

Abstract
Clonal progression to myelodysplastic syndrome (MDS) or acute myeloid leukemia (AML) remains a dreaded complication for a subset of patients with bone marrow failure (BMF). Recognizing risk factors for the development of MDS or AML would inform individualized treatment decisions and identify patients who may benefit from early or upfront hematopoietic stem cell transplantation. Now that next-generation DNA sequencing is available in the clinical laboratory, research has focused on the implications of germ line and somatic mutations for diagnosing and monitoring patients with BMF. Most germ line genetic BMF disorders are characterized by a high propensity to develop MDS or AML. Many affected patients lack the physical stigmata traditionally associated with the inherited marrow failure syndromes. Although any single inherited marrow failure disorder is rare, multiplexed genetic sequencing that allows simultaneous evaluation of marrow failure genes en masse demonstrated that, as a group, these inherited disorders compose a significant subset (5% to 10%) of patients with BMF. Early diagnosis of a germ line genetic marrow failure disorder allows individualized monitoring and tailored therapy. Recent studies of somatic variants in marrow failure revealed a high frequency of clonal hematopoiesis with the acquisition of mutations in genes associated with MDS or AML. Investigation of somatic mutations in marrow failure revealed important insights into the mechanisms promoting clonal disease but also raised additional questions. This review will focus on the evaluation and implications of germ line and somatic mutations for the development of clonal disorders in patients with BMF. Challenges and limitations of clinical genetic testing will be explored.
The development of myelodysplastic syndrome (MDS) or acute myeloid leukemia (AML) poses a serious clinical complication of bone marrow failure (BMF) and remains a significant cause of mortality.1,2 Approximately 15% of patients with acquired aplastic anemia (AA)3 develop MDS or AML within 10 years. The risk of progression to MDS or AML is much higher in patients with genetic marrow failure disorders.4 The distinction between hypocellular MDS and BMF or AA remains challenging, particularly in children. MDS arising in childhood is typically hypocellular, in contrast to the increased marrow cellularity with ineffective hematopoiesis typically noted with MDS in older adults.5 The marrow dysplasias that characterize some of the genetic marrow failure disorders at baseline may raise concern for possible MDS. Although the presence of specific clonal cytogenetic abnormalities may provide important diagnostic indicators of MDS, many cases of MDS in adults and the majority of cases of MDS in children lack apparent chromosomal abnormalities. Because next-generation DNA sequencing is available in the clinic, the diagnostic evaluation and risk stratification of BMF and AA is rapidly evolving. Early identification of patients at high risk for clonal progression would inform medical monitoring and early intervention to improve prognosis. The germ line and somatic genetics of BMF will be discussed with a focus on diagnostic strategies. The clinical challenges accompanying these exciting new advances will be explored. This discussion focuses primarily on multilineage marrow failure disorders. For single-lineage marrow failure disorders, the reader is referred to other excellent recent reviews.
BMF: germ line genetics
The observation of familial cases of AA and syndromic marrow failure, many of which carry a high risk of MDS or AML, led to the recognition of genetic causes of AA (Table 1). In the past, germ line predisposition to BMF was believed to be very rare. Diagnosis previously relied heavily on physical stigmata classically associated with specific inherited marrow failure syndromes (Table 2). It is now recognized that a significant subset of patients with a germ line predisposition to marrow failure, MDS, or AML lack abnormal physical findings and have a negative family history for these disorders.6 Recent genomic screening of patients with idiopathic BMF or MDS identified germ line genetic BMF disorders in 5% to 10% of cases.6,7 Reliance on physical stigmata or family history will miss a significant subset of patients with germ line predisposition to BMF. The importance of diagnosing a germ line marrow failure disorder is summarized in Table 3. Many familial marrow failure cases remain genetically undefined despite extensive evaluation.
Table1. Germ line genetic bone marrow failure disorders.
| Disorder | Gene | Inheritance |
|---|---|---|
| Congenital amegakaryocytic thrombocytopenia | MPL | AR |
| Diamond Blackfan anemia | RPL5 | AD |
| RPL11 | AD | |
| RPL15 | AD | |
| RPL23 | AD | |
| RPL26 | AD | |
| RPL27 | AD | |
| RPL31 | AD | |
| RPL35a | AD | |
| RPL36 | AD | |
| RPS7 | AD | |
| RPS10 | AD | |
| RPS15 | AD | |
| RPS17 | AD | |
| RPS19 | AD | |
| RPS24 | AD | |
| RPS26 | AD | |
| RPS27 | AD | |
| RPS27A | AD | |
| RPS28 | AD | |
| RPS29 | AD | |
| GATA1 | X-linked | |
| Dyskeratosis congenita | ACD/TPP1 | AR, AD |
| CTC1 | AR | |
| DKC1 | X-linked | |
| NOLA2/NHP2 | AR | |
| NOLA3/NOP10 | AR | |
| PARN | AR, AD | |
| RTEL1 | AR, AD | |
| TERC | AD | |
| TERT | AR, AD | |
| TINF2 | AD | |
| WRAP53/TCAB1 | AR | |
| Fanconi anemia | FANCA | AR |
| FANCB | X-linked | |
| FANCC | AR | |
| FANCD1/BRCA2 | AR | |
| FANCD2 | AR | |
| FANCE | AR | |
| FANCF | AR | |
| FANCG | AR | |
| FANCI | AR | |
| FANCJ/BACH1/ | AR | |
| BRIP1 | ||
| FANCL | AR | |
| FANCN/PALB2 | AR | |
| FANCO/RAD51C | AR | |
| FANCP/SLX4 | AR | |
| FANCQ/ERCC4 | AR | |
| FANCR/Rad51 | AD | |
| FANCS/BRCA1 | AR | |
| FANCT/UBE2T | AR | |
| FANCU/XRCC2 | AR | |
| FANCV/REV7 | AR | |
| GATA2 spectrum disorders | GATA2 | AD |
| Shwachman-Diamond syndrome | SBDS | AR |
| SRP72 | SRP72 | AD |
Only multilineage bone marrow failure disorders are included here.
Table 2. Clinical and laboratory features of genetic bone marrow failure disorders.
| Disorder | Major physical features | Laboratory tests |
|---|---|---|
| Fanconi anemia | Short stature, skin hyper-/hypo-pigmentation; limb or hand, skeletal, facial dysmorphologies; renal, gonadal, neurocognitive/central nervous system, cardiopulmonary, gastrointestinal anomalies | Chromosomal breakage or cell cycle arrest induced by mitomycin C or diepoxybutane |
| Dyskeratosis congenita or telomere biology disorders | Nail dystrophy, lacey or reticular skin pigmentation, oral leukoplakia, liver cirrhosis, short stature, pulmonary fibrosis, vascular anomalies, hyperhidrosis, and ophthalmologic, hair (early graying, early hair loss) dental, central nervous system, gastrointestinal, facial cardiac, genitourinary anomalies | Telomere lengths less than first percentile |
| GATA2 spectrum disorders | Warts, atypical mycobacterial infections, lymphedema, deafness, pulmonary alveolar proteinosis | Monocytopenia*; low B, T, and natural killer cells*; low immunoglobulin levels* |
| Shwachman-Diamond syndrome | Steatorrhea, thoracic dysplasias, jeune syndrome, metaphyseal dysostosis, short stature | Low serum trypsinogen (age <3 y), low serum pancreatic isoamylase (age ≥3 y) |
| Diamond Blackfan anemia | Thumb anomalies, short stature, cleft lip or palate, Pierre Robin syndrome, facial dysmorphologies, and cardiac and genitourinary anomalies | Elevated erythrocyte adenosine deaminase† |
This list is not comprehensive. Absence of physical stigmata does not rule out these disorders.
