

Abstract
The Nuclear receptor-binding SET Domain (NSD) family of histone H3 lysine 36 methyl transferases is comprised of NSD1, NSD2 (MMSET/WHSC1), and NSD3 (WHSC1L1). These enzymes recognize and catalyze methylation of histone lysine marks to regulate chromatin integrity and gene expression. The growing number of reports demonstrating that alterations or translocations of these genes fundamentally affect cell growth and differentiation leading to developmental defects illustrates the importance of this family. In addition, overexpression, gain of function somatic mutations and translocations of NSDs are associated with human cancer and can trigger cellular transformation in model systems. Here we review the functions of NSD family members and the accumulating evidence that these proteins play key roles in tumorigenesis. Since epigenetic therapy is an important emerging anti-cancer strategy, understanding the function of NSD family members may lead to the development of novel therapies.
Introduction
Histone lysine methyl transferases (HMTases) catalyze the transfer of up to three methyl groups to specific lysine (K) residues on the tails of histones H3 and H4 that are critical for chromatin maintenance and the fine regulation of gene expression. Histone marks created by lysine HMTases are associated with either active transcription (such as H3K4me or H3K36me2) or repressed transcription (such as H3K27me or H2K9me) (Lachner et al. 2003; Barski et al. 2007). The global activities of these enzymes keep genes in a state poised for rapid activation or repression. Methylated histones are sensed and linked to downstream biological functions by an array of methyl-lysine binding proteins. Thus lysine HMTases play important roles in many downstream cellular processes such as DNA replication, DNA damage response, cell cycle progression, cytokinesis, and transcriptional regulation of important developmental and tumor suppressor genes. The human genome encodes more than fifty predicted lysine methyltransferases, and for many of these genes dysregulation has been reported to play a causative role in human disease (Kouzarides 2007; Morishita and di Luccio 2011). Here we review the most recently reported functions of nuclear receptor-binding SET domain (NSD) family members with a particular attention paid to their roles in cancer.
NSD family members are structurally conserved histone H3 lysine 36 mono- and di-methyltransferases
The NSD family of HMTases are a phylogenetically distinct subfamily of lysine-HMTases comprised of: NSD1, NSD2 (MMSET/WHSC1), and NSD3 (WHSC1L1) (Morishita and di Luccio 2011). The full length members of the NSD family are large multi-domain proteins that contain the evolutionarily conserved catalytic SET [Su(var)3–9, Enhancer-of-zeste, Trithorax] domain that is further subdivided into pre-SET, SET, and post-SET domains (Fig. 1) (Dillon et al. 2005; Herz et al. 2013). In addition, full-length NSD family members have two PWWP (proline-tryptophan-tryptophan-proline) domains that are critical for binding to methylated histone H3 as well as DNA and plant homeodomain (PHD) zinc fingers important for interactions with other methylated histones (Figure 1) (Baker et al. 2008; Pasillas et al. 2011; Sankaran et al. 2016).
Figure 1.
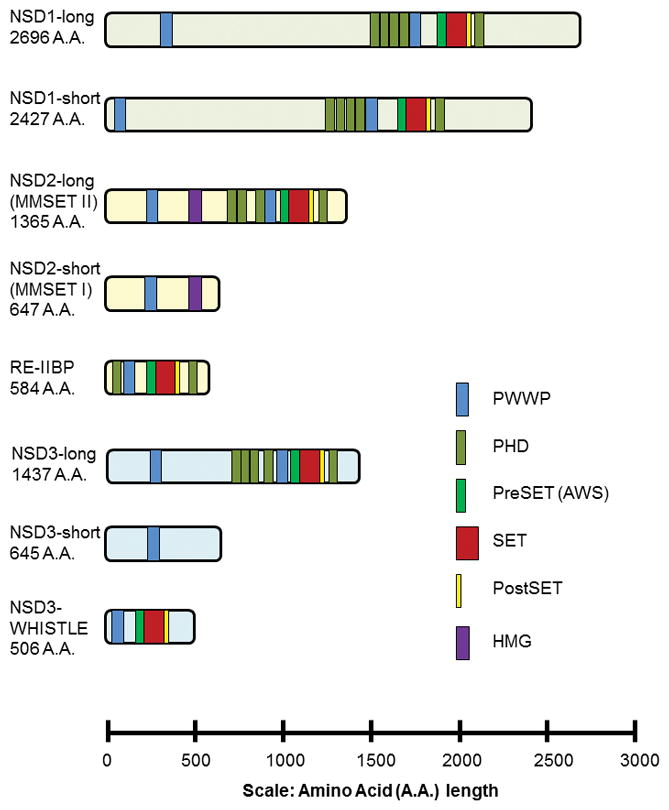
Conserved structural domains in the NSD family of histone lysine methyl transferases. Protein domain assignments were calculated using SMART (a Simple Modular Architecture Research Tool) and the following UniProtKB entries: NSD1-long, Q96L73-1; NSD1-short, Q96L73-2; NSD2-Long, O96028-1; NSD2-short, O96028-3; RE-IIBP, O96028-4; NSD3-long, Q9BZ95-1 and NSD3-short, Q9BZ95-3. NSD3-WHISTLE domain assignments are from Kim et al., 2006.
NSD1, NSD2 and NSD3 function to mono- and dimethylate histone H3 on lysine 36 (H3K36) (Kim et al. 2006; Kim et al. 2008; Marango et al. 2008; Li et al. 2009b; Nimura et al. 2009; Lucio-Eterovic et al. 2010; Kuo et al. 2011; Qiao et al. 2011; Rahman et al. 2011). Histone H3K36 is found in nonmethylated and mono, di and tri methylated forms (me1, me2, me3, respectively), and methylation of H3K36 is associated with transcription of active euchromatin (Rao et al. 2005; Wagner and Carpenter 2012). To date, eight different mammalian H3K36 HMTases have been identified with distinct preferences for which methylation state of H3K36 they recognize and modify in vitro and/or in vivo (Wagner and Carpenter 2012). In addition, the various methylated forms of H3K36 may have different biological functions depending on the organism or cellular context. In worms and humans K36me3 has been shown to link transcription with splicing (Kolasinska-Zwierz et al. 2009; Sims and Reinberg 2009; Spies et al. 2009). In flies H3K36me3 functions in dosage compensation (Larschan et al. 2007). In yeast H3K36me2 and H3K36me3 have been implicated in transcription frequency and elongation that is coupled to histone acetylation (Kaplan et al. 2003; Carrozza et al. 2005; Joshi and Struhl 2005; Kizer et al. 2005; Morris et al. 2005; Shilatifard 2006; Lee and Shilatifard 2007; Xu et al. 2008b; Li et al. 2009a). In Arabidopsis and chicken H3K36me2 and H3K36me3 mark actively transcribed chromatin (Bannister et al. 2005; Xu et al. 2008a). In humans there is a preference for K36me1 at active promoters, and this mark is detected in active regions of the beta-globin locus (Kim et al. 2007). Furthermore, at the human globin genes, H3K36me3 is broadly associated with transcription (Kim et al. 2007).
Evidence suggests that NSD1, NSD2 and/or NSD3 normally play nonredundant roles during development since genetic deletion of either NSD1 or NSD2 is lethal in mice (Rayasam et al. 2003; Nimura et al. 2009). Thus, alterations or amplifications of NSD1, NSD2 and/or NSD3 that dysregulate H3K36 methylation marks have profound effects on cell growth and differentiation and are linked to numerous developmental defects. In addition, overexpression, gain of function somatic mutations and translocation of NSDs have been reported to frequently occur in a variety of cancers.
Identification and Function of NSD
The human nuclear receptor SET domain-containing 1 (NSD1, KMT3B) gene is comprised of 24 exons on chromosome 5q35. NSD1 has two protein isoforms: a predominant 2427 amino acid (AA) short form and a less abundant 2696 AA long form that occurs by retention of the intron between exons 2 and 3 (Figure 1) (Lucio-Eterovic et al. 2010). NSD1 was originally identified to contain two nuclear steroid receptor interaction domains that regulate the function of retinoic acid, thyroid, retinoid X, and estrogen nuclear receptors (Huang et al. 1998). Subsequent studies revealed that the SET domain of NSD1 methylated H3K36 and H4K20 in vitro (Rayasam et al. 2003). However, more recent experiments suggest that the enzyme is specific for H3K36 (Bender et al. 2006; Bell et al. 2007; Stabell et al. 2007; Li et al. 2009b). Importantly, reports using defined nucleosome substrates with various forms of methylated histone H3 at lysine 36 demonstrate that NSD1 is a dimethylase specific for H3K36 (Li et al. 2009b; Qiao et al. 2011). Furthermore, depletion of NSD1 reduced the levels of H3K36me1, 2, and 3 suggesting that NSD1 is a mono/dimethylase and that this modification serves as a substrate for trimethylation by the HMTase SETD2 (Lucio-Eterovic et al. 2010). In addition, depletion of NSD1 reduced RNA polymerase II (RNAPII) promoter occupancy and inhibited the transition of RNAPII from an initiation to an elongation-competent state (Lucio-Eterovic et al. 2010).
The crystal structure for NSD1 has been solved and serves as a model for the other NSD family proteins (Qiao et al. 2011; Graham et al. 2016). This structure reveals that an autoinhibitory loop blocks binding of NSD1 to the histone peptide as well as the entrance to the lysine-binding channel. As nucleosomal DNA contacts the NSD1 post-SET loop the active conformation is stabilized. This may explain the preference of NSD family members for nucleosomal H3K36 substrates compared to octamers and why NSD family HMTases methylate histone lysine residues other than H3K36 when octamers, recombinant histones or peptides are used as substrates (Li et al. 2009b; Kudithipudi et al. 2014; Morishita et al. 2014).
