

Abstract
Use of human induced pluripotent stem cells (iPSC) or embryonic stem cells (ESC) for cell replacement therapies holds great promise. Several limitations including low yields and heterogeneous populations of differentiated cells hinder the progress of stem cell therapies. A fate restricted immortalized multipotent otic progenitor (iMOP) cell line was generated to facilitate efficient differentiation of large numbers of functional hair cells and spiral ganglion neurons (SGN) for inner ear cell replacement therapies. Starting from dissociated cultures of single iMOP cells, protocols that promote cell cycle exit and differentiation by growth factor (bFGF) withdrawal were described. A significant decrease in proliferating cells after bFGF withdrawal was confirmed using an EdU cell proliferation assay. Concomitant with a decrease in proliferation, successful differentiation resulted in expression of molecular markers and morphological changes. Immunostaining of Cdkn1b (p27KIP) and Cdh1 (E-cadherin) in iMOP-derived otospheres was used as an indicator for differentiation into inner ear sensory epithelia while immunostaining of Cdkn1b and Tubb3 (neuronal β-tubulin) was used to identify iMOP-derived neurons. Use of iMOP cells provides an important tool for understanding cell fate decisions made by inner ear neurosensory progenitors and will help develop protocols for generating large numbers of iPSC or ESC-derived hair cells and SGNs. These methods will accelerate efforts for generating otic cells for replacement therapies.
Keywords: Inner ear, iMOP, immortalized multipotent otic progenitor, differentiation, hair cell, spiral ganglia neurons, supporting cell, cell fate
Introduction
The organs of the inner ear, the cochlea, utricle, saccule and three semicircular canals, mediate the ability to hear and balance. Within the cochlea, hair cells convert sounds into electrical signals that are relayed to the spiral ganglion neurons (SGN). The SGNs fire action potentials to propagate neural signals through the auditory circuit. Genetic mutations, ototoxic drugs and exposure to loud sounds contribute to hair cell and SGN death that result in hearing loss1-4. Once lost, these cells are not replaced. Use of iPSC and ESC to generate nascent hair cells or SGNs holds great promise for inner ear cell replacement therapies5-8. A flurry of progress has shown that pluripotent stem cells and inner ear derived progenitors can differentiate into hair cells and SGNs at various stages of maturity. Mammalian embryonic stem cells (ESC) and induced pluripotent stem cells (iPSC) can be used to generate functional hair cells and SGNs9-11. Stem cells and progenitor cells derived from the mammalian inner ear have also been shown to form hair cells and neurons with properties of their in vivo cellular counterparts 12-16.
Use of iPSC or ESC-derived otic progenitors to replace lost hair cells and SGNs requires efficient differentiation. Improper differentiation or continued proliferation of engrafted stem-derived progenitors in the inner ear can exacerbate inner ear function and pose a tumorigenic risk such as teratomas formation in the inner ear17. There is a clear need for developing culture conditions and understanding differentiation of otic progenitors. One strategy in developing these methods is to recapitulate cell fate decisions made by neurosensory progenitors during inner ear development. Protocols that prevent proliferation and direct otic progenitors into hair cells or SGNs will help improve safety as well as efficacy of replacement therapies.
During development, the inner ear begins with the thickening of surface ectoderm in a restricted region between rhombomeres 5 and 6 to become the otic placode. As the otic placode invaginates to form an otic cup, a collection of cells in the anterior region of the otic cup gives rise to the neural-sensory-competent domain (NSD), which contains precursors of hair cells and neurons of the inner ear18. Fate mapping studies from mouse, chicken and zebrafish developing inner ear suggest multiple populations of neurosensory progenitors that give rise to the sensory hair cells, surrounding supporting cells and otic neurons19-22. The high mobility group transcription factor, Sox2, has been implicated in sensory cell specification and used as a marker for inner ear progenitors23,24. Hypomorphic mutations that decrease Sox2 expression levels in the inner ear result in the loss of the hair cells, supporting cells and SGNs in the cochlea25,26.
To study otic progenitor cells undergoing cell fate decisions, a fate restricted immortalized multipotent otic progenitor (iMOP) cell line from Sox2 expressing cochlear progenitors was previously established. iMOP cells can continually proliferate as colony forming cells known as otospheres and have the capacity to differentiate into hair cells, supporting cells and SGNs27. Understanding the capacity of iMOP cells to differentiate into distinct otic lineages allows application of these findings to efficiently generate iPSC or ESC-derived hair cells and SGNs. Efficient differentiation protocols will open new avenues for cell replacement therapies of inner ear diseases that are recalcitrant to conventional treatments. A crucial issue in generating otic cells by in vitro cell culture is to have differentiation markers that help determine if cells are undergoing differentiating. Cdkn1b (p27KIP) has been extensively used as an early marker for differentiation in developing inner ear, however, expression of Cdkn1b in iMOP cells and how it correlates to differentiation has not been addressed. In this study, the current culture conditions and how Cdkn1b expression correlates to other markers of iMOP differentiation are described.
Protocol
1. Maintaining self-renewal in iMOP cells
1.1) Prepare iMOP culture media: DMEM/F12, 1X B27 supplement, 25 μg/ml carbenecillin and 20 ng/ml bFGF.
- 1.2) Make 50 ml of iMOP culture media using sterile reagents.
- 1.2.1) Warm up 49 ml of DMEM/F12 in a 50 ml conical in a 37°C water bath.
- 1.2.2) Thaw 50X B27 supplement and filter-sterilized 100 mg/ml carbenecillin aliquots for 5 minutes in a 37°C water bath. Thaw out 100 μg/ml bFGF aliquot at room temperature (RT).
- 1.2.3) Add 1 ml 50X B27, 10 μl of 100 μg/ml bFGF and 12.5 μl of 100 mg/ml carbenecillin into DMEM/F12.
1.3) Add 2 ml of fresh media to iMOP cells every other day without removing any media. Culture iMOP cells at 37°C with 5% CO2. Passage cells after 5-7 days in culture as listed in the steps below.
1.4) Transfer culture to a 15 ml conical tube using a 10 ml pipette tip.
1.5) Make 1 mM EDTA in HBSS by diluting 0.1 ml of 0.5 M EDTA pH 8.0 into 50 ml of HBSS. Pre-warm 1 mM EDTA made in HBSS in a 37°C water bath.
1.6) Harvest cells by gravity sedimentation or centrifugation at 200g for 5 min. at RT. For gravity sedimentation, place the 15 ml conical containing the cultures in the 37°C incubator for 5-10 min. After that time, the otospheres should collect at the bottom of the 15 ml conical. Note: Excessive centrifugal force can cause cell damage.
1.7) Carefully aspirate spent media using a 2 ml aspirating pipette without disturbing the cell pellet.
1.8) Add 0.5 ml of pre-warmed 1 mM EDTA HBSS solution to cell pellet. Using a P1000 pipette, gently pipet up and down 2-3 times. Place conical tube at 37°C and incubate for <5 min to facilitate dissociation into single cells.
1.9) Gently swirl the cell solution to determine if otospheres are dissociated. If no otospheres sediment to the bottom of the conical, the cells are dissociated. The time of incubation may vary. Note: Prolonged incubation with EDTA will result in excessive cell death.
1.10) Remove cells from 37°C, add 2 ml of media to neutralize and dilute the EDTA. Collect the cells by centrifugation at 200g for 5 min at RT.
