

Abstract
Transcription factors (TFs) and microRNAs (miRNAs) regulate gene expression: TFs by influencing messenger RNA (mRNA) transcription and miRNAs by influencing mRNA translation and transcript degradation. Additionally, miRNAs and TFs alter each other’s expression, making it difficult to ascertain the effect either one has on target gene (TG) expression. In this investigation, we use a two-way interaction model with the TF and miRNA as independent variables to investigate whether miRNAs and TFs work together to influence TG expression levels in colon cancer subjects. We utilized known Transcription factor binding sites (TFBSs) and validated miRNA targets to determine potential miRNA-TF-TG interactions, restricting interactions to those with a TF previously associated with altered risk of colorectal cancer death. We analyzed interactions using normal colonic mucosa expression as well as differential expression, which is measured as colonic carcinoma expression minus normal colonic mucosa expression. We analyzed 3518 miRNA-TF-TG triplets using normal mucosa expression and 617 triplets using differential expression. Normal colonic RNA-Seq data were available for 168 individuals; of these, 159 also had carcinoma RNA-Seq data. Thirteen unique miRNA-TF-TG interactions, comprising six miRNAs, four TFs, and 11 TGs, were statistically significant after adjustment for multiple comparisons in normal colonic mucosa, and 14 unique miRNA-TF-TG interactions, comprising two miRNAs, two TFs, and 13 TGs, were found for carcinoma-normal differential expression. Our results show that TG expression is influenced by both miRNAs as well as TFs, and the influence of one regulator impacts the effect of the other on the shared TG expression.
Keywords: miRNA, transcription factor, target gene, colon cancer
1 Introduction
Gene expression regulation encompasses a myriad of biological molecules and processes working together to influence expression levels and eventual protein production. Transcription factors (TFs) regulate messenger RNA (mRNA) transcription by binding to cis-regulatory DNA elements called Transcription factor binding sites (TFBSs), and either enhancing or repressing mRNA transcription1. MicroRNAs (miRNAs) work downstream of TFs, regulating mRNA expression post-transcriptionally via translational repression and mRNA destabilization and subsequent transcript degradation2,3. This is accomplished by 6–8 nucleotides in the 5’ UTR of the miRNA, known as the seed region, binding to the 3’ UTR region of the mRNA2,4,5. An individual miRNA may regulate a multitude of mRNAs, and a given mRNA may be regulated by many different miRNAs6. Additionally, TFs influence miRNA expression, and miRNAs may repress TF expression7,8. The complex relationships between miRNAs, TFs, and target genes (TG) are known as feedback loops (FBLs) and feed-forward loops (FFLs)4,7. Typically, FBLs occur when a TF activates or represses a miRNA, which in turn represses the TF; the miRNA and TF each regulate independent sets of TGs9. FFLs are those where a regulator, such as a TF, controls the expression of a specific TG both directly, through promoting or enhancing its transcription, as well as indirectly through another regulator, such as a miRNA that also regulates that TG9. FFLs and FBLs are typically described as either ‘coherent’ or ‘incoherent’. FBLs are considered coherent when the TF and miRNA have the same effect (repression) on one another, and incoherent when the TF increases miRNA transcription, and the miRNA in turn represses the TF4. FFLs are considered coherent when the TF and indirect effector (in this instance a miRNA) have the same effect on the TG; when the TF and miRNA have opposing effects on the TG, this relationship is classified as an incoherent FFL9. FFLs illustrate the complexity of networks contributing to mRNA expression levels. Even in FBLs, miRNAs and TFs are highly connected, as the direct regulation of the TF by the miRNA has an indirect effect on all of the target genes regulated by the TF1,8. The likelihood that observed levels of mRNA expression are the result of a combination of regulatory effects7 makes only identifying specific miRNA-TG or TF-TG interactions difficult, and possibly unwarranted.
It is probable that biological interactions between miRNAs, TFs, and TGs as they occur in vivo may be better modeled when considering these molecules’ activity concurrently rather than identifying simple interactions between specific TFs and TGs, or miRNAs and TGs. Additionally, little is known about the range of action the majority of miRNAs have. In order to expand the existing information on miRNA involvement in the carcinogenic process, a large-scale, discovery approach is needed to investigate jointly the global miRNA and mRNA activity. In this study, we use a two-way interaction model with the TF and miRNA as independent variables to investigate whether miRNAs and TFs work together in a FFL fashion to influence TG expression levels in colon cancer subjects. We hypothesize that TG expression is the product of combined TF and miRNA influence.
2 Materials and Methods
2.1 Study Participants
Study participants were recruited as part of two population-based case-control studies that included all incident colon and rectal cancer between 30 to 79 years of age who resided in Utah or were of the Kaiser Permanente Medical Care Program (KPMCP) in Northern California (Table 1). Participants were non-Hispanic white (NHW), Hispanic, or African American for the colon cancer study and also included participants of Asian race for the rectal portion of the study10,11. Case diagnosis was verified by tumor registry data as a first primary adenocarcinoma of the colon and were diagnosed between October 1991 and September 1994 and for rectal were diagnosed between May 1997 and May 2001. Detailed study methods have been described12. All participants signed an informed consent. The Institutional Review Boards at the University of Utah and at KPMCP approved the study.
Table 1.
Description of study participants.
| Site | N1 | % | |
|---|---|---|---|
| Proximal | 81 | 48.2 | |
| Distal | 87 | 51.8 | |
| Sex | |||
| Male | 93 | 55.4 | |
| Female | 75 | 44.6 | |
| Age | |||
| Mean (SD) | 65.1 | 10.1 | |
| Race | |||
| Non-Hispanic White | 116 | 69.1 | |
| Hispanic | 11 | 6.6 | |
| African American | 7 | 4.2 | |
| Unknown | 34 | 20.2 | |
| AJCC Stage | |||
| 1 | 39 | 23.4 | |
| 2 | 53 | 31.7 | |
| 3 | 53 | 31.7 | |
| 4 | 22 | 13.2 | |
| Tumor Phenotype | |||
| TP53 mutated | 70 | 41.7 | |
| KRAS mutated | 48 | 28.6 | |
| BRAF mutated | 21 | 13.3 | |
| CIMP High | 45 | 26.8 | |
| MSI | 29 | 17.3 | |
This table describes the participants that had normal colonic RNA-Seq data available; 159 cases from this population had colonic carcinoma data, which was used to perform the analyses for differential tissue expression.
2.2 RNA Processing
Formalin-fixed paraffin embedded (FFPE) tissue from the initial biopsy or surgery was used to extract RNA. Carcinoma tissue and adjacent normal mucosa were used to make RNA. Cells were dissected from 1–4 sequential sections on aniline blue stained slides using an H&E slide for reference. Total RNA was extracted, isolated, and purified using the RecoverAll Total Nucleic Acid isolation kit (Ambion); RNA yields were determined using a NanoDrop spectrophotometer. RNA was subsequently used for both miRNA and mRNA analyses.
