR-H3 features are highly heritable [13,14] indicate that even strong genetically determined biases on individual V, D, and J gene usage (and thus their non-random combination during V(D)J rearrangement), could also be directly linked to variation observed within CDR-H3. This is an important point given that CDR-H3 variation has classically been considered independent of the germline [13,14].
In addition to effects of IG polymorphism on gene usage, functional CDR variants can also be directly encoded in the genome. For example, across the ~267 coding alleles catalogued in IMGT for functional and ORF IGHV genes, 60% of the 382 polymorphic positions are non-synonymous (Table 1), including sites located in CDR-H1 and CDR-H2 with predicted relevance to Ab functional residue diversity (Box 1; Figure IB). Although the CDR-H3 loop, formed at the V(D)J junction, is the most diverse region of an antibody and is a principal determinant of specificity [47,48], there is a growing appreciation for the importance of residues outside of CDR-H3 in antigen recognition and binding [15,49–51]. For example, recent analyses have shown that the median length of CDR-H2, which is solely encoded by germline V gene sequence, is substantially longer than that of CDR-H3, and typically forms the same number of interactions with antigen [52]. Specifically, analyses of antigen binding residues (ABRs; which roughly correspond to CDRs, but differ slightly in their boundaries) have shown that Abs contain a median of 6, 6, and 4 contact residues in the heavy chain CDR-H3, H2, and H1 ABR regions, respectively. In addition, the overall percentage of energetically important Ag-binding residues within each ABR follows the same rank order, with circa 31%, 23% and 14% for H3, H2 and H1, respectively. Similar trends were noted for light chain ABRs as well [52]. In addition, considering that many known non-synonymous sites reside outside of CDRs (Table 1), it is worth highlighting the fact that there are also examples demonstrating indirect effects of framework region variants on Ag-binding [53,54].
The identification of shared Ab immune response signatures across individuals
A critical question is whether the germline effects on the repertoire outlined above can also partially account for inter-individual variation of the Ab-mediated response in disease and clinical phenotypes. The initial observation from RepSeq studies that essentially no antibody clones were shared among individuals, including MZ twins, posed a challenge to comparative Ab repertoire analysis: how could correlates of protection be identified in the Ab repertoire if every individual was responding with different antibodies? However, an answer began to emerge with the observation that in multiple settings, including viral and bacterial infection, different individuals have been shown to respond to a given antigen with antibodies that share convergent amino acid signatures [13,49,55–57,54,58]. These convergent antibodies are often encoded by common V genes or sets of V genes, and specific amino acid residues in their CDRs enable them to converge upon a common binding solution against a shared antigen. Critically, in some cases evaluated, convergent signatures include amino acid residues that are directly encoded in the germline. The occurrence of such convergent antibody responses highlight the potential for tracking common immune responses across individuals, and understanding the role of genetic factors, even when each individual creates unique antibodies. Importantly, the implications of this line of thinking could be broad, as IG gene biases have been observed in contexts other than infection, including autoimmunity and cancer [59,60]. Moreover, IG gene biases may also extend to usage patterns of D and J genes, light chain genes, and heavy and light chain V gene pairing frequencies [56,61,62].
Structural residues critical for Ag binding and involved in biased gene usage are encoded in the germline and exhibit population variability
There are now many instances for which functional contributions of biased IG genes have been traced back to specific germline encoded residues, including sites that are polymorphic in the human population [15,16,50,53–55,63–65]. These examples illuminate a direct role of the IG germline in disease-associated Ab responses. In the case of stem-directed broadly neutralizing Abs (BnAbs) against influenza hemagglutinin (HA), the most prevalent Abs use the heavy chain gene IGHV1-69 [66,67,67–70]. These IGHV1-69 BnAbs recognize an overlapping epitope of group 1 influenza A viruses and only amino acids from IGHV make contact with HA. Importantly, of the 14 known alleles at IGHV1-69, only those encoding a critical phenylalanine at position 54 (F54) within CDR-H2 have a major role in shaping the BnAbs response [16,15,55,71]. Although IGHV1-69 F54-encoding alleles are dominant, there is a growing list of additional HA-directed BnAbs that also show IG germline biases [51,56,72–74], including those also known to be polymorphic with respect to coding variants and CNVs.