Variable.
In a majority of patients.
Table 3. Rationale for diagnostic testing of genetic marrow failure disorders.
| Diagnostic testing for genetic marrow failure disorders is necessary to: |
|---|
| Screen for cancer |
| Inform treatment choice |
| Tailor transplantation regimen |
| Ensure potential stem cell backup with cancer therapy |
| Avoid affected related donors |
| Provide counseling for affected families |
| Discuss preimplantation genetic diagnostic options |
Most of the inherited BMF syndromes are associated with a predisposition toward cancer, particularly a high risk of MDS or AML, in contrast to the low incidence of MDS in children and young adults in the general population. MDS or AML may be the feature that first brings the patient with an inherited BMF disorder to medical attention. There is a growing recognition that germ line genetic predisposition accounts for a subset of seemingly de novo cases of MDS that present at a young age. A recent study of 426 pediatric patients with primary MDS and 82 cases of secondary MDS identified germ line GATA2 mutations in 7% of all primary MDS cases and 15% of advanced MDS cases.8 Germ line GATA2 mutations were identified in 37% of pediatric MDS patients with monosomy 7. In the adolescent age group with monosomy 7 MDS, 72% harbored germ line GATA2 mutations.8
This article will discuss genetic disorders predisposing to marrow failure and MDS or AML with a focus on diagnostic strategies and pitfalls in the genomics era.
GATA2 spectrum disorders
Clinical features
Heterozygous mutations in the GATA2 gene result in autosomal dominant (AD) disorders associated with marrow failure, MDS, and AML.9 The broad clinical phenotypes of GATA2 mutations are summarized in Table 2. Presentations may vary widely among affected members of the same family. The bone marrow is often hypocellular and may have striking multilineage dysplasia particularly of the megakaryocytes.10 Increased reticulin fibrosis may also be observed.
Genetic and molecular features
The GATA2 gene encodes a zinc finger transcription factor, which plays an important role in hematopoiesis and lymphatic development.9 The mutational spectrum of GATA2 includes missense, nonsense, frameshift, and partial or complete gene deletion. Mutations within intron 4 disrupt an intronic enhancer sequence and reduce GATA2 expression. GATA2 mutations associated with BMF result in loss of function and GATA2 haploinsufficiency.
Diagnosis
Sequencing of the coding regions and the intronic enhancer region as well as array comparative genomic hybridization (CGH) analysis for gene deletions are clinically available. Whole exome sequencing may miss the intronic enhancer mutations and deletions. Targeted panels designed to detect somatically acquired mutations in tumor tissue may be suboptimal to use for screening for germ line GATA2 mutations, depending on the regions targeted and coverage.
Fanconi anemia
Clinical features
Although Fanconi anemia (FA)11,12 is classically characterized by physical anomalies (Table 2), many patients with FA lack apparent physical stigmata. FA is characterized by BMF, increased risk of MDS or AML, and solid tumors such as squamous cell carcinomas of the head, neck, gastrointestinal tract, and female genital tract. MDS, AML, or solid tumors may be the sole medical complication that brings the patient to medical attention. Approximately 25% of patients with FA lack clinical stigmata. FA patients are exquisitely sensitive to agents that damage DNA, especially DNA cross-linking agents, alkylators, and ionizing radiation. The high risks of treatment-related morbidity and mortality led to the development of reduced-intensity conditioning for allogeneic stem cell transplantation and tailored therapies for treating malignancies.
Genetic and molecular features
FA is a multigenic disorder with at least 21 genes currently identified. Most FA genes are autosomal recessive (AR) except for FANCB, which is X-linked, and FANCR/RAD51, which is AD. Approximately two-thirds of patients can be categorized into the most common FA subtypes: FANCA, FANCC, and FANCG. A few of the FA subtypes (eg, FANCO, FANCR,and FANCS) are exceedingly rare and have not yet been associated with BMF in the patients described. Whether FANCM or XRCC2 causes FA is unclear. The FA genes function coordinately to repair DNA damage. Although the mechanisms whereby the FA pathway contributes to DNA repair are still incompletely understood, the FA genes participate in the repair of stalled DNA replication forks by unhooking DNA interstrand cross-links and promoting homologous recombination. Sensitivity to endogenous and exogenous aldehydes, which can cause DNA damage, has also been demonstrated when the FA pathway is impaired. Additional functions of FA genes in cellular stress signaling have also been identified. FA is genetically heterogeneous and includes large numbers of private and founder mutations. A small subset of FA patients lack identifiable mutations.
Diagnosis
The gold standard for clinical diagnosis of FA remains the demonstration of increased chromosomal breaks and radials after treatment with the clastogens mitomycin C or diepoxybutane. Diagnostic testing is typically performed on peripheral blood lymphocytes stimulated with phytohemagglutinin. A false-negative result may occur because of somatic mosaicism due to reversion of the FA mutation in hematopoietic and/or lymphoid cells. In such cases, diagnosis may be made by testing fibroblasts, typically cultured from skin punch biopsies.
Recent studies have explored the role of genetic testing in diagnosing FA. Because of the large number of genes and different types of mutations, one proposal was to integrate DNA sequencing, array CGH, and RNA analysis for FA diagnosis.13 Identification of FA mutations, including deletions, was also achieved through deep targeted next-generation sequencing alone.6 To ascertain whether novel mutations are pathogenic, a combination of genetic analysis and functional analysis may be integrated. Identifying subtypes through genetic analysis informs clinical management of those subtypes associated with an especially high risk of a range of solid tumors and leukemias.
Dyskeratosis congenita and telomere biology disorders
Clinical features
Individuals with mutations in genes that function in telomere maintenance are at increased risk for BMF, MDS, and AML. In the era before causative genes and molecular pathways were identified, recognition of this set of disorders relied on the observation of nail dystrophy, oral leukoplakia, and abnormal reticulated or mottled skin pigmentation, but all three may be absent. Patients presenting with only nonhematologic manifestations (Table 2) typically remain at increased risk for the development of cytopenias when exposed to stressors. These additional organ system abnormalities result in increased risk of regimen-related toxicities with cancer treatments or hematopoietic stem cell transplantation and necessitate specialized reduced-intensity regimens. Disease anticipation has been described in dyskeratosis congenita (DC) and telomere biology disorders (TBDs)14,15 whereby the clinical symptoms become progressively more severe and manifest at an earlier age in successive generations through the inheritance of progressively shorter telomeres.