2.3 miRNA: Microarray Analysis
The Agilent Human miRNA Microarray V19.0 was used. The microarray contains probes for 2006 unique human miRNAs as described previously. Data were required to pass stringent QC parameters established by Agilent that included tests for excessive background fluorescence, excessive variation among probe sequence replicates on the array, and measures of the total gene signal on the array to assess low signal. If samples failed to meet quality standards for any of these parameters, the sample was re-labeled, hybridized to arrays, and re-scanned. If a sample failed QC assessment a second time, the sample was deemed to be of poor quality and the sample was excluded from analysis. Our previous analysis has shown that the repeatability associated with this microarray was extremely high (r=0.98)12, and that comparison of miRNA expression levels obtained from the Agilent microarray to those obtained from qPCR had an agreement of 100% in terms of directionality of findings and that the fold change calculated for the miRNA expression difference between carcinoma and normal colonic mucosa was almost identical13.
To normalize differences in miRNA expression that could be attributed to the array, amount of RNA, location on array, or factors that could erroneously influence miRNA expression levels, total gene signal was normalized by multiplying each sample by a scaling factor which was the median of the 75th percentiles of all the samples divided by the individual 75th percentile of each sample14.
2.4 mRNA: RNA-Seq Library Construction and Processing
One hundred and eighty-seven samples were originally successfully run for normal colonic mucosa and 169 were run for colonic carcinoma tissue; in total 209 subjects had RNA-Seq successfully performed for either colonic carcinoma or normal colonic mucosa. These samples were taken from the study subjects used for miRNA analysis and were extracted, isolated and purified as previously described15. RNA library construction was done with the Illumina TruSeq Stranded Total RNA Sample Preparation Kit with Ribo-Zero. The samples were then fragmented and primed for cDNA synthesis, adapters were then ligated onto the cDNA, and the resulting samples were then amplified using PCR; the amplified library was then purified using Agencount AMPure XP beads. A more detailed description of the methods can be found in our previous work16. Illumina TruSeq v3 single read flow cell and a 50 cycle single-read sequence run was performed on an Illumina HiSeq instrument. Reads were aligned to a sequence database containing the human genome (build GRCh37/hg19, February 2009 from genome.ucsc.edu) and alignment was performed using novoalign v2.08.01. Counts were calculated for each exon and UTR of the genes using a list of gene coordinates obtained from http://genome.ucsc.edu. Total gene counts were determined. We dropped genes that were not expressed in our data or had limited expression for the majority of samples16.
2.5 Statistical Analysis
Our statistical analysis builds on our previous analysis of TF and miRNA pairs and survival in 168 colon cancer subjects17. In that analysis, we assessed 154 TFs with survival and identified 30 TFs with significant associations17. In this study, as a means of focusing our investigation on interactions that involve biologically important TFs, we restrict the analysis to miRNA-TF-TG triplets that include only these 30 TFs. Of the 30 TFs, 27 were associated with expression of 65 miRNAs in normal colonic mucosa, resulting in 719 TF-miRNA pairs. In this analysis, we further explored these TFs and miRNAs interactions in both normal colonic mucosa expression and carcinoma expression minus normal mucosa expression, which is hereafter referred to as differential expression, to test our hypothesis that TF and miRNAs work together to alter TG expression. One hundred and sixty eight subjects had normal colonic RNA-Seq data and miRNA data available that passed quality control. Of these, 159 subjects also had paired colonic carcinoma RNA-Seq and miRNA data.
2.6 miRNA-TF-TG Triplet Identification
The gene assembly used was GRCh37/hg19 for all coordinates. The UCSC Table Browser18 was utilized to obtain TFBSs as well at match ensembl IDs to known gene names. Determining triplets of TFs, miRNAs, and TGs required multiple steps and various databases. First, TFBS coordinates were obtained using the ‘Regulation’ group, the ‘Txn Factor ChIP’ table, and the ‘wgEncodeTegTfbsClusteredV3’ table19. This was then compared to coordinates of primary-microRNAs (pri-miRNAs), downloaded from miRBase v19 archived files. TF-miRNA pairs were made when a TFBS occurred +/− 300 base pairs (bps) from the start or end of the pri-miRNA transcript. This criterion was chosen based on a study done by Koudritsky and Domany, who found a concentrated proportion of TFBSs occur 300 bps upstream of the transcription start site, leading to the determination of a ‘proximal region’ as the interval +/−300 bps straddling the TSS for a given gene20. Additionally, enhancers, including TFBSs, are known to occur downstream of and within the gene itself21,22 and this prompted us to apply the same +/−300 bps to the end of the pri-miRNA transcript as well. Pri-miRNAs were then matched to mature miRNAs, using the same coordinate file from miRBase. Using miRTarBase v623, we identified target genes for the mature miRNAs whose corresponding pri-miRNA fell within a TFBS. These genes have been validated as targets for these miRNAs through different laboratory methods; all experimentally validated targets, identified by any experimental method, were used. Finally, TFBSs were downloaded from UCSC using the same table as the first step, however it was intersected with ‘Genes and Gene Predictions’ group ‘knownGenes’ table to get gene names for corresponding transcript IDs. As these results are in the ensembl ID format, a second table, ‘ensembleToGeneName’ was used to find the gene names for these genes. The TGs were then paired up to TFs if a TFBS occurred +/−300 bps from a TG start or end. Triplets were defined where a given TFBS overlapped with a pri-miRNA start site, whose mature miRNA end product has as a validated target a mRNA, whose transcription start or end also overlapped with the same TF. This was meant to represent TF regulation of the miRNA and TG, and miRNA regulation of the TG. In total, we identified 10,170 potential miRNA-TF-TG triplets meeting these criteria. These triplets model FFLs, in that they include a TF that binds near a TG as well as a miRNA, and a miRNA that has been shown to target the same TG. All coordinate matching was done using R scripting, and triplet determination was done using SQL commands.
2.7 Dataset Determination
We utilized identified miRNA-TF-TG triplets, and we restricted the TGs to those with a minimum fold change in expression between carcinoma and normal mucosa of 50% as had been done previously to the TFs and miRNAs. Using this list of triplets, we restricted the analysis to those 30 TFs and 65 miRNAs previously identified with survival. This resulted in 5707 triplets (i.e. miRNA-TF-TG) that included 11TFs, 17 miRNAs, 2901 TGs in normal mucosa expression and 1267 triplets that included 8 TFs, 11 miRNAs, and 955 TGs for differential expression.
2.8 TF-TG Linear Regression Analysis
We fit least squares linear regression model between TF-TG using the reads per kilobase per million (RPKMs) expression levels. This provided a main-effect reference association for TGs, and served to limit the associations analyzed in the interaction analysis. P-values were generated using the bootstrap method to generate a distribution of 10,000 F statistics derived by resampling the residuals with replacement from the null hypothesis of no association between TF and TG expression using the ‘boot’ package in R24. The linear models were adjusted for age, center and sex. Multiple testing corrections were made using an false discovery rate of 0.05 or less25. This analysis determined 11 TFs and 1983 TGs in normal mucosa expression, and 7 TFs and 483 TGs in differential expression, were significantly associated.