Interestingly, there are additional instances of biased IGHV1-69 allele usage in other disease contexts, with both overlapping and contrasting patterns to that observed for influenza. For example, F54 alleles are predominately observed by IGHV1-69 associated with CLL, whereas alleles encoding a leucine (L54) at this position are primarily used by non-neutralizing anti-gp41 Abs in HIV-1 [63,64]. Moreover, it has been shown that IGHV1-69 F54 alleles, in comparison to L54 alleles, have lower usage in the memory B cell pool [10,15]. This observation may be similar to trends noted for IGHV4-34, which is also significantly underrepresented in the memory compartment of healthy individuals [10], presumed to reflect a selective pressure against autoreactive Abs [75,76].
Other polymorphic positions in the framework regions of IGHV1-69, in conjunction with CDR-H2 54, have also recently been shown to influence Ab binding of MERs-CoV [53] and the Staphylococcus aureus NEAT2 (NEAr iron Transporter 2) domain [54]. In the example of NEAT2, neutralizing Abs encoded by IGHV1-69 alleles carrying an arginine (R) at position 50 in place of glycine (G) showed significantly reduced NEAT2 binding [54]. Interestingly, based on publically available data, the frequencies of critical alleles within polymorphic positions of IGHV1-69 vary across populations (Box 1; Figure IC).
A strategy for defining relationships between IG polymorphisms, expressed Ab signatures, and functional outcomes
Considering the evidence above, we argue that the antigen-specific Ab repertoire is likely influenced by host genotype. Although the genetic basis for repertoire and germline gene biases have not been comprehensively investigated, several recent studies provide a strategy for systematically integrating data on IG polymorphism and Ab responses at the population and molecular level to provide unique insight into Ab signatures associated with disease.
We have begun to explore this idea in detail at the IGHV1-69 locus in the context of influenza vaccination [15]. Providing strong proof-of-concept, by initially focusing on observed IGHV1-69 allelic usage bias against a critical broadly neutralizing epitope, we genotyped the IGHV1-69 Phe54/Leu54 allele and copy number frequencies in a cohort of 85 H5N1 vaccinees, including 18 individuals with accompanying Ab repertoire data [15]. Drawing directly on aspects of repertoire heritability reviewed above, we found robust connections between these polymorphisms and repertoire gene usage in both the unmutated IgM (naive) and IgG memory repertoires, with IGHV1-69 germline gene usage increasing with the number of copies of F54 alleles. In addition to usage frequencies, IGHV1-69 genotype also associated with IGHV1-69 B cell expansion, SHM, and Ig class switching. It is important to note that, these genotype effects extended to levels of circulating anti-HA stem BnAbs post-vaccination, with individuals carrying only germline-encoded CDR-H2 L54 alleles having lower IGHV1-69 BnAbs. Furthermore, with direct repertoire sequencing of all subjects, we were able to specifically demonstrate that IGHV1-69 convergent anti-BnAb sequence group members were only effectively elicited in carriers of the F54 allele. These results are bolstered by similar observations recently made by two other groups that also carried out IGHV1-69 F54/L54 allele genotyping in their cohorts [16,55]. Altogether, these data demonstrate that genetically determined baseline differences in the Ab repertoire can set the stage for disease-related responses.
A crucial aspect of this story (which is expected to emerge in other cases as well) is that the frequency of IGHV1-69 F54 alleles and CNV varies considerably across populations [7,15]. Specifically, the number of individuals that would be predicted to lack the capacity to generate effective IGHV1-69 BnAbs was much higher in some populations. However, we and others have shown that individuals lacking IGHV1-69 F54 alleles likely utilize other germline genes in place of IGHV1-69 [51,55]. This finding in particular both highlights the complexity of the Ab response, and demonstrates that the integration of genotyping information can help provide a more nuanced interpretation of the signatures discovered in the expressed repertoire. Moreover, it suggests that efforts should be made to study these complex responses in larger and more diverse cohorts, including individuals from presently understudied populations.
