

Abstract
IMPORTANCE
Players of American football may be at increased risk of long-term neurological conditions, particularly chronic traumatic encephalopathy (CTE).
OBJECTIVE
To determine the neuropathological and clinical features of deceased football players with CTE.
DESIGN, SETTING, AND PARTICIPANTS
Case series of 202 football players whose brains were donated for research. Neuropathological evaluations and retrospective telephone clinical assessments (including head trauma history) with informants were performed blinded. Online questionnaires ascertained athletic and military history.
EXPOSURES
Participation in American football at any level of play.
MAIN OUTCOMES AND MEASURES
Neuropathological diagnoses of neurodegenerative diseases, including CTE, based on defined diagnostic criteria; CTE neuropathological severity (stages I to IV or dichotomized into mild [stages I and II] and severe [stages III and IV]); informant-reported athletic history and, for players who died in 2014 or later, clinical presentation, including behavior, mood, and cognitive symptoms and dementia.
RESULTS
Among 202 deceased former football players (median age at death, 66 years [interquartile range, 47–76 years]), CTE was neuropathologically diagnosed in 177 players (87%; median age at death, 67 years [interquartile range, 52–77 years]; mean years of football participation, 15.1 [SD, 5.2]), including 0 of 2 pre–high school, 3 of 14 high school (21%), 48 of 53 college (91%), 9 of 14 semiprofessional (64%), 7 of 8 Canadian Football League (88%), and 110 of 111 National Football League (99%) players. Neuropathological severity of CTE was distributed across the highest level of play, with all 3 former high school players having mild pathology and the majority of former college (27 [56%]), semiprofessional (5 [56%]), and professional (101 [86%]) players having severe pathology. Among 27 participants with mild CTE pathology, 26 (96%) had behavioral or mood symptoms or both, 23 (85%) had cognitive symptoms, and 9 (33%) had signs of dementia. Among 84 participants with severe CTE pathology, 75 (89%) had behavioral or mood symptoms or both, 80 (95%) had cognitive symptoms, and 71 (85%) had signs of dementia.
CONCLUSIONS AND RELEVANCE
In a convenience sample of deceased football players who donated their brains for research, a high proportion had neuropathological evidence of CTE, suggesting that CTE may be related to prior participation in football.
Chronic traumatic encephalopathy (CTE) is a progressive neurodegeneration associated with repetitive head trauma.1–8 In 2013, based on a report of the clinical and pathological features of 68 men with CTE (including 36 football players from the current study), criteria for neuropathological diagnosis of CTE and a staging scheme of pathological severity were proposed.6 Two clinical presentations of CTE were described; in one, the initial features developed at a younger age and involved behavioral disturbance, mood disturbance, or both; in the other, the initial presentation developed at an older age and involved cognitive impairment.9 In 2014, a methodologically rigorous approach to assessing clinicopathological correlation in CTE was developed using comprehensive structured and semistructured informant interviews and online surveys conducted by a team of behavioral neurologists and neuropsychologists.10 In 2015, the neuropathological criteria for diagnosis of CTE were refined by a panel of expert neuropathologists organized by the National Institute of Neurological Disorders and Stroke and the National Institute of Biomedical Imaging and Bioengineering (NINDS-NIBIB).8
Using the NINDS-NIBIB criteria to diagnose CTE and the improved methods for clinicopathological correlation, the purpose of this study was to determine the neuropathological and clinical features of a case series of deceased football players neuropathologically diagnosed as having CTE whose brains were donated for research.
Methods
Study Recruitment
In 2008, as a collaboration among the VA Boston Healthcare System, Bedford VA, Boston University (BU) School of Medicine, and Sports Legacy Institute (now the Concussion Legacy Foundation [CLF]), a brain bank was created to better understand the long-term effects of repetitive head trauma experienced through contact sport participation and military-related exposure. The purpose of the brain bank was to comprehensively examine the neuropathology and clinical presentation of brain donors considered at risk of development of CTE. The institutional review board at Boston University Medical Campus approved all research activities. The next of kin or legally authorized representative of each brain donor provided written informed consent. No stipend for participation was provided. Inclusion criteria were based entirely on exposure to repetitive head trauma (eg, contact sports, military service, or domestic violence), regardless of whether symptoms manifested during life. Playing American football was sufficient for inclusion. Because of limited resources, more strict inclusion criteria were implemented in 2014 and required that football players who died after age 35 years have at least 2 years of college-level play. Donors were excluded if postmortem interval exceeded 72 hours or if fixed tissue fragments representing less than half the total brain volume were received (eFigure in the Supplement).