NSD1 is critical for normal murine embryonic development. Homozygous loss-of-function embryos are able to gastrulate and initiate mesoderm formation but fail at embryonic day 6.5 (Rayasam et al. 2003). Defects of NSD1 are linked to Sotos syndrome (aberrant NSD1 expression is present in 80% of patients) as well as unique variants of Weaver or Beckwith-Wiedemann syndrome (Kurotaki et al. 2002; Douglas et al. 2003; Baujat et al. 2004; Gibson et al. 2012). Characterized as overgrowth disorders, these patients experience pre/post-natal overgrowth, enhanced bone age, neurodevelopmental delay, and an enhanced risk for cancer (Rahman 2005). A genomic analysis of 435 Sotos patient samples revealed nonsense, deletion/insertion/duplication, splice site, and missense NSD1 mutations (Waggoner et al. 2005). While half of the point mutations (nonsense and missense) occurred at CpG sites, missense mutations were primarily in exons that code for functional domains (i.e., PHD, PWWP, and SET domains) (Waggoner et al. 2005). Though not experimentally confirmed, there is evidence of NSD1 gene dosage effects linked to significant phenotypic outcomes beyond Sotos syndrome. Microdeletions lead to bone overgrowth/macrocephaly while microduplications are mirrored by stunted growth and microcephaly (Dikow et al. 2013; Rosenfeld et al. 2013). NSD1 knockout mouse are embryonic lethal (Rayasam et al. 2003). However, mice carrying a heterozygous 1.5-Mb deletion of 36 genes on mouse chromosome 13 that corresponds to the human chromosome 5q35.2–q35.3 region where NSD1 is located have been characterized (Migdalska et al. 2012). Though these mice did not exhibit the phenotypic overgrowth observed in Sotos syndrome, they did display impaired long-term memory and renal abnormalities (Migdalska et al. 2012).
In addition to histone H3, non-histone targets of NSD1 have been reported. The C-terminus of NSD1 contains a unique PHD finger region termed the PHDvC5HCH domain that interacts with the transcription repressor NIzp1 and mutations in the NSD1 PHDvC5HCH domain may interfere with NIsp1 transcription repression (Nielsen et al. 2004; Berardi et al. 2016). Also, upon activation of NFκB, NSD1 has been reported to co-immunoprecipitate with the NFκB p65 subunit, and be required for inducible p65 methylation at lysine residues K218 and K221 (Lu et al. 2010). However, whether p65 is a direct substrate of NSD1 has yet to be confirmed in vitro. These functions may contribute to the overgrowth phenotype observed when NSD1 is mutated in Sotos syndrome.
NSD1 Role in Cancer
Aberrant NSD1 expression has been associated with many cancer pathologies and tumors occur in 3% of patients diagnosed with Sotos syndrome (Tatton-Brown et al. 2005). In addition, NSD1 was inactivated via CpG island-promoter hypermethylation in neuroblastomas and gliomas (Berdasco et al. 2009). This transcriptional silencing was associated with diminished methylation of H3K36 and H4K20. In prostate tumors, enhanced NSD1 expression was associated with metastases, while reduced expression was associated with cancers having biochemical recurrence (Bianco-Miotto et al. 2010). Also, NSD1 expression was upregulated in patient-derived metastatic melanoma cell lines compared to primary epidermal melanocytes. However, during the progression from non-metastatic to metastatic melanoma expression of NSD1 was downregulated (de Souza et al. 2012). Furthermore, a transposon screen for frequently occurring mutations in skin tumors of mice revealed that NSD1 is among those genes with significantly decreased expression during tumor development (Quintana et al. 2013). Taken together these studies suggest that increased NSD1 expression may promote oncogenic initiation through enhanced methylation of H3K36, which activates genes normally silenced by H3K27me3 (e.g., MEIS1 and Hox) (Wang et al. 2007; Berdasco et al. 2009). Subsequently, metastatic progression may activate negative feedback of NSD1 in a tissue and/or stage specific manner. Further studies are required to elucidate these potential roles of NSD1 in cancer.
A NUP98-NSD1 fusion protein has been observed in childhood acute myeloid leukemia (AML) (Jaju et al. 2001; Cerveira et al. 2003; La Starza et al. 2004). This recurrent translocation at t(5;11)(q35;p15.5) contains the FG-repeat domain of NUP98, a nucleoporin protein family member that can interact with the histone acetyltransferase CBP/p300 and the C-terminal of NSD1 that retains the five PHD fingers, the Cys-His rich domain (C5HCH), one PWWP domain, and the catalytic SET domain. NUP98-NSD1 is the most frequent (4%–5% of cases) fusion reported in pediatric AML and is associated with poor prognosis (Shiba et al. 2013). Retroviral infection of NUP98-NSD1 enhanced expression of HoxA7, HoxA7, HoxA9, and Meis1 proto-oncogenes (Wang et al. 2007). In addition, marrow progenitors transduced with NUP98-NSD1 induced AML when transplanted into lethally irradiated mice (Wang et al. 2007). Mechanistically, the NUP98-NSD1 fusion drives methylation of H3K36 and histone acetylation. This persistent methylation prevents transcriptional repression of the Hox-A locus by EZH2 causing progenitor immortalization (Wang et al. 2007). Though the NUP98-NSD1 fusion protein occurs in a low percentage of patients, the incidence of concurrence between NUP98-NSD1 and FLT3 (FMS-like tyrosine kinase 3) internal tandem duplication (ITD) mutants is high (70%) in human AML cases (Thanasopoulou et al. 2014). Co-infection of progenitor cells expressing both NUP98-NSD1 and FLT3-ITD mutation resulted in a strikingly decreased latency of development of AML in mice compared to those reconstituted with NUP98-NSD1 alone (Thanasopoulou et al. 2014). Furthermore, experiments using immortalized bone marrow progenitor cells demonstrate that NUP98-NSD1 promotes the expression and activation of FLT3-ITD, suggesting a potent cooperation between NUP98-NSD1 and FLT3-ITD during leukemic transformation that may be therapeutically targeted with SET domain or FLT3 inhibitors (Thanasopoulou et al. 2014).
In summary, NSD1 is an H3K36 specific mono and dimethyltransferase that promotes transcription and is critical for normal growth and development. Aberrant expression of NSD1 drives the pathobiology of Sotos syndrome and tumorigenesis. Though much work has been done regarding the structure and function of NSD1, questions remain. For instance, while normal and cancer cell lines endogenously express both the short and long form of NSD1 the specific functions of these protein isoforms have not been well characterized. In addition, work remains to determine what cellular context or mechanisms regulate the oncogenic properties of NSD1 and how transcription of NSD1 may be altered during metastasis.
Identification and Function of NSD2
NSD2 (also known as WHSC1 and MMSET) was identified by its presence in a chromosomal region found to be deleted in the Wolf-Hirschhorn syndrome (WHS) (Stec et al. 1998), and separately by its rearrangement with the immunoglobulin locus in a subset of cases of the plasma cell malignancy, multiple myeloma (Chesi et al. 1998). Since then, NSD2 has been found to play a role in many facets of development and malignancy. This 90kb, 25-exon gene on chromosome 4p16.3 encodes two main isoforms, NSD2-short (MMSET-I) and NSD2-long (MMSET-II), and an intronic transcript that encodes response element II binding protein (RE-IIBP). The 1365 amino acid full-length species is composed of two PWWP domains, a high mobility group (HMG) DNA binding domain, four PHD zinc fingers, and a SET domain (Fig. 1), while the 647 amino acid short species lacks all but the initial PWWP and HMG domains (Stec et al. 1998).
NSD2 catalyzes the mono and dimethylation of H3K36 (Martinez-Garcia et al. 2011; Morishita et al. 2014). The first N-terminal PWWP domain of NSD2 specifically binds to H3K36me2 to stabilize NSD2 at chromatin, and the catalytic SET domain of NSD2 propagates this gene-activating mark to adjacent nucleosomes (Waggoner et al. 2005; Kuo et al. 2011; Martinez-Garcia et al. 2011; Morishita et al. 2014; Sankaran et al. 2016). Similar to NSD1, the NSD2 post-SET domain is attached to the catalytic SET domain via an autoinhibitory loop region and inhibition is relieved upon nucleosome binding (Qiao et al. 2011; Poulin et al. 2016a). Furthermore, NSD2 has been reported to preferentially catalyze H3K36 dimethylation compared to H3K36 monomethylation (Li et al. 2009b; Kuo et al. 2011; Poulin et al. 2016b).
NSD2 is broadly expressed and its importance in development is highlighted by its involvement in Wolf-Hirschhorn malformation syndrome (WHS). The full syndrome is characterized by brain defects associated with developmental delay and epilepsy as well as cranio-facial anomalies, growth delay, heart defects, and midline fusion abnormalities (Stec et al. 1998; Bergemann et al. 2005). Variable deletions in the short arm of chromosome 4 (4p16.3) are typical of WHS, and NSD2 is the only gene in this region that is deleted in almost every case. Partial or full hemizygosity of NSD2 appears to be necessary but not sufficient for the development of WHS, as the deletion of other genes nearby contributes to the constellation of abnormalities that make up the syndrome (Bergemann et al. 2005; Andersen et al. 2013). Mice with a homozygous NSD2 SET domain deletion do not survive past 10 days of age, and heterozygous mice develop significant developmental defects that imitate WHS (Nimura et al. 2009). Furthermore, ChIP experiments on ES cells of these mice revealed that NSD2 binds to several genes associated with development including Sall1, Sall4, and Nanog. Additionally, WHS patients often have antibody deficiencies, suggesting a role for NSD2 in B cell development. This theory is supported by the ability of NSD2 to recruit the DNA damage responsive, p53-binding protein 1 (53BP1), which is critical for class switch recombination and also implicates NSD2 in DNA repair (Hajdu et al. 2011; Pei et al. 2011). Indeed, it was recently reported that NSD2 regulates the expression of DNA repair genes and may play a critical function in multiple myeloma chemoresistance (Shah et al. 2016). Cancer cells expressing high levels of NSD2 repaired DNA damage at a much faster rate than cells with low levels of NSD2, allowing them to survive and proliferate even while being treated with DNA-damaging chemotherapy.