2.9 Interaction Analysis
Finally, we examined the impact of interaction between the TF and miRNA upon the TG from the triplets that remain significant to this point (3518 triplets in normal mucosa expression, 617 triplets in differential expression). We used the bootstrap method to evaluate the statistical impact of the interaction term in the linear model also containing the TF and miRNA main effects. Thirteen triplets (6 miRNAs, 4 TFs, 11 TGs) in normal mucosa expression and 14 triplets (2 miRNAs, 2 TFs, 13 TGs) in differential expression had significant interactions between the TF and miRNA. We transformed the miRNA, TF and TG to standard normal to calculated standardized beta coefficients in order to compare results across triplets after FDR correction.
2.10 miRNA-TF Linear Regression
Previously, we performed a linear regression between TFs and miRNAs in normal colonic mucosa. In this analysis, we include the relevant results for normal colonic expression from our previous study, and we performed linear regressions between the same TFs and miRNAs, using the method described above, for differential expression. The beta coefficients derived from these analyses are labeled as ‘miRNA-TF Beta Co.’ in Tables 2 and 3. This was done to determine the effect of one regulator upon the other, as these associations are not detected in the interaction analysis.
Table 2.
Significant microRNA (miRNA)-Transcription Factor(TF)-Target Gene (TG) interactions identified in normal colonic mucosa.
| miRNA | Mean Expression | miRNA Beta Co. |
TF | Mean Expression | TF Beta Co. |
TG | Mean Expression | miRNA*TF (interaction) Beta Co.3 |
miRNA- TF Beta Co.4 |
P-Value | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Tumor1 | Normal | Tumor1 | Normal | Tumor1 | Normal | Raw | FDR | |||||||
| hsa-miR-1258 | 1.75 | 3.92 | 0.03 | ZNF2632 | 95.39 | 44.41 | 0.68 | CPM2 | 33.18 | 122.94 | 0.28 | −0.23 | <.0001 | 0.0177 |
| hsa-miR-145-5p | 134.57 | 207.91 | 0.18 | CTCF | 135.97 | 72.45 | 0.72 | IGF1R2 | 365.93 | 187.47 | 0.15 | 0.21 | 0.0003 | 0.0228 |
| hsa-miR-145-5p | 134.57 | 207.91 | 0.18 | ELF1 | 250.60 | 119.97 | 0.77 | IGF1R2 | 365.93 | 187.47 | 0.15 | 0.21 | <.0001 | 0.0177 |
| hsa-miR-150-5p | 15.27 | 40.22 | −0.17 | CTCF | 135.97 | 72.45 | 0.75 | VEGFA2 | 971.21 | 369.26 | −0.20 | 0.24 | 0.0008 | 0.0354 |
| hsa-miR-150-5p | 15.27 | 40.22 | −0.16 | ELF1 | 250.60 | 119.97 | 0.81 | VEGFA2 | 971.21 | 369.26 | −0.23 | 0.22 | <.0001 | 0.0177 |
| hsa-miR-193b-3p | 8.76 | 5.08 | −0.12 | CTCF | 135.97 | 72.45 | 0.70 | RRM22 | 172.47 | 37.43 | −0.28 | 0.24 | 0.0003 | 0.0228 |
| hsa-miR-193b-3p | 8.76 | 5.08 | −0.13 | RBBP5 | 79.50 | 39.15 | 0.83 | HELLS | 268.00 | 82.11 | −0.26 | 0.27 | 0.0002 | 0.0212 |
| hsa-miR-193b-3p | 8.76 | 5.08 | −0.07 | RBBP5 | 79.50 | 39.15 | 0.94 | MAPK1IP1L2 | 270.04 | 167.11 | −0.14 | 0.27 | 0.0002 | 0.0212 |
| hsa-miR-193b-3p | 8.76 | 5.08 | −0.12 | RBBP5 | 79.50 | 39.15 | 0.74 | RAD512 | 20.32 | 9.74 | −0.23 | 0.27 | 0.0007 | 0.0338 |
| hsa-miR-193b-3p | 8.76 | 5.08 | −0.04 | RBBP5 | 79.50 | 39.15 | 0.82 | RCC1 | 137.97 | 46.22 | −0.21 | 0.27 | 0.0006 | 0.0319 |
| hsa-miR-193b-3p | 8.76 | 5.08 | −0.05 | RBBP5 | 79.50 | 39.15 | 0.87 | TJP2 | 412.82 | 182.83 | −0.19 | 0.27 | 0.0004 | 0.0265 |
| hsa-miR-330-3p | 2.71 | 5.56 | −0.08 | ELF12 | 250.60 | 119.97 | 0.77 | PTPLAD12 | 216.84 | 55.26 | −0.19 | −0.19 | 0.0005 | 0.0295 |
| hsa-miR-4469 | 0.97 | 2.40 | −0.09 | ELF12 | 250.60 | 119.97 | 0.83 | MRPS272 | 140.87 | 57.30 | −0.14 | −0.26 | 0.0009 | 0.0368 |
This table has been split into four groupings based on the directionality of the different beta coefficients. This has been done to assist in interpretation and following the discussion.
Tumor expression is shown for additional information only, and is not used in the calculation of any results in this table.
A seed match was identified between this TG and the miRNA.
This beta coefficient was generated from the two-way interaction analysis.
This beta coefficient was calculated in a separate linear regression between the miRNA and TF.
Table 3.
Significant microRNA (miRNA)-Transcription Factor (TF)-Target Gene (TG) interactions identified in differential colonic expression between colonic carcinoma tissue and normal colonic mucosa.