Genetic and molecular features
Currently 11 genes causing DC have been identified (Table 1). X-linked, AD, and AR patterns of inheritance have been described. These genes are involved in telomere maintenance as part of the telomerase complex, in localization of telomerase to the telomere, or in telomere stabilization. Telomeres are specialized protein-DNA complexes at the ends of chromosomes that prevent DNA fusions or activation of stress response pathways that would result from exposed free DNA ends. Telomere shortening is associated with aging. Telomeres of patients with DC or TBD are significantly shorter that those of age-matched controls. A correlation between telomere length and disease severity has been observed.16 Some DC genes also function in ribosome homeostasis.17
Diagnosis
Clinical testing for age-specific telomere length is available and provides a functional screen for DC or TBD. Telomeres of patients with DC or TBD are typically significantly shorter than those of patients with other BMF disorders. Short telomeres are frequently seen in the granulocyte lineage and in isolation are not indicative of a genetic telomeropathy. Telomere lengths in patients with BMF resulting from any of a variety of causes may be shorter compared with those in healthy controls. A pattern of very short telomeres falling below the first percentile for age in at least 3 different leukocyte subsets is highly suspicious for an underlying genetic disorder of telomere maintenance and warrants further evaluation.18,19 Sequencing of the DC genes is clinically available using either single gene tests or multiplexed targeted next-generation sequencing panels. False-negative results may occur because of somatic reversion in the blood, in which case the diagnosis may be made from nonhematopoietic tissue.20 A subset of patients who present with clinical features of DC or TBD and very short telomeres lack identifiable mutations, suggesting that additional genes have yet to be identified. Current diagnostic strategies integrate clinical assessment, functional testing for telomere length, and mutational analysis.
Shwachman-Diamond syndrome
Clinical features
Shwachman-Diamond syndrome (SDS) 21 was originally recognized by the combination of steatorrhea as a result of exocrine pancreatic insufficiency and BMF with predisposition to MDS or AML. Pancreatic dysfunction is often asymptomatic, so the diagnosis is easily missed.22 Physical anomalies and poor growth may also provide important clues to the aid the diagnosis (Table 2). SDS may be diagnosed in seemingly healthy siblings of a proband with SDS.
Genetic and molecular features
SDS is an AR inherited disorder. Biallelic mutations in the SBDS gene are identified in more than 90% of SDS patients. The SBDS protein has been implicated in ribosome biogenesis, mitotic spindle stabilization, and stress responses, and it plays a role intrinsic to hematopoietic cells and in bone marrow stromal function. SBDS binds to the 60S ribosomal subunit and promotes the dissociation of EIF6 through the GTPase EFL1 during ribosomal maturation.
Mutations in the SBDS gene appear to result from a gene conversion event with a nearby highly conserved pseudogene that can pose diagnostic challenges. The most common mutation is a missense mutation in the splice donor consensus sequence in intron 2 (c.258+2T>C) and is found on at least 1 allele in almost all SDS patients reported to date. This is a hypomorphic mutation that allows expression of a small amount of SBDS protein. Typically, reduced SBDS protein expression levels are observed. Missense mutations may also affect SBDS protein function.
The bone marrow may be normocellular early in life but typically becomes hypocellular during childhood. Marrow dysmorphologies are common and include myeloid hypogranularity with reduced nuclear segmentation, small megakaryocytes, and mild erythroid dysplasias. Although SDS is typically associated with neutropenia, patients commonly develop thrombocytopenia, anemia, pancytopenia, or AA.
Diagnosis
SBDS genetic testing is clinically available. Mutations may be missed if the length of the gene conversion extends beyond the exon-directed primers. Analytic pipelines for next-generation sequencing panels or whole exome sequencing must avoid mistakenly mapping pathologic SBDS variants to the pseudogene. Analysis of SBDS protein levels may be helpful in the evaluation of variants of unknown clinical significance but is not clinically available. Serum trypsinogen and serum pancreatic isoamylase provide sensitive tests for even subclinical exocrine pancreatic dysfunction, which is currently one of the clinical diagnostic hallmarks of SDS. Normal fecal elastase levels do not rule out the diagnosis of SDS.
SRP72
Clinical features
Heterozygous mutations have been reported in SRP7223 in 2 unrelated pedigrees of familial AA and MDS. In the first family, affected individuals also had hearing loss, although the proband had possible labyrinthitis.
Genetic and molecular features
SRP72 segregates in an AD pattern. SRP72 is a component of a signal recognition particle involved in nascent protein processing and trafficking. The mechanism whereby SRP72 mutations affect protein function in hematopoiesis remains unknown.
Diagnosis
Clinical genetic sequencing is available. Variants of unknown clinical significance are challenging in the absence of functional assays.
Congenital amegakaryocytic thrombocytopenia
Clinical features
Congenital amegakaryocytic thrombocytopenia (CAMT)24 typically presents early in life with thrombocytopenia with later development of AA. Although a few cases of MDS or AML have been reported in patients with CAMT, it is currently unknown whether CAMT is associated with an increased risk of MDS or AML.
Genetic and molecular features
CAMT is caused by AR mutations in the c-MPL gene which encodes the thrombopoietin receptor. The thrombopoietin receptor is expressed on the surface of hematopoietic stem cells and platelet progenitor cells and functions in hematopoiesis and megakaryopoiesis.
Diagnosis
Genetic testing for c-MPL mutations is clinically available. c-MPL testing targeted for somatically acquired mutations in myeloproliferative disorders should not be used to screen for CAMT.
Diamond Blackfan anemia
Clinical features
Diamond Blackfan anemia (DBA)25 is characterized by red cell aplasia typically but not always presenting in the first year of life. The risk of MDS or AML and solid tumors such as soft tissue sarcomas is elevated in DBA. Additional physical anomalies are associated with DBA but may be absent (Table 2). A few cases of AA have been described in patients with DBA.
Molecular and genetic features
DBA is caused by AD mutations in genes encoding ribosomal proteins. Approximately 25% of mutations involve gene deletions. Another genetic variant of DBA is caused by X-linked mutations in GATA1.
Diagnosis
DBA is diagnosed by clinical presentation and bone marrow examination showing red cell aplasia. Multiplexed panels for DBA mutations as well as array CGH for deletion analysis are clinically available. Approximately 25% of DBA patients lack identifiable mutations. Ethidium bromide staining of ribosomal RNA (rRNA) or northern blotting for rRNA intermediates may be helpful to assess whether variants impair rRNA maturation in the research setting.