NSD2 Role in Cancer
NSD2 was initially described as a gene rearranged and linked to regulatory sequences of the immunoglobulin heavy chain gene in t(4;14) multiple myeloma (Chesi et al. 1998; Stec et al. 1998), NSD2 and its translocations, amplifications, and mutations were subsequently identified in a wide spectrum of malignancies. In multiple myeloma the t(4;14) translocation is present in 15%–20% of cases, resulting in overexpression of NSD2 and FGFR3 (Chesi et al. 1998; Stec et al. 1998; Finelli et al. 1999). However, NSD2 is purported to be the primary oncogenic driver, as approximately 30% of cases harboring this translocation have normal expression of FGFR3 alongside NSD2 overexpression (Santra et al. 2003). Furthermore, knockdown of NSD2 expression in t(4;14)+ multiple myeloma cell lines reduces proliferation, cell cycle progression, and DNA repair, while increasing apoptosis and adhesion (Lauring et al. 2008; Brito et al. 2009; Martinez-Garcia et al. 2011; Huang et al. 2013; Shah et al. 2016). These phenotypic changes are driven by redistribution of activating and repressive chromatin marks that in turn affect gene expression. Overexpression of NSD2 globally increases levels of H3K36 dimethylation (Martinez-Garcia et al. 2011; Zheng et al. 2012; Popovic et al. 2014; Sankaran et al. 2016). Interestingly, NSD2 also affects enhancer of zeste homolog 2 (EZH2), which is responsible for the reciprocal repressive chromatin mark, histone 3 lysine 27 trimethylation (H3K27me3). The spread of H3K36me2 from NSD2 overexpression leads to global reduction in H3K27me3 and restricts EZH2 to small islands of chromatin where it then hypermethylates H3K27me3 (Zheng et al. 2012; Popovic et al. 2014). The transcriptional disturbance that results from NSD2 overexpression primarily involves inappropriate activation of genes, but there are some genes that are inappropriately repressed due to these pockets of EZH2. This effect appears to be important for the survival of cells overexpressing NSD2, as they are sensitive to inhibition of EZH2. One contribution to this sensitivity is that inhibition of EZH2 decreases c-MYC protein levels, a fundamentally upregulated gene in multiple myeloma (Popovic et al. 2014). In t(4;14)+ multiple myeloma cells EZH2 represses miR-126, a microRNA which targets the c-MYC transcript (Min et al. 2012). Therefore, EZH2 inhibitors derepress miR-126, allowing it to reduce c-MYC protein levels and slow proliferation. However, c-MYC is not the sole cause of high NSD2 mediated oncogenesis. Transcriptional profiling of t(4;14)+ multiple myeloma indicates that NSD2 regulates the expression of genes in apoptosis, DNA repair, cell cycle control, and cell motility.
In addition to multiple myeloma, NSD2 overexpression is detected in gastric, colon, lung, and skin cancer (Hudlebusch et al. 2011a). It may play a major role in neuroblastoma and breast, bladder, and prostate tumors, where overexpression is associated with worse prognosis (Hudlebusch et al. 2011b). In many of these cancers NSD2 expression is positively correlated with EZH2 expression, and as opposed to the situation in t(4;14)+ multiple myeloma, global levels of H3K27me3 and H3K36me2 are both increased (Asangani et al. 2013). In prostate cancer EZH2 functions upstream of NSD2, repressing several microRNAs, including miR-203, miR-26, and miR-31, that target NSD2 (Asangani et al. 2013). However, it is the knockdown of NSD2 that abrogates the ability of prostate cancer cells to proliferate, form colonies, migrate, and invade (Ezponda et al. 2012). Furthermore, enforced expression of NSD2 in non-transformed prostate epithelial cells promotes migration and invasion and leads to epithelial-mesenchymal transition (EMT). NSD2 directly binds the TWIST1 locus and upregulates expression of this EMT factor, which plays a key role in the aggressive biological behavior of advanced prostate cancer.
The NSD2 E1099K Point Mutation in Lymphoid Malignancies
In cases of t(4;14)+ multiple myeloma and some acute lymphoblastic leukemia (ALL) where TWIST1 is also overexpressed, NSD2 harbors a recurrent point mutation in the SET domain (Oyer et al. 2014). The significance of the guanine to alanine substitution that results in a glutamic acid to lysine switch at amino acid 1099 (E1099K) of NSD2 was first noted following examination of the Broad Institute’s Cancer Cell Line Encyclopedia (CCLE) that revealed numerous ALL cell lines with the mutation, which has since been identified in 10–20% of relapsed pediatric ALL, 10% of mantle cell lymphoma (MCL), and 10% of chronic lymphocytic leukemia (CLL) (Fabbri et al. 2011; Barretina et al. 2012; Beà et al. 2013; Loh et al. 2013; Oyer et al. 2014). Rare mutations are found in glioblastoma, lung cancer, multiple myeloma Though the mechanism remains to be determined, the E1099K mutation was revealed to result in hyperactive NSD2, leading to increased H3K36me2 and decreased H3K27me3, similar to the epigenetic profile of t(4;14)+ multiple myeloma (Oyer et al. 2014). Due to the common occurrence of this mutation in relapsed pediatric ALL, especially cases harboring other oncogenic lesions such as the TEL-AML1 and E2A-PBX1 fusions, E1099K appears to be an important factor in progression of these malignancies rather than initiation (Jaffe et al. 2013; Loh et al. 2013).
In summary, NSD2 plays a significant role in normal development and malignancy. Haploinsufficiency of NSD2 leads to severe developmental defects, such as cardiac lesions and midline abnormalities associated with WHS. In malignancy, NSD2 has prolific effects resulting from translocation, overexpression, and activating mutations of the gene. The t(4;14) translocation in multiple myeloma has been extensively characterized and indicates that high levels of NSD2 drive oncogenic phenotypes by spreading H3K36me2 throughout the genome and altering gene expression profiles. The relationship between NSD2 and EZH2 is crucial in myeloma as the reciprocal H3K27me3 mark deposited by EZH2 is reduced and restricted to certain regions by NSD2 overexpression. In other malignancies, such as prostate cancer, this relationship is different, as EZH2 appears to function upstream of NSD2, though it is no less important. Inhibition or knockdown of NSD2 or EZH2 in both situations abrogates critical oncogenic pathways and phenotypes. However, there is still much to learn about NSD2 in these malignancies. While NSD2 itself is already an attractive therapeutic candidate, thoroughly characterizing its binding partners will tell us how NSD2 functions both endogenously and in malignancy and provide more targets for designing therapies for NSD2-misregulated cancers. The recurrent E1099K mutation that results in a hyperactive NSD2 is also not fully understood. Its global effects on chromatin mimic NSD2 overexpression, but it must be investigated further to identify its local effects on chromatin, gene expression, and oncogenicity. Additionally, as it appears that this mutation is more likely to contribute to relapse after therapy, it will be important to understand its role in drug-resistance. Finally, we must understand the mechanism by which E1099K and other activating mutations alter the function of NSD2 in order to design directed therapies.
Identification and Function of NSD3
Using database searches for sequences similar to NSD1 and NSD2, NSD3 (also named Wolf-Hirschhorn syndrome candidate 1-like 1, WHSC1L1) was independently identified by two groups in 2001 (Angrand et al. 2001; Stec et al. 2001). The NSD3 gene contains 24 exons that span approximately 112 kb of genomic DNA on chromosome 8p11.2 and is predicted to have 12 transcript variants. Three protein products of NSD3 have been reported and characterized to date: Long, Short and WHISTLE.
The NSD3-long transcript encodes a 1437-amino-acid protein containing 2 PWWP domains, 5 PHD-type zinc finger motifs, a SAC (SET-associated Cys-rich) domain, and a SET domain (Fig. 1) (Angrand et al. 2001; Stec et al. 2001). NSD3-long is highly expressed in brain heart and skeletal muscle and to a lesser degree in liver and lung (Angrand et al. 2001). The sequence of NSD3-long from amino acids 703 to 1409 is highly conserved between NSD1 (68% identical) and NSD2 (55% identical) and contains the PHD, second PWWP and SET domain (Angrand et al. 2001). Similar to NSD1 and NSD2, the carboxyl terminal region of NSD3-long that contains the catalytic preSET, SET and postSET domains is able to recognize and methylate histone H3 and H4 targets in vitro (Allali-Hassani et al. 2014; Morishita et al. 2014). However, unlike NSD2 a clear HMG box is missing from NSD3-long and the fourth PHD-type zinc finger contains an insertion of 49 amino acids (Stec et al. 2001). In addition, evidence suggests that the conserved PHD5 domain of NSD3-long is functionally distinct in its histone binding properties from the PHD5 domain of NSD1 or 2 and targets NSD3 to specific genomic regions in vivo (He et al. 2013). The NSD3-short transcript encodes a protein of 645 amino acids (Stec et al. 2001). NSD3-long and NSD3-short have an identical N-terminal 620 amino acid sequence, but NSD3-short lacks a catalytic SET domain and only contains the N-terminal PWWP domain that binds to histone H3 when it is methylated on lysine 36 (Stec et al. 2001; Vermeulen et al. 2010; Wu et al. 2011). The NSD3-long and NSD3-short transcripts are coexpressed in many tissues (Stec et al. 2001).
WHISTLE (WHSC1-like 1 isoform 9 with methyltransferase activity to lysine) was identified by homology searching and functional assay to be the shortest isoform of NSD3 to retain a SET domain and methyltransferase activity (Kim et al. 2006). WHISTLE consists of 506 amino acids and was found expressed in testis and in bone marrow mononuclear cells of AML and ALL patients (Kim et al. 2006). It contains the PWWP, SET and PostSET domains and was reported to facilitate transcription repression by promoting methylation of histone H3K4 and H3K27 (Kim et al. 2006; Allali-Hassani et al. 2014).
NSD3 has been identified as an essential methyltransferase for neural crest gene expression during specification (Jacques-Fricke and Gammill 2014). NSD3 is expressed in pre-migratory and migratory neural crest cells and is necessary for expression of the neural plate border gene Msx1, as well as the key neural crest transcription factors Sox10, Snail2, Sox9, and FoxD3 (Jacques-Fricke and Gammill 2014). In addition, recent reports indicate that neuronal ten-eleven translocation (TET) hydroxylase 3 interacts with and activates NSD3 to stimulate H3K36 trimethylation and transcription of neuronal genes in retinal cells (Perera et al. 2015). Thus, NSD3 may promote context specific chromatin remodeling and gene activation that is necessary for neural crest migration and retinal network formation during development.
NSD3 Role in cancer
Since its discovery, amplification and overexpression of NSD3 has been a consistently identified feature in many cancer types (Angrand et al. 2001; Mahmood et al. 2013). For example, integrated DNA-RNA analyses of regional amplifications and deletions coupled with gene-expression profiling and have identified the 8p11–12 NSD3 locus as an amplicon commonly expressed in both pancreatic ductal adenocarcinoma and non-small-cell lung cancer (Tonon et al. 2005). In addition, a bioinformatics screen to identify putative cancer driver genes amplified across TCGA datasets discovered frequent NSD3 amplifications in bladder, breast, liver, lung, ovarian, head and neck and colorectal cancer samples (Chen et al. 2014). Amplification of the 8p11–12 region has also been reported to be present in about 15% of primary human breast cancer samples, correlate with histological grade and is associated with poor prognosis (Angrand et al. 2001; Yang et al. 2010; Chen et al. 2014). Furthermore, NSD3 expression and protein level are increased in breast, lung, pancreatic and colorectal cell lines and immunohistochemical analysis indicates that NSD3 is increased in primary breast carcinoma, bladder cancer, lung cancer and liver cancer (Kang et al. 2013; Mahmood et al. 2013; Chen et al. 2014).