| miRNA | Mean Expression | miRNA Beta Co. |
TF | Mean Expression | TF Beta Co. |
TG | Mean Expression | miRNA*TF (interaction) Beta Co.1 |
miRNA- TF Beta Co.2 |
P-Value | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Tumor | Norma l |
Tumor | Normal | Tumor | Normal | Raw | FDR | |||||||
| hsa-miR-23a-3p | 175.27 | 84.70 | −0.11 | ELF1 | 250.60 | 119.97 | 0.44 | ADAM283 | 70.82 | 180.84 | 0.29 | 0.23 | 0.0004 | 0.0224 |
| hsa-miR-23a-3p | 175.27 | 84.70 | 0.01 | ELF1 | 250.60 | 119.97 | 0.72 | CNOT63 | 190.11 | 91.72 | −0.22 | 0.23 | 0.0002 | 0.0224 |
| hsa-miR-23a-3p | 175.27 | 84.70 | 0.02 | ELF1 | 250.60 | 119.97 | 0.77 | GNB2 | 182.96 | 95.79 | −0.20 | 0.23 | 0.0003 | 0.0224 |
| hsa-miR-23a-3p | 175.27 | 84.70 | 0.06 | ELF1 | 250.60 | 119.97 | 0.70 | IMPDH2 | 318.23 | 111.06 | −0.23 | 0.23 | 0.0002 | 0.0224 |
| hsa-miR-23a-3p | 175.27 | 84.70 | 0.10 | ELF1 | 250.60 | 119.97 | 0.66 | LDHA | 781.68 | 233.10 | −0.21 | 0.23 | 0.0006 | 0.0308 |
| hsa-miR-23a-3p | 175.27 | 84.70 | 0.02 | ELF1 | 250.60 | 119.97 | 0.80 | MCFD23 | 180.53 | 72.21 | −0.17 | 0.23 | 0.0007 | 0.0332 |
| hsa-miR-23a-3p | 175.27 | 84.70 | 0.08 | ELF1 | 250.60 | 119.97 | 0.57 | MYC | 330.10 | 68.27 | −0.28 | 0.23 | <.0001 | 0.0224 |
| hsa-miR-23a-3p | 175.27 | 84.70 | 0.03 | ELF1 | 250.60 | 119.97 | 0.75 | PDIA63 | 386.70 | 133.67 | −0.19 | 0.23 | 0.0004 | 0.0224 |
| hsa-miR-23a-3p | 175.27 | 84.70 | 0.08 | GABPA | 56.95 | 31.56 | 0.47 | MYC | 330.10 | 68.27 | −0.27 | 0.24 | 0.0003 | 0.0224 |
| hsa-miR-23a-3p | 175.27 | 84.70 | −0.03 | ELF1 | 250.60 | 119.97 | 0.71 | CCT53 | 249.64 | 82.36 | −0.25 | 0.23 | <.0001 | 0.0224 |
| hsa-miR-23a-3p | 175.27 | 84.70 | −0.001 | ELF1 | 250.60 | 119.97 | 0.69 | GMPS3 | 188.58 | 61.07 | −0.22 | 0.23 | 0.0002 | 0.0224 |
| hsa-miR-23a-3p | 175.27 | 84.70 | −0.05 | ELF1 | 250.60 | 119.97 | 0.62 | NACC13 | 152.39 | 73.87 | −0.23 | 0.23 | 0.0004 | 0.0224 |
| hsa-miR-23a-3p | 175.27 | 84.70 | −0.04 | ELF1 | 250.60 | 119.97 | 0.74 | SSRP13 | 234.90 | 80.96 | −0.22 | 0.23 | 0.0003 | 0.0224 |
| hsa-miR-4469 | 0.97 | 2.40 | 0.05 | ELF13 | 250.60 | 119.97 | 0.60 | TMEM170B3 | 28.31 | 16.98 | −0.24 | −0.23 | 0.0008 | 0.0353 |
This table has been split into four groupings based on the directionality of the different beta coefficients. This has been done to assist in interpretation and following the discussion.
This beta coefficient was generated from the two-way interaction analysis.
This beta coefficient was calculated in a separate linear regression between the miRNA and TF.
A seed match was identified between this TG and the miRNA.
2.11 Bioinformatics Analysis: Seed Pairing and Functional Analysis
To assist in determining directionality of interactions, we analyzed the 3’ UTRs of mRNAs for all TGs as well as TFs with negative miRNA-TF-beta coefficients with 5’ miRNA seed sequences for matches. While our RNA-Seq data is aligned to the hg19 (GRCh37), as we utilized miRTarBase to identify validated miRNA-TG interactions, and this repository includes various experiments spanning years, we evaluated mRNA FASTA sequences for both the GRCh38 and GRCh37 alignments. A more detailed description of this process can be found in our previous work26. Ensembl was used to identify biological processes identified by the Gene Ontology (GO). Cytoscape was used to visualize miRNA-TF-TG interactions in normal colonic mucosa and differential expression (Figures 1 and 2). We used the miRNA-TF-beta coefficient to describe the relationship between miRNAs and TFs, the TF-beta coefficient to describe the relationship between TFs and TGs, and the miRNA-beta coefficient to describe the relationship between miRNAs and TGs. MiRNAs are shown as gray squares, TFs are shown as dark blue circles, and TGs are shown as light blue triangles. Positive beta coefficients are denoted with green arrows (→), while negative beta coefficients are denoted with red stops (-|). In such circumstances where a positive miRNA-beta coefficient as well as a seed match was identified between a miRNA and TG, we have shown this interaction with a red stop. These instances are clearly noted in Tables 2 and 3 with footnotes. GO biological processes terms were identified using Ensembl’s BioMart, with the GRCh37 assembly. To identify pathways that might be more meaningful to this set of genes, only pathways that were mapped to more than one gene were included.
Fig 1. MiRNA-TF-TG Interactions in Normal Colonic Mucosa.
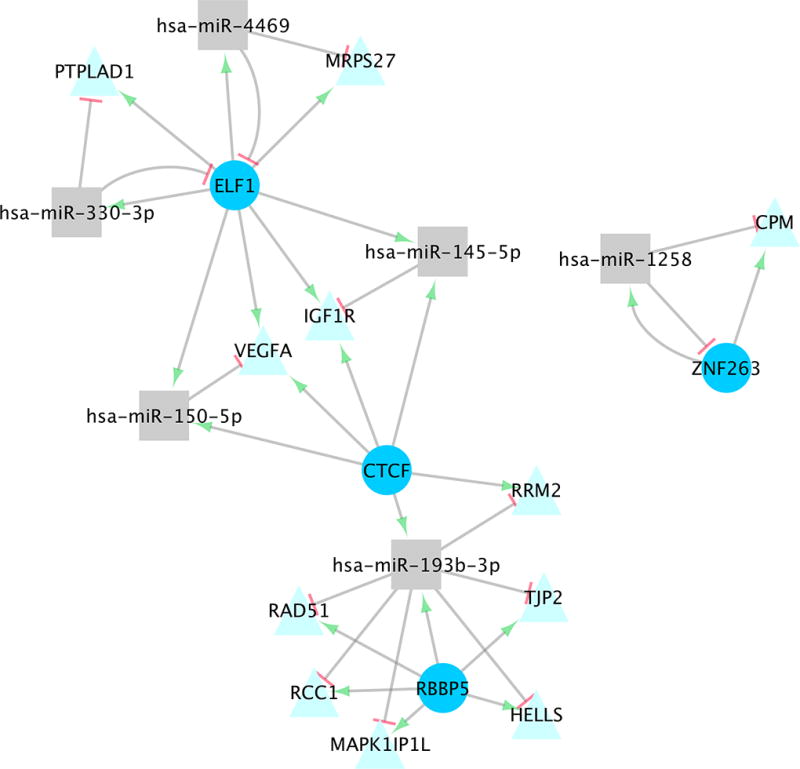
Significant miRNA-TF-TG interactions identified for normal colonic mucosa expression.
Fig 2. MiRNA-TF-TG Interactions Altered Between Colonic Carcinoma and Normal Colonic Mucosa.