BMF: somatic genetics
Intense interest has focused on somatic genetic studies of clonal hematopoiesis and on acquired mutations analyzed by cytogenetics, single nucleotide polymorphism array (SNP array) karyotyping, and next-generation deep sequencing.26,27 The implications of clonal hematopoiesis for disease pathogenesis and progression to MDS or AML in marrow failure is being actively investigated. The potential role of immunologic targeting of the bone marrow to promote clonal evolution in AA has recently been reviewed and will not be discussed here.26 Studies of clonal hematopoiesis in acquired AA and in inherited marrow failure disorders are considered separately.
Acquired AA
Paroxsymal nocturnal hemoglobinura
Paroxsymal nocturnal hemoglobinura (PNH)28 is a clonal disorder characterized by acquired somatic mutations in the PIGA gene resulting in the absence of glycosyl phosphatidylinositol–anchored cell surface proteins. Individuals with PNH are at increased risk of developing AA, and PNH clones are frequently observed in patients presenting with AA, but why these patients are prone to developing PNH clones remains unclear. Data thus far do not support an intrinsic growth or proliferative advantage of the PNH clone. It has been hypothesized that PNH clones may be spared from putative immunologic targeting of the glycosyl phosphatidylinositol anchor or its associated proteins. Thus far, no increased leukemia risk has been observed in patients with PNH clones. The clinical implications of PNH clones in AA largely depend on whether the clones are symptomatic or reach sufficient size to become a concern for potential thrombosis.29,30 Studies suggest that the presence of a PNH clone may be a good prognostic marker associated with response to immunosuppressive therapy (IST) and that PNH clones usually are not observed in inherited marrow failure syndromes.31,32
Cytogenetics
It is often difficult to predict the clinical implications of cytogenetic clones developing in AA patients. Clinical significance of cytogenetic abnormalities is informed by the marrow morphology, cell counts, specific type of clone, presence of additional cytogenetic abnormalities, and progression over time. Because metaphases may be sparse and may arise from lymphocytes rather than from early progenitor or myeloid lineages, fluorescent in situ hybridization for high-risk cytogenetic clones such as monosomy 7/del7q is also recommended. Trisomy 8 and monosomy 7/del7q are frequent cytogenetic abnormalities that develop over time in patients with AA. Individuals with AA andtrisomy8canrespondtoIST.In1series,trisomy 8 was associated with a low risk of progression to MDS or AML,1 although a recent study reported progression to AML in 2 of 6 AA patients with isolated trisomy 8.32 A low risk of malignant progression has been described with del13q in association with concomitant but separate PNH clones.33 Monosomy 7/del7q is associated with a high risk of progression to MDS or AML,1,34,35 particularly when the clone is persistent in the context of falling blood counts, marrow morphologic dysplasia, additional cytogenetic abnormalities in the same clone, or increasing blasts. Some isolated chromosomal abnormalities such as –Y, +8, or del20q without other features of MDS may be found in seemingly healthy individuals and are generally not considered indicative of MDS by themselves. Robust long-term data are lacking on whether the presence of such an isolated nonspecific clonal cytogenetic abnormality at presentation of otherwise seemingly acquired AA carries a higher risk of long-term clonal evolution, and some centers opt to treat these patients the same way they treat AA patients with normal karyotypes.
SNP-A karyotyping
SNP-A karyotyping detects abnormalities in interphase chromosomes and can detect small deletions and copynumber–neutral loss of heterozygosity (copy-neutral LOH), which are missed by karyotype analysis. SNP-A analysis combined with metaphase karyotype analysis of 93 AA patients identified chromosomal aberrations in 19% of the patients.36 Malignant evolution was observed in 14 (15%) of 93 AA patients, with earlier detection of monosomy 7 using SNP-A. Recurrent observation of copy-neutral LOH of chromosome 6p in the region of the HLA locus in up to 13% of AA patients raised the possibility that uniparental HLA expression might affect immune response in disease pathogenesis.26,27
Next-generation deep sequencing
A targeted next-generation sequencing screen of 219 genes recurrently mutated in hematologic malignancies identified somatic mutations in 9 of 39 AA patients. The variant allele fraction was <10% in 7 of the 9 patients.37 Whole exome sequencing of paired bone marrow and skin samples from 22 AA patients identified somatic mutations in 16 patients (72.7%), many of whom were children.38
A larger study of paired bone marrow and skin or buccal mucosa samples from 150 patients with AA without morphologic MDS used two-tiered targeted sequencing of BMF, MDS, and AML genes to identify 32 somatic mutations in 29 patients (19%), excluding PIGA mutations.34 Mutated genes included ASXL1 (n = 12), DNMT3A (n = 58), and BCOR (n = 6). Patients with somatic mutations had a longer duration of disease (37 vs 8 months; P < .4) relative to patients without mutations. Progression to MDS was seen in 17 patients (11%) over the course of 9 to 260 months (median disease duration, 86 months) of whom 2 progressed to AML. At the time of MDS diagnosis, 10 had normal cytogenetics, 4 had monosomy 7, 1 had trisomy 15, 1 had trisomy 8, and 1 had a complex karyotype. None of the patients with isolated BCOR mutations developed MDS. Seven of 12 patients with ASXL1 mutations progressed to MDS, including 2 of 3 with <10% ASXL1 mutation burden. All 3 patients with large DNMT3A clones progressed to MDS, 2 of whom developed monosomy 7. The 5 patients with small (<10%) DNMT3A clones had not developed MDS at the time of the study. Of the 17 MDS patients, 11 (65%) had somatic mutations detected at a median of 12 months prior to MDS diagnosis. The remaining 6 MDS patients had no detectable somatic mutations at the time of the sequence analysis. With the caveats that this was not a longitudinal study and that samples were not obtained at a uniform time relative to diagnosis or treatment, 11 (38%) of 38 patients with somatic mutations evolved to MDS vs 6 (6%) of 121 cases of MDS in patients without targeted mutations (P < .001). Thus, although there was a correlation between somatic mutation burden and MDS risk, significant individual variation was observed.
