

SUMMARY
Neonatal cyanosis resulting from a fetal methaemoglobin variant is rare. Most such variants are only described in a few published case reports. We present the case of a newborn with unexplained persistent cyanosis, ultimately determined to have a γ-chain mutation causing Hb FM-Fort Ripley. This neonatal haemoglobinopathy can be challenging to diagnose, as significant oxygen desaturation may result from barely detectable levels of the mutant haemoglobin and co-oximetry studies may show a falsely normal methaemoglobin level. Our analysis of the infant’s haemoglobin included high-performance liquid chromatography, cellulose acetate electrophoresis and citrate agar electrophoresis, which showed trace amounts of a suspected variant. Ultimately, the diagnosis was made through a novel application of next-generation sequencing (NGS). NGS-based diagnostic approaches are becoming increasingly available to clinicians, and our case provides a framework and evidence for the utilisation of such testing paradigms in the diagnosis of a rare cause of neonatal cyanosis.
BACKGROUND
The differential diagnosis for neonatal cyanosis is broad, and includes disorders of the respiratory system, cardiac anomalies and haemoglobin disorders causing excess deoxyhaemoglobin or meth-aemoglobin.1,2 Neonatal haemoglobinopathies can pose diagnostic challenges, due in part to the dynamic physiology of human haemoglobin in the perinatal period. In newborns, fetal haemoglobin (Hb F), which is composed of two α-chains and two γ-chains, is the predominant haemoglobin. Before birth, there is a switch from predominantly γ-chain to β-chain synthesis, which allows for the increased formation of adult haemoglobin (Hb A) comprising two α-chains and two β-chains. Mutations that affect any of these three globin chains can lead to haemoglobin variants manifesting clinically as cyanosis, but the presentation and natural history will vary depending on which globin chain is affected. Mutations in the γ-chain typically present in otherwise healthy newborns as early and transient cyanosis that eventually resolves as Hb F is replaced by Hb A.3–10
We report a case of Hb FM-Fort Ripley, a fetal M-haemoglobin variant3,4,11,12 that was diagnosed using next-generation sequencing (NGS). The case broadens the clinical description of this disorder and illustrates the utility of modern genetic tools to diagnose neonatal haemoglobin disorders that are difficult to establish using traditional biochemical assays.
CASE PRESENTATION
A full-term, 3.125 kg baby girl of Indian descent was noted to be cyanotic and unresponsive at 1 h of life. The prenatal course had been uneventful and the infant appeared healthy at birth. She was emergently intubated and transferred to the neonatal intensive care unit, where her initial arterial blood gases showed metabolic acidosis (pH 7.22, pCO2=32, base deficit=−13, lactate=17), which resolved over the next few hours. She was extubated to nasal continuous positive airway pressure after 1 day and quickly weaned to low-flow nasal cannula with supplemental oxygen. However, she remained visibly cyanotic without any other sign of respiratory distress, and continued to require supplemental oxygen throughout the first several weeks of life, to maintain oxygen saturation >85%.
The family history (figure 1A) revealed that a paternal cousin had neonatal cyanosis of unknown aetiology that resolved after the first few weeks of life without intervention. Haemoglobinopathy work up on both parents showed normal haemoglobin high-performance liquid chromatography (HPLC). The father had microcytosis and red blood cell (RBC) indices suggestive of an α-thalassaemia trait. The patient’s only sibling, a brother, had RBC indices suggestive of α-thalassaemia trait as well. Neither the sibling nor the father had a history of cyanosis.
Figure 1.
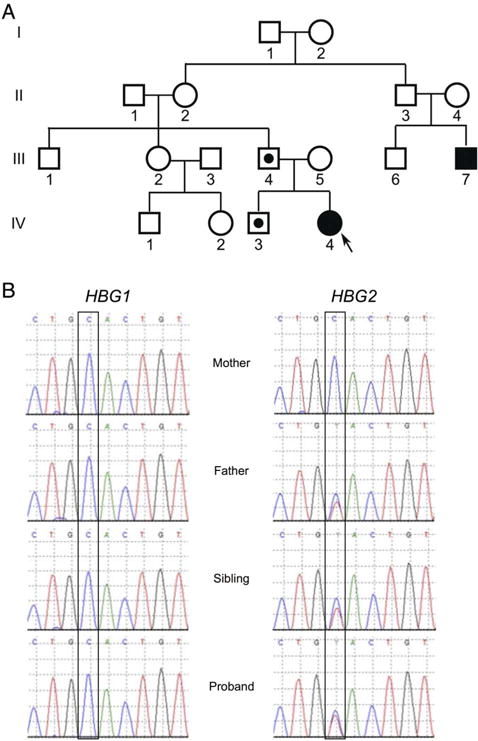
The proband has a paternally inherited mutation in HGB2 that results in Hb FM-Fort Ripley. (A) Family pedigree with IV-4 representing the proband. Black filled symbols represent individuals who presented with neonatal cyanosis. The father (III-4) and sibling (IV-3) of the proband have red blood cell indices suggestive of an α-thalassaemia trait, which is indicated by a star. (B) Chromatograms from the indicated individuals showing HBG1 and HBG2. Long-range PCR using primers from Crowley et al was performed followed by dideoxy sequencing.