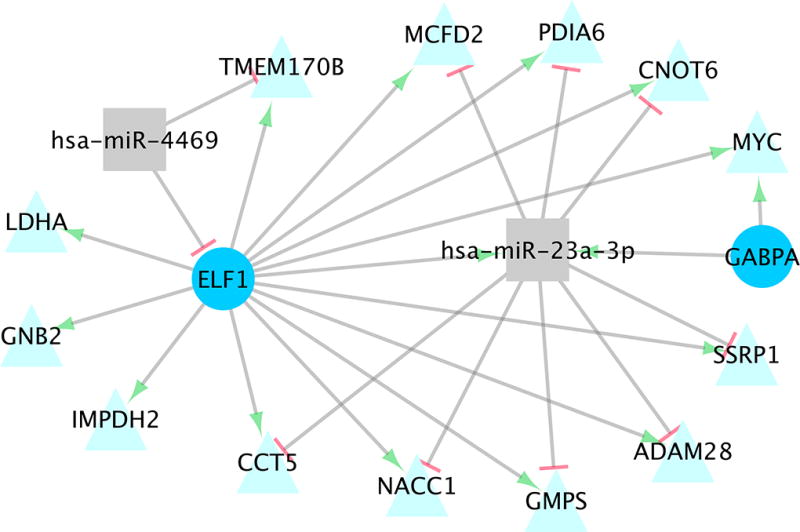
Significant miRNA-TF-TG interactions identified for differential expression between colonic carcinoma tissue and normal colonic mucosa.
3 Results
The study population is described in Table 1. The mean age of the study population was 65.1 years. A little over half, 55.4%, of the population was male; 44.6% was female. The majority of the population was NHW, 6.6% were Hispanic and 4.2% were African American. About 20% of the population had unknown race; 33 of 34 cases in this category were not interviewed. Cases comprised 48.2% proximal tumors and 51.8% distal tumors; 41.7% of tumors were TP53 mutated, 28.6 were KRAS mutated, 26.8 were CIMP-High, 17.3% were Microsatellite Unstable (MSI) and 13.3% were BRAF mutated. Almost a quarter of cases, 23.4%, were classified as AJCC stage 1, 31.7% as stage 2, 31.7% as stage 3 and 13.2% as stage.
Thirteen unique miRNA-TF-TG interactions were statistically significant after adjustment for multiple comparisons in normal colonic mucosa; these interactions comprised six miRNAs, four TFs, and 11 TGs (Table 2, Figure 1). Three of these interactions, which included miRNAs hsa-miR-1258 and 145-5p, had positive interaction-betas, indicating that as TF expression increased, the relationship between the miRNA and TG became more positive, meaning that changes in expression were in the same direction. The remaining eight interactions, comprising miRNAs hsa-miR-150-5p, 193b-5p, 330-3p, 4469, had negative interaction-betas, indicating that as TF expression increased, the relationship between the miRNA and TG became more negative. These four miRNAs also had negative beta coefficients between TGs and miRNAs, reflecting an inverse relationship between the miRNA and TG. All of the interactions had positive TF betas, reflecting a potential transcription activation of the TG by the TF. Eight of the 11 miRNA-TF/TG interactions with negative beta coefficients in normal colonic mucosa that were tested for seed matches had a confirmed match (Table 2).
There were 14 unique miRNA-TF-TG interactions found for differential expression, comprising two miRNAs, two TFs, and 13 TGs (Table 3, Figure 2). Thirteen of these interactions, including miRNAs 23a-3p and 4469, had negative interaction-beta coefficients, and one, between hsa-miR-23a-3p, ELF1 (TF), and ADAM28 (TG) had a positive interaction-beta coefficient. A negative miRNA-beta coefficient was found between hsa-miR-23a-3p and ADAM28, CCT5, GMPS, NACC1, and SSRP1; all other interactions had positive a miRNA-beta coefficient. A negative miRNA-TF-beta coefficient also was seen between hsa-miR-4469 and ELF1 (TF). All five of the miRNA-TF and miRNA-TG pairs tested for seed matches had a confirmed match using both alignments (Table 3).
4 Discussion
Thirteen unique miRNA-TF-TG interactions were significantly associated in normal colonic mucosa, and 14 interactions were significantly associated for differential expression between colonic carcinoma tissue and normal colonic mucosa. MiRNAs associated with TG and TF expression in normal colonic mucosa were distinct from those associated with TG and TF differential expression. Additionally, the TGs associated with miRNAs in normal colonic mucosa were unique from those associated with differential miRNA expression.
A majority of GO biological processes represented by the genes associated with normal and differential miRNA expression were the same. Of the 21 biological processes corresponding with the TGs associated with normal colonic miRNA expression and the 26 biological processes corresponding with the TGs associated with differential miRNA expression, 17 were in common between both sets of processes (Table 4). These pathways included DNA transcription and repair, process involving cell proliferation, and metabolic processes among others. This illustrates the complexity of gene regulation, and how the same cellular responses can be regulated by discrete groups of TGs, which are in turn influenced by various effector molecules such as miRNAs and TFs.
Table 4.
Biological Pathways regulated by Target Genes (TGs) participating in significant miRNA-TF-TG interactions in normal colonic mucosa and differential colonic mucosa.
| Normal TG | Differential TG | Biological Pathway1 |
|---|---|---|
| CCT5, MCFD2, PDIA6 | cellular protein metabolic process | |
| RAD51 | MYC | cellular response to DNA damage stimulus |
| RAD51 | SSRP1 | DNA repair |
| RRM2 | SSRP1 | DNA replication |
| GNB2, MYC | energy reserve metabolic process | |
| RCC1, RRM2 | G1/S transition of mitotic cell cycle | |
| CNOT6, MYC, SSRP1 | gene expression | |
| GMPS, IMPDH2 | GMP biosynthetic process | |
| HELLS | IMPDH2 | lymphocyte proliferation |
| HELLS, RCC1 | mitosis | |
| HELLS, IGF1R, VEGFA | MYC | negative regulation of apoptotic process |
| VEGFA | MYC | negative regulation of transcription from RNA polymerase II promoter |
| RRM2 | GMPS, IMPDH2 | nucleobase-containing small molecule metabolic process |
| RRM2 | IMPDH2, LDHA | oxidation-reduction process |
| IGF1R, VEGFA | positive regulation of cell migration | |
| IGF1R, VEGFA | CNOT6, MYC, NACC1 | positive regulation of cell proliferation |
| VEGFA | MYC | positive regulation of epithelial cell proliferation |
| VEGFA | MYC | positive regulation of mesenchymal cell proliferation |
| IGF1R, VEGFA | positive regulation of protein kinase B signaling | |
| VEGFA | MYC | positive regulation of transcription from RNA polymerase II promoter |
| CCT5, PDIA6 | protein folding | |
| RAD51 | NACC1 | protein homooligomerization |
| CPM | ADAM28 | proteolysis |
| GMPS, IMPDH2 | purine nucleobase metabolic process | |
| GMPS, IMPDH2 | purine nucleotide biosynthetic process | |
| GMPS, IMPDH2 | purine ribonucleoside monophosphate biosynthetic process | |
| CNOT6, MYC, SSRP1 | regulation of transcription, DNA-dependent | |
| RRM2 | GMPS, GNB2, IMPDH2, LDHA | small molecule metabolic process |
| HELLS | CNOT6, MYC, NACC1 | transcription, DNA-templated |
| RCC1 | SSRP1 | viral process |
Pathways that were regulated by more than one TG were included in the table. Lines shaded in gray contain pathways that were identified for more than one TG in both normal and differential colonic mucosa.