A subsequent comprehensive study35 of 439 AA patients from the United States and Japan conducted targeted deep sequencing analysis of 106 genes implicated in myeloid malignancies combined with SNP-A. In addition, whole exome sequencing was performed for 52 patients, and longitudinal analyses were conducted for 82 patients. Somatic mutations were identified by targeted analysis in 156 (36%) of 439 patients. Whole exome analysis revealed somatic mutations in 25 (48%) of 52 AA patients. Combining the results of all methods, clonal hematopoiesis was seen in 85% of the 52 samples. More than 1 mutation was found in 56 (36%) of 156 patients. The mean allele burden (proportion of the mutant allele) was 9.3%, with 75% of mutations having an allelic fraction of less than 10% in contrast to the higher frequency of larger clones noted in MDS.39 Most of the detected mutations (58 of 79 mutations) were already present at diagnosis at a lower allele frequency. In 6 of 6 patients examined, the somatic mutation was present in multiple hematopoietic lineages, including hematopoietic stem cells, common myeloid progenitors, and myeloid erythroid progenitors, which is consistent with the acquisition of the mutation within a stem cell compartment. Somatic mutations were observed in younger patients, an age group wherein spontaneous somatic acquired mutations are rare.40
Seventy-seven percent of mutations involved a small number of genes: BCOR and BCORL1 (9.3% of patients), PIGA (7.5%), DNMT3A (8.4%), and ASXL1 (6.2%). Other MDS-related genes were also involved but at much lower frequencies. A comparison of the spectrum of commonly mutated genes in AA vs MDS vs age-associated clonal hematopoiesis of indeterminate potential41-43 revealed both shared and distinct patterns (Figure 1). DNMT3A and ASXL1 are commonly mutated in AA, MDS, and age-associated clonal hematopoiesis of indeterminate potential. PIGA and BCOR/BCOR1 were commonly mutated in AA but less frequently in MDS. Genes frequently mutated in MDS such as TET2, JAK2, RUNX1, and TP53, along with cohesin factors and splicing factors were relatively less common in AA. The mutational patterns in AA also differed from that observed with clonal hematopoiesis of indeterminate potential in which frequently affected genes include DNMT3A, TET2, ASXL1, TP53, SF3B1, JAK2, and SRSF2.
Figure 1.
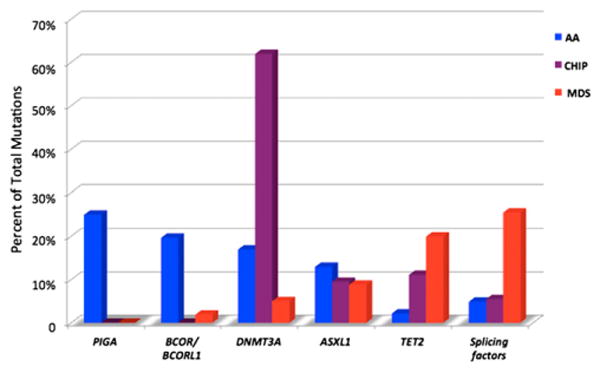
Relative mutation frequencies in AA, clonal hematopoiesis of indeterminate potential (CHIP), and MDS. Modified from Ogawa.27
When considering somatic mutations as a whole, there was no correlation between the overall presence of somatic mutations and response to IST, risk of MDS or AML, or survival. However, specific mutations were associated with different clinical outcomes. Mutations in PIGA, BCOR,and BCOR1 correlated with a positive response to IST, whereas mutations in ASXL1 and DNMT3A correlated with a lower response to IST, lower survival particularly in younger patients, and progression to MDS or AML. Significant patient variability provided a cautionary note to extrapolating these results prospectively to individual patients. Longitudinal sequencing of serial samples revealed broad variability in the temporal patterns of clones in individual patients. Although most clones remained stable at a low frequency, dominant clones emerged with increasing size over time in patients progressing to MDS or AML. Of note, increasing clone size was also seen in patients who maintained stable blood counts without progression to MDS or AML within the time frame of follow-up.
In a study of 13 AA patients who developed monosomy 7,44 only 4 of 13 patients carried somatic mutations in myeloid malignancy genes, although all patients demonstrated clonal hematopoiesis. Shorter telomere lengths relative to stable AA controls preceded the emergence of monosomy 7 after IST.44 Short telomere lengths at diagnosis and before treatment have been associated with increased risk of relapse, clonal evolution, and reduced survival.45
Inherited BMF
The latency and variability of the development of MDS or AML in patients with germ line genetic BMF disorders is consistent with the notion that additional events are required for malignant transformation.
Cytogenetics
Monosomy 7 and trisomy 8 are frequent cytogenetic abnormalities and should raise suspicion for a possible germ line GATA2 mutation in the pediatric population.8,10 Abnormal cytogenetic findings were identified in 16 (58%) of 2810 and 47 (84%) of 568 patients with GATA2 mutations.
Analysis of bone marrow samples from 57 patients with FA identified somatic chromosomal gains and losses in 35 patients.46 Clonal cytogenetic abnormalities arising in FA patients who progressed to MDS or AML include 1q+, 3q+, –7/del7q, and 11q–.46,47 1q+ and del20q were also observed in patients with or without MDS or AML.
Del20q11 and isochromosome 7q10 frequently arise in SDS and may persist as isolated abnormalities without progression to MDS or AML.48 The location of the SBDS gene within 7q led to the proposal that duplication of the SBDS gene may be beneficial. The deleted region of 20q includes the EIF6 gene, leading to the hypothesis that haploinsufficiency of EIF6 may compensate for the loss of SBDS, which functions to remove EIF6 from the nascent 60S ribosomal subunit during ribosome maturation. Monosomy 7 or del7q are commonly seen in patients with MDS or AML, typically in association with additional chromosomal abnormalities.
Next-generation deep sequencing
Data are sparse regarding acquired somatic mutations in inherited marrow failure syndromes. Targeted sequencing of exons 12 and 13 of ASXL1 identified mutations in 14 (29%) of 48 patients with germ line GATA2 mutations, including 4 patients who progressed to chronic myelomonocytic leukemia.49 However, 27 of 37 patients who developed MDS lacked ASXL1 mutations. Whole exome sequencing of paired tumor and matched normal samples from 19 patients with germ line RUNX1 or GATA2 mutations were analyzed for somatic mutations.50 The median number of somatic single nucleotide variants/indels per patient (n 5 13) was similar to that observed in de novo AML. The pattern of genes mutated in patients with familial MDS or AML differed from that observed in de novo AML.
Targeted sequencing of RUNX1 together with chromosomal analyses revealed mutations, translocations, and deletions in 6 (18%) of 34 FA patients.46 All 6 FA patients with RUNX1 mutations had MDS or AML. Sequencing of FA patients with AA, MDS, or AML for genes recurrently mutated in MDS or AML revealed MLL-PTD mutations in 2 patients, and single patients with FLT3-ITD, NRAS, ERG amplification, and ZFP36L2-PRDM16 translocation. No mutations were noted in TP53, TET2, CBL, NPM1, FLT3-TKD, and CEBP alpha. Copy-neutral LOH was not seen, which suggests that homologous recombination is not a common mechanism in FA. The majority of the chromosomal abnormalities were the result of unbalanced translocations leading to regional chromosomal gain or loss, consistent with a model of increased DNA double-strand breaks followed by nonhomologous end joining. No gene reversion of FANC mutations were noted in MDS and AML FA patients.