Building on findings in these initial studies [15,16,55], we propose a framework for integrating genotypic information into future studies of the Ab response in wellness and disease (Figure 2, Key Figure). The general strategy is as follows: (1) identify IG gene biases observed in a disease-related or epitope-specific response; (2) characterize this response at the population level by performing comprehensive genotyping of coding, non-coding, and gene copy number variants at and around the locus of interest (and others if there is rationale); (3) perform repertoire sequencing and analysis of the response in all relevant B cell subsets to identify all antibody convergence groups with allele bias; and (4) evaluate genotype-phenotype linkages of the functional Ab response and specific antibody convergence groups.
Figure 2 (Key Figure). A new paradigm for integrating genotypic information into the study of the Ab mediated response in disease and clinical phenotypes.
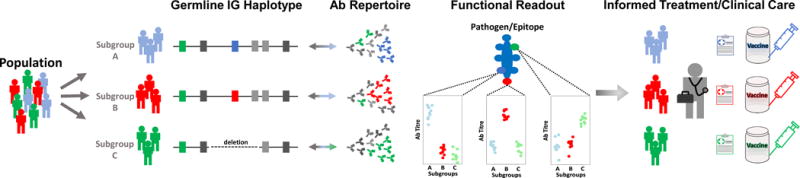
In the proposed paradigm, a population cohort is partitioned into subgroups based on functional genotypes/haplotypes that are directly associated with subgroup-specific signatures in the expressed repertoire and other relevant phenotypes (e.g., Ab titer; clinical outcome) associated with the Ab response to a given antigen/epitope. This partitioning can be used to inform tailored clinical care and treatment (e.g., vaccination regime).
Concluding Remarks
We see a growing body of evidence to support the link between IG polymorphism and phenotype that may have important clinical applications. The most obvious of these correlations include potential effects of CNV and SNPs in non-translated and translated IG gene regions on the size, type and distribution of Abs expressed within repertoires of naive and memory B cell subsets. Some of these polymorphisms could be expected to more broadly impact variation in protective antibody responses [77] and quality of the memory B cell pool. We anticipate that IG polymorphism will contribute to differences in expression of common (public) and unique (private) antibody signatures that are associated with protective responses in disease and in response to vaccination. We propose a model for the future in which cataloging these public signatures for biased gene use, V(D)J associations, SHMs and heavy-light chain pairing in the context of IG germline variation should begin to provide us with information to advance our understanding of the immunogenetic potential of an individual’s baseline naive repertoire (Figure 2, Key Figure), particularly when more complete datasets of biased Ab signatures to specific epitopes become available. Based on existing genetic data, it is probable that similar IG haplotypes will associate with overlapping signatures in baseline repertoire profiles even if not to the degree of repertoire similarity observed in MZ twins. This IG polymorphism, as we and others have begun to show, may further influence the evolution of antigen-experienced B cells and plasma cells, where other genetic polymorphisms in the IG loci and environmental exposures come into play in continuing to shape affinity, epitope specificity and fate. In addition, class-switched memory B cell compartments will vary over time [37], and could be quantitated in the type and size of clonotypes with both public and private signatures against immunodominant epitopes.
Together, this knowledge should pave the way to using molecular and genetic signatures for mapping an individual’s exposure history, current wellness state, and immune potential against future antigenic threats. For example, characterization of genotypes that specifically lead to common BnAb signatures in the repertoire should be useful for tailoring vaccines to responsive genotypes with the goal of achieving 100% “universal vaccine” responsiveness at the population level (Figure 2, Key Figure). In addition, such information could lead to advances in the use of anti-idiotypic antibody and chimeric antigen receptor (CAR) T cell therapies that are directed against germline gene expressing B cell clonotypes that are directly involved in auto-immune disease and hematologic malignancies [78,79]. We face tall hurdles to moving this paradigm forward, the greatest being the completion of a comprehensive human IG haplotype map for the more effective application of genomic tools that represent variation across a more diverse set of human populations [26]. However, with ever expanding advances in immunologic and genomic technologies, we believe that such integrative approaches are within our reach, and have the potential to transform our understanding of Ab mediated immune responses in the clinical and research arenas.