Ten of the 13 significant interactions identified in normal colonic mucosa involved a negative interaction-beta coefficient, signifying that when either miRNA or TF expression increased, the effect of the other regulator on the TG expression was lessened. These interactions included the miRNAs hsa-miR-150-5p, 193b-3p, 330-3p, and 4469, and the TFs ELF1, CTCF and RBBP5. All ten of these interactions included positive TF-beta coefficients, signifying that as TF expression increased, so did the TG expression; this indicates that the TFs enhance TG transcription. Two of these interactions, which included miRNA-TF interactions between ELF1 and hsa-miR-330-3p and ELF1 and hsa-miR-4469, displayed a negative miRNA-TF-beta and miRNA-beta coefficients. This suggests that either the TF represses miRNA transcription, or the miRNA promotes TF degradation, and that the miRNA promotes TG degradation. A negative interaction-beta coefficient suggests that as one effector increases, the effect on the TG is lessened; it is possible, then, that if miRNA expression increased, TF expression was repressed, leading to lessened TG expression. Seed pairing identified between ELF1 and both hsa-miR-330-3p and 4469 strengthens this hypothesis. Given that ELF1 had a TFBS within +/−300 bps of these miRNAs’ primary sequences, it is also possible that the TF represses the miRNA in turn, as is the case in some coherent FFLs4. Seed matches were also identified between hsa-miR-330-3p and PTPLAD1 as well as between hsa-miR-4469 and MRPS27, supporting the negative miRNA-beta coefficients for these interactions and indicating these miRNAs repress these TGs.
The other eight interactions that involved a negative interaction-beta coefficient displayed positive miRNA-TF-beta coefficients, most likely indicating miRNA transcription enhancement by the TF, or indirect effects on the miRNA expression. All 10 interactions with negative interaction-beta coefficients also displayed negative miRNA-beta coefficients, indicating TG repression by miRNAs. Seed pairing was identified in six of these interactions, between MAPK1IP1L and hsa-miR-193b-3p, MRPS27 and hsa-miR-4469, PTPLAD1 and hsa-miR-330-3p, RAD51 and hsa-miR-193b-3p, RRM2 and hsa-miR-193b-3p, and VEGFA and hsa-miR-150-5p, supporting the theory that these interactions involve miRNA repression of the TG.
The three miRNA-TF-TG interactions identified in normal colonic mucosa that displayed positive interaction-betas involved miRNAs hsa-miR-1258 and 145-5p and TFs ZNF263, CTCF, and ELF1. A negative miRNA-TF-beta was identified between hsa-miR-1258 and ZNF263, and a seed match was identified between hsa-miR-1258 and the 3’ UTR of ZNF263, suggesting that hsa-miR-1258 represses ZNF263. The TF-beta for this interaction was positive, indicating that the TF enhances transcription of the target gene, CPM. The miRNA-beta coefficient was also positive, although very slight (0.03); as miRNAs act in the majority of documented circumstances as translational repressors, it is unlikely that the miRNA enhances CPM gene expression. It is possible that miR-1258 does indeed repress CPM expression, however the upregulation of CPM expression by ZNF263 outweighs this effect; a seed match identified between miR-1258 and CPM supports this hypothesis. The other two interactions with positive interaction beta coefficients identified in normal colonic mucosa involved the miRNA hsa-miR-145-5p, TFs ELF1 and CTCF, and the TG IGF1R. These interactions also displayed positive miRNA- and TF-beta coefficients, as well as a positive miRNA-TF-beta coefficient. MiRNA transcription enhancement by CTCF or ELF1 is supported by the positive miRNA-TF-beta coefficient, and TG enhancement is supported by the positive TF-beta coefficient. A seed match was identified between miR-145-5p and IGF1R, suggesting that this miRNA represses this TG, however as IGF1R is upregulated by CTCF and ELF1, the net effect on its expression is still positive.
The 14 miRNA-TF-TG interactions identified in colonic differential expression comprised two miRNAs, hsa-miR-23a-3p and 4469, two TFs, ELF1 and GABPA, and 13 TGs. Only one of these interactions (between hsa-miR-23a-3p, ELF1, and ADAM28) involved a positive interaction-beta coefficient. Both ELF1 and miR-23a-3p had greater carcinoma expression than normal colonic expression, while ADAM28 had higher normal colonic mucosa expression. This interaction included a negative miRNA-beta coefficient, indicating repression of ADAM28 and mRNA degradation by miR-23a-3p, which was supported by a seed match identified between the nucleotides 2–7 in 5’ end of the miRNA and the 3’ UTR of ADAM28. This interaction also involved a positive TF-beta coefficient and a positive miRNA-TF-beta coefficient, indicating that differential expression of ADAM28 and hsa-miR-23a-3p increased as differential expression of ELF1 increased. As the normal colonic mucosa is used as the referent tissue, and differential expression is calculated as the carcinoma tissue expression minus the normal mucosa expression, an increase in differential expression indicates higher expression in the carcinoma tissue. Taken altogether, this interaction suggests that ELF1 increases miR-23a-3p expression, which in turn suppresses ADAM28 expression in carcinoma tissue. As ELF1 likely increases ADAM28 expression as well, as is indicated by the positive TF-beta coefficient, this interaction models an incoherent FFL.
The rest of the miRNA-TF-TG interactions identified in colonic differential expression involved negative interaction-beta coefficients, indicating that as either TF or miRNA expression increased, the effect on TG expression by the other effector was reduced. The only interaction to involve a negative miRNA-TF-beta coefficient (from the miRNA-TF linear regression) was between hsa-miR-4469, the TF ELF1, and the TG TMEM170B. The negative miRNA-TF-beta coefficient suggests that either the miRNA represses TF expression, by way of mRNA degradation, or that ELF1 represses hsa-miR-4469 transcription. A seed match was identified between ELF1 and hsa-miR-4469, supporting the hypothesis of miRNA degradation of ELF1. A positive TF-beta coefficient indicates that ELF1 enhances TMEM170B. The negative interaction-beta coefficient combined with the negative miRNA-TF-beta coefficient suggests that as hsa-miR-4469 increases, ELF1 is degraded, and subsequently TMEM170B levels are reduced. A positive miRNA-beta coefficient was observed for this interaction. There are some reported instances of TG upregulation by miRNAs, however this has only been reported in certain cellular states and whether a given miRNA up or downregulates a TG appears to differ depending on the specific miRNA, TG and tissue27–30. It is possible that this interaction illustrates indirect effects8,31, such as miR-4469 repressing a repressor of TMEM170B, or possibly downregulation of miR-4469 by another TF. It is also possible the effect of miR-4469 is less than the enhancement by ELF1, and as ELF1 increases both miR-4469 and TMEM170B, we detect the net positive effect on TG expression.