INVESTIGATIONS
An extensive work up was undertaken to determine the cause of the patient’s persistent cyanosis. Chest radiographs, echocardiogram and head ultrasound were normal. Blood culture was negative and complete blood count revealed normal haemoglobin (15.1 g/dL), haematocrit (46.3%), mean corpuscular volume (113) and peripheral blood smear. Haemoglobinopathy was suspected based on discordance between pulse oximetry readings, which were consistently <90%, and the arterial partial pressure of oxygen (PaO2), which was >250 mm Hg (while on fraction of inspired oxygen, FiO2 of 0.7). Methaemoglobin level was normal (0.4–0.6%, obtained from two independent samples). Oxygen dissociation studies showed a normal sigmoidal dissociation curve. HPLC on day of life two revealed Hb A and Hb F in normal proportions for age. However, an unknown haemoglobin peak with a retention time of 4.6 min, comprising 1.9% of the total haemoglobin, was observed (figure 2A). The variant haemoglobin migrated near the Hb C position on alkaline electrophoresis and likely comigrated with another haemoglobin on citrate agar since a novel band was not seen (figure 2B). This unknown variant was virtually undetectable by day of life 69 (figure 2A).
Figure 2.
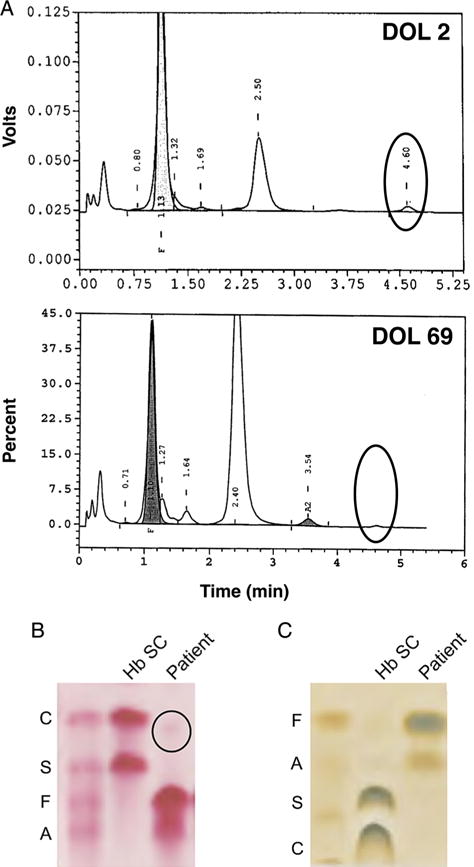
High-performance liquid chromatography (HPLC) analysis and haemoglobin electrophoresis identifing a putative haemoglobin variant. (A) HPLC retention plots showing day of life (DOL) 2 (top) and DOL 69 (bottom). An unidentified peak, representing 1.9% of total haemoglobin on DOL 2 but undetectable on DOL 69, is indicated. (B) A gel from cellulose acetate haemoglobin electrophoresis showing a sample corresponding to DOL 2. A faint band identified in the patient’s sample is indicated. (C) A gel from citrate agar electrophoresis showing a sample corresponding to DOL 2.
The paternal cousin’s history of neonatal cyanosis raised the possibility of an underlying genetic aetiology. Therefore, NGS was utilised to sequence all the globin genes from our patient. A paternally inherited mutation in HGB2 (c.277C>T) that results in a missense mutation (p.H93Y) was identified in our patient (figure 1B). This mutation results in Hb FM-Fort Ripley, one of the six known haemoglobin-M γ-globin variants associated with neonatal cyanosis.3,4,11 The patient’s sibling also inherited the mutation (Discussion).
DIFFERENTIAL DIAGNOSIS
Several fetal haemoglobinopathies that present with unexplained cyanosis have been described. The names, by convention, reflect the site where the variant was discovered:
-
▸
Hb F-Viseu
-
▸
Hb F-Cincinnati
-
▸
Hb FM-Osaka
-
▸
Hb F-Circleville
-
▸
Hb F-Toms River
-
▸
Hb FM-Fort Ripley
TREATMENT
No specific treatment was provided beyond supportive care. We gave consideration to a simple blood transfusion with adult donor blood, but by that point the patient’s cyanosis was resolving.