Outstanding Questions Box.
-
-
How large of an effect does IG polymorphism have on the development of the baseline naïve repertoire, and what types of genetic variation (CNV; coding variants; regulatory variants) matter most?
-
-
Do effects of IG genetic variants on the Ab repertoire correspond to known biases in disease and/or clinically relevant Ab responses?
-
-
What can population-level data on genetic and expressed Ab repertoire signatures tell us about an individual’s exposure history, current wellness state, and immune potential against future antigenic threats?
-
-
Can we leverage integrated population-level datasets to inform clinical care, and more effective vaccine and therapeutic strategies?
Trends Box.
-
-
Genetic variation in human populations affects how individuals are able to mount functional antibody responses
-
-
Some alleles encode convergent binding motif signatures that predispose successful responses against specific infections and vaccinations
-
-
Given the complexity of the IG loci and the diversity of the antibody repertoire, links between IG polymorphism and antibody repertoire variability have not been thoroughly explored
-
-
We present a strategy to mine genotype-repertoire-disease associations
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lefranc M-P, L G. The Immunoglobulin Factsbook. Academic Press; 2001. [Google Scholar]
- 2.Tonegawa S. Somatic generation of antibody diversity. Nature. 1983;302:575–581. doi: 10.1038/302575a0. [DOI] [PubMed] [Google Scholar]
- 3.Schroeder HW. Similarity and divergence in the development and expression of the mouse and human antibody repertoires. Dev Comp Immunol. 2006;30:119–135. doi: 10.1016/j.dci.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 4.Chimge NO, et al. Determination of gene organization in the human IGHV region on single chromosomes. Genes Immun. 2005;6:186–93. doi: 10.1038/sj.gene.6364176. [DOI] [PubMed] [Google Scholar]
- 5.Li H, et al. Genetic diversity of the human immunoglobulin heavy chain VH region. Immunol Rev. 2002;190:53–68. doi: 10.1034/j.1600-065x.2002.19005.x. [DOI] [PubMed] [Google Scholar]
- 6.Kidd MJ, et al. The inference of phased haplotypes for the immunoglobulin H chain V region gene loci by analysis of VDJ gene rearrangements. J Immunol. 2012;188:1333–40. doi: 10.4049/jimmunol.1102097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Watson CT, et al. Complete haplotype sequence of the human immunoglobulin heavy-chain variable, diversity, and joining genes and characterization of allelic and copy-number variation. Am J Hum Genet. 2013;92:530–46. doi: 10.1016/j.ajhg.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sasso EH, et al. Ethnic differences in polymorphism of an immunoglobulin VH3 gene. J Clin Invest. 1995;96:1591–1600. doi: 10.1172/JCI118198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scheepers C, et al. Ability To Develop Broadly Neutralizing HIV-1 Antibodies Is Not Restricted by the Germline Ig Gene Repertoire. J Immunol. 2015;194:4371–8. doi: 10.4049/jimmunol.1500118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Glanville J, et al. Naive antibody gene-segment frequencies are heritable and unaltered by chronic lymphocyte ablation. Proc Natl Acad Sci U S A. 2011;108:20066–71. doi: 10.1073/pnas.1107498108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boyd SD, et al. Individual variation in the germline Ig gene repertoire inferred from variable region gene rearrangements. J Immunol. 2010;184:6986–6992. doi: 10.4049/jimmunol.1000445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kidd MJ, et al. DJ Pairing during VDJ Recombination Shows Positional Biases That Vary among Individuals with Differing IGHD Locus Immunogenotypes. J Immunol. 