1.11) Aspirate media out diluted EDTA using a 2 ml aspirating pipette and add 5 ml of 1X PBS to wash the cells.
1.12) Spin cells down at 200g for 5 min at RT and aspirate out 1X PBS using a 2 ml aspirating pippete.
1.13) Resuspend the cells using a P1000 pipette in 0.5 ml of iMOP culture media by gently pipetting up and down 2-3 times.
1.14) Count cells using a microfluidic particle counter with the appropriate cassettes28. Dilute cells 1:100 in media and add 75 ul of cell solution into the cassette for counting.
1.15) Plate 1 X 106 cells in a 6 cm dish in iMOP culture media (∼1:10 dilution).
1.16) Passage iMOPs every 5-7 days. Note: Cells that form large otospheres or attach to the bottom of the tissue culture dish will differentiate. Cells will also die if the cultures are over confluent.
2. Freezing and thawing iMOP cells
2.1) Prepare synthetic freezing medium by thawing and equilibrating the solution to 4°C prior to freezing cells.
2.2) Collect cells from a confluent 6 cm dish of iMOP cells. Use a 10 ml pipette and transfer cells (∼6-8 × 106 cells) into a 15 ml conical.
2.3) Harvest cells by gravity sedimentation or centrifugation at 200g for 5 min at RT. If the otospheres are small, cells will not settle by gravity sedimentation. Centrifuge at 200 g for 5 min. at RT to harvest cells. Optional: Count cells using steps 1.7-1.14 to determine total cell numbers.
2.4) Aspirate spent media using a 2ml aspirating pipette while leaving the loose cell pellet behind.
2.5) Add 0.25 ml of synthetic freezing media to resuspend cells at a density of ∼5 × 105 to 3 × 106 cells/ml. Gently pipette cells with a P1000 pipette.
2.6) Transfer the cell suspension to cryogenic vials using a P1000 pipette with 1 ml filtered tip.
2.7) Place the vials in an alcohol free freezing container and place the freezing container at -80°C to reduce the temperature 1°C per minute until the temperature reaches -80°C.
2.8) For long-term storage of cells, transfer the vials to the vapor phase of a liquid nitrogen storage tank.
2.9) To thaw out cells for culture, equilibrate cryogenic vial containing frozen cells at -80°C overnight.
2.10) Pre-warm iMOP culture media in a 37°C water bath.
2.11) Thaw the frozen vial quickly by swirling the bottom of the vial in a 37°C water bath.
2.12) Add 1 ml of pre-warmed iMOP culture media to thawed cells once the last ice crystal disappears.
2.13) Transfer cells into a 15 ml conical and add an additional 4 ml of iMOP culture media
2.14) Spin the 15 ml conical for 5 min. at 200g and aspirate the spent media.
2.15) Resuspend the cells in 2 ml of iMOP culture media and plate cells in a 6 cm dish. Incubate cultures at 37°C with 5% CO2 for expansion.
3. Differentiation of iMOP cells into sensory epithelia
Media Preparation
3.1) Prepare 50 ml of iMOP sensory epithelia differentiation media: DMEM/F12, 1X B27 supplement, 25 μg/ml carbenecillin.
- 3.2) Make 50 ml of iMOP sensory epithelia differentiation media.
- 3.2.1) Warm up 49 ml of DMEM/F12 in a 50 ml conical in a 37°C water bath.
- 3.2.2) Thaw 50 B27 supplement and 100 mg/ml carbenecillin aliquots for 5 min. in a 37°C water bath.
- 3.2.3) Add 1 ml 50X B27 and 12.5 μl of 100 mg/ml carbenecillin into DMEM/F12.
Plating iMOP cells for sensory epithelia differentiation
3.3) To harvest, dissociate, resuspend and count cells repeat steps 1.4-1.14.
3.4) Plate 1 × 106 cells in a 6 cm dish at Day -3 using iMOP culture media.
3.5) On Day 0, transfer cultures using a 10 ml pipette into a 15 ml conical.
3.6) Collect the otospheres by gravity sedimentation as stated in step 1.6.
3.7) Aspirate out spent media using a 2 ml aspirating pipette and leave the otospheres on the bottom of the conical.
3.8) Gently add in 2 ml of sensory epithelia differentiation media.
3.9) Transfer otospheres to a 6 cm dish using a large bore 10 ml pipette. Note: Mechanical shearing from harsh pipetting can dissociate cells from the otospheres.
3.10) Add 2 ml of fresh sensory epithelial differentiation media to cultures every other day. If necessary, cells can be collected by gravity sedimentation and media replaced.
3.11) Collect otospheres at Day 10 by transferring to a 15 ml conical and allowing the otospheres to sediment as stated in step 1.6.
3.12) Aspirate the spent media using a 2 ml aspirating pipette and leave the otospheres undisturbed.
3.13) Fix otospheres by incubating in 4% formaldehyde in 1X PBS for 15 min at RT. Remove formaldehyde solution, wash otospheres with 1X PBS and subject the otosphere to immunostaining27.
3.14) Acquire epifluorescence images using an inverted microscope setup equipped with a 16 bit CCD camera and either a 20X 0.75 air or a 40X 1.3 NA oil immersion objective. Collect fluorescence from different color channels using the listed (excitation and emission)wavelengths: blue (377±25nm/447±30nm), green (475±25 nm/540±25 nm), red (562±20nm/and625±20nm) and infrared (628±20nm/and 692±20nm).
4. EdU incorporation assay
Determining proliferation of iMOP cells after bFGF withdrawal
4.1) On Day -3, plate 1 × 106 iMOP cells in a 6 cm dish.
4.2) Three days later on Day 0, harvest, dissociate, resuspend and count cells by repeating steps 1.4-1.14.
4.3) Plate 2.5 × 105 cells in iMOP culture media and 5 × 105 cells in sensory epithelia differentiation media in a single well from a 6 well dish.
- 4.4) On Day 3, determine the percentage of cells in S phase by EdU incorporation using the Click-iT EdU Alexa Fluor 488 assay kit.
- 4.4.1) Add EdU stock directly to the iMOP cultures to obtain a final concentration of 1 μM EdU in the culture media.
- 4.4.2) Incubate EdU in iMOP cultures for 2 hours and incubate at 37°C with 5% CO2.
- 4.4.3) Harvest, dissociate and collect cells repeat steps 1.4-1.12.
- 4.4.4) Add in 4% formaldehyde in 1X PBS for 15 min. at RT to fix cells.
- 4.4.5) Harvest cells by centrifugation at 200g for 5 min at RT.
- 4.4.6) Remove formaldehyde solution and properly dispose.
- 4.4.7) Label nuclei of cells with Hoechst and incorporated EdU with Alexafluor 488 azide according to manufacturer's protocol. All washes are done with 1X PBS, 3% BSA and 0.1% Tween 20.
- 4.4.8) Wash cells with 1X PBS twice.
- 4.4.9) Mount cells on a slide using antifade reagent and place a 1.5 cover glass over the sample.
- 4.4.10) Acquire fluorescent images of labeled cells using an epifluorescence microscopy.
5. Neuronal Differentiation of iMOP Cells
Media Preparations
- 5.1) Make 50 ml of neuronal differentiation media: Neurobasal media, 1X B27 supplement, 2 mM L-Glutamine.