OUTCOME AND FOLLOW-UP
The patient was discharged home at 1-month of life with supplemental oxygen via nasal cannula, which was weaned over the next month. She is now healthy and thriving without recurrence of cyanosis.
DISCUSSION
Hb FM-Fort Ripley was first described in 1989, after a newborn presented with unexplained but transient cyanosis;3 a second case was described in 1992.4 The haemoglobinopathy results from H93Y mutation in the γ-chain, believed to alter the susceptibility of the abnormal haemoglobin to reduction by nicotinamide adenine dinucleotide reductase, leading to accumulation of haeme moieties bearing iron in the oxidised state (‘met’, Fe3+), which cannot effectively bind and release oxygen.13 This hypothesis is based on the phenotypes of other variant haemoglobins with similar active site amino acid substitutions and spectrophotometric analysis.4,14 The Hb FM-Fort Ripley mutation has an autosomal dominant inheritance pattern with incomplete penetrance,11 which is consistent with our patient’s family history.
Our case highlights the difficulty of diagnosing Hb FM-Fort Ripley using traditional laboratory tests. The clinical presentation of persistent cyanosis can be out of proportion to the very small fraction of variant haemoglobin detected by HPLC or electrophoresis, and the diagnosis missed by a ‘normal’ methaemoglobin spectrophotometric assay. Previous cases reported detecting only small amounts of an abnormal haemoglobin, between 4% and 9% of total haemoglobin.3,4 Similarly, we found only a small fraction of the variant haemoglobin (1.9%) by HPLC and electrophoresis. Co-oximeter measurement of methaemoglobin relies on detection of a characteristic 630–635 nm absorbance peak.13 This peak is present in acquired forms of methaemoglobinaemia (ie, wild type Hb A or F locked in the oxidised state due to a reducing enzyme deficiency or exposure to an oxidising toxin), but not always in congenital fetal methaemoglobinaemias (ie, due to a γ-chain mutation)15,16 such as Hb FM-Fort Ripley.
Others have suggested that Hb FM-Fort Ripley’s relative instability may impair in vitro detection,3 which—if true—could also have affected our methaemoglobin measurement. Alternatively, our readings may have been correct, and Hb FM-Fort Ripley may cause disease through a mechanism other than accumulation of methaemoglobin. Other authors have proposed an interaction in vivo between Hb FM-Fort Ripley and wild type adult haemoglobin that affects the oxygen affinity of the latter.3,4 Other fetal methaemoglobin variants have been shown to reduce co-operativity, which could amplify the effect of a small fraction of abnormal subunits.17
NGS did not exist when Hb FM-Fort Ripley was first described in the literature, and our case illustrates the utility and efficiency of employing this technology in the setting of rare haemoglobinopathies. Simultaneous sequencing of all of the globin genes allowed this case to reach a prompt and satisfying conclusion. Falling prices and increased insurance coverage are making such testing more accessible.18 We suggest, when faced with inconclusive results after routine haemoglobin testing in cases of neonatal cyanosis consistent with Hb FM disease, that NGS-based testing be utilised if possible.
Learning points.
-
▸
Fetal haemoglobinopathies affecting the γ-chain can present as unexplained neonatal cyanosis.
-
▸
The abnormal haemoglobin can be difficult to detect using traditional laboratory approaches.
-
▸
The natural history for these disorders is resolution within approximately the first month of life as fetal haemoglobin is replaced with adult haemoglobin.
-
▸
Next-generation sequencing may be the most efficient approach to diagnosing a fetal haemoglobinopathy, when the clinical data support the diagnosis.
Acknowledgments
The authors are grateful to Dr Peter Nagy, who provided valuable guidance interpreting laboratory data.
Footnotes
Contributors TAH provided direct care to the patient, interpreted the data, drafted the manuscript, drafted figures and approved the final version. EMH provided direct care to the patient, interpreted the data, revised the manuscript and approved the final version. SNW performed genetic analyses and interpreted the data, drafted figures, revised the manuscript and approved the final version. ROF performed and interpreted gel electrophoresis and HPLC, revised the manuscript and approved the final version. RS provided direct care to the patient, interpreted the data, helped conceive and revised the manuscript, and approved the final version. MTL provided direct care to the patient, interpreted the data, helped conceive of and revised the manuscript, and approved the final version.
Competing interests None declared.
Patient consent Obtained.
Provenance and peer review Not commissioned; externally peer reviewed.