2015;196:1158–64. doi: 10.4049/jimmunol.1501401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang C, et al. B-cell repertoire responses to varicella-zoster vaccination in human identical twins. Proc Natl Acad Sci U S A. 2015;112:500–5. doi: 10.1073/pnas.1415875112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rubelt F, et al. Individual heritable differences result in unique Lymphocyte receptor repertoires of naïve and antigen-experienced cells. Nat Commun. 2016;6:1–12. doi: 10.1038/ncomms11112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Avnir Y, et al. IGHV1-69 polymorphism modulates anti-influenza antibody repertoires, correlates with IGHV utilization shifts and varies by ethnicity. Sci Rep. 2016;6:20842. doi: 10.1038/srep20842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wheatley aK, et al. H5N1 Vaccine-Elicited Memory B Cells Are Genetically Constrained by the IGHV Locus in the Recognition of a Neutralizing Epitope in the Hemagglutinin Stem. J Immunol. 2015;195:602–10. doi: 10.4049/jimmunol.1402835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Watson CT, et al. Sequencing of the human IG light chain loci from a hydatidiform mole BAC library reveals locus-specific signatures of genetic diversity. Genes Immun. 2014;16:24–34. doi: 10.1038/gene.2014.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pallarès N, et al. The Human Immunoglobulin Heavy Variable Genes. Exp Clin Immunogenet. 1999;16:36–60. doi: 10.1159/000019095. [DOI] [PubMed] [Google Scholar]
- 19.Lefranc MP, et al. IMGT(R), the international ImMunoGeneTics information system(R) 25 years on. Nucleic Acids Res. 2014;43:D413–D422. doi: 10.1093/nar/gku1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pallares N, et al. The human immunoglobulin lambda variable (IGLV) genes and joining (IGLJ) segments. Experimental and Clinical Immunogenetics. 1998;15:8–18. doi: 10.1159/000019054. [DOI] [PubMed] [Google Scholar]
- 21.Barbié V, Lefranc MP. The human Immunoglobulin Kappa Variable (IGKV) Genes and Joining (IGKJ) Segments. Exp Clin Immunogenet. 1998;15:171–183. doi: 10.1159/000019068. [DOI] [PubMed] [Google Scholar]
- 22.Wang Y, et al. Many human immunoglobulin heavy-chain IGHV gene polymorphisms have been reported in error. Immunol Cell Biol. 2008;86:111–5. doi: 10.1038/sj.icb.7100144. [DOI] [PubMed] [Google Scholar]
- 23.Gadala-Maria D, et al. Automated analysis of high-throughput B-cell sequencing data reveals a high frequency of novel immunoglobulin V gene segment alleles. Proc Natl Acad Sci. 2015;112:E862–70. doi: 10.1073/pnas.1417683112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Corcoran MM, et al. Production of individualized V gene databases reveals high levels of immunoglobulin genetic diversity. Nat Commun. 2016;7:13642. doi: 10.1038/ncomms13642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Y, et al. Genomic screening by 454 pyrosequencing identifies a new human IGHV gene and sixteen other new IGHV allelic variants. Immunogenetics. 2011;63:259–265. doi: 10.1007/s00251-010-0510-8. [DOI] [PubMed] [Google Scholar]
- 26.Watson CT, Breden F. The immunoglobulin heavy chain locus: genetic variation, missing data, and implications for human disease. Genes Immun. 2012;13:363–73. doi: 10.1038/gene.2012.12. [DOI] [PubMed] [Google Scholar]
- 27.Milner EC, et al. Polymorphism and utilization of human VH Genes. Ann N Y Acad Sci. 1995;764:50–61. doi: 10.1111/j.1749-6632.1995.tb55806.x. [DOI] [PubMed] [Google Scholar]
- 28.Shin EK, et al. Polymorphism of the human immunoglobulin variable region segment V1-4.1. Immunogenetics. 1993;38:304–306. doi: 10.1007/BF00188810. [DOI] [PubMed] [Google Scholar]
- 29.Bottaro a, et al. Pulsed-field electrophoresis screening for immunoglobulin heavy-chain constant-region (IGHC) multigene deletions and duplications. Am J Hum Genet. 1991;48:745–56. [PMC free article] [PubMed] [Google Scholar]