- 5.1.1) Thaw bottle of 50X B27 and 200 mM L-Glutamine in 37°C water bath for 5 min.
- 5.1.2) Add 1ml 50X B27 and 0.5 ml 200 mM L-Glutamine to 48.5 ml of Neurobasal media
Coating coverglass
5.2) Place 12 mm round 1.5 glass coverslips in a sterile 10 cm plate and add 70% EtOH to the plate to sterilized and clean the coverslips
5.3) Gently agitate the coverslips to ensure they are covered in ethanol. Leave the plate for 10 minutes at RT.
5.4) Rinse the coverslips 3 times with 1X PBS to wash out the remaining ethanol.
5.5) Rinse the coverslip once with H20 to wash out remaining 1X PBS.
5.6) Aspirate the H20 using a 2 ml aspirating pipette and let the coverslips dry.
5.7) Expose the coverslips to UV light in the tissue culture hood for 15 min. Store coverslips in a sterile environment if not immediately used.
5.8) Place one 12 mm round coverslip in each well of a 24 well dish. Shake plate gently to ensure that the coverslips lie flat on the bottom of the well.
5.9) Thaw 2 mg/ml poly-D-lysine stock solution at RT and dilute to a 10 μg/ml poly-D-lysine concentration in 1X PBS.
5.10) Leave the plate in the 37°C incubator for 1 hr.
5.11) Wash the wells 3 times with sterile 1X PBS.
5.12) Add 0.25 ml of 10 μg/ml laminin working solution into a single well of a 24 multiwell dish. Incubate the plate at 37°C incubator overnight.
5.13) Aspirate out the laminin solution using a 2 ml aspirating pipette.
5.14) Wash the coverslip 3 times with 1X PBS by adding in 1 ml of 1X PBS with a P1000 pipette and removing the 1X PBS by aspirating with a 2 ml aspirating pippette. Leave 1X PBS from last wash in the well until cells are ready to be plated.
Plating iMOP cells for neuronal differentiation
5.15) To initiate neuronal differentiation, harvest, dissociate and count cells by repeating steps 1.4-1.14
5.16) Plate 1 × 106 cells in a 6 cm dish at Day -3.
5.17) On Day 0, harvest, dissociate and count cells by repeating steps 1.4-1.14.
5.18) Seed 1 × 105 – 1.5 × 105 iMOP cells into 0.5 ml pre-warmed neuronal differentiation media per well in a 24 multiwell dish.
5.19) Aspirate and add pre-warmed neuronal differentiation media to cultures every other day.
5.20) Fix iMOP-derived neurons in 4% formaldehyde for 15 min. at RT on Day 7. Immunostain the cells as previously described27.
5.21) Acquire epifluorescence images as listed in step 3.14.
Representative Results
bFGF withdrawal decreases proliferation in iMOP cells
To decrease the proliferative capacity of iMOP cells and initiate differentiation of iMOP cells, bFGF was withdrawn from the cultures. To confirm that growth factor withdrawal decreases proliferation, EdU incorporation was employed as a proliferation assay. The percentage of cells that incorporated EdU from otospheres cultured with iMOP culture media (containing bFGF) and otospheres cultured in sensory epithelia media (without bFGF) was compared. Otosphere cultures were pulsed with the nucleotide analog EdU, harvested and fixed. Incorporated EdU nucleotides were fluorescently labeled with Alexafluor 488 azide using click-chemistry. Cells from otospheres grown in the absence of bFGF showed decreased incorporation of EdU relative to cells cultured with bFGF (Fig 1A-F). As the size of the otosphere increased, it was increasingly difficult to visualize all the cells from the otosphere with an epifluorescent microscope. To address this, cells were dissociated and mounted so that they can be unambiguously visualized en mass. Even after dissociation and fixation, the cells re-aggregated. To maintain the cells in a dissociated state, detergents that prevented the cells from re-aggregating were tested. Tween 20 was one of the detergents that prevented re-aggregation of the cells. Dissociated cells were washed with 0.1% Tween 20 and mounted onto slides. EdU labeled cells were then visualized by epifluorescence microscopy (Fig 1G,H). Representative results from individual experiments (n=5) showed a significant decrease in the percentage of EdU labeled cells from 48% to 19% after 3 days after bFGF withdrawal (p<0.005). These results confirm a dramatic drop in the percentage of S phase cells and proliferative potential of iMOP cells after bFGF withdrawal.
Figure 1. EdU incorporation as an assay to determine proliferative state of iMOP cultures.
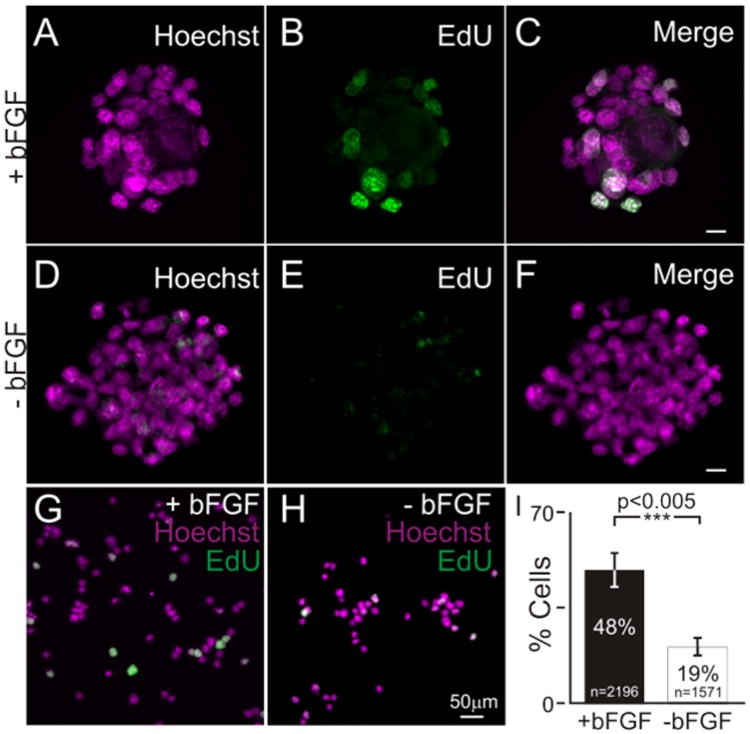
iMOP-derived otospheres cultured in the presence of bFGF. Nuclei of cells were labeled with (A) Hoechst and (B) EdU Alexafluor 488. (C) Merged image of Hoechst and EdU fluorescence. Otospheres cultured 3 days in media lacking bFGF. Cells from cultures were labeled with (D) Hoechst and (E) EdU Alexafluor 488. (F) Merged images of Hoechst and EdU labeling. Merged fluorescence images of Hoechst (magenta) and EdU (green) labeled cells after dissociating otospheres and washing with 1X PBS containing 0.1% Tween 20. Cells were from otospheres cultured (G) in the presence of bFGF or (H) absence of bFGF. Length of scale bars are 10 μm unless indicated. (I) Percent of EdU labeled cells from iMOP cells cultured in the presence or absence of bFGF. The number of individual cells analyzed was denoted within the bar graph and error bars were depicted as standard error of the mean (SEM). Cell counts were from n=5 independent experiments. The Student's t-test was done to determine statistical significance.
iMOP-derived sensory epithelia express Cdkn1b and show morphological changes
A timeline of iMOP sensory epithelia differentiation protocol is shown (Fig. 2A). Otospheres from proliferative cultures were dissociated into single cells and allowed to recover for 3 days in iMOP culture media. This method helps enrich for proliferating cells and selects against post-mitotic cells in our starting cultures. After 3 days, newly formed otospheres were seeded in sensory epithelia differentiation media and allowed to undergo unguided differentiation for 10 days. Bright field images of typical cultures containing otospheres at different time points after seeding showed a general increase in otosphere size (Fig 2B-E).