Challenges to the clinical application of genetic and genomic testing
Diagnosis
The diagnosis of a germ line genetic BMF disorder carries profound clinical implications (Table 3). AA presenting in a patient with physical anomalies, short stature, poor growth, or clinical history suggestive of a germ line BMF disorder warrants further evaluation. A family history of cytopenias, BMF, physical stigmata associated with BMF disorders, early onset cancer, and excessive toxicity with chemotherapy or radiation raises suspicion of a germ line marrow failure disorder. Importantly, the absence of a suggestive family history does not rule out an inherited marrow failure syndrome. Variable phenotypes are observed even among individuals who share the same inherited BMF gene mutation within a given family. Disease anticipation in disorders of telomere maintenance or de novo mutations may result in a negative family history. In the past, diagnostic testing was performed for individual genes on the basis of clinical suspicion. However, this approach misses a significant subset of patients with an inherited disorder; with the availability of next-generation targeted sequencing panels, an entire set of BMF genes may now be screened simultaneously and more economically than was possible for each potential individual gene. Although mutations in a single BMF gene are rare, screening for BMF disorders as a whole identifies germ line mutations in 5% to 10% of young AA and MDS patients.
In addition to clinical history, family history, and physical examination, laboratory testing should screen for clues to a potential germ line marrow failure disorder. Because BMF can progress with time, a history of previously normal blood counts does not rule out an inherited marrow failure disorder. Some patients with inherited BMF disorders experience an improvement in blood counts over time. However, an unexplained decrease or increase in blood counts warrants evaluation for possible MDS. Because FA patients may lack physical findings, chromosomal breakage testing for FA is recommended for all pediatric patients with BMF, and may also be considered for older patients. Telomere length analysis provides a screen for telomeropathies. Analysis for immunologic abnormalities within lymphocyte subsets to evaluate B cells, T-cell subsets, natural killer cell numbers, and immunoglobulin levels may provide clues to mutations in GATA2 or SBDS or a telomeropathy. Serum trypsinogen (age <3 years) or pancreatic isoamylase (age ≥3 years) are usually low in patients with SDS, even with subclinical exocrine pancreatic dysfunction. Erythrocyte adenosine deaminase is typically elevated in patients with DBA, although a normal erythrocyte adenosine deaminase level does not rule out this diagnosis. Elevated fetal hemoglobin may be seen with inherited BMF disorders.
Challenges of genetic testing
Although genetic testing constitutes a huge advance in our ability to diagnose cryptic or ambiguous genetic BMF disorders, many challenges remain. Some mutations, such as inversions, translocations, and deletions, are easily missed by sequencing analysis. Some regions of the genome are poorly covered because of the limitations of sequencing technologies, such as highly GC-rich regions or poor exome capture. The presence of pseudogenes that share a high degree of homology with the bona fide gene creates difficulties with correct alignment of sequence reads. This is particularly problematic for the SBDS gene for which pathogenic variants seem to be introduced by gene conversion with its neighboring pseudogene with which it shares 97% sequence homology. For recessive disorders, variant orientation in cis vs trans must be determined, and testing of parents may be helpful in this regard. For genes that may be mutated in the germ line or acquired somatically in hematopoietic cells, sequencing of an alternate nonhematopoietic tissue such as skin fibroblasts may be necessary. Lymphocyte contamination of saliva or buccal samples is problematic in this regard. Genes that do not encode a protein product are often filtered out in analytic pipelines. Some genes have pathogenic mutations located in noncoding regions, for example, the GATA2 intronic enhancer, that are not captured by standard exon-directed sequencing strategies that typically cover only short flanking regions within adjacent introns. When screening for germ line mutations in BMF or MDS genes, it may be inappropriate to rely on panels designed and used for analyzing somatic mutations in MDS and AML samples for germ line testing, depending on the genomic targeting design and analysis.
One of the major challenges of diagnostic genetic testing is the interpretation of novel variants or variants for which no functional data are available. Where feasible, the effect of a variant on gene function may be tested (for example, chromosomal breakage for FA genes or telomere length for DC genes). Determining the appropriate functional assay for assessing variant pathogenicity is sometimes challenging. Furthermore, assessing hypomorphic variants that partially impair gene function but are not pathogenic in the heterozygous state poses additional challenges. Demonstrating that a variant tracks with the phenotype across multiple unrelated families provides genetic evidence that the variant is pathogenic. The limited availability of publically accessible, comprehensive, and critically reviewed clinical and functional annotation of BMF genetic variants poses a barrier to their clinical interpretation.
Risk stratification
Identification of germ line BMF disorders associated with a high risk of MDS or AML allows close monitoring of blood counts and bone marrow examinations. Clinical outcomes are generally superior if hematopoietic stem cell transplantation is initiated before leukemia develops. Diagnosis also allows therapy to be tailored to avoid excessive treatment-related toxicity and mortality.
For AA, it was initially hypothesized that analysis of somatic mutations might identity AA patients at high risk for progression to MDS or AML. The clinical significance of clonal hematopoiesis was called into question following reports of CHIP51 involving mutations in myeloid malignancy genes in healthy older people.40-43 CHIP has a low predictive value for development of MDS or AML. As noted previously, although some somatic mutations are associated with increased risk of progression from AA to MDS or AML, the clinical course of any individual patient with these high-risk mutations is variable with unpredictable fluctuations in the variant allele fraction and clinical progression of even high-risk clones. Clonal hematopoiesis was detected in 67% of seemingly healthy individuals who carried constitutional RUNX1 mutations, a genetic syndrome associated with increased risk of AML.50 The prognostic significance of such somatic mutations remains to be clarified. Considerations in the diagnostic evaluation and monitoring of BMF are outlined in Figure 2. A multifaceted assessment with testing for germ line BMF disorders, serial blood counts, marrow morphology, karyotyping, fluorescent in situ hybridization and, where available, SNP-A karyotyping and deep sequencing together with assessment of the risks and benefits of available treatments must be integrated to inform prognosis and medical management.
Figure 2.
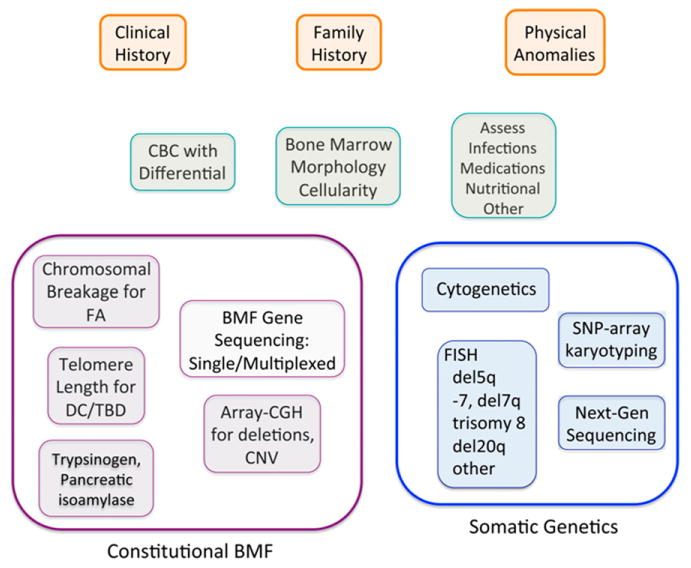
Integration of germ line and somatic genetics into diagnostic evaluation of bone marrow failure. Clinical presentation serves as a guide for initial diagnostic evaluation for BMF and assessment for potential causes such as infection, medication, or underlying disorders. The orange boxes outline the initial clinical assessment. The green boxes outline the initial laboratory evaluation to ascertain whether a patient has bone marrow failure. The purple boxes summarize some of the general laboratory and genetic tests for germ line marrow failure disorders. The blue boxes outline somatic genetic evaluation. CBC, complete blood count; CNV, copy number variants; FISH, fluorescent in situ hybridization.