Mounting evidence suggests that NSD3 activates signaling pathways that promote cell cycle progression and proliferation. Although a NSD3 knockout mouse has not yet been reported, NSD3 has been identified to cooperate with oncogenic KRAS to drive tumorigenesis in a pancreatic cancer model mouse (Mann et al. 2012). Furthermore, transduction of either NSD3-short or NSD3-long into the mammary epithelial cell line MCF10A cells has been reported to increase proliferation, soft agar colony formation and cause abnormal acini formation (Yang et al. 2010). By contrast, knockdown of NSD3 reduced proliferation and promoted cell death in 8p11–12-amplified breast cancer cells and significantly decreased anchorage-dependent and anchorage-independent growth of pancreatic adenocarcinoma and small-cell lung cancer-derived cell lines (Yang et al. 2010; Mahmood et al. 2013). In addition, knockdown of NSD3 expression induced G2/M cell cycle arrest and suppressed proliferation of breast, bladder and lung cancer cell lines (Zhou et al. 2010; Kang et al. 2013). Expression profile analysis demonstrated that NSD3 affected the expression of a number of genes known to play crucial roles in cell cycle progression (Kang et al. 2013).
While the mechanism by which NSD3 regulates mechanism governing the cell cycle and proliferation is not well characterized, evidence suggests that NSD3 amplification may promote transcription of a subset of genes involved in proliferation pathways. NSD3 can form complexes with H3K4-specific demethylase LSD2 and H3K9-specific methyltransferase G9a and may cooperate with these enzymes to coordinate the dynamics of H3K4 and H3K36 methylation to promote transcription elongation for a subset of genes (Fang et al. 2010). NSD3 promoted expression of transcription factors Iroquois homeobox 3 (IRX3) and TBIL1X known to regulate WNT-signaling (Yang et al. 2010). Furthermore, knockdown of NSD3 in MCF10A cells or breast cancer cell lines with amplified 8p11–12 resulted in increased expression of SRFp1 a negative regulator of WNT-signaling and decreased expression of TGFBI leading to profound loss of growth and survival of these cells (Yang et al. 2010). Taken together, these results suggest that NSD3 is a putative cancer driver gene.
In addition to amplification, chromosomal translocations resulting in NSD3 fusion products have been described in myelodysplastic syndrome (MDS), AML and nuclear protein in testis (NUT) carcinoma. NUP98-NSD3 fusion transcripts associated with t(8;11)(p11.2;p15) have been reported in one patient with AML, one patient with therapy-related MDS and one patient with radiation-associated MDS, and both NSD3-long and NSD3-short isoforms have been detected to be fused to NUP98 in leukemic cell lines (Rosati et al. 2002; Romana et al. 2006; Taketani et al. 2009). In addition, NSD3 was detected as a fusion oncoprotein with the NUT gene in the rare and aggressive NUT midline carcinoma (NMC) (French et al. 2014; Harms et al. 2015; Kuroda et al. 2015; Suzuki et al. 2015). A t(8;15)(p12;q15) translocation was identified as responsible for the NSD3-NUT fusion in a patient-derived NMC cell line, and knockdown of the NSD3-NUT fusion revealed that this fusion protein functioned to block differentiation and promote proliferation (French et al. 2014).
NSD3 as an effector of BRD4 in cancer
The bromodomain and extraterminal domain (BET) family of transcriptional activators are promising therapeutic targets for cancers, particularly AML, due to their role in maintaining the expression of key oncogenes (Dawson et al. 2011; Mertz et al. 2011; Zuber et al. 2011). NSD3 can bind the ET domain of BET proteins and associates with BRD4 in nuclear lysates (Rahman et al. 2011; French et al. 2014; Crowe et al. 2016). The ET domain of BRD4 interacts with amino acids 100 – 263 of NSD3 a region immediately prior to the N-terminal PWWP domain located at amino acids 270 – 333 (Fig. 2) (Shen et al. 2015). The corresponding regions in NSD1 and NSD2 have significant homology to NSD3, suggesting this may be a common element responsible for BRD4 interaction with NSD family members (Fig. 2). In addition, the NSD3-NUT fusion binds to BRD4 and BRD bromodomain inhibitors induce differentiation and arrest proliferation of t(8;15)(p12;q15) cells (French et al. 2014). Recent evidence suggests that interaction with NSD3-short may be required for the AML maintenance function of BRD4 (Shen et al. 2015). The NSD3-short isoform has been reported to be an adaptor protein that links BRD4 to the CHD8 chromatin remodeler. BRD4, NSD3, and CHD8 co-localize across the AML genome, and each is released from super-enhancer regions upon treatment with BET inhibitors such as JQ1 (Shen et al. 2015). Furthermore, genetic targeting of NSD3 or CHD8 mimics the phenotypic and transcriptional effects of pharmacological BRD4 inhibition (Shen et al. 2015). Thus, BET inhibitors may function by evicting BRD4-NSD3-CHD8 complexes from chromatin to suppress transcription.
Figure 2.
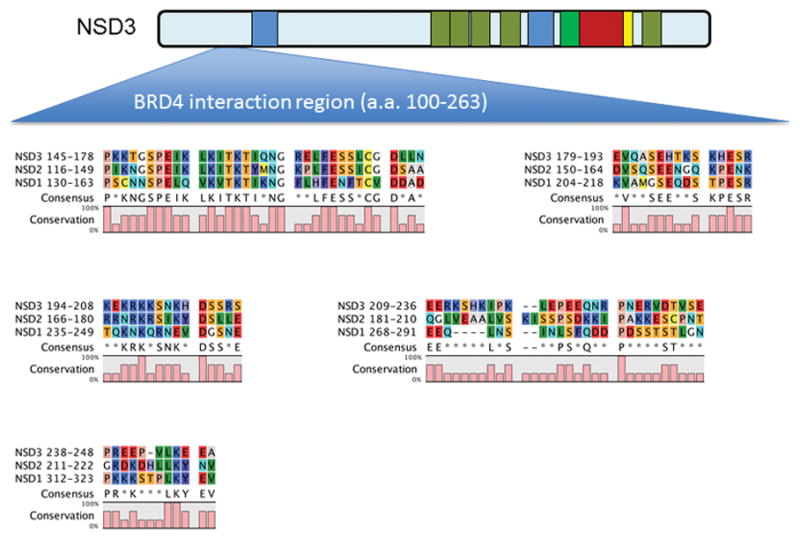
Sequence alignment of the region of NSD3 required for BRD4 interaction with NSD1 and NSD2 reveals conserved regions N-terminal to the 1st PWWP domain. The region in NSD3 identified as being required for interaction with BRD4 (amino acids 100–263) was found to have significant homology to the corresponding region in NSD1 and NSD2. Sequence analysis was performed using CLC sequence viewer (Qiagen).
Concluding Remarks
The NSD HMTases are key regulators of development. Mutations, amplifications or translocations of these genes lead to developmental defects and cancer. Differences between the function and tissue specificity of the many isoforms of each NSD family member are poorly understood, and additional research remains critical to improve our understanding for how dysregulation of NSD family members may lead to cancer. For instance, NSD family members have no known sequence specific DNA binding properties, and the precise mechanism that directs these lysine-HMTs to specific chromatin loci is unclear. Thus, identification of partner proteins that may guide NSD family members to specific promoter/enhancer regions is important to understand how these proteins may be directed to specific functional contexts. The recent report that BRD4 may direct NSD3 to chromatin as well as other reports indicating NSD2 associates with BRD4 raises the intriguing possibility that bromodomains (acetyl-lysine side chains) on Histone H3 may recruit BET family members such as BRD4 to enhancer and promoter regions that in turn recruit NSD family proteins to direct H3 K36 methylation, inhibit EZH2-mediated H3 K27 methylation and promote transcription initiation and elongation (Fig. 3) (Min et al. 2012; Shen et al. 2015). In addition, identifying how activating mutations such as E1099K in NSD2 may alter enzymatic activity to drive increased H3K36 dimethylation is critical to understand the biological effect of this mutation in oncogenesis. Preliminary epigenetic and transcriptional profiling of E1099K in ALL cells indicates there are both similarities and differences between E1099K NSD2 and overexpression of NSD2 (unpublished data). Elucidating the downstream effects of E1099K may provide new insight into the mechanism by which aberrant lysine HMTase activity promotes tumorigenesis.
Figure 3.
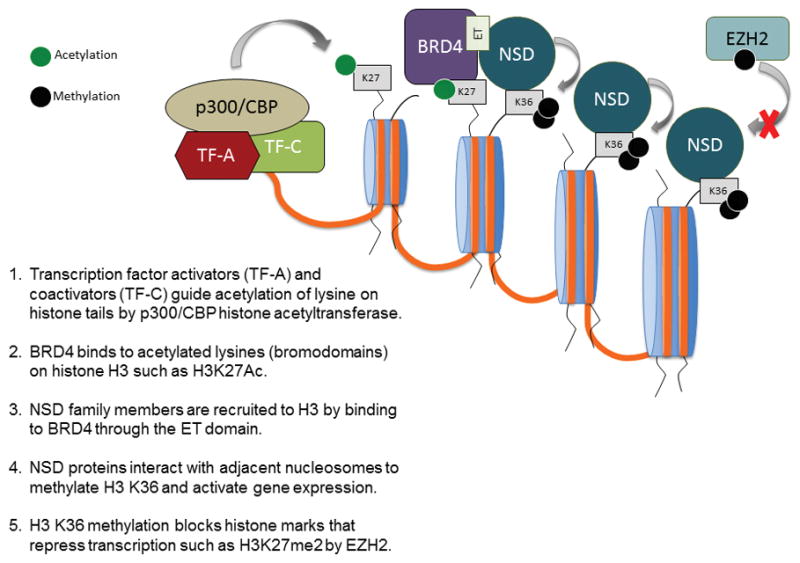
Proposed mechanism for targeting of NSD family proteins to specific chromatin loci.