Figure 2. General schematic of sensory epithelia differentiation.
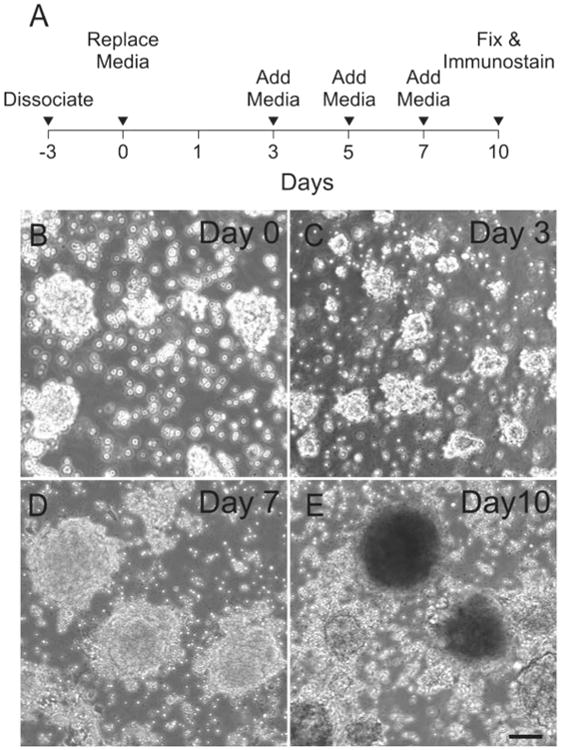
(A) Timeline for differentiating iMOP cells into sensory epithelia. The line graph denotes the time of cell dissociation and media changes. (B) Typical phase contrast images of otospheres on Day 0, (C) 3, (D) 7 and (E) 10 during unguided sensory epithelia differentiation. Length of scale bar is 100 μm.
Since brightfield microscopy cannot reveal many of the changes that occur during differentiation of iMOP cells, immunostaining with molecular markers was used to highlight morphological and molecular features of the differentiated cells27. To determine if expression of Cdkn1b (p27KIP) can be used as a marker for differentiation, otospheres from proliferative or sensory epithelia differentiated iMOP cultures were compared. Fluorescent markers were visualized and captured by epifluorescence microscopy. Representative images of otospheres from proliferative cultures showed low expression of Cdkn1b outside the nuclei with almost no phalloidin staining (Fig 3 A-D). Otospheres cultured in sensory epithelia differentiation media displayed increased expression of nuclear Cdkn1b concomitant with the appearance of phalloidin labeling in the peripheral edges of cells (Fig 3 E-F). During sensory epithelia differentiation, the morphological changes in actin filaments parallel the appearance of Cdh1 expression in adhesion sites between cells (Fig 3 I-L). In differentiated iMOP otospheres, phalloidin and Cdh1 (E-cadherin) highlighted morphological changes in actin filaments and regions of cell-cell adhesion, similar to previous results27. In our protocol, the increase in Cdkn1b expression along with the changes in morphological features highlighted by phalloidin and Cdh1 serve as indicators of sensory epithelia differentiation in iMOP cells.
Figure 3. Expression of markers indicative of cell cycle arrest and differentiation in iMOP-derived sensory epithelia.
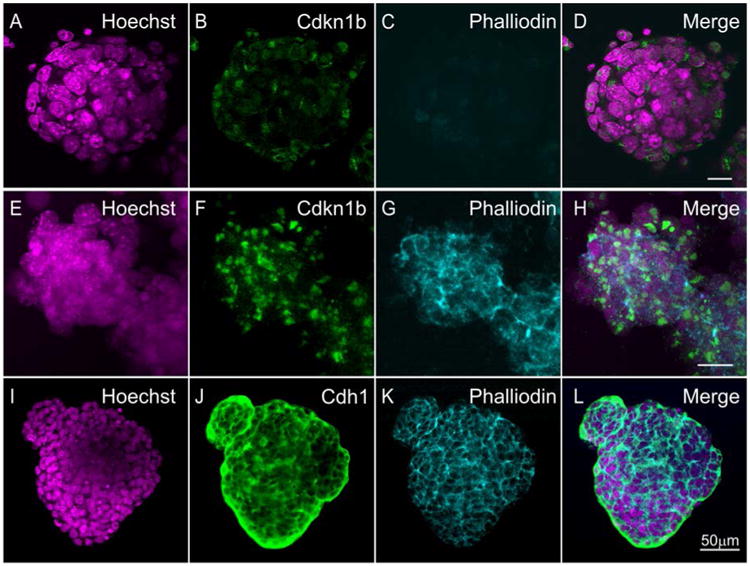
Otospheres of iMOP cells cultured in iMOP culture media. Nuclei of cells were labeled with (A) Hoechst and (B) Cdkn1b antibody. Filamentous actin was labeled with (C) phalloidin. (D) The merged image of a typical otospheres containing proliferating iMOP cells. Otospheres from iMOP cells were cultured in sensory epithelia culture media for 10 days. Cells from otopsheres were marked with (E) Hoechst, (F) Cdkn1b and (G) phalloidin. (H) Merged image of otospheres showing increased Cdkn1b and phalloidin labeling. Otospheres differentiated into sensory epithelia were also labeled (I) Hoechst, (J) Cdh1, (K) phalloidin. (L) The merged image shows typical strong Cdh1 labeling in the otospheres. Length of scale bars are 10 μm unless indicated.
iMOP-derived neurons express Cdkn1b and Tubb3
A schematic of iMOP neuronal differentiation protocol is shown (Fig 4A). Similar to the aforementioned protocol, iMOP cells were dissociated and allowed to recovery for 3 days in iMOP culture media. Otospheres were harvested, dissociated into single cells and seeded onto poly-D-lysine and laminin coated coverslips. The cells were allowed to differentiate for 7 days. Representative bright field images at time points displayed progressive morphological changes as they differentiated into iMOP-derived neurons (Fig 4B-E). By Day 7, long neurites can be seen extending from cell bodies (Fig 4E arrowheads). To determine the changes in expression levels of Cdkn1b during neuronal differentiation, proliferating iMOP cells and iMOP-derived neurons were compared. Otospheres were labeled with Hoechst and Cdkn1b antibodies. iMOP cells from proliferating otospheres had few cells with low nuclear Cdkn1b expression (Fig 4 F-H). Furthermore, iMOP-derived neurons showed an increase in the number of cells with nuclear Cdkn1b expression 7 days after neuronal differentiation (Fig 4 I-K). To determine if the Cdkn1b cells were adopting a neuronal lineage, labeling of iMOP-derived neurons with the neuronal marker Tubb3 was done. Representative images of iMOP-derived neurons showed co-labeling of Cdkn1b and Tubb3 (Fig 5A-D). Magnification and quantification of neurites from these cells showed an average 1.5 neurites associated with each Tubb3 labeled cell (n=60) (Fig 5 E-G). In our neuronal differentiation protocol, immunostaining with Cdkn1b and Tubb3 can be used as an indicator of differentiation into bipolar or pseudo-unipolar iMOP-derived neurons.