- 30.Luo S, et al. Estimating Copy Number and Allelic Variation at the Immunoglobulin Heavy Chain Locus Using Short Reads. PLoS Comput Biol. 2016;12:1–21. doi: 10.1371/journal.pcbi.1005117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Trowsdale J, Knight JC. Major histocompatibility complex genomics and human disease. Annu Rev Genomics Hum Genet. 2013;14:301–23. doi: 10.1146/annurev-genom-091212-153455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Parham P, Moffett A. Variable NK cell receptors and their MHC class I ligands in immunity, reproduction and human evolution. Nat Rev Immunol. 2013;13:133–44. doi: 10.1038/nri3370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Georgiou G, et al. The promise and challenge of high-throughput sequencing of the antibody repertoire. Nat Biotechnol. 2014;32:158–68. doi: 10.1038/nbt.2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boyd SD, Joshi SA. High-Throughput DNA Sequencing Analysis of Antibody Repertoires. 2014;2:1–13. doi: 10.1128/microbiolspec.AID-0017-2014. [DOI] [PubMed] [Google Scholar]
- 35.Yaari G, Kleinstein SH. Practical guidelines for B-cell receptor repertoire sequencing analysis. Genome Med. 2015;7:121. doi: 10.1186/s13073-015-0243-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jackson KJL, et al. The shape of the lymphocyte receptor repertoire: Lessons from the B cell receptor. Front Immunol. 2013;4:1–12. doi: 10.3389/fimmu.2013.00263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Galson JD, et al. In-depth assessment of within-individual and inter-individual variation in the B cell receptor repertoire. Front Immunol. 2015;6:1–13. doi: 10.3389/fimmu.2015.00531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Laserson U, et al. High-resolution antibody dynamics of vaccine-induced immune responses. Proc Natl Acad Sci U S A. 2014;111:4928–33. doi: 10.1073/pnas.1323862111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vollmers C, et al. Genetic measurement of memory B-cell recall using antibody repertoire sequencing. Proc Natl Acad Sci U S A. 2013;110:13463–8. doi: 10.1073/pnas.1312146110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sasso EH, et al. xpression of the immunoglobulin VH gene 51p1 is proportional to its germline gene copy number. J Clin Invest. 1996;97:2074–80. doi: 10.1172/JCI118644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sharon E, et al. Genetic variation in MHC proteins is associated with T cell receptor expression biases. Nat Genet. 2016;48:995–1002. doi: 10.1038/ng.3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Feeney AJ, et al. A defective Vkappa A2 allele in Navajos which may play a role in increased susceptibility to haemophilus influenzae type b disease. J Clin Invest. 1996;97:2277–2282. doi: 10.1172/JCI118669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Feeney AJ. Genetic and epigenetic control of V gene rearrangement frequency. Adv Exp Med Biol. 2009;650:73–81. doi: 10.1007/978-1-4419-0296-2_6. [DOI] [PubMed] [Google Scholar]
- 44.Choi NM, et al. Deep sequencing of the murine IgH repertoire reveals complex regulation of nonrandom V gene rearrangement frequencies. J Immunol. 2013;191:2393–402. doi: 10.4049/jimmunol.1301279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Volpi SA, et al. Germline deletion of Igh 3′ regulatory region elements hs 5, 6, 7 (hs5–7) affects B cell-specific regulation, rearrangement, and insulation of the Igh locus. J Immunol. 2012;188:2556–2566. doi: 10.4049/jimmunol.1102763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Verma-Gaur J, et al. Noncoding transcription within the Igh distal VH region at PAIR elements affects the 3D structure of the Igh locus in pro-B cells. Proc Natl Acad Sci. 2012;109:17004–17009. doi: 10.1073/pnas.1208398109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xu JL, Davis MM. Diversity in the CDR3 Region of V H Is Sufficient for Most Antibody Specificities. Immunity. 2000;13:37–45. doi: 10.1016/s1074-7613(00)00006-6. [DOI] [PubMed] [Google Scholar]