The prominent role of translocations, upregulation, and activating mutations of NSD family members in driving tumor progression and aggressiveness, suggests that specific lysine-HMTase inhibitors are a promising therapeutic approach to suppress cancer growth. In support of this, abolition of the methyltransferase activity of NUP98-NSD1 by point mutation suppresses Hox-A gene activation and myeloid progenitor immortalization (Wang et al. 2007). In addition, knockdown of NSD2 in several model systems has indicated the potential utility of NSD2 inhibitors for downregulating critical tumor cell phenotypes. Targeting the SET domain is an attractive strategy for development of NSD family inhibitors, since the lysine HMTase activity is considered most likely to drive oncogenic reprogramming (Kuo et al. 2011; Martinez-Garcia et al. 2011). Recently, a high throughput luminescence-based assay using the NSD1 SET domain was used to screen for specific inhibitors of NSD1 (Drake et al. 2014). The HMTase inhibitor suramin was identified in this screen but had minimal selectivity for NSD1 over other histone methyltransferases and other compounds identified by this screen inactivated NSD1 through non-specific mechanisms. Alternatively, targeting domains outside of the catalytic SET domain may be a useful strategy for developing NSD inhibitors. The PHD zinc fingers 2, 3, and 4, as well as PWWP domain 2 of NSD2 are critical for the H3K36me2 upregulation in t(4;14)+ multiple myeloma (Popovic et al. 2014). The fourth PHD finger plays a role in the NSD2/EZH2 interplay, as its deletion abrogates the reduction of H3K27me3 typically observed with NSD2 overexpression in myeloma. Furthermore, recent reports that BET inhibitors evict NSD3-BRD4 from chromatin raise the attractive hypothesis that these compounds may also inhibit interactions between other BRD and NSD family members to suppress aberrant gene activation and limit tumor growth. While evidence suggests that genetic and epigenetic alterations cooperate in the stepwise initiation and progression of cancers, only epigenetic aberrations can be reversed. Thus, as we work to further our understanding of NSD family members, we are hopeful that inhibition of these epigenetic regulators will play a central role in the next generation of cancer therapy.
References
- Allali-Hassani A, Kuznetsova E, Hajian T, Wu H, Dombrovski L, Li Y, Graslund S, Arrowsmith CH, Schapira M, Vedadi M. A Basic Post-SET Extension of NSDs Is Essential for Nucleosome Binding In Vitro. Journal of biomolecular screening. 2014;19:928–935. doi: 10.1177/1087057114525854. [DOI] [PubMed] [Google Scholar]
- Andersen EF, Carey JC, Earl DL, Corzo D, Suttie M, Hammond P, South ST. Deletions involving genes WHSC1 and LETM1 may be necessary, but are not sufficient to cause Wolf-Hirschhorn Syndrome. European journal of human genetics : EJHG. 2013;22:464–470. doi: 10.1038/ejhg.2013.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angrand PO, Apiou F, Stewart AF, Dutrillaux B, Losson R, Chambon P. NSD3, a new SET domain-containing gene, maps to 8p12 and is amplified in human breast cancer cell lines. Genomics. 2001;74:79–88. doi: 10.1006/geno.2001.6524. [DOI] [PubMed] [Google Scholar]
- Asangani Ia, Ateeq B, Cao Q, Dodson L, Pandhi M, Kunju LP, Mehra R, Lonigro RJ, Siddiqui J, Palanisamy N, et al. Characterization of the EZH2-MMSET histone methyltransferase regulatory axis in cancer. Molecular cell. 2013;49:80–93. doi: 10.1016/j.molcel.2012.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker LA, Allis CD, Wang GG. PHD fingers in human diseases: Disorders arising from misinterpreting epigenetic marks. Mutation Research - Fundamental and Molecular Mechanisms of Mutagenesis. 2008;647:3–12. doi: 10.1016/j.mrfmmm.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannister AJ, Schneider R, Myers FA, Thorne AW, Crane-Robinson C, Kouzarides T. Spatial distribution of di- and tri-methyl lysine 36 of histone H3 at active genes. The Journal of biological chemistry. 2005;280:17732–17736. doi: 10.1074/jbc.M500796200. [DOI] [PubMed] [Google Scholar]
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehár J, Kryukov GV, Sonkin D, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Baujat G, Rio M, Rossignol S, Sanlaville D, Lyonnet S, Le Merrer M, Munnich A, Gicquel C, Cormier-Daire V, Colleaux L. Paradoxical NSD1 mutations in Beckwith-Wiedemann syndrome and 11p15 anomalies in Sotos syndrome. American journal of human genetics. 2004;74:715–720. doi: 10.1086/383093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beà S, Valdés-Mas R, Navarro A, Salaverria I, Martín-Garcia D, Jares P, Giné E, Pinyol M, Royo C, Nadeu F, et al. Landscape of somatic mutations and clonal evolution in mantle cell lymphoma. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:18250–18255. doi: 10.1073/pnas.1314608110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell O, Wirbelauer C, Hild M, Scharf AN, Schwaiger M, MacAlpine DM, Zilbermann F, van Leeuwen F, Bell SP, Imhof A, et al. Localized H3K36 methylation states define histone H4K16 acetylation during transcriptional elongation in Drosophila. The EMBO journal. 2007;26:4974–4984. doi: 10.1038/sj.emboj.7601926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender LB, Suh J, Carroll CR, Fong Y, Fingerman IM, Briggs SD, Cao R, Zhang Y, Reinke V, Strome S. MES-4: an autosome-associated histone methyltransferase that participates in silencing the X chromosomes in the C. elegans germ line. Development. 2006;133:3907–3917. doi: 10.1242/dev.02584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berardi A, Quilici G, Spiliotopoulos D, Corral-Rodriguez MA, Martin-Garcia F, Degano M, Tonon G, Ghitti M, Musco G. Structural basis for PHDVC5HCHNSD1-C2HRNizp1 interaction: implications for Sotos syndrome. Nucleic acids research. 2016 doi: 10.1093/nar/gkw103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berdasco M, Ropero S, Setien F, Fraga MF, Lapunzina P, Losson R, Alaminos M, Cheung NK, Rahman N, Esteller M. Epigenetic inactivation of the Sotos overgrowth syndrome gene histone methyltransferase NSD1 in human neuroblastoma and glioma. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:21830–21835. doi: 10.1073/pnas.0906831106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergemann AD, Cole F, Hirschhorn K. The etiology of Wolf-Hirschhorn syndrome. Trends in genetics : TIG. 2005;21:188–195. doi: 10.1016/j.tig.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Bianco-Miotto T, Chiam K, Buchanan G, Jindal S, Day TK, Thomas M, Pickering MA, O’Loughlin MA, Ryan NK, Raymond WA, et al. Global levels of specific histone modifications and an epigenetic gene signature predict prostate cancer progression and development. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2010;19:2611–2622. doi: 10.1158/1055-9965.EPI-10-0555. [DOI] [PubMed] [Google Scholar]
- Brito JLR, Walker B, Jenner M, Dickens NJ, Brown NJM, Ross FM, Avramidou A, Irving JaE, Gonzalez D, Davies FE, et al. MMSET deregulation affects cell cycle progression and adhesion regulons in t(4;14) myeloma plasma cells. Haematologica. 2009;94:78–86. doi: 10.3324/haematol.13426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrozza MJ, Li B, Florens L, Suganuma T, Swanson SK, Lee KK, Shia WJ, Anderson S, Yates J, Washburn MP, et al. Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell. 2005;123:581–592. doi: 10.1016/j.cell.2005.10.023. [DOI] [PubMed] [Google Scholar]
- Cerveira N, Correia C, Doria S, Bizarro S, Rocha P, Gomes P, Torres L, Norton L, Borges BS, Castedo S, et al. Frequency of NUP98-NSD1 fusion transcript in childhood acute myeloid leukaemia. Leukemia. 2003;17:2244–2247. doi: 10.1038/sj.leu.2403104. [DOI] [PubMed] [Google Scholar]
- Chen Y, McGee J, Chen X, Doman TN, Gong X, Zhang Y, Hamm N, Ma X, Higgs RE, Bhagwat SV, et al. Identification of druggable cancer driver genes amplified across TCGA datasets. PloS one. 2014;9:e98293. doi: 10.1371/journal.pone.0098293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesi M, Nardini E, Lim RS, Smith KD, Kuehl WM, Bergsagel PL. The t(4;14) translocation in myeloma dysregulates both FGFR3 and a novel gene, MMSET, resulting in IgH/MMSET hybrid transcripts. Blood. 1998;92:3025–3034. [PubMed] [Google Scholar]
- Crowe BL, Larue RC, Yuan C, Hess S, Kvaratskhelia M, Foster MP. Structure of the Brd4 ET domain bound to a C-terminal motif from gamma-retroviral integrases reveals a conserved mechanism of interaction. Proceedings of the National Academy of Sciences of the United States of America. 2016;113:2086–2091. doi: 10.1073/pnas.1516813113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan WI, Robson SC, Chung CW, Hopf C, Savitski MM, et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature. 2011;478:529–533. doi: 10.1038/nature10509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Souza CF, Xander P, Monteiro AC, Silva AG, da Silva DC, Mai S, Bernardo V, Lopes JD, Jasiulionis MG. Mining gene expression signature for the detection of pre-malignant melanocytes and early melanomas with risk for metastasis. PloS one. 2012;7:e44800. doi: 10.1371/journal.pone.0044800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dikow N, Maas B, Gaspar H, Kreiss-Nachtsheim M, Engels H, Kuechler A, Garbes L, Netzer C, Neuhann TM, Koehler U, et al. The phenotypic spectrum of duplication 5q35.2-q35.3 encompassing NSD1: is it really a reversed Sotos syndrome? American journal of medical genetics Part A. 2013;161A:2158–2166. doi: 10.1002/ajmg.a.36046. [DOI] [PubMed] [Google Scholar]