Figure 4. General schematic of neuronal differentiation.
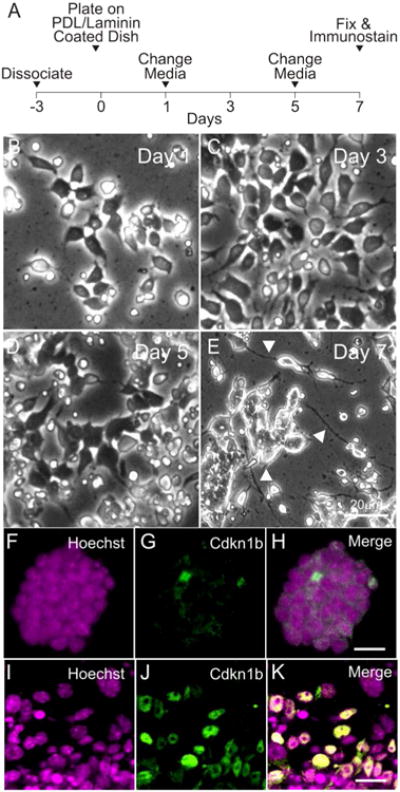
(A) Timeline for differentiating iMOP cells into neurons. Phase contrast images of iMOP cells undergoing neuronal differentiation at (B) Day 1, (C) 3, (C) 5 and (E) 7. Fluorescence images of iMOP cells cultured as otospheres in iMOP culture media with (F) Hoechst and (G) Cdkn1b. (H) Merged image of cells labeled with Hoechst and Cdkn1b. Fluorescence images of iMOP cells 7 days after neuronal differentiation. Nuclei of cells were labeled with (I) Hoechst and (J) Cdkn1b. (K) Merged image of cells labeled with Hoechst and Cdkn1b. Length of scale bars are 10 μm unless noted.
Figure 5. Expression of molecular markers indicative of cell cycle exit and neuronal differentiation.
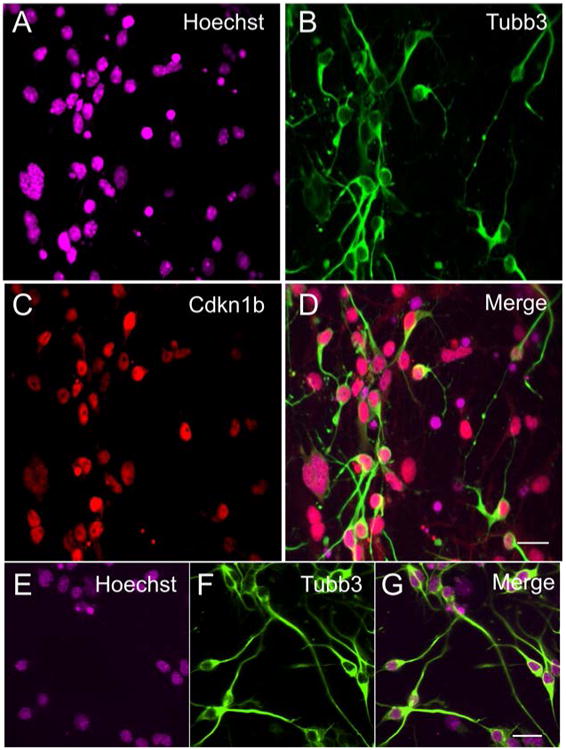
iMOP cells 7 days after neuronal differentiation. Nuclei were labeled with (A) Hoechst and (B) Cdkn1b antibodies. The cellular morphology was highlighted with (C) Tubb3 (neuronal β-tubulin) antibodies. (D) Merged image with labeling of Cdkn1b and Tubb3 in individual cells. Magnified image of iMOP-derived neurons with nuclei labeled with (E) Hoechst and neuronal processes labeled with (F) Tubb3 antibody. (G) Merged image of bipolar or pseudo-unipolar morphology displayed by iMOP-derived neurons. Length of scale bars are 20 μm.
Nuclear Cdkn1b expression levels increase after initiating differentiation
The morphological changes in otospheres show qualitative changes in iMOP cells as they undergo differentiation. To attain quantitative results from the immunofluorescence images to denote early differentiation events, nuclear fluorescence intensity of Cdkn1b from individual proliferating iMOP cells (n=239) and iMOP cells undergoing sensory epithelia differentiation (n=246) were measured. To normalize Cdkn1b fluorescence signals from independent experiments, the ratio of Cdkn1b to Hoechst fluorescence from individual cells was determined. Histograms of the normalized fluorescence intensity of Cdkn1b from proliferating iMOP cells (black) and sensory epithelia differentiating iMOP cells (red) were plotted (Fig 6A). An increase in the number of high Cdkn1b expressing cells was observed as a rightward shift of the histogram for sensory epithelia differentiation (red) relative to the histogram for proliferating iMOP cells (black). To determine the increase in the percentage of cells expressing Cdkn1b, a threshold for normalized Cdkn1b fluorescence intensity units (0.65) was set (arrowhead) and the percentage of cells above the threshold was determined. After sensory epithelia differentiation, iMOP cells displayed an increase from 25.3% to 67.1% of cells expressing Cdkn1b (Fig 6B). The same quantitative analysis was applied to iMOP-derived neurons. When comparing proliferative iMOP cells (n=525) and 7 day iMOP-derived neurons (n=583) a rightward shift in the histogram associated with iMOP-derived neurons was observed relative to the histogram for proliferating iMOP cells (Fig 6C). Determining the percentage of cells above the threshold revealed an increase in Cdkn1b expressing cells from 23.5% to 85.5% (Fig 6D). Using the described protocols, quantitative analysis of Cdkn1b expression confirmed an increase in the number of cells that upregulate Cdkn1b during sensory epithelia and neuronal differentiation. The increased numbers of Cdkn1b expressing cells can be used to confirm differentiation of iMOP cells in our protocols.
Figure 6. Single cell quantitative fluorescence intensity analysis of Cdkn1b.