References
- 1.Steinhorn RH. Evaluation and management of the cyanotic neonate. Clin Pediatr Emerg Med. 2008;9:169–75. doi: 10.1016/j.cpem.2008.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sarkar S, Rosenkrantz TS. Neonatal polycythemia and hyperviscosity. Semin Fetal Neonatal Med. 2008;13:248–55. doi: 10.1016/j.siny.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 3.Priest JR, Watterson J, Jones RT, et al. Mutant fetal hemoglobin causing cyanosis in a newborn. Pediatrics. 1989;83:734–6. [PubMed] [Google Scholar]
- 4.Molchanova TP, Wilson JB, Gu LH, et al. A Second Observation of the Fetal Methehoglobin Variant HB F-M-Fort Ripley or α 2 G γ 2 92 (F8)HIS→TYR. Hemoglobin. 1992;16:389–98. doi: 10.3109/03630269209005690. [DOI] [PubMed] [Google Scholar]
- 5.Kohli Kumar M, Zwerdling T, Rucknagel DL. Hemoglobin F-Cincinnati, α2Gγ2 41(C7) Phe → Ser in a newborn with cyanosis. Am J Hematol. 1995;49:43–7. doi: 10.1002/ajh.2830490108. [DOI] [PubMed] [Google Scholar]
- 6.Dainer E, Shell R, Miller R, et al. Neonatal cyanosis due to a novel fetal hemoglobin: Hb F-Circleville [Ggamma63(E7)His—>Leu, CAT>CTT] Hemoglobin. 2008;32:596–600. doi: 10.1080/03630260802507915. [DOI] [PubMed] [Google Scholar]
- 7.Crowley MA, Mollan TL, Abdulmalik OY, et al. A hemoglobin variant associated with neonatal cyanosis and anemia. N Engl J Med. 2011;364:1837–43. doi: 10.1056/NEJMoa1013579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Viana MB, Belisário AR. De novo alpha 2 hemoglobin gene (HBA2) mutation in a child with hemoglobin M Iwate and symptomatic methemoglobinemia since birth. Rev Bras Hematol Hemoter. 2014;36:230–4. doi: 10.1016/j.bjhh.2014.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zimmermann-Baer U, Capalo R, Dutly F, et al. Neonatal cyanosis due to a new (G) γ-globin variant causing low oxygen affinity: Hb F-Sarajevo [(G)γ102(G4)Asn→Thr, AAC>ACC] Hemoglobin. 2012;36:109–13. doi: 10.3109/03630269.2012.655872. [DOI] [PubMed] [Google Scholar]
- 10.Bento C, Magalhães Maia T, Carvalhais I, et al. Transient neonatal cyanosis associated with a new Hb F variant: Hb F viseu. J Pediatr Hematol Oncol. 2013;35:e77–80. doi: 10.1097/MPH.0b013e3182667be3. [DOI] [PubMed] [Google Scholar]
- 11.Hain R, Chitayat D, Cooper R, et al. Hb FM-Fort Ripley: confirmation of autosormal dominant inheritance and diagnosis by PCR and direct nucleotide sequencing. Hum Mutat. 1994;3:239–42. doi: 10.1002/humu.1380030310. [DOI] [PubMed] [Google Scholar]
- 12.Glader BE. Hemoglobin Fm-Fort-Ripley—another lesson from the neonate. Pediatrics. 1989;83:792–3. [PubMed] [Google Scholar]
- 13.Wright RO, Lewander WJ, Woolf AD. Methemoglobinemia: etiology, pharmacology, and clinical management. Ann Emerg Med. 1999;34:646–56. doi: 10.1016/s0196-0644(99)70167-8. [DOI] [PubMed] [Google Scholar]
- 14.Gerald PS, Efron ML. Chemical studies of several varieties of Hb M. Proc Natl Acad Sci USA. 1961;47:1758–67. doi: 10.1073/pnas.47.11.1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Speakman ED, Boyd JC, Bruns DE. Measurement of methemoglobin in neonatal samples containing fetal hemoglobin. Clin Chem. 1995;41:458–61. [PubMed] [Google Scholar]
- 16.Haymond S, Cariappa R, Eby CS, et al. Laboratory assessment of oxygenation in methemoglobinemia. 2005;51:434–44. doi: 10.1373/clinchem.2004.035154. [DOI] [PubMed] [Google Scholar]
- 17.Shih TB, Imai K, Tyuma I, et al. Further-studies on the functional-properties of hemoglobin-M Hyde-Park. Hemoglobin. 1980;4:125–47. doi: 10.3109/03630268009042380. [DOI] [PubMed] [Google Scholar]
- 18.Bick D, Dimmock D. Whole exome and whole genome sequencing. Curr Opin Pediatr. 2011;23:594–600. doi: 10.1097/MOP.0b013e32834b20ec. [DOI] [PubMed] [Google Scholar]