The other 12 interactions identified using differential expression with negative interaction-beta coefficients can be split into two groups based on the directionality of the other beta coefficients. The first group, containing four interactions including only miRNA hsa-miR-23a-3p and TF ELF1, all had negative miRNA-beta coefficients, indicating degradation of the TGs (CCT5, GMP5, NACC1, and SSRP1) by miR-23a-3p. Seed pairing was found between hsa-miR-23a-3p and CCT5, GMP5, and NACC1, supporting the theory of TG degradation. This group of interactions displayed positive TF-beta coefficients, indicating TG transcription enhancement by ELF1. ELF1 and miR-23a-3p also have a positive miRNA-TF-beta coefficient; this, combined with the negative interaction-beta coefficient and negative miRNA-beta coefficient, support the theory that ELF1 increases miR-23a-3p, which in turn represses TGs through mRNA degradation. This relationship reflects an incoherent FFL, in which the TF increases both TG and miRNA expression, and the miRNA acts as a buffer for gene expression by repressing the same TG that the TF enhances, and generally serve to maintain steady levels of gene expression4. The other eight interactions with negative interaction-beta coefficients had positive miRNA-beta coefficients, TF-beta coefficients, and miRNA-TF-beta coefficients. This suggests that the TFs (ELF1 and GABPA) increase miR-23a-3p transcription, as well as TG (namely CNOT6, GNB2, IMPDH2, LDHA, MCFD2, MYC, and PDIA6) transcription. A positive miRNA-beta coefficient suggests that increases in miRNA expression result in corresponding increases in TG expression; however the negative interaction-beta coefficient suggests that as miRNA expression increases, the effect of the TF on the TG is reduced. This contradicts the positive TF-beta coefficient, which indicates that as TF expression increases, so does TG expression. It is possible that the miRNA does in fact target the TG, however the rate of mRNA degradation does not outweigh the increase in transcription caused by the TF. Such a scenario would be classified as an incoherent FFL. Three of these interactions, which involved positive miRNA-beta coefficients between hsa-miR-23a-3p and CNOT6, MCFD2 and PDIA6, did have seed matches identified. Additionally, the miRNA-beta coefficients, which were 0.1, 0.2 and 0.3 respectively, were fairly small, indicating very little change in expression in the TG relative to the miRNA. It may be then, that the miRNA does repress the TG expression via degradation in some of these interactions, acting as a buffer for mRNA expression, which has been reported as a principle objective of miRNA-TF interactions8. It is also possible that the positive miRNA-beta coefficient reflects downstream effects that were not directly identified in the interaction, such as miRNA-induced repression of a repressor of the TG.
This study was performed using FFPE tissue. While it is true that these samples have a greater risk of being fragmented, our paired data allows us to control for expression changes that may be due to differences in collection and storage across samples and we have achieved a high level of replication for the microarray platform with our FFPE data32. Additionally, obtaining RNA from FFPE is the only way that large studies such as this can be done to obtain RNA for both miRNA and mRNA. We were only able to look at mRNA gene expression, and as such we are only able to detect evidence of mRNA degradation and not necessarily partial miRNA binding, which results in translation inhibition without degradation of the transcript. As such, we have potentially missed important interactions resulting in decreased protein production. We encourage others to validate these findings. Negative beta coefficients may reflect repression of either molecule on the other and as such these findings should be replicated in laboratory experiments to verify directionality and whether the findings represent direct or indirect effects. In interactions involving a miRNA, we were able to look for seed pairings, which when present support the theory that it is the miRNA that is degrading the gene (be it a TF or TG), however it is possible that other influencing factors haven’t been identified. It is also possible that other definitions of seeds would have resulted in seed matches, however as many of our tested pairs did have an identified match, we do not believe this significantly limited our investigation. Additionally, mRNA degradation typically results from longer, continuous seed pairing2, which is what we search for, and this is appropriate for our mRNA dataset. We chose to look only at miRNA-TF-TG triplets (FFLs) rather than interactions in which the miRNA and TF do not share TGs (FBLs). These interactions may be less common than FBLs, and in this decision may have omitted important interactions from the analysis, however we do not feel that this detracts from our findings. Our objective in this study was to elucidate the intricate relationship between TFs and miRNAs, and how they influence each other’s effect on TG expression. This approach can be applied to different scenarios and other data types to elucidate other important biological interactions. We did not take consider intronic miRNAs, which may have altered where we looked for TFBS overlap. As we performed miRNA-TF linear regressions that confirm significant associations in expression between miRNAs and TFs identified by coordinate overlap we don’t believe that our criteria have misidentified miRNA-TF associations, however it is possible that we excluded meaningful interactions. Additionally, while we chose our TFBS overlap with miRNA and TG transcripts deliberately and with evidentiary support, the number of base pairs is somewhat arbitrary and different overlap criteria may yield different results. Our dataset, while relatively small (N=168), is still larger than most studies containing miRNA and mRNA data. We consider our dataset to be an asset to this investigation, as we were able to investigate paired miRNA and mRNA data with expression from both tumor and paired normal samples for a large number of individuals, however it is possible that, given the study size and the adjustment for multiple comparisons, we may have missed detecting relevant associations. Furthermore, many exogenous and endogenous factors that we did not take in to consideration in this investigation are able to influence both miRNA and mRNA expression8. While we do not consider other influences on miRNA and mRNA expression, such as competing endogenous RNAs, we do not feel that this detracts from our overall aim of showing the miRNAs and TFs work in concert to regulate TG expression.
Our results illustrate the complex relationship between regulator molecules and gene expression. These results show how different types of regulators influence expression levels of a single TG, and how these regulators in fact influence each other, as well as each other’s effect on shared targets. We combined expression data, known TFBSs, and compared miRNA and mRNA FASTA sequences for seed matches to obtain a better picture of TG regulation. We employed an interaction model to better elucidate how miRNAs and TFs work together to influence TG expression. This approach enabled our investigation, which utilized RNA-Seq and microarray platforms and therefore has the advantage of a discovery approach, to outline a more comprehensive picture of molecular interactions. Many interactions displayed a positive miRNA-beta coefficient, which would typically suggest TG upregulation by the miRNA. However, when taken in the context of an observed positive TF-beta coefficient, a negative interaction-beta coefficient and identified seed pairing between the miRNA and TG, we can infer that the TF, miRNA and TG are likely participating in an incoherent FFL. Should others replicate individual findings reported in this study, this large-scale, discovery approach for investigating miRNA-TF-TG interactions would be supported, enabling the expansion of information available in current databases at a much faster rate.
Acknowledgments
The contents of this manuscript are solely the responsibility of the authors and do not necessarily represent the official view of the National Cancer Institute. We would like to acknowledge Dr. Bette Caan and the Kaiser Permanente Medical Research Program for sample contributions, Erika Wolff and Michael Hoffman at the University of Utah for miRNA processing, Brett Milash and the Bioinformatics Shared Resource of the Huntsman Cancer Institute and University of Utah for miRNA and mRNA bioinformatics data processing, Sandie Edwards at the University of Utah for her efforts in overall study monitoring and tumor tissue collection, and Daniel Pellatt at the University of Utah for his assistance with statistical analysis.