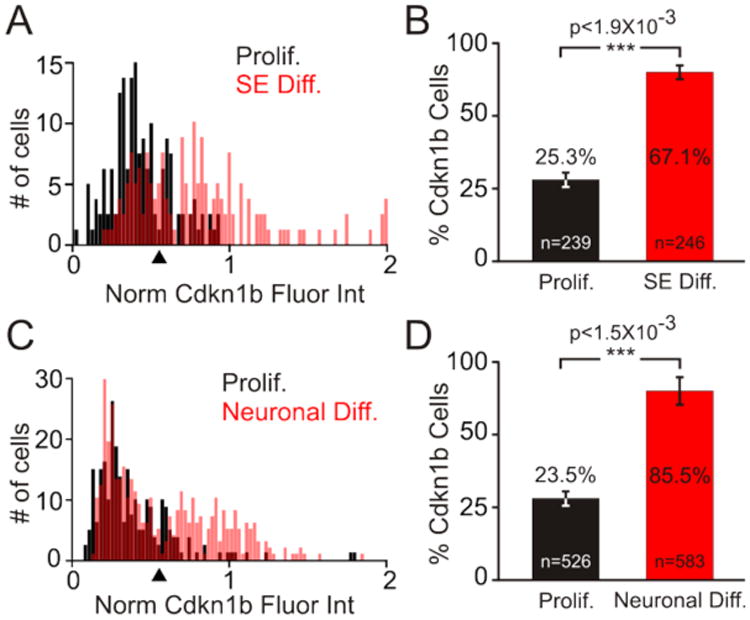
(A) Normalized fluorescence intensity of Cdkn1b expression was determined by calculating the ratio of Cdkn1b and Hoechst fluorescence intensity from individual cells. The normalized fluorescence intensity was plotted relative to cell numbers as a histogram. Normalized fluorescence intensity of Cdkn1b from proliferating iMOP cells cultured in the presence of bFGF (black) and iMOP otospheres differentiated into sensory epithelia (SE) (red) are shown. A threshold was set at 0.65 normalized fluorescence intensity units (arrowhead) (B) Percentage of iMOP cells expressing Cdkn1b above the threshold value, from proliferating (black) and sensory epithelia differentiated cultures (red). (C) Normalized fluorescence intensity of Cdkn1b expression from proliferating iMOP cells (black) and neuronal differentiated iMOP cells (red) were plotted and a threshold set (arrowhead) as described above. (D) Percentage of iMOP cells expressing Cdkn1b from proliferating (black) and neuronal differentiating cultures (red). Individual cells used for analysis were denoted in each bar graph. Results were compiled from different experiments (n=3) and error bars depicted as SEM. The Student t-test was used to determine the statistical significance.
Discussion
Variables that affect iMOP cultures
A protocol for maintaining self-renewal and promoting differentiation of a novel iMOP cell line is described. Several critical steps that help with routine expansion and differentiation of iMOP cells are noted. One critical step in culturing iMOP cells is to maintain an active concentration of bFGF in the cultures to promote self-renewal. bFGF is reportedly highly labile at 37°C when culturing cells29,30. Growth of cultures can be significantly affected by spontaneous differentiation due to bFGF instability. In the current protocol, fresh media is constantly supplied to the cultures in order to maintain bFGF levels above 5 ng/ml without perturbing the otospheres. Stabilization of bFGF levels using controlled release from glycolic and lactic acid polyester microspheres could reduce the frequency of media changes needed to maintain steady levels of bFGF in media31. Addition of the steadily releasable bFGF will reduce the need to frequently add media to maintain bFGF levels in iMOP cultures.
Another critical step in maintaining viability of iMOP cells is to limit exposure to dissociation reagents and mechanical shearing. In the current protocol, dissociation is accomplished by incubation with 1 mM EDTA. Curbing excessive incubation time with EDTA, reducing excessive mechanical shearing caused by vigorous pipetting and limiting multiple centrifugation steps when passaging iMOP cells will help increase cell viability. Attention given to these steps can effectively maintain viability of large numbers of cells during expansion of cultures.
During the routine culture of iMOP cells, several methods for dissociating the cells were tested. Mechanical shearing from pipetting can be used to dissociate otospheres, but does not uniformly or consistently produce single cells. Use of commercially available reagents for cellular dissociation of iMOP cells can effectively generate single cells from otospheres, but these reagents affect the adhesive properties of cells to reform otospheres and to adhere to treated surfaces. In order to use commercial reagents, multiple washes were required to remove the dissociation reagents before employing differentiation protocols.
In the current differentiation protocols, both sensory epithelia and neuronal differentiation conditions resulted in cell death that was observed in the form of single cells or clumps of dead cells. Apoptotic cell death could be due to the lack of appropriate differentiation or survival cues during the prolonged culture in vitro. Both differentiation conditions contained B27 supplement to promote survival of cells. Although B27 supplement has been shown to promote survival of primary hippocampal neurons32, it may not be sufficient for promoting survival of iMOP cells. Addition of compounds such as the ROCK inhibitor may help maintain and increase survival of iMOP cells during routine culture and in vitro differentiation. ROCK inhibitor has been extensively used in pluripotent stem cell cultures to promote survival of cells in serum free cultures without affecting differentiation33. Addition of ROCK inhibitor to the culture media can help increase cell survival during long differentiation protocols without affecting the differentiation potential of iMOP cells. Increased cell survival will allow development of protocols to efficiently generate large numbers of mature hair cells or SGNs.
Increased Cdkn1b expression as an early marker of iMOP differentiation
During early cochlear development, the initial event of cell cycle exit precedes differentiation for hair cells, supporting cells and SGNs34. Terminal mitosis of neuronal progenitors spreads in a wave starting from the base and progressing towards the apex of the cochlea35. In an opposing gradient, terminal mitosis of prosensory progenitors that give rise to hair cells and supporting cells progresses from the apex to base of the cochlea34,36. Although the temporal expression of Cdkn1b correlates well to the post-mitotic state, it is most likely not the sole factor that promotes cell cycle exit in the developing cochlea. In support of this, Cdkn1b mutant animals can still develop post-mitotic hair cells37. Instead, Cdkn1b can be used as an early indicator to mark the onset of differentiation. In unguided iMOP differentiation cultures, an early marker for differentiation will assist in developing protocols to promote early stages of differentiation before obvious morphological features can be observed.
Similar to otic development, cycle exit initiates differentiation and results in increased Cdkn1b levels in iMOP cells. In proliferating iMOP cells, only low level expression of Cdkn1b can be observed. Other cyclin dependent kinase inhibitors such as Cdkn1a (p21CIP) may be responsible for normal cell cycle progression in iMOP cells. During sensory epithelia and neuronal differentiation of iMOP cells, single cell quantitative analysis revealed a subset of cells with increase expression of Cdkn1b in differentiating iMOP cultures (Fig 6A, C). Expression of Cdkn1b in these cells are an indication of iMOP cells undergoing terminal differentiation.
iMOP cells as a cellular platform for studying cell fate determination
Since iMOP cells can transition from a progenitor state to different otic lineages in vitro, they can be utilized to study otic cell fate determination. Currently, the described protocol used unguided differentiation procedures to generate different otic cell types. The iMOP system allows for addition of bioactive molecules, compounds and defined genes that guide iMOP cells towards the mature hair cell, supporting cell and SGN lineages. One use of iMOP cellular platform is to test for candidate transcriptions factors (TF) that promote differentiation. Expression of key TF can be used to drive hair cell or neuronal differentiation. Expression of Cdkn1b along with established markers or morphological features of hair cells and SGNs can be used to determine the contribution of candidate TFs to distinct otic lineages38,39.
Candidate TFs such as Atoh1 are essential for hair cell development and have been repurposed to promote hair cell differentiation in the damaged cochlea40,41. Introduction of Atoh1 into iMOP cells may help promote hair cell differentiation. In the developing cochlea, both Ngn1 and NeuroD1 are required for proper SGN differentiation in vivo42-44. Expression of Ngn1 or NeuroD1 in iMOP cells may promote SGN differentiation. These experiments are similar to generating induced neuronal cells (iN), where expression of the TFs individually or in combination can convert fibroblasts or in pluripotent stem cells into neurons45-48. Introduction of TFs that are normally expressed during development of hair cells or SGNs is a good strategy for guiding iMOP cells towards the hair cell or SGN lineage.