Learning Objectives.
To describe recent advances in our understanding of germ line and somatic mutations in BMF and clonal progression to MDS or AML
To recognize the challenges of genetic testing for germ line and somatic mutations in BMF
To understand the indications and caveats of genetic screening strategies for diagnosing and monitoring patients with BMF
Acknowledgments
The author apologizes to all those whose publications could not be included because of space limitations. Recent review articles are cited to guide the reader to additional published studies.
Footnotes
Conflict-of-interest disclosure: A.S. has received an honorarium from GlaxoSmithKline and is in discussion regarding a possible clinical trial for aplastic anemia with Novartis.
Off-label drug use: None disclosed.
References
- 1.Maciejewski JP, Risitano A, Sloand EM, Nunez O, Young NS. Distinct clinical outcomes for cytogenetic abnormalities evolving from aplastic anemia. Blood. 2002;99(9):3129–3135. doi: 10.1182/blood.v99.9.3129. [DOI] [PubMed] [Google Scholar]
- 2.Kim SY, Le Rademacher J, Antin JH, et al. Myelodysplastic syndrome evolving from aplastic anemia treated with immunosuppressive therapy: efficacy of hematopoietic stem cell transplantation. Haematologica. 2014;99(12):1868–1875. doi: 10.3324/haematol.2014.108977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Camitta BM, Thomas ED, Nathan DG, et al. Severe aplastic anemia: a prospective study of the effect of early marrow transplantation on acute mortality. Blood. 1976;48(1):63–70. [PubMed] [Google Scholar]
- 4.Shimamura A, Alter BP. Pathophysiology and management of inherited bone marrow failure syndromes. Blood Rev. 2010;24(3):101–122. doi: 10.1016/j.blre.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Glaubach T, Robinson LJ, Corey SJ. Pediatric myelodysplastic syndromes: they do exist! J Pediatr Hematol Oncol. 2014;36(1):1–7. doi: 10.1097/MPH.0000000000000046. [DOI] [PubMed] [Google Scholar]
- 6.Zhang MY, Keel SB, Walsh T, et al. Genomic analysis of bone marrow failure and myelodysplastic syndromes reveals phenotypic and diagnostic complexity. Haematologica. 2015;100(1):42–48. doi: 10.3324/haematol.2014.113456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Keel SB, Scott A, Sanchez-Bonilla M, et al. Genetic features of myelodysplastic syndrome and aplastic anemia in pediatric and young adult patients. Haematologica. doi: 10.3324/haematol.2016.149476. [published online ahead of print on 14 July 2016] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wlodarski MW, Hirabayashi S, Pastor V, et al. EWOG-MDS. Prevalence, clinical characteristics, and prognosis of GATA2-related myelodysplastic syndromes in children and adolescents. Blood. 2016;127(11):1387–1397. doi: 10.1182/blood-2015-09-669937. [DOI] [PubMed] [Google Scholar]
- 9.Hsu AP, McReynolds LJ, Holland SM. GATA2 deficiency. Curr Opin Allergy Clin Immunol. 2015;15(1):104–109. doi: 10.1097/ACI.0000000000000126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ganapathi KA, Townsley DM, Hsu AP, et al. GATA2 deficiency associated bone marrow disorder differs from idiopathic aplastic anemia. Blood. 2015;125(1):56–70. doi: 10.1182/blood-2014-06-580340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Longerich S, Li J, Xiong Y, Sung P, Kupfer GM. Stress and DNA repair biology of the Fanconi anemia pathway. Blood. 2014;124(18):2812–2819. doi: 10.1182/blood-2014-04-526293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bogliolo M, Surrallés J. Fanconi anemia: a model disease for studies on human genetics and advanced therapeutics. Curr Opin Genet Dev. 2015;33:32–40. doi: 10.1016/j.gde.2015.07.002. [DOI] [PubMed] [Google Scholar]
- 13.Chandrasekharappa SC, Lach FP, Kimble DC, et al. NISC Comparative Sequencing Program. Massively parallel sequencing, aCGH, and RNA-Seq technologies provide a comprehensive molecular diagnosis of Fanconi anemia. Blood. 2013;121(22):e138–e148. doi: 10.1182/blood-2012-12-474585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Savage SA, Bertuch AA. The genetics and clinical manifestations of telomere biology disorders. Genet Med. 2010;12(12):753–764. doi: 10.1097/GIM.0b013e3181f415b5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Townsley DM, Dumitriu B, Young NS. Bone marrow failure and the telomeropathies. Blood. 2014;124(18):2775–2783. doi: 10.1182/blood-2014-05-526285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alter BP, Rosenberg PS, Giri N, Baerlocher GM, Lansdorp PM, Savage SA. Telomere length is associated with disease severity and declines with age in dyskeratosis congenita. Haematologica. 2012;97(3):353–359. doi: 10.3324/haematol.2011.055269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ruggero D, Shimamura A. Marrow failure: a window into ribosome biology. Blood. 2014;124(18):2784–2792. doi: 10.1182/blood-2014-04-526301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alter BP, Baerlocher GM, Savage SA, et al. Very short telomere length by flow fluorescence in situ hybridization identifies patients with dyskeratosis congenita. Blood. 2007;110(5):1439–1447. doi: 10.1182/blood-2007-02-075598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alter BP, Giri N, Savage SA, Rosenberg PS. Telomere length in inherited bone marrow failure syndromes. Haematologica. 2015;100(1):49–54. doi: 10.3324/haematol.2014.114389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jongmans MC, Verwiel ET, Heijdra Y, et al. Revertant somatic mosaicism by mitotic recombination in dyskeratosis congenita. Am J Hum Genet. 2012;90(3):426–433. doi: 10.1016/j.ajhg.2012.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Myers KC, Davies SM, Shimamura A. Clinical and molecular pathophysiology of Shwachman-Diamond syndrome: an update. Hematol Oncol Clin North Am. 2013;27(1):117–128. doi: 10.1016/j.hoc.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Myers KC, Bolyard AA, Otto B, et al. Variable clinical presentation of Shwachman-Diamond syndrome: update from the North American Shwachman-Diamond Syndrome Registry. J Pediatr. 2014;164(4):866–870. doi: 10.1016/j.jpeds.2013.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kirwan M, Walne AJ, Plagnol V, et al. Exome sequencing identifies autosomal-dominant SRP72 mutations associated with familial aplasia and myelodysplasia. Am J Hum Genet. 2012;90(5):888–892. doi: 10.1016/j.ajhg.2012.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ballmaier M, Germeshausen M. Congenital amegakaryocytic thrombocytopenia: clinical presentation, diagnosis, and treatment. Semin Thromb Hemost. 2011;37(6):673–681. doi: 10.1055/s-0031-1291377. [DOI] [PubMed] [Google Scholar]