- 48.Mahon CM, et al. Comprehensive interrogation of a minimalist synthetic CDR-H3 library and its ability to generate antibodies with therapeutic potential. J Mol Biol. 2013;425:1712–1730. doi: 10.1016/j.jmb.2013.02.015. [DOI] [PubMed] [Google Scholar]
- 49.Thomson CA, et al. Germline V-genes sculpt the binding site of a family of antibodies neutralizing human cytomegalovirus. EMBO J. 2008;27:2592–2602. doi: 10.1038/emboj.2008.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bryson S, et al. Structures of Preferred Human IgV Genes-Based Protective Antibodies Identify How Conserved Residues Contact Diverse Antigens and Assign Source of Specificity to CDR3 Loop Variation. J Immunol. 2016;196:4723–4730. doi: 10.4049/jimmunol.1402890. [DOI] [PubMed] [Google Scholar]
- 51.Fu Y, et al. A broadly neutralizing anti-influenza antibody reveals ongoing capacity of haemagglutinin-specific memory B cells to evolve. 2016;7:12780. doi: 10.1038/ncomms12780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kunik V, Ofran Y. The indistinguishability of epitopes from protein surface is explained by the distinct binding preferences of each of the six antigen-binding loops. Protein Eng Des Sel. 2013;26:599–609. doi: 10.1093/protein/gzt027. [DOI] [PubMed] [Google Scholar]
- 53.Ying T, et al. Junctional and allele-specific residues are critical for MERS-CoV neutralization by an exceptionally potent germline-like antibody. Nat Commun. 2015;6:8223. doi: 10.1038/ncomms9223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yeung YA, et al. Germline-encoded neutralization of a Staphylococcus aureus virulence factor by the human antibody repertoire. Nat Commun. 2016;7:13376. doi: 10.1038/ncomms13376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pappas L, et al. Rapid development of broadly influenza neutralizing antibodies through redundant mutations. Nature. 2014;516:418–422. doi: 10.1038/nature13764. [DOI] [PubMed] [Google Scholar]
- 56.Joyce MG, et al. Vaccine-Induced Antibodies that Neutralize Group 1 and Group 2 Influenza A Viruses. Cell. 2016;166:609–623. doi: 10.1016/j.cell.2016.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Parameswaran P, et al. Article Convergent Antibody Signatures in Human Dengue. Cell Host Microbe. 2013;13:691–700. doi: 10.1016/j.chom.2013.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Strauli NB, Hernandez RD. Statistical inference of a convergent antibody repertoire response to influenza vaccine. Genome Med. 2016;8:60. doi: 10.1186/s13073-016-0314-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Johansen JN, et al. Intrathecal BCR transcriptome in multiple sclerosis versus other neuroinflammation: Equally diverse and compartmentalized, but more mutated, biased and overlapping with the proteome. Clin Immunol. 2015;160:211–225. doi: 10.1016/j.clim.2015.06.001. [DOI] [PubMed] [Google Scholar]
- 60.Bomben R, et al. Expression of mutated IGHV3-23 genes in chronic lymphocytic leukemia identifies a disease subset with peculiar clinical and biological features. Clin Cancer Res. 2010;16:620–628. doi: 10.1158/1078-0432.CCR-09-1638. [DOI] [PubMed] [Google Scholar]
- 61.Forconi F, et al. The IGHV1-69/IGHJ3 recombinations of unmutated CLL are distinct from those of normal B cells Brief report The IGHV1-69/IGHJ3 recombinations of unmutated CLL are distinct from those of normal B cells. 2013;119:2106–2109. doi: 10.1182/blood-2011-08-375501. [DOI] [PubMed] [Google Scholar]
- 62.Zhu D, et al. Biased immunoglobulin light chain use in the Chlamydophila psittaci negative ocular adnexal marginal zone lymphomas. Am J Hematol. 2013;88:379–384. doi: 10.1002/ajh.23416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hwang KK, et al. IGHV1-69 B cell chronic lymphocytic leukemia antibodies cross-react with HIV-1 and hepatitis C virus antigens as well as intestinal commensal bacteria. PLoS One. 2014;9:e90725. [Google Scholar]