Conditions that promote differentiation of large number of iMOP-derived hair cells and SGNs will provide a robust cellular platform for disease modelling, small molecule screening and toxicology testing that can be accomplished quickly and cost-effectively before labor-intensive tests in rodents are initiated. The protocol described presents a simple and versatile system that can be utilized for studying otic progenitor differentiation and is amenable to large scale experiments.
Table 1. Reagents used for culture and differentiation of iMOP cells.
| Name of the Reagent/Equipment | Company | Catalog Number | Comments/Description (optional) |
|---|---|---|---|
| Tissue Culture Treated 24 Multiwell Plate | Thermo Fisher Scientific | 130188 | |
| Tissue Culture Treated 60 mm Dish | Thermo Fisher Scientific | 130181 | |
| Moxi Z Mini Automated Cell Counter | Orflo | MXZ001 | |
| Moxi Z Cassette Type S | Orflo | MXC002 | |
| DMEM/F12 | Life Technologies | 11320-033 | |
| Neurobasal Medium | Life Technologies | 10010-023 | |
| Phosphate Buffered Saline (PBS) pH 7.4 | Life Technologies | 14025-092 | |
| Hank's Balanced Salt Solution (HBSS) | Life Technologies | 17504-044 | Stored as 1 ml aliquots |
| B27 Supplement (50X) Serum Free | Life Technologies | 25030-081 | Stored as 5 ml aliquots |
| L-Glutamine(200 mM) | Life Technologies | 23017-015 | Stored as 1 mg/ml 100 μl aliquots |
| Recombinant Murine Fibroblast Growth Factor, basic (bFGF) | Peprotech | 450-33 | Resuspended per manufacturerer and stored as 20 mg/ml 20 μl aliquots |
| Carbenicillin, Disodium Salt | Fisher | BP2648-1 | |
| Natural Mouse Laminin | Life Technologies | P36934 | |
| Poly-D-Lysine | Life Technologies | 11320-082 | |
| CoolCell LX Alcohol-Free Cell Freezing Containers | BioCision | BCS-405 | |
| Cryogenic Vials (2 ml) | Corning | 430654 | |
| 1.5 Thickness Glass Coverslip (Round 12 mm) | Electron Microscopy Sciences | 72230-01 | |
| Prolong Gold Antifade Mountant | VWR | 47743-736 | Stored as 10 mg/ml 100 μl aliquots |
Acknowledgments
The work was supported in part by the Duncan and Nancy MacMillan Faculty Development Chair Endowment Fund (K.Y.K.), Busch Biomedical Research Grant (K.Y.K.) and the Rutgers Faculty Development Grant (K.Y.K.).
Footnotes
A complete version of this article that includes the video component is available at http://dx.doi.org/10.3791/53692.
Disclosures: The authors have nothing to disclose.
Contributor Information
Azadeh Jadali, Email: Jadali@dls.rutgers.edu, Cell Biology & Neuroscience, Rutgers University, Piscataway, NJ.
Zhichao Song, Email: Song@dls.rutgers.edu, Cell Biology & Neuroscience, Rutgers University, Piscataway, NJ.
Alejandra S. Ruiz-Laureano, Email: Laureano@dls.rutgers.edu, Cell Biology & Neuroscience Rutgers University Piscataway, NJ.
Alana Toro-Ramos, Email: Toro@dls.rutgers.edu, Cell Biology & Neuroscience, Rutgers University, Piscataway, NJ.
Kelvin Y. Kwan, Email: Kwan@dls.rutgers.edu, Cell Biology & Neuroscience, Rutgers University, Piscataway, NJ.
References
- 1.Petit C, Richardson GP. Linking genes underlying deafness to hair-bundle development and function. Nature neuroscience. 2009;12(6):703–710. doi: 10.1038/nn.2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kujawa SG, Liberman MC. Adding insult to injury: cochlear nerve degeneration after “temporary” noise-induced hearing loss. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2009;29(45):14077–14085. doi: 10.1523/JNEUROSCI.2845-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kujawa SG, Liberman MC. Acceleration of age-related hearing loss by early noise exposure: evidence of a misspent youth. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2006;26(7):2115–2123. doi: 10.1523/JNEUROSCI.4985-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huth ME, Ricci AJ, Cheng AG. Mechanisms of aminoglycoside ototoxicity and targets of hair cell protection. International journal of otolaryngology. 2011;2011(937861) doi: 10.1155/2011/937861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brigande JV, Heller S. Quo vadis, hair cell regeneration? Nature neuroscience. 2009;12(6):679–685. doi: 10.1038/nn.2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Groves AK. The challenge of hair cell regeneration. Exp Biol Med (Maywood) 2010;235(4):434–446. doi: 10.1258/ebm.2009.009281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Okano T, Kelley MW. Stem cell therapy for the inner ear: recent advances and future directions. Trends in amplification. 2012;16(1):4–18. doi: 10.1177/1084713812440336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ronaghi M, Nasr M, Heller S. Concise review: Inner ear stem cells--an oxymoron, but why? Stem Cells. 2012;30(1):69–74. doi: 10.1002/stem.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen W, et al. Restoration of auditory evoked responses by human ES-cell-derived otic progenitors. Nature. 2012;490(7419):278–282. doi: 10.1038/nature11415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koehler KR, Mikosz AM, Molosh AI, Patel D, Hashino E. Generation of inner ear sensory epithelia from pluripotent stem cells in 3D culture. Nature. 2013 doi: 10.1038/nature12298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oshima K, et al. Mechanosensitive hair cell-like cells from embryonic and induced pluripotent stem cells. Cell. 2010;141(4):704–716. doi: 10.1016/j.cell.2010.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Diensthuber M, Oshima K, Heller S. Stem/progenitor cells derived from the cochlear sensory epithelium give rise to spheres with distinct morphologies and features. Journal of the Association for Research in Otolaryngology : JARO. 2009;10(2):173–190. doi: 10.1007/s10162-009-0161-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Doetzlhofer A, White P, Lee YS, Groves A, Segil N. Prospective identification and purification of hair cell and supporting cell progenitors from the embryonic cochlea. Brain research. 2006;1091(1):282–288. doi: 10.1016/j.brainres.2006.02.071. [DOI] [PubMed] [Google Scholar]
- 14.Doetzlhofer A, White PM, Johnson JE, Segil N, Groves AK. In vitro growth and differentiation of mammalian sensory hair cell progenitors: a requirement for EGF and periotic mesenchyme. Developmental biology. 2004;272(2):432–447. doi: 10.1016/j.ydbio.2004.05.013. [DOI] [PubMed] [Google Scholar]
- 15.Martinez-Monedero R, Yi E, Oshima K, Glowatzki E, Edge AS. Differentiation of inner ear stem cells to functional sensory neurons. Developmental neurobiology. 2008;68(5):669–684. doi: 10.1002/dneu.20616. [DOI] [PubMed] [Google Scholar]
- 16.Oshima K, Senn P, Heller S. Isolation of sphere-forming stem cells from the mouse inner ear. Methods Mol Biol. 2009;493:141–162. doi: 10.1007/978-1-59745-523-7_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nishimura K, Nakagawa T, Sakamoto T, Ito J. Fates of murine pluripotent stem cell-derived neural progenitors following transplantation into mouse cochleae. Cell transplantation. 2012;21(4):763–771. doi: 10.3727/096368911X623907. [DOI] [PubMed] [Google Scholar]