- Dillon SC, Zhang X, Trievel RC, Cheng X. The SET-domain protein superfamily: protein lysine methyltransferases. Genome biology. 2005;6:227. doi: 10.1186/gb-2005-6-8-227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas J, Hanks S, Temple IK, Davies S, Murray A, Upadhyaya M, Tomkins S, Hughes HE, Cole TR, Rahman N. NSD1 mutations are the major cause of Sotos syndrome and occur in some cases of Weaver syndrome but are rare in other overgrowth phenotypes. American journal of human genetics. 2003;72:132–143. doi: 10.1086/345647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake KM, Watson VG, Kisielewski A, Glynn R, Napper AD. A sensitive luminescent assay for the histone methyltransferase NSD1 and other SAM-dependent enzymes. Assay and drug development technologies. 2014;12:258–271. doi: 10.1089/adt.2014.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezponda T, Popovic R, Shah MY, Martinez-Garcia E, Zheng Y, Min D-J, Will C, Neri a, Kelleher NL, Yu J, et al. The histone methyltransferase MMSET/WHSC1 activates TWIST1 to promote an epithelial–mesenchymal transition and invasive properties of prostate cancer. Oncogene. 2012:2882–2890. doi: 10.1038/onc.2012.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabbri G, Rasi S, Rossi D, Trifonov V, Khiabanian H, Ma J, Grunn A, Fangazio M, Capello D, Monti S, et al. Analysis of the chronic lymphocytic leukemia coding genome: role of NOTCH1 mutational activation. J Exp Med. 2011;208:1389–1401. doi: 10.1084/jem.20110921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang R, Barbera AJ, Xu Y, Rutenberg M, Leonor T, Bi Q, Lan F, Mei P, Yuan GC, Lian C, et al. Human LSD2/KDM1b/AOF1 regulates gene transcription by modulating intragenic H3K4me2 methylation. Molecular cell. 2010;39:222–233. doi: 10.1016/j.molcel.2010.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finelli P, Fabris S, Zagano S, Baldini L, Intini D, Nobili L, Lombardi L, Maiolo aT, Neri a. Detection of t(4;14)(p16.3;q32) chromosomal translocation in multiple myeloma by double-color fluorescent in situ hybridization. Blood. 1999;94:724–732. [PubMed] [Google Scholar]
- French CA, Rahman S, Walsh EM, Kuhnle S, Grayson AR, Lemieux ME, Grunfeld N, Rubin BP, Antonescu CR, Zhang S, et al. NSD3-NUT fusion oncoprotein in NUT midline carcinoma: implications for a novel oncogenic mechanism. Cancer discovery. 2014;4:928–941. doi: 10.1158/2159-8290.CD-14-0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson WT, Hood RL, Zhan SH, Bulman DE, Fejes AP, Moore R, Mungall AJ, Eydoux P, Babul-Hirji R, An J, et al. Mutations in EZH2 cause Weaver syndrome. American journal of human genetics. 2012;90:110–118. doi: 10.1016/j.ajhg.2011.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham SE, Tweedy SE, Carlson HA. Dynamic Behavior of the Post-SET Loop Region of NSD1: Implications for Histone Binding and Drug Development. Protein science : a publication of the Protein Society. 2016 doi: 10.1002/pro.2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajdu I, Ciccia A, Lewis SM, Elledge SJ. Wolf-Hirschhorn syndrome candidate 1 is involved in the cellular response to DNA damage. Proc Natl Acad Sci U S A. 2011;108:13130–13134. doi: 10.1073/pnas.1110081108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harms A, Herpel E, Pfarr N, Penzel R, Heussel CP, Herth FJ, Dienemann H, Weichert W, Warth A. NUT carcinoma of the thorax: Case report and review of the literature. Lung cancer. 2015;90:484–491. doi: 10.1016/j.lungcan.2015.10.001. [DOI] [PubMed] [Google Scholar]
- He C, Li F, Zhang J, Wu J, Shi Y. The methyltransferase NSD3 has chromatin-binding motifs, PHD5-C5HCH, that are distinct from other NSD (nuclear receptor SET domain) family members in their histone H3 recognition. The Journal of biological chemistry. 2013;288:4692–4703. doi: 10.1074/jbc.M112.426148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herz HM, Garruss A, Shilatifard A. SET for life: biochemical activities and biological functions of SET domain-containing proteins. Trends in biochemical sciences. 2013;38:621–639. doi: 10.1016/j.tibs.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang N, vom Baur E, Garnier JM, Lerouge T, Vonesch JL, Lutz Y, Chambon P, Losson R. Two distinct nuclear receptor interaction domains in NSD1, a novel SET protein that exhibits characteristics of both corepressors and coactivators. The EMBO journal. 1998;17:3398–3412. doi: 10.1093/emboj/17.12.3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z, Wu H, Chuai S, Xu F, Yan F, Englund N, Wang Z, Zhang H, Fang M, Wang Y, et al. NSD2 Is recruited through Its PHD domain to oncogenic gene loci to drive multiple myeloma. Cancer research. 2013;73:6277–6288. doi: 10.1158/0008-5472.CAN-13-1000. [DOI] [PubMed] [Google Scholar]
- Hudlebusch HR, Santoni-Rugiu E, Simon R, Ralfkiær E, Rossing HH, Johansen JV, Jørgensen M, Sauter G, Helin K. The histone methyltransferase and putative oncoprotein MMSET is overexpressed in a large variety of human tumors. Clinical Cancer Research. 2011a;17:2919–2933. doi: 10.1158/1078-0432.CCR-10-1302. [DOI] [PubMed] [Google Scholar]
- Hudlebusch HR, Skotte J, Santoni-Rugiu E, Zimling ZG, Lees MJ, Simon R, Sauter G, Rota R, De Ioris MA, Quarto M, et al. MMSET is highly expressed and associated with aggressiveness in neuroblastoma. Cancer research. 2011b;71:4226–4235. doi: 10.1158/0008-5472.CAN-10-3810. [DOI] [PubMed] [Google Scholar]
- Jacques-Fricke BT, Gammill LS. Neural crest specification and migration independently require NSD3-related lysine methyltransferase activity. Molecular biology of the cell. 2014;25:4174–4186. doi: 10.1091/mbc.E13-12-0744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe JD, Wang Y, Chan HM, Zhang J, Huether R, Kryukov GV, Bhang H-eC, Taylor JE, Hu M, Englund NP, et al. Global chromatin profiling reveals NSD2 mutations in pediatric acute lymphoblastic leukemia. Nature genetics. 2013;45:1386–1391. doi: 10.1038/ng.2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaju RJ, Fidler C, Haas OA, Strickson AJ, Watkins F, Clark K, Cross NCP, Cheng JF, Aplan PD, Kearney L, et al. A novel gene, NSD1, is fused to NUP98 in the t(5;11)(q35;p15.5) in de novo childhood acute myeloid leukemia. Blood. 2001;98:1264–1267. doi: 10.1182/blood.v98.4.1264. [DOI] [PubMed] [Google Scholar]
- Joshi AA, Struhl K. Eaf3 chromodomain interaction with methylated H3-K36 links histone deacetylation to Pol II elongation. Molecular cell. 2005;20:971–978. doi: 10.1016/j.molcel.2005.11.021. [DOI] [PubMed] [Google Scholar]
- Kang D, Cho HS, Toyokawa G, Kogure M, Yamane Y, Iwai Y, Hayami S, Tsunoda T, Field HI, Matsuda K, et al. The histone methyltransferase Wolf-Hirschhorn syndrome candidate 1-like 1 (WHSC1L1) is involved in human carcinogenesis. Genes, chromosomes & cancer. 2013;52:126–139. doi: 10.1002/gcc.22012. [DOI] [PubMed] [Google Scholar]
- Kaplan CD, Laprade L, Winston F. Transcription elongation factors repress transcription initiation from cryptic sites. Science. 2003;301:1096–1099. doi: 10.1126/science.1087374. [DOI] [PubMed] [Google Scholar]
- Kim A, Kiefer CM, Dean A. Distinctive signatures of histone methylation in transcribed coding and noncoding human beta-globin sequences. Molecular and cellular biology. 2007;27:1271–1279. doi: 10.1128/MCB.01684-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J-Y, Kee HJ, Choe N-W, Kim S-M, Eom G-H, Baek HJ, Kook H, Kook H, Seo S-B. Multiple-myeloma-related WHSC1/MMSET isoform RE-IIBP is a histone methyltransferase with transcriptional repression activity. Molecular and cellular biology. 2008;28:2023–2034. doi: 10.1128/MCB.02130-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SM, Kee HJ, Eom GH, Choe NW, Kim JY, Kim YS, Kim SK, Kook H, Kook H, Seo SB. Characterization of a novel WHSC1-associated SET domain protein with H3K4 and H3K27 methyltransferase activity. Biochemical and biophysical research communications. 2006;345:318–323. doi: 10.1016/j.bbrc.2006.04.095. [DOI] [PubMed] [Google Scholar]
- Kizer KO, Phatnani HP, Shibata Y, Hall H, Greenleaf AL, Strahl BD. A novel domain in Set2 mediates RNA polymerase II interaction and couples histone H3 K36 methylation with transcript elongation. Molecular and cellular biology. 2005;25:3305–3316. doi: 10.1128/MCB.25.8.3305-3316.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolasinska-Zwierz P, Down T, Latorre I, Liu T, Liu XS, Ahringer J. Differential chromatin marking of introns and expressed exons by H3K36me3. Nature genetics. 2009;41:376–381. doi: 10.1038/ng.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T. SnapShot: Histone-modifying enzymes. Cell. 2007;128:802. doi: 10.1016/j.cell.2007.02.018. [DOI] [PubMed] [Google Scholar]
- Kudithipudi S, Lungu C, Rathert P, Happel N, Jeltsch A. Substrate specificity analysis and novel substrates of the protein lysine methyltransferase NSD1. Chemistry & biology. 2014;21:226–237. doi: 10.1016/j.chembiol.2013.10.016. [DOI] [PubMed] [Google Scholar]
- Kuo AJ, Cheung P, Chen K, Zee BM, Kioi M, Lauring J, Xi Y, Park BH, Shi X, Garcia BA, et al. NSD2 Links Dimethylation of Histone H3 at Lysine 36 to Oncogenic Programming. Molecular cell. 2011;44:609–620. doi: 10.1016/j.molcel.2011.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroda S, Suzuki S, Kurita A, Muraki M, Aoshima Y, Tanioka F, Sugimura H. Cytological Features of a Variant NUT Midline Carcinoma of the Lung Harboring the NSD3-NUT Fusion Gene: A Case Report and Literature Review. Case reports in pathology. 2015;2015:572951. doi: 10.1155/2015/572951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurotaki N, Imaizumi K, Harada N, Masuno M, Kondoh T, Nagai T, Ohashi H, Naritomi K, Tsukahara M, Makita Y, et al. Haploinsufficiency of NSD1 causes Sotos syndrome. Nature genetics. 2002;30:365–366. doi: 10.1038/ng863. [DOI] [PubMed] [Google Scholar]