- 25.Vlachos A, Ball S, Dahl N, et al. Participants of Sixth Annual Daniella Maria Arturi International Consensus Conference. Diagnosing and treating Diamond Blackfan anaemia: results of an international clinical consensus conference. Br J Haematol. 2008;142(6):859–876. doi: 10.1111/j.1365-2141.2008.07269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marsh JC, Mufti GJ. Clinical significance of acquired somatic mutations in aplastic anaemia. Int J Hematol. 2016;104(2):159–167. doi: 10.1007/s12185-016-1972-8. [DOI] [PubMed] [Google Scholar]
- 27.Ogawa S. Clonal hematopoiesis in acquired aplastic anemia. Blood. 2016;128(3):337–347. doi: 10.1182/blood-2016-01-636381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brodsky RA. Paroxysmal nocturnal hemoglobinuria. Blood. 2014;124(18):2804–2811. doi: 10.1182/blood-2014-02-522128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pu JJ, Mukhina G, Wang H, Savage WJ, Brodsky RA. Natural history of paroxysmal nocturnal hemoglobinuria clones in patients presenting as aplastic anemia. Eur J Haematol. 2011;87(1):37–45. doi: 10.1111/j.1600-0609.2011.01615.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scheinberg P, Young NS. How I treat acquired aplastic anemia. Blood. 2012;120(6):1185–1196. doi: 10.1182/blood-2011-12-274019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.DeZern AE, Symons HJ, Resar LS, Borowitz MJ, Armanios MY, Brodsky RA. Detection of paroxysmal nocturnal hemoglobinuria clones to exclude inherited bone marrow failure syndromes. Eur J Haematol. 2014;92(6):467–470. doi: 10.1111/ejh.12299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hosokawa K, Sugimori N, Katagiri T, et al. Increased glycosylphosphatidylinositol-anchored protein-deficient granulocytes define a benign subset of bone marrow failures in patients with trisomy 8. Eur J Haematol. 2015;95(3):230–238. doi: 10.1111/ejh.12484. [DOI] [PubMed] [Google Scholar]
- 33.Hosokawa K, Katagiri T, Sugimori N, et al. Favorable outcome of patients who have 13q deletion: a suggestion for revision of the WHO ‘MDS-U’ designation. Haematologica. 2012;97(12):1845–1849. doi: 10.3324/haematol.2011.061127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kulasekararaj AG, Jiang J, Smith AE, et al. Somatic mutations identify a subgroup of aplastic anemia patients who progress to myelodysplastic syndrome. Blood. 2014;124(17):2698–2704. doi: 10.1182/blood-2014-05-574889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yoshizato T, Dumitriu B, Hosokawa K, et al. Somatic Mutations and Clonal Hematopoiesis in Aplastic Anemia. N Engl J Med. 2015;373(1):35–47. doi: 10.1056/NEJMoa1414799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Afable MG, II, Wlodarski M, Makishima H, et al. SNP array-based karyotyping: differences and similarities between aplastic anemia and hypocellular myelodysplastic syndromes. Blood. 2011;117(25):6876–6884. doi: 10.1182/blood-2010-11-314393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lane AA, Odejide O, Kopp N, et al. Low frequency clonal mutations recoverable by deep sequencing in patients with aplastic anemia. Leukemia. 2013;27(4):968–971. doi: 10.1038/leu.2013.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Babushok DV, Perdigones N, Perin JC, et al. Emergence of clonal hematopoiesis in the majority of patients with acquired aplastic anemia. Cancer Genet. 2015;208(4):115–128. doi: 10.1016/j.cancergen.2015.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Haferlach T, Nagata Y, Grossmann V, et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia. 2014;28(2):241–247. doi: 10.1038/leu.2013.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Welch JS, Ley TJ, Link DC, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012;150(2):264–278. doi: 10.1016/j.cell.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jaiswal S, Fontanillas P, Flannick J, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371(26):2488–2498. doi: 10.1056/NEJMoa1408617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Genovese G, Kähler AK, Handsaker RE, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014;371(26):2477–2487. doi: 10.1056/NEJMoa1409405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xie M, Lu C, Wang J, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med. 2014;20(12):1472–1478. doi: 10.1038/nm.3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dumitriu B, Feng X, Townsley DM, et al. Telomere attrition and candidate gene mutations preceding monosomy 7 in aplastic anemia. Blood. 2015;125(4):706–709. doi: 10.1182/blood-2014-10-607572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Scheinberg P, Cooper JN, Sloand EM, Wu CO, Calado RT, Young NS. Association of telomere length of peripheral blood leukocytes with hematopoietic relapse, malignant transformation, and survival in severe aplastic anemia. JAMA. 2010;304(12):1358–1364. doi: 10.1001/jama.2010.1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Quentin S, Cuccuini W, Ceccaldi R, et al. Myelodysplasia and leukemia of Fanconi anemia are associated with a specific pattern of genomic abnormalities that includes cryptic RUNX1/AML1 lesions. Blood. 2011;117(15):e161–e170. doi: 10.1182/blood-2010-09-308726. [DOI] [PubMed] [Google Scholar]
- 47.Cioc AM, Wagner JE, MacMillan ML, DeFor T, Hirsch B. Diagnosis of myelodysplastic syndrome among a cohort of 119 patients with fanconi anemia: morphologic and cytogenetic characteristics. Am J Clin Pathol. 2010;133(1):92–100. doi: 10.1309/AJCP7W9VMJENZOVG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pressato B, Valli R, Marletta C, et al. Cytogenetic monitoring in Shwachman-Diamond syndrome: a note on clonal progression and a practical warning. J Pediatr Hematol Oncol. 2015;37(4):307–310. doi: 10.1097/MPH.0000000000000268. [DOI] [PubMed] [Google Scholar]
- 49.West RR, Hsu AP, Holland SM, Cuellar-Rodriguez J, Hickstein DD. Acquired ASXL1 mutations are common in patients with inherited GATA2 mutations and correlate with myeloid transformation. Haematologica. 2014;99(2):276–281. doi: 10.3324/haematol.2013.090217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Churpek JE, Pyrtel K, Kanchi KL, et al. Genomic analysis of germ line and somatic variants in familial myelodysplasia/acute myeloid leukemia. Blood. 2015;126(22):2484–2490. doi: 10.1182/blood-2015-04-641100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Steensma DP, Bejar R, Jaiswal S, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood. 2015;126(1):9–16. doi: 10.1182/blood-2015-03-631747. [DOI] [PMC free article] [PubMed] [Google Scholar]