- 64.Williams WB, et al. Diversion of HIV-1 vaccine-induced immunity by gp41-microbiota cross-reactive antibodies. Science (80-) 2015;349:aab1253–aab1253. doi: 10.1126/science.aab1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liu L, Lucas AH. IGH V3-23*01 and its allele V3-23*03 differ in their capacity to form the canonical human antibody combining site specific for the capsular polysaccharide of Haemophilus influenzae type b. Immunogenetics. 2003;55:336–338. doi: 10.1007/s00251-003-0583-8. [DOI] [PubMed] [Google Scholar]
- 66.Throsby M, et al. Heterosubtypic neutralizing monoclonal antibodies cross-protective against H5N1 and H1N1 recovered from human IgM+ memory B cells. PLoS One. 2008;3:e3942. doi: 10.1371/journal.pone.0003942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wrammert J, et al. Broadly cross-reactive antibodies dominate the human B cell response against 2009 pandemic H1N1 influenza virus infection. J Exp Med. 2011;208:181–93. doi: 10.1084/jem.20101352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ekiert DC, et al. Antibody recognition of a highly conserved influenza virus epitope. Science. 2009;324:246–251. doi: 10.1126/science.1171491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kashyap AK, et al. Combinatorial antibody libraries from survivors of the Turkish H5N1 avian influenza outbreak reveal virus neutralization strategies. Proc Natl Acad Sci U S A. 2008;105:5986–5991. doi: 10.1073/pnas.0801367105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Corti D, et al. A neutralizing antibody selected from plasma cells that binds to group 1 and group 2 influenza A hemagglutinins. Science (80-) 2011;333:850–856. doi: 10.1126/science.1205669. [DOI] [PubMed] [Google Scholar]
- 71.Lingwood D, et al. Structural and genetic basis for development of broadly neutralizing influenza antibodies. Nature. 2012;489:566–570. doi: 10.1038/nature11371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nakamura G, et al. An in vivo human-plasmablast enrichment technique allows rapid identification of therapeutic influenza A antibodies. Cell Host Microbe. 2013;14:93–103. doi: 10.1016/j.chom.2013.06.004. [DOI] [PubMed] [Google Scholar]
- 73.Kallewaard NL, et al. Structure and Function Analysis of an Antibody Recognizing All Influenza A Subtypes. Cell. 2016;166:596–608. doi: 10.1016/j.cell.2016.05.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wu Y, et al. A potent broad-spectrum protective human monoclonal antibody crosslinking two haemagglutinin monomers of influenza A virus. Nat Commun. 2015;6:7708. doi: 10.1038/ncomms8708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pugh-Bernard AE, et al. Regulation of inherently autoreactive VH4-34 B cells in the maintenance of human B cell tolerance. J Clin Invest. 2001;108:1061–1070. doi: 10.1172/JCI12462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cappione AJ, et al. Lupus IgG VH4.34 antibodies bind to a 220-kDa glycoform of CD45/B220 on the surface of human B lymphocytes. J Immunol. 2004;172:4298–4307. doi: 10.4049/jimmunol.172.7.4298. [DOI] [PubMed] [Google Scholar]
- 77.Lee J, et al. Molecular-level analysis of the serum antibody repertoire in young adults before and after seasonal influenza vaccination. Nat Med. 2016;22:1456–1464. doi: 10.1038/nm.4224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fesnak AD, et al. Engineered T cells: the promise and challenges of cancer immunotherapy. Nat Rev Cancer. 2016;16:566–81. doi: 10.1038/nrc.2016.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chang DK, et al. Humanized mouse G6 anti-idiotypic monoclonal antibody has therapeutic potential against IGHV1-69 germline gene-based B-CLL. MAbs. 2016;8:787–798. doi: 10.1080/19420862.2016.1159365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Auton A, et al. A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]