- 18.Wu DK, Kelley MW. Molecular mechanisms of inner ear development. Cold Spring Harbor perspectives in biology. 2012;4(8):a008409. doi: 10.1101/cshperspect.a008409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiang H, et al. Lineage analysis of the late otocyst stage mouse inner ear by transuterine microinjection of a retroviral vector encoding alkaline phosphatase and an oligonucleotide library. PloS one. 2013;8(7):e69314. doi: 10.1371/journal.pone.0069314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sapede D, Dyballa S, Pujades C. Cell lineage analysis reveals three different progenitor pools for neurosensory elements in the otic vesicle. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2012;32(46):16424–16434. doi: 10.1523/JNEUROSCI.3686-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Satoh T, Fekete DM. Clonal analysis of the relationships between mechanosensory cells and the neurons that innervate them in the chicken ear. Development. 2005;132(7):1687–1697. doi: 10.1242/dev.01730. [DOI] [PubMed] [Google Scholar]
- 22.Raft S, et al. Cross-regulation of Ngn1 and Math1 coordinates the production of neurons and sensory hair cells during inner ear development. Development. 2007;134(24):4405–4415. doi: 10.1242/dev.009118. [DOI] [PubMed] [Google Scholar]
- 23.Neves J, Parada C, Chamizo M, Giraldez F. Jagged 1 regulates the restriction of Sox2 expression in the developing chicken inner ear: a mechanism for sensory organ specification. Development. 2011;138(4):735–744. doi: 10.1242/dev.060657. [DOI] [PubMed] [Google Scholar]
- 24.Mak AC, Szeto IY, Fritzsch B, Cheah KS. Differential and overlapping expression pattern of SOX2 and SOX9 in inner ear development. Gene expression patterns : GEP. 2009;9(6):444–453. doi: 10.1016/j.gep.2009.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kiernan AE, et al. Sox2 is required for sensory organ development in the mammalian inner ear. Nature. 2005;434(7036):1031–1035. doi: 10.1038/nature03487. [DOI] [PubMed] [Google Scholar]
- 26.Puligilla C, Dabdoub A, Brenowitz SD, Kelley MW. Sox2 induces neuronal formation in the developing mammalian cochlea. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2010;30(2):714–722. doi: 10.1523/JNEUROSCI.3852-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kwan KY, Shen J, Corey DP. C-MYC transcriptionally amplifies SOX2 target genes to regulate self-renewal in multipotent otic progenitor cells. Stem cell reports. 2015;4(1):47–60. doi: 10.1016/j.stemcr.2014.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dittami GM, Sethi M, Rabbitt RD, Ayliffe HE. Determination of mammalian cell counts, cell size and cell health using the Moxi Z mini automated cell counter. Journal of visualized experiments : JoVE. 2012;64 doi: 10.3791/3842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Levenstein ME, et al. Basic fibroblast growth factor support of human embryonic stem cell self-renewal. Stem Cells. 2006;24(3):568–574. doi: 10.1634/stemcells.2005-0247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Furue MK, et al. Heparin promotes the growth of human embryonic stem cells in a defined serum-free medium. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(36):13409–13414. doi: 10.1073/pnas.0806136105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lotz S, et al. Sustained levels of FGF2 maintain undifferentiated stem cell cultures with biweekly feeding. PloS one. 2013;8(2):e56289. doi: 10.1371/journal.pone.0056289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brewer GJ, Torricelli JR, Evege EK, Price PJ. Optimized survival of hippocampal neurons in B27-supplemented Neurobasal, a new serum-free medium combination. Journal of neuroscience research. 1993;35(5):567–576. doi: 10.1002/jnr.490350513. [DOI] [PubMed] [Google Scholar]
- 33.Watanabe K, et al. A ROCK inhibitor permits survival of dissociated human embryonic stem cells. Nature biotechnology. 2007;25(6):681–686. doi: 10.1038/nbt1310. [DOI] [PubMed] [Google Scholar]
- 34.Ruben RJ. Development of the inner ear of the mouse: a radioautographic study of terminal mitoses. Acta oto-laryngologica. 1967;(220):221–244. [PubMed] [Google Scholar]
- 35.Matei V, et al. Smaller inner ear sensory epithelia in Neurog 1 null mice are related to earlier hair cell cycle exit. Developmental dynamics : an official publication of the American Association of Anatomists. 2005;234(3):633–650. doi: 10.1002/dvdy.20551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee YS, Liu F, Segil N. A morphogenetic wave of p27Kip1 transcription directs cell cycle exit during organ of Corti development. Development. 2006;133(15):2817–2826. doi: 10.1242/dev.02453. [DOI] [PubMed] [Google Scholar]
- 37.Chen P, Segil N. p27(Kip1) links cell proliferation to morphogenesis in the developing organ of Corti. Development. 1999;126(8):1581–1590. doi: 10.1242/dev.126.8.1581. [DOI] [PubMed] [Google Scholar]
- 38.Hasson T, et al. Unconventional myosins in inner-ear sensory epithelia. The Journal of cell biology. 1997;137(6):1287–1307. doi: 10.1083/jcb.137.6.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barclay M, Ryan AF, Housley GD. Type I vs type II spiral ganglion neurons exhibit differential survival and neuritogenesis during cochlear development. Neural development. 2011;6(33) doi: 10.1186/1749-8104-6-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bermingham NA, et al. Math1: an essential gene for the generation of inner ear hair cells. Science. 1999;284(5421):1837–1841. doi: 10.1126/science.284.5421.1837. [DOI] [PubMed] [Google Scholar]
- 41.Izumikawa M, et al. Auditory hair cell replacement and hearing improvement by Atoh1 gene therapy in deaf mammals. Nat Med. 2005;11(3):271–276. doi: 10.1038/nm1193. [DOI] [PubMed] [Google Scholar]
- 42.Kim WY, et al. NeuroD-null mice are deaf due to a severe loss of the inner ear sensory neurons during development. Development. 2001;128(3):417–426. doi: 10.1242/dev.128.3.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu M, et al. Essential role of BETA2/NeuroD1 in development of the vestibular and auditory systems. Genes & development. 2000;14(22):2839–2854. doi: 10.1101/gad.840500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ma Q, Anderson DJ, Fritzsch B. Neurogenin 1 null mutant ears develop fewer, morphologically normal hair cells in smaller sensory epithelia devoid of innervation. Journal of the Association for Research in Otolaryngology : JARO. 2000;1(2):129–143. doi: 10.1007/s101620010017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pang ZP, et al. Induction of human neuronal cells by defined transcription factors. Nature. 2011;476(7359):220–223. doi: 10.1038/nature10202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vierbuchen T, et al. Direct conversion of fibroblasts to functional neurons by defined factors. Nature. 2010;463(7284):1035–1041. doi: 10.1038/nature08797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang Y, et al. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron. 2013;78(5):785–798. doi: 10.1016/j.neuron.2013.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Blanchard JW, et al. Selective conversion of fibroblasts into peripheral sensory neurons. Nature neuroscience. 2015;18(1):25–35. doi: 10.1038/nn.3887. [DOI] [PMC free article] [PubMed] [Google Scholar]