- La Starza R, Gorello P, Rosati R, Riezzo A, Veronese A, Ferrazzi E, Martelli MF, Negrini M, Mecucci C. Cryptic insertion producing two NUP98/NSD1 chimeric transcripts in adult refractory anemia with an excess of blasts. Genes, chromosomes & cancer. 2004;41:395–399. doi: 10.1002/gcc.20103. [DOI] [PubMed] [Google Scholar]
- Lachner M, O’Sullivan RJ, Jenuwein T. An epigenetic road map for histone lysine methylation. Journal of cell science. 2003;116:2117–2124. doi: 10.1242/jcs.00493. [DOI] [PubMed] [Google Scholar]
- Larschan E, Alekseyenko AA, Gortchakov AA, Peng S, Li B, Yang P, Workman JL, Park PJ, Kuroda MI. MSL complex is attracted to genes marked by H3K36 trimethylation using a sequence-independent mechanism. Molecular cell. 2007;28:121–133. doi: 10.1016/j.molcel.2007.08.011. [DOI] [PubMed] [Google Scholar]
- Lauring J, Abukhdeir AM, Konishi H, Garay JP, Gustin JP, Wang Q, Arceci RJ, Matsui W, Park BH. The multiple myeloma associated MMSET gene contributes to cellular adhesion, clonogenic growth, and tumorigenicity. Blood. 2008;111:856–864. doi: 10.1182/blood-2007-05-088674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JS, Shilatifard A. A site to remember: H3K36 methylation a mark for histone deacetylation. Mutation research. 2007;618:130–134. doi: 10.1016/j.mrfmmm.2006.08.014. [DOI] [PubMed] [Google Scholar]
- Li B, Jackson J, Simon MD, Fleharty B, Gogol M, Seidel C, Workman JL, Shilatifard A. Histone H3 lysine 36 dimethylation (H3K36me2) is sufficient to recruit the Rpd3s histone deacetylase complex and to repress spurious transcription. The Journal of biological chemistry. 2009a;284:7970–7976. doi: 10.1074/jbc.M808220200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Trojer P, Xu CF, Cheung P, Kuo A, Drury WJ, Qiao Q, Neubert TA, Xu RM, Gozani O, et al. The target of the NSD family of histone lysine methyltransferases depends on the nature of the substrate. Journal of Biological Chemistry. 2009b;284:34283–34295. doi: 10.1074/jbc.M109.034462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh ML, Ma X, Rusch M, Wu G, Harvey RC, Wheeler DA, Hampton OA, Carroll WL, Chen I-M, Gerhard DS, et al. Comparison Of Mutational Profiles Of Diagnosis and Relapsed Pediatric B-Acute Lymphoblastic Leukemia: A Report From The COG ALL Target Project. Blood. 2013;122:824. [Google Scholar]
- Lu T, Jackson MW, Wang B, Yang M, Chance MR, Miyagi M, Gudkov AV, Stark GR. Regulation of NF-kappaB by NSD1/FBXL11-dependent reversible lysine methylation of p65. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:46–51. doi: 10.1073/pnas.0912493107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucio-Eterovic AK, Singh MM, Gardner JE, Veerappan CS, Rice JC, Carpenter PB. Role for the nuclear receptor-binding SET domain protein 1 (NSD1) methyltransferase in coordinating lysine 36 methylation at histone 3 with RNA polymerase II function. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:16952–16957. doi: 10.1073/pnas.1002653107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmood SF, Gruel N, Nicolle R, Chapeaublanc E, Delattre O, Radvanyi F, Bernard-Pierrot I. PPAPDC1B and WHSC1L1 are common drivers of the 8p11–12 amplicon, not only in breast tumors but also in pancreatic adenocarcinomas and lung tumors. The American journal of pathology. 2013;183:1634–1644. doi: 10.1016/j.ajpath.2013.07.028. [DOI] [PubMed] [Google Scholar]
- Mann KM, Ward JM, Yew CC, Kovochich A, Dawson DW, Black MA, Brett BT, Sheetz TE, Dupuy AJ, et al. Australian Pancreatic Cancer Genome I. Sleeping Beauty mutagenesis reveals cooperating mutations and pathways in pancreatic adenocarcinoma. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:5934–5941. doi: 10.1073/pnas.1202490109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marango J, Shimoyama M, Nishio H, Meyer JA, Min DJ, Sirulnik A, Martinez-Martinez Y, Chesi M, Bergsagel PL, Zhou MM, et al. The MMSET protein is a histone methyltransferase with characteristics of a transcriptional corepressor. Blood. 2008;111:3145–3154. doi: 10.1182/blood-2007-06-092122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Garcia E, Popovic R, Min DJ, Sweet SMM, Thomas PM, Zamdborg L, Heffner A, Will C, Lamy L, Staudt LM, et al. The MMSET histone methyl transferase switches global histone methylation and alters gene expression in t(4;14) multiple myeloma cells. Blood. 2011;117:211–220. doi: 10.1182/blood-2010-07-298349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertz JA, Conery AR, Bryant BM, Sandy P, Balasubramanian S, Mele DA, Bergeron L, Sims RJ., 3rd Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:16669–16674. doi: 10.1073/pnas.1108190108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migdalska AM, Van Der Weyden L, Ismail O, Rust AG, Rashid M, White JK, Sánchez-Andrade G, Lupski JR, Logan DW, Arends MJ, et al. Generation of the Sotos syndrome deletion in mice. Mammalian Genome. 2012;23:749–757. doi: 10.1007/s00335-012-9416-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min D-J, Ezponda T, Kim MK, Will CM, Martinez-Garcia E, Popovic R, Basrur V, Elenitoba-Johnson KS, Licht JD. MMSET stimulates myeloma cell growth through microRNA-mediated modulation of c-MYC. Leukemia. 2012;27:686–694. doi: 10.1038/leu.2012.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishita M, di Luccio E. Cancers and the NSD family of histone lysine methyltransferases. Biochimica et biophysica acta. 2011;1816:158–163. doi: 10.1016/j.bbcan.2011.05.004. [DOI] [PubMed] [Google Scholar]
- Morishita M, Mevius D, di Luccio E. In vitro histone lysine methylation by NSD1, NSD2/MMSET/WHSC1 and NSD3/WHSC1L. BMC structural biology. 2014;14:25. doi: 10.1186/s12900-014-0025-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris SA, Shibata Y, Noma K, Tsukamoto Y, Warren E, Temple B, Grewal SI, Strahl BD. Histone H3 K36 methylation is associated with transcription elongation in Schizosaccharomyces pombe. Eukaryotic cell. 2005;4:1446–1454. doi: 10.1128/EC.4.8.1446-1454.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen AL, Jørgensen P, Lerouge T, Cerviño M, Chambon P, Losson R. Nizp1, a novel multitype zinc finger protein that interacts with the NSD1 histone lysine methyltransferase through a unique C2HR motif. Molecular and cellular biology. 2004;24:5184–5196. doi: 10.1128/MCB.24.12.5184-5196.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimura K, Ura K, Shiratori H, Ikawa M, Okabe M, Schwartz RJ, Kaneda Y. A histone H3 lysine 36 trimethyltransferase links Nkx2–5 to Wolf-Hirschhorn syndrome. Nature. 2009;460:287–291. doi: 10.1038/nature08086. [DOI] [PubMed] [Google Scholar]
- Oyer Ja, Huang X, Zheng Y, Shim J, Ezponda T, Carpenter Z, Allegretta M, Okot-Kotber CI, Patel JP, Melnick a, et al. Point mutation E1099K in MMSET/NSD2 enhances its methyltranferase activity and leads to altered global chromatin methylation in lymphoid malignancies. Leukemia. 2014;28:198–201. doi: 10.1038/leu.2013.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasillas MP, Shah M, Kamps MP. NSD1 PHD domains bind methylated H3K4 and H3K9 using interactions disrupted by point mutations in human sotos syndrome. Human Mutation. 2011;32:292–298. doi: 10.1002/humu.21424. [DOI] [PubMed] [Google Scholar]
- Pei H, Zhang L, Luo K, Qin Y, Chesi M, Fei F, Bergsagel PL, Wang L, You Z, Lou Z. MMSET regulates histone H4K20 methylation and 53BP1 accumulation at DNA damage sites. Nature. 2011;470:124–128. doi: 10.1038/nature09658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera A, Eisen D, Wagner M, Laube SK, Kunzel AF, Koch S, Steinbacher J, Schulze E, Splith V, Mittermeier N, et al. TET3 is recruited by REST for context-specific hydroxymethylation and induction of gene expression. Cell reports. 2015;11:283–294. doi: 10.1016/j.celrep.2015.03.020. [DOI] [PubMed] [Google Scholar]
- Popovic R, Martinez-Garcia E, Giannopoulou EG, Zhang Q, Zhang Q, Ezponda T, Shah MY, Zheng Y, Will CM, Small EC, et al. Histone methyltransferase MMSET/NSD2 alters EZH2 binding and reprograms the myeloma epigenome through global and focal changes in H3K36 and H3K27 methylation. PLoS genetics. 2014;10:e1004566. doi: 10.1371/journal.pgen.1004566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulin MB, Schneck JL, Matico RE, Hou W, McDevitt PJ, Holbert M, Schramm VL. Nucleosome Binding Alters the Substrate Bonding Environment of Histone H3 Lysine 36 Methyltransferase NSD2. Journal of the American Chemical Society. 2016a;138:6699–6702. doi: 10.1021/jacs.6b01612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulin MB, Schneck JL, Matico RE, McDevitt PJ, Huddleston MJ, Hou W, Johnson NW, Thrall SH, Meek TD, Schramm VL. Transition state for the NSD2-catalyzed methylation of histone H3 lysine 36. Proceedings of the National Academy of Sciences of the United States of America. 2016b;113:1197–1201. doi: 10.1073/pnas.1521036113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao Q, Li Y, Chen Z, Wang M, Reinberg D, Xu RM. The structure of NSD1 reveals an autoregulatory mechanism underlying histone H3K36 methylation. The Journal of biological chemistry. 2011;286:8361–8368. doi: 10.1074/jbc.M110.204115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana RM, Dupuy AJ, Bravo A, Casanova ML, Alameda JP, Page A, Sanchez-Viera M, Ramirez A, Navarro M. A transposon-based analysis of gene mutations related to skin cancer development. The Journal of investigative dermatology. 2013;133:239–248. doi: 10.1038/jid.2012.245. [DOI] [PubMed] [Google Scholar]
- Rahman N. Mechanisms predisposing to childhood overgrowth and cancer. Current opinion in genetics & development. 2005;15:227–233. doi: 10.1016/j.gde.2005.04.007. [DOI] [PubMed] [Google Scholar]