Clinical data were collected into a Federal Interagency Traumatic Brain Injury Research–compliant database. Since tracking began in 2014, for 98 (81%) brain donations to the VA-BU-CLF Brain Bank, the next of kin approached the brain bank near the time of death. The remaining brain donors were referred by medical examiners (11 [9%]), recruited by a CLF representative (7 [6%]), or participated in the Brain Donation Registry during life (5 [4%]) (eFigure in the Supplement).
Clinical Evaluation
Retrospective clinical evaluations were performed using online surveys and structured and semistructured postmortem telephone interviews between researchers and informants. Researchers conducting these evaluations were blinded to the neuropathological analysis, and informants were interviewed before receiving the results of the neuropathological examination. A behavioral neurologist, neuroscientist, or neuropsychologist (J.M., D.H.D., T.M.S., M.L.A., or R.A.S.) obtained a detailed history, including a timeline of cognitive, behavioral, mood, and motor symptomology. Additionally, other neuropsychiatric symptoms, exposures and symptoms consistent with posttraumatic stress disorder, features of a substance use disorder, neurodegenerative diagnoses made in life (Alzheimer disease [AD], frontotemporal dementia, vascular dementia, dementia with Lewy bodies, Parkinson disease, CTE, or dementia of unknown etiology), headaches that impaired function, symptoms and diagnoses made in life of sleep disorders, and causes of death were assessed. Clinicians qualitatively summarized the participants’ clinical presentation (eg, presence and course of symptoms, functional independence) into a narrative and presented the case to a multidisciplinary consensus team of clinicians, during which it was determined whether the participant met criteria for dementia. To resolve discrepancies in methods that evolved over time, only clinical variables ascertained after January 2014 using a standardized informant report were included because of the larger subset of participants recruited during this time frame (n = 125).
Prior to January 2014, demographics, educational attainment, athletic history (type of sports played, level, position, age at first exposure, and duration), military history (branch, location of service, and duration of combat exposure), and traumatic brain injury (TBI) history (including number of concussions) were queried during the telephone interview. Beginning in January 2014, demographics, educational attainment, and athletic and military history were queried using an online questionnaire. Informant-reported race was collected as part of demographic information so that neuropathological differences across race could be assessed. To be considered a National Football League (NFL) athlete, a participant must have played in at least 1 regular-season NFL game. Professional position and years of play were verified using available online databases (http://www.pro-football-reference.com, http://databasefootball.com, http://www.justsportsstats.com). History of TBI was queried using informant versions of the Ohio State University TBI Identification Method Short Form11 and 2 questionnaires adapted from published studies that address military-related head injuries and concussions.12,13 With the addition of these questionnaires, informants were read a formal definition of concussion prior to being asked about concussion history, which was not the case prior to January 2014.
Neuropathological Evaluation
Pathological processing and evaluation were conducted using previously published methods.14,15 Brain volume and macroscopic features were recorded during initial processing. Twenty-two sections of paraffin-embedded tissue were stained for Luxol fast blue, hematoxylin and eosin, Bielschowsky silver, phosphorylated tau (ptau) (AT8), α-synuclein, amyloid-β, and phosphorylated transactive response DNA binding protein 43 kDa (pTDP-43) using methods described previously.16 In some cases, large coronal slabs of the cerebral hemispheres were also cut at 50 μm on a sledge microtome and stained as free-floating sections using AT8 or CP-13.16,17
A neuropathological diagnosis was made using criteria for CTE recently defined by the 2015 NINDS-NIBIB Consensus Conference8 and well-established criteria for other neuropathological diseases, including AD,18,19 Lewy body disease,20 frontotemporal lobar degeneration,21–25 and motor neuron disease.26,27 Neuropathological criteria for CTE require at least 1 perivascular ptau lesion consisting of ptau aggregates in neurons, astrocytes, and cell processes around a small blood vessel; these pathognomonic CTE lesions are most often distributed at the depths of the sulci in the cerebral cortex and are distinct from the lesions of aging-related tau astrogliopathy.8 Supportive features for the diagnosis of CTE include ptau pretangles and neurofibrillary tangles (NFTs) in superficial cortical layers (layers II/III) of the cerebral cortex; pretangles, NFTs or extracellular tangles in CA2 and CA4 of the hippocampus; subpial ptau astrocytes at the glial limitans; and dot-like ptau neurites.8
Chronic traumatic encephalopathy ptau pathology was classified into 4 stages using previously proposed criteria.6 Briefly, stage I CTE is characterized by 1 or 2 isolated perivascular epicenters of ptau NFTs and neurites (ie, CTE lesions) at the depths of the cerebral sulci in the frontal, temporal, or parietal cortices. In stage II, 3 or more CTE lesions are found in multiple cortical regions and superficial NFTs are found along the sulcal wall and at gyral crests. Multiple CTE lesions, superficial cortical NFTs, and diffuse neurofibrillary degeneration of the entorhinal and perirhinal cortices, amygdala, and hippocampus are found in stage III CTE. In stage IV CTE, CTE lesions and NFTs are densely distributed throughout the cerebral cortex, diencephalon, and brain stem with neuronal loss, gliosis, and astrocytic ptau pathology. Chronic traumatic encephalopathy pathology in stages I and II is considered to be mild and in stages III and IV is considered to be severe.
Neuropathological evaluation was blinded to the clinical evaluation and was reviewed by 4 neuropathologists (V.A., B.H., T.D.S., and A.M.); any discrepancies in the neuropathological diagnosis were solved by discussion and consensus of the group. In addition to diagnoses, the density of ptau immunoreactive NFTs, neurites, diffuse amyloid-β plaques, and neuritic amyloid-β plaques; vascular amyloid-β; pTDP-43; and α-synuclein immunoreactive Lewy bodies were measured semiquantitatively (0–3, with 3 being most severe) across multiple brain regions.
Descriptive statistics were generated using SPSS software version 20 (IBM Inc).
Results
Among the 202 deceased brain donors (median age at death, 66 years [interquartile range [IQR], 47–76 years]), CTE was neuropathologically diagnosed in 177 (87%; median age at death, 67 years [IQR, 52–77 years]; mean years of football participation, 15.1 [SD, 5.2]; 140 [79%] self-identified as white and 35 [19%] self-identified as black), including 0 of 2 pre-high school, 3 of 14 high school (21%), 48 of 53 college (91%), 9 of 14 semiprofessional (64%), 7 of 8 Canadian Football League (88%), and 110 of 111 NFL (99%) players.
The median age at death for participants with mild CTE pathology (stages I and II) was 44 years (IQR, 29–64 years) and for participants with severe CTE pathology (stages III and IV) was 71 years (IQR, 64–79 years). The most common cause of death for participants with mild CTE pathology was suicide (12 [27%]) and for those with severe CTE pathology was neurodegenerative (ie, dementia-related and parkinsonian-related causes of death) (62 [47%]). The severity of CTE pathology was distributed across the highest level of play, with all former high school players having mild pathology (3 [100%]) and the majority of former college (27 [56%]), semiprofessional (5 [56%]), Canadian Football League (6 [86%]), and NFL (95 [86%]) players having severe pathology. The mean duration of play for participants with mild CTE pathology was 13 years (SD, 4.2 years) and for participants with severe CTE pathology was 15.8 years (SD, 5.3 years) (Table 1).
Table 1.
Demographic and Exposure Characteristics of 177 American Football Players Diagnosed With CTE, Stratified by Neuropathological Severitya
| Characteristics | No. (%) of Brain Donorsb | ||
|---|---|---|---|
| Mild CTE (n = 44) |
Severe CTE (n = 133) |
Total (n = 177) |
|
| Men | 44 (100) | 133 (100) | 177 (100) |
| Race | |||
| White | 35 (80) | 105 (79) | 140 (79) |
| Black | 8 (18) | 27 (20) | 35 (19) |
| Pacific Islander | 0 | 1 (1) | 1 (1) |
| Asian | 0 | 0 | 0 |
| Other | 0 | 0 | 0 |
| Unknown | 1 (2) | 0 | 1 (1) |
| Age at death, median (IQR), y | 44 (29–64) | 71 (64–79) | 67 (52–77) |
| Cause of death | |||
| Neurodegenerativec | 7 (16) | 62 (47) | 69 (39) |
| Cardiovascular disease | 5 (11) | 29 (22) | 34 (19) |
| Suicide | 12 (27) | 6 (5) | 18 (10) |
| Cancer | 2 (5) | 10 (8) | 12 (7) |
| Motor neuron disease | 4 (9) | 7 (5) | 11 (6) |
| Unintentional overdose | 3 (7) | 4 (3) | 7 (4) |
| Injury | 2 (5) | 3 (2) | 5 (3) |
| Other | 9 (21) | 12 (9) | 21 (12) |
| Concussion count, median (IQR)d | |||
| Definition provided (n = 99) | 90 (22–150) | 50.5 (12–163) | 70 (12–150) |
| No definition provided (n = 61) | 2.5 (0–5) | 8 (1–19) | 5 (1–13) |
| Age at first exposure to football, median (IQR), y | 10 (8–14) | 13 (10–14) | 12 (10–14) |
| Duration of play, mean (SD), y | 13 (4.2) | 15.8 (5.3) | 15.1 (5.2) |
| Highest level of play | |||
| Youth | 0 | 0 | 0 |
| High school | 3 (7) | 0 | 3 (2) |
| College | 21 (48) | 27 (20) | 48 (27) |
| Semiprofessional | 4 (9) | 5 (4) | 9 (5) |
| Canadian Football League | 1 (2) | 6 (5) | 7 (4) |
| National Football League | 15 (34) | 95 (71) | 110 (62) |
| Primary position at highest level of play | |||
| Offensive lineman | 8 (18) | 29 (22) | 37 (21) |
| Defensive lineman | 8 (18) | 27 (20) | 35 (20) |
| Running back | 4 (9) | 27 (20) | 31 (18) |
| Linebacker | 12 (27) | 14 (11) | 26 (15) |
| Defensive back | 4 (9) | 18 (14) | 22 (12) |
| Quarterback | 2 (5) | 11 (8) | 13 (7) |
| Tight end | 1 (2) | 6 (5) | 7 (4) |
| Wide receiver | 3 (7) | 1 (1) | 4 (2) |
| Kicker or punter | 2 (5) | 0 | 2 (1) |
| Other special teams | 0 | 0 | 0 |
| Military veteran | 5 (11) | 40 (30) | 45 (25) |
Abbreviations: CTE, chronic traumatic encephalopathy; IQR, interquartile range.
Mild CTE (CTE neuropathological stages I and II) is characterized by sparse to frequent perivascular CTE lesions at the sulcal depths of the cerebral cortex. Severe CTE (CTE neuropathological stages III and IV) consists of multiple CTE lesions in the cerebral cortex and moderate to severe neurofibrillary degeneration of medial temporal lobe, diencephalon, and brain stem.
Data are expressed as No. (%) unless otherwise indicated.
Includes dementia-related and parkinsonian-related causes of death.
Median estimates of the number of concussions reported per participant. Beginning in 2014, informants were read a formal definition of concussion prior to being asked about concussion history.
In all cases, perivascular clusters of ptau immunoreactive NFTs diagnostic for CTE (ie, CTE lesions)8 were found in the cerebral cortex (Figure 1 and Figure 2). In cases with mild CTE pathology (stages I and II), isolated perivascular CTE lesions were found at the sulcal depths of the cerebral cortex, most commonly in the superior and dorsolateral frontal cortices, but also in the lateral temporal, inferior parietal, insula, and septal cortices (Figure 1). Neurofibrillary tangles were sparse in other cortical regions, and there was no diffuse neurofibrillary degeneration of the medial temporal lobe structures (Figure 1, open arrowheads). Neurofibrillary tangles were also found in the locus coeruleus, substantia nigra, and substantia innominata (Figure 3) in mild CTE. In cases with severe CTE pathology, perivascular CTE lesions were large and confluent (Figure 2). Neurofibrillary tangles were widely distributed in the superficial laminae of cortical regions and there was severe neurofibrillary degeneration of the medial temporal lobe structures, including the hippocampus, amygdala, and entorhinal cortex (Figure 2, black arrowheads, and Figure 3). Neurofibrillary tangles were also frequent in the thalamus, nucleus basalis of Meynert, substantia innominata, substantia nigra, and locus coeruleus in severe CTE (Figure 3).
Figure 1. Representative Images of Phosphorylated Tau Pathology at CTE Pathological Stages I and II.
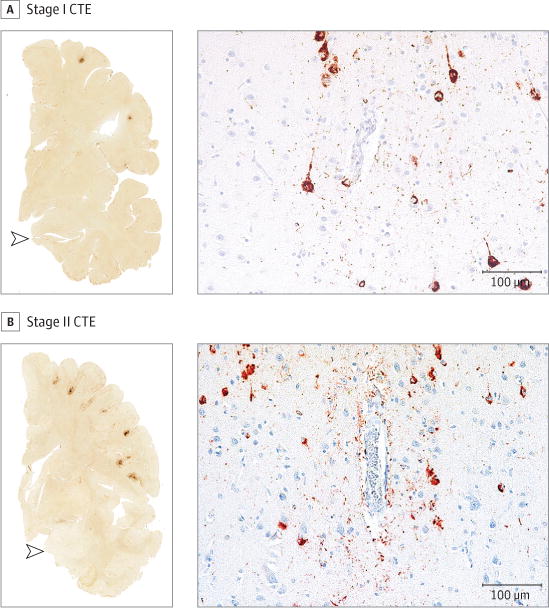
CTE indicates chronic traumatic encephalopathy; NFT, neurofibrillary tangle; ptau, phosphorylated tau. For all images, 10-μm paraffin-embedded tissue sections were immunostained with microscopic mouse monoclonal antibody for phosphorylated tau (AT8) (Pierce Endogen). Positive ptau immunostaining appears dark red, hematoxylin counterstain; calibration bar indicates 100 μm. Stage I CTE is characterized by 1 or 2 isolated perivascular epicenters of ptau NFTs and neurites (ie, CTE lesions) at the depths of the cortical sulci. In stage II, 3 or more cortical CTE lesions are found. All hemispheric tissue section images are 50-μm sections immunostained with mouse monoclonal antibody CP-13, directed against phosphoserine 202 of tau (courtesy of Peter Davies, PhD, Feinstein Institute for Medical Research; 1:200); this is considered to be an early site of tau phosphorylation in NFT formation.28 Positive ptau immunostaining appears dark brown. A, Former college football player with stage I CTE. Two perivascular ptau CTE lesions are evident at sulcal depths of the frontal cortex; there is no neurofibrillary degeneration in the medial temporal lobe (open arrowhead). Perivascular CTE lesion: neurofibrillary tangles and dot-like and threadlike neurites encircle a small blood vessel. B, Former NFL player with stage II CTE. There are multiple perivascular ptau CTE lesions at depths of sulci of the frontal cortex; there is no neurofibrillary degeneration in the medial temporal lobe (open arrowhead). Perivascular CTE lesion: a cluster of NFTs and large dot-like and threadlike neurites surround a small blood vessel.
Figure 2. Representative Images of Phosphorylated Tau Pathology at CTE Pathological Stages III and IV.
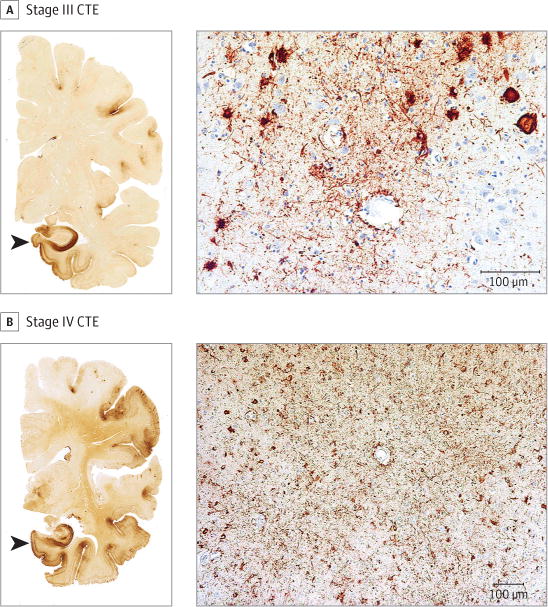
CTE indicates chronic traumatic encephalopathy; NFT, neurofibrillary tangle; ptau, phosphorylated tau. For all images, 10-μm paraffin-embedded tissue sections were immunostained with microscopic mouse monoclonal antibody for phosphorylated tau (AT8) (Pierce Endogen). Positive ptau immunostaining appears dark red, hematoxylin counterstain; calibration bar indicates 100 μm. In stage III CTE, multiple CTE lesions and diffuse neurofibrillary degeneration of the medial temporal lobe are found. In stage IV CTE, CTE lesions and NFTs are widely distributed throughout the cerebral cortex, diencephalon, and brain stem.6 All hemispheric tissue section images are 50-μm sections immunostained with mouse monoclonal antibody CP-13, directed against phosphoserine 202 of tau (courtesy of Peter Davies, PhD, Feinstein Institute for Medical Research; 1:200); this is considered to be an early site of tau phosphorylation in NFT formation.28 Positive ptau immunostaining appears dark brown. A, Former NFL player with stage III CTE. There are multiple large CTE lesions in the frontal cortex and insula; there is diffuse neurofibrillary degeneration of hippocampus and entorhinal cortex (black arrowhead). Perivascular CTE lesion: a dense collection of NFTs and large dot-like and threadlike neurites enclose several small blood vessels. B, Former NFL player with stage IV CTE. There are large, confluent CTE lesions in the frontal, temporal, and insular cortices and there is diffuse neurofibrillary degeneration of the amygdala and entorhinal cortex (black arrowhead). Perivascular CTE lesion: a large accumulation of NFTs, many of them ghost tangles, encompass several small blood vessels.
Figure 3. Phosphorylated Tau Pathology for Each Brain Region by CTE Neuropathological Stage.
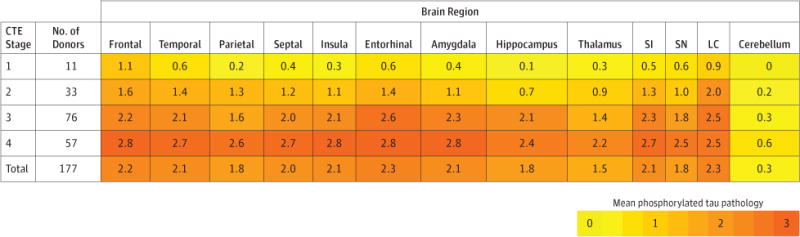
CTE indicates chronic traumatic encephalopathy; NFT: neurofibrillary tangle, SI: substantia innominata, SN: substantia nigra; LC: locus coeruleus. Cerebellum indicates dentate nucleus of the cerebellum. In each region, 0 = no NFTs (yellow); 1 = 1 NFT per 20× field (orange); 2 = 2 to 3 NFTs per 20× field (amber); and 3 = ≥ 4 NFTs per 20× field (red). The color scale is based on the distribution of all values, not by each individual stage. Values represent means of phosphorylated tau pathology among participants in each stage.
Deposition of amyloid-β was present in a subset of participants at all stages of CTE pathology, predominantly as diffuse amyloid-β plaques, but neuritic amyloid-β plaques and amyloid angiopathy were also present. In stage IV CTE, amyloid-β deposition occurred in 52 cases (91%). Deposition of TDP-43 and α-synuclein were found in all stages of CTE pathology; TDP-43 deposition occurred in 47 (83%) and α-synuclein deposition occurred in 23 (40%) stage IV CTE cases (Table 2).
Table 2.
Neuropathological Findings in 177 American Football Players, Stratified by Severity of Phosphorylated Tau Pathology (CTE Stage)a
| CTE Stage | No. of Brain Donors | Age at Death, Median (IQR), y | Neuropathological Features, No. (%) | Other Neuropathological Diagnoses, No. (%) | Pure CTE, No. (%) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Aß | DP | NP | AA | TDP-43 | as | AD | LBD | FTLD TDP-43 | FTLD-Tau | MND | ||||
| 1 | 11 | 36 (25–56) | 2 (18) | 2 (18) | 1 (9) | 1 (9) | 2 (18) | 1 (9) | 0 | 1 (9) | 1 (9) | 0 | 0 | 8 (73) |
| 2 | 33 | 49 (29–65) | 8 (24) | 8 (24) | 5 (15) | 7 (21) | 10 (30) | 3 (9) | 1 (3) | 2 (6) | 1 (3) | 1 (3) | 4 (12) | 21 (64) |
| 3 | 76 | 67 (57–78) | 45 (59) | 41 (54) | 25 (33) | 29 (38) | 26 (34) | 16 (21) | 4 (5) | 15 (20) | 1 (1) | 3 (4) | 6 (8) | 42 (55) |
| 4 | 57 | 76 (69–82) | 52 (91) | 52 (91) | 42 (74) | 32 (56) | 47 (83) | 23 (40) | 18 (32) | 16 (28) | 5 (9) | 2 (4) | 1 (2) | 27 (47) |
| Total | 177 | 67 (53–78) | 107 (61) | 103 (58) | 73 (41) | 69 (39) | 85 (48) | 43 (24) | 23 (13) | 34 (19) | 8 (5) | 6 (3) | 11 (6) | 98 (55) |
Abbreviations: AA, amyloid angiopathy; Aß, amyloid-β; AD, Alzheimerdisease; as, a-synuclein immunopositive Lewy bodies; CTE, chronic traumatic encephalopathy diagnosed neuropathologically; DP, diffuse Aß plaques; FTLD-tau, frontotemporal lobar degeneration-tau; FTLDTDP-43,frontotemporal lobar degeneration TDP-43; IQR, interquartile range; LBD, Lewy body disease; MND, motor neuron disease; NP, neuritic Aß plaques; TDP-43, TDP-43 immunopositive neurites or inclusions.
Pure CTE is defined as CTE with no neuropathological evidence of other comorbid neurodegenerative disease.
Stage I CTE is characterized by 1 or 2 perivascular CTE lesions at the depths of the cerebral sulci in the cerebral cortex. In stage II,3 or more CTE lesions are found in multiple cortical regions. In stage III CTE, many CTE lesions, superficial cortical neurofibrillary tangles, and diffuse neurofibrillary degeneration of the entorhinal and perirhinal cortices, amygdala, and hippocampus are found. In stage IV CTE, CTE lesions and neurofibrillary tangles are densely distributed throughout the cerebral cortex, diencephalon, and brain stem with neuronal loss, gliosis, and astrocytic phosphorylated tau pathology.
Among the 25 football players without CTE, 9 showed no pathological abnormalities and 7 showed nonspecific changes; eg, hemosiderin-laden macrophages (n = 7) and axonal injury (n = 5). Other diagnoses included vascular pathology (n = 4), unspecified tauopathy not meeting criteria for CTE (n = 3), AD (n = 2), argyrophilic grain disease (n = 1), and Lewy body disease (n = 1).
Data on informants were collected beginning in 2014. The median number of participating informants was 2 (IQR, 1–3) per participant. Among all of the interviews, 71 (64%) included a spouse/partner, 56 (51%) included an adult child, 27 (24%) included a sibling, 16 (14%) included a parent, 13 (12%) included a non–first-degree relative, 8 (7.2%) included a neighbor or friend, and 4 included other informants. Among the informants who knew the participant the longest, the mean relationship length was 45.8 years (SD, 1.5 