References
- 1.Nazarov PV, Reinsbach SE, Muller A, et al. Interplay of microRNAs, transcription factors and target genes: linking dynamic expression changes to function. Nucleic Acids Res. 2013;41(5):2817–2831. doi: 10.1093/nar/gks1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bartel DP. MicroRNAs: Target Recognition and Regulatory Functions. Cell. 2009;136(2):215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Macfarlane LA, Murphy PR. MicroRNA: Biogenesis, Function and Role in Cancer. Curr Genomics. 2010;11(7):537–561. doi: 10.2174/138920210793175895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Martinez NJ, Walhout AJ. The interplay between transcription factors and microRNAs in genome-scale regulatory networks. Bioessays. 2009;31(4):435–445. doi: 10.1002/bies.200800212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Friedman RCFK, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Research. 2009;19(1):92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 7.Arora S, Rana R, Chhabra A, Jaiswal A, Rani V. miRNA-transcription factor interactions: a combinatorial regulation of gene expression. Molecular genetics and genomics : MGG. 2013;288(3–4):77–87. doi: 10.1007/s00438-013-0734-z. [DOI] [PubMed] [Google Scholar]
- 8.Bracken CP, Scott HS, Goodall GJ. A network-biology perspective of microRNA function and dysfunction in cancer. Nature reviews Genetics. 2016;17(12):719–732. doi: 10.1038/nrg.2016.134. [DOI] [PubMed] [Google Scholar]
- 9.Zhang HM, Kuang S, Xiong X, Gao T, Liu C, Guo AY. Transcription factor and microRNA co-regulatory loops: important regulatory motifs in biological processes and diseases. Brief Bioinform. 2015;16(1):45–58. doi: 10.1093/bib/bbt085. [DOI] [PubMed] [Google Scholar]
- 10.Slattery ML, Potter J, Caan B, et al. Energy balance and colon cancer--beyond physical activity. Cancer research. 1997;57(1):75–80. [PubMed] [Google Scholar]
- 11.Slattery ML, Caan BJ, Benson J, Murtaugh M. Energy balance and rectal cancer: an evaluation of energy intake, energy expenditure, and body mass index. Nutr Cancer. 2003;46(2):166–171. doi: 10.1207/S15327914NC4602_09. [DOI] [PubMed] [Google Scholar]
- 12.Slattery ML, Herrick JS, Pellatt DF, et al. MicroRNA profiles in colorectal carcinomas, adenomas and normal colonic mucosa: variations in miRNA expression and disease progression. Carcinogenesis. 2016;37(3):245–261. doi: 10.1093/carcin/bgv249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pellatt DF, Stevens JR, Wolff RK, et al. Expression Profiles of miRNA Subsets Distinguish Human Colorectal Carcinoma and Normal Colonic Mucosa. Clin Transl Gastroenterol. 2016;7:e152. doi: 10.1038/ctg.2016.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Agilent Technologies I. Agilent GeneSpring User Manual. [Accessed July 16, 2015];2013 [Google Scholar]
- 15.Slattery ML, Herrick JS, Mullany LE, et al. An evaluation and replication of miRNAs with disease stage and colorectal cancer-specific mortality. Int J Cancer. 2015;137(2):428–438. doi: 10.1002/ijc.29384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Slattery ML, Pellatt DF, Mullany LE, Wolff RK, Herrick JS. Gene expression in colon cancer: A focus on tumor site and molecular phenotype. Genes, chromosomes & cancer. 2015;54(9):527–541. doi: 10.1002/gcc.22265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mullany LE, Herrick JS, Wolff RK, Stevens JR, Samowitz W, Slattery ML. Transcription factor-microRNA associations and their impact on colorectal cancer survival. Mol Carcinog. 2017;56(11):2512–2526. doi: 10.1002/mc.22698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Karolchik D, Hinrichs AS, Furey TS, et al. The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 2004;32(Database issue):D493–496. doi: 10.1093/nar/gkh103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rosenbloom KR, Sloan CA, Malladi VS, et al. ENCODE data in the UCSC Genome Browser: year 5 update. Nucleic Acids Res. 2013;41(Database issue):D56–63. doi: 10.1093/nar/gks1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koudritsky M, Domany E. Positional distribution of human transcription factor binding sites. Nucleic Acids Res. 2008;36(21):6795–6805. doi: 10.1093/nar/gkn752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Visel A, Rubin EM, Pennacchio LA. Genomic views of distant-acting enhancers. Nature. 2009;461(7261):199–205. doi: 10.1038/nature08451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pennacchio LA, Bickmore W, Dean A, Nobrega MA, Bejerano G. Enhancers: five essential questions. Nature reviews Genetics. 2013;14(4):288–295. doi: 10.1038/nrg3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chou CH, Chang NW, Shrestha S, et al. miRTarBase 2016: updates to the experimentally validated miRNA-target interactions database. Nucleic Acids Res. 2016;44(D1):D239–247. doi: 10.1093/nar/gkv1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Davison AC, Hinkley DV. Bootstrap methods and their application. Cambridge ; New York, NY, USA: Cambridge University Press; 1997. [Google Scholar]
- 25.Benjamini Y, Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society. 1995;57(1):289–300. [Google Scholar]
- 26.Mullany LE, Herrick JS, Wolff RK, Slattery ML. MicroRNA Seed Region Length Impact on Target Messenger RNA Expression and Survival in Colorectal Cancer. PloS one. 2016;11(4):e0154177. doi: 10.1371/journal.pone.0154177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vasudevan S. Posttranscriptional upregulation by microRNAs. Wiley Interdiscip Rev RNA. 2012;3(3):311–330. doi: 10.1002/wrna.121. [DOI] [PubMed] [Google Scholar]
- 28.Vasudevan S, Tong Y, Steitz JA. Switching from repression to activation: microRNAs can up-regulate translation. Science. 2007;318(5858):1931–1934. doi: 10.1126/science.1149460. [DOI] [PubMed] [Google Scholar]
- 29.Orom UA, Nielsen FC, Lund AH. MicroRNA-10a binds the 5'UTR of ribosomal protein mRNAs and enhances their translation. Mol Cell. 2008;30(4):460–471. doi: 10.1016/j.molcel.2008.05.001. [DOI] [PubMed] [Google Scholar]
- 30.Valinezhad Orang A, Safaralizadeh R, Kazemzadeh-Bavili M. Mechanisms of miRNA-Mediated Gene Regulation from Common Downregulation to mRNA-Specific Upregulation. Int J Genomics. 2014;2014:970607. doi: 10.1155/2014/970607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lai X, Wolkenhauer O, Vera J. Understanding microRNA-mediated gene regulatory networks through mathematical modelling. Nucleic Acids Res. 2016;44(13):6019–6035. doi: 10.1093/nar/gkw550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Slattery ML, Wolff E, Hoffman MD, Pellatt DF, Milash B, Wolff RK. MicroRNAs and colon and rectal cancer: differential expression by tumor location and subtype. Genes, chromosomes & cancer. 2011;50(3):196–206. doi: 10.1002/gcc.20844. [DOI] [PMC free article] [PubMed] [Google Scholar]