- Rahman S, Sowa ME, Ottinger M, Smith JA, Shi Y, Harper JW, Howley PM. The Brd4 extraterminal domain confers transcription activation independent of pTEFb by recruiting multiple proteins, including NSD3. Molecular and cellular biology. 2011;31:2641–2652. doi: 10.1128/MCB.01341-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao B, Shibata Y, Strahl BD, Lieb JD. Dimethylation of histone H3 at lysine 36 demarcates regulatory and nonregulatory chromatin genome-wide. Molecular and cellular biology. 2005;25:9447–9459. doi: 10.1128/MCB.25.21.9447-9459.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayasam GV, Wendling O, Angrand PO, Mark M, Niederreither K, Song L, Lerouge T, Hager GL, Chambon P, Losson R. NSD1 is essential for early post-implantation development and has a catalytically active SET domain. The EMBO journal. 2003;22:3153–3163. doi: 10.1093/emboj/cdg288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romana SP, Radford-Weiss I, Ben Abdelali R, Schluth C, Petit A, Dastugue N, Talmant P, Bilhou-Nabera C, Mugneret F, Lafage-Pochitaloff M, et al. NUP98 rearrangements in hematopoietic malignancies: a study of the Groupe Francophone de Cytogenetique Hematologique. Leukemia. 2006;20:696–706. doi: 10.1038/sj.leu.2404130. [DOI] [PubMed] [Google Scholar]
- Rosati R, La Starza R, Veronese A, Aventin A, Schwienbacher C, Vallespi T, Negrini M, Martelli MF, Mecucci C. NUP98 is fused to the NSD3 gene in acute myeloid leukemia associated with t(8;11)(p11.2;p15) Blood. 2002;99:3857–3860. doi: 10.1182/blood.v99.10.3857. [DOI] [PubMed] [Google Scholar]
- Rosenfeld JA, Kim KH, Angle B, Troxell R, Gorski JL, Westemeyer M, Frydman M, Senturias Y, Earl D, Torchia B, et al. Further Evidence of Contrasting Phenotypes Caused by Reciprocal Deletions and Duplications: Duplication of NSD1 Causes Growth Retardation and Microcephaly. Molecular syndromology. 2013;3:247–254. doi: 10.1159/000345578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaran SM, Wilkinson AW, Elias JE, Gozani O. A PWWP Domain of Histone-Lysine N-Methyltransferase NSD2 Binds to Dimethylated Lys-36 of Histone H3 and Regulates NSD2 Function at Chromatin. The Journal of biological chemistry. 2016;291:8465–8474. doi: 10.1074/jbc.M116.720748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santra M, Zhan F, Tian E, Barlogie B, Jr, JS Brief report A subset of multiple myeloma harboring the t ( 4 ; 14 )( p16 ; q32 ) translocation lacks FGFR3 expression but maintains an IGH/MMSET fusion transcript. 2003;101:2374–2376. doi: 10.1182/blood-2002-09-2801. [DOI] [PubMed] [Google Scholar]
- Shah MY, Martinez-Garcia E, Phillip JM, Chambliss AB, Popovic R, Ezponda T, Small EC, Will C, Phillip MP, Neri P, et al. MMSET/WHSC1 enhances DNA damage repair leading to an increase in resistance to chemotherapeutic agents. Oncogene. 2016 doi: 10.1038/onc.2016.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen C, Ipsaro JJ, Shi J, Milazzo JP, Wang E, Roe JS, Suzuki Y, Pappin DJ, Joshua-Tor L, Vakoc CR. NSD3-Short Is an Adaptor Protein that Couples BRD4 to the CHD8 Chromatin Remodeler. Molecular cell. 2015;60:847–859. doi: 10.1016/j.molcel.2015.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiba N, Ichikawa H, Taki T, Park MJ, Jo A, Mitani S, Kobayashi T, Shimada A, Sotomatsu M, Arakawa H, et al. NUP98-NSD1 gene fusion and its related gene expression signature are strongly associated with a poor prognosis in pediatric acute myeloid leukemia. Genes, chromosomes & cancer. 2013;52:683–693. doi: 10.1002/gcc.22064. [DOI] [PubMed] [Google Scholar]
- Shilatifard A. Chromatin modifications by methylation and ubiquitination: implications in the regulation of gene expression. Annual review of biochemistry. 2006;75:243–269. doi: 10.1146/annurev.biochem.75.103004.142422. [DOI] [PubMed] [Google Scholar]
- Sims RJ, 3rd, Reinberg D. Processing the H3K36me3 signature. Nature genetics. 2009;41:270–271. doi: 10.1038/ng0309-270. [DOI] [PubMed] [Google Scholar]
- Spies N, Nielsen CB, Padgett RA, Burge CB. Biased chromatin signatures around polyadenylation sites and exons. Molecular cell. 2009;36:245–254. doi: 10.1016/j.molcel.2009.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stabell M, Larsson J, Aalen RB, Lambertsson A. Drosophila dSet2 functions in H3-K36 methylation and is required for development. Biochemical and biophysical research communications. 2007;359:784–789. doi: 10.1016/j.bbrc.2007.05.189. [DOI] [PubMed] [Google Scholar]
- Stec I, van Ommen GJ, den Dunnen JT. WHSC1L1, on human chromosome 8p11.2, closely resembles WHSC1 and maps to a duplicated region shared with 4p16.3. Genomics. 2001;76:5–8. doi: 10.1006/geno.2001.6581. [DOI] [PubMed] [Google Scholar]
- Stec I, Wright TJ, van Ommen GJ, de Boer PA, van Haeringen A, Moorman AF, Altherr MR, den Dunnen JT. WHSC1, a 90 kb SET domain-containing gene, expressed in early development and homologous to a Drosophila dysmorphy gene maps in the Wolf-Hirschhorn syndrome critical region and is fused to IgH in t(4;14) multiple myeloma. Hum Mol Genet. 1998;7:1071–1082. doi: 10.1093/hmg/7.7.1071. [DOI] [PubMed] [Google Scholar]
- Suzuki S, Kurabe N, Ohnishi I, Yasuda K, Aoshima Y, Naito M, Tanioka F, Sugimura H. NSD3-NUT-expressing midline carcinoma of the lung: first characterization of primary cancer tissue. Pathology, research and practice. 2015;211:404–408. doi: 10.1016/j.prp.2014.10.013. [DOI] [PubMed] [Google Scholar]
- Taketani T, Taki T, Nakamura H, Taniwaki M, Masuda J, Hayashi Y. NUP98-NSD3 fusion gene in radiation-associated myelodysplastic syndrome with t(8;11)(p11;p15) and expression pattern of NSD family genes. Cancer genetics and cytogenetics. 2009;190:108–112. doi: 10.1016/j.cancergencyto.2008.12.008. [DOI] [PubMed] [Google Scholar]
- Tatton-Brown K, Douglas J, Coleman K, Baujat G, Chandler K, Clarke a, Collins a, Davies S, Faravelli F, Firth H, et al. Multiple mechanisms are implicated in the generation of 5q35 microdeletions in Sotos syndrome. Journal of medical genetics. 2005;42:307–313. doi: 10.1136/jmg.2004.027755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thanasopoulou A, Tzankov A, Schwaller J. Potent co-operation between the NUP98-NSD1 fusion and the FLT3-ITD mutation in acute myeloid leukemia induction. Haematologica. 2014;99:1465–1471. doi: 10.3324/haematol.2013.100917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonon G, Wong KK, Maulik G, Brennan C, Feng B, Zhang Y, Khatry DB, Protopopov A, You MJ, Aguirre AJ, et al. High-resolution genomic profiles of human lung cancer. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:9625–9630. doi: 10.1073/pnas.0504126102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeulen M, Eberl HC, Matarese F, Marks H, Denissov S, Butter F, Lee KK, Olsen JV, Hyman AA, Stunnenberg HG, et al. Quantitative interaction proteomics and genome-wide profiling of epigenetic histone marks and their readers. Cell. 2010;142:967–980. doi: 10.1016/j.cell.2010.08.020. [DOI] [PubMed] [Google Scholar]
- Waggoner DJ, Raca G, Welch K, Dempsey M, Anderes E, Ostrovnaya I, Alkhateeb A, Kamimura J, Matsumoto N, Schaeffer GB, et al. NSD1 analysis for Sotos syndrome: insights and perspectives from the clinical laboratory. Genetics in medicine : official journal of the American College of Medical Genetics. 2005;7:524–533. doi: 10.1097/01.GIM.0000178503.15559.d3. [DOI] [PubMed] [Google Scholar]
- Wagner EJ, Carpenter PB. Understanding the language of Lys36 methylation at histone H3. Nature Reviews Molecular Cell Biology. 2012;13:115–126. doi: 10.1038/nrm3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GG, Cai L, Pasillas MP, Kamps MP. NUP98-NSD1 links H3K36 methylation to Hox-A gene activation and leukaemogenesis. Nature cell biology. 2007;9:804–812. doi: 10.1038/ncb1608. [DOI] [PubMed] [Google Scholar]
- Wu H, Zeng H, Lam R, Tempel W, Amaya MF, Xu C, Dombrovski L, Qiu W, Wang Y, Min J. Structural and histone binding ability characterizations of human PWWP domains. PloS one. 2011;6:e18919. doi: 10.1371/journal.pone.0018919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Zhao Z, Dong A, Soubigou-Taconnat L, Renou JP, Steinmetz A, Shen WH. Di- and tri- but not monomethylation on histone H3 lysine 36 marks active transcription of genes involved in flowering time regulation and other processes in Arabidopsis thaliana. Molecular and cellular biology. 2008a;28:1348–1360. doi: 10.1128/MCB.01607-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu LN, Wang X, Zou SQ. Effect of histone deacetylase inhibitor on proliferation of biliary tract cancer cell lines. World journal of gastroenterology. 2008b;14:2578–2581. doi: 10.3748/wjg.14.2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang ZQ, Liu G, Bollig-Fischer A, Giroux CN, Ethier SP. Transforming properties of 8p11–12 amplified genes in human breast cancer. Cancer research. 2010;70:8487–8497. doi: 10.1158/0008-5472.CAN-10-1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Sweet SMM, Popovic R, Martinez-Garcia E, Tipton JD, Thomas PM, Licht JD, Kelleher NL. Total kinetic analysis reveals how combinatorial methylation patterns are established on lysines 27 and 36 of histone H3. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:13549–13554. doi: 10.1073/pnas.1205707109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Thomsen R, Kahns S, Nielsen AL. The NSD3L histone methyltransferase regulates cell cycle and cell invasion in breast cancer cells. Biochemical and biophysical research communications. 2010;398:565–570. doi: 10.1016/j.bbrc.2010.06.119. [DOI] [PubMed] [Google Scholar]
- Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, Magoon D, Qi J, Blatt K, Wunderlich M, et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011;478:524–528. doi: 10.1038/nature10334. [DOI] [PMC free article] [PubMed] [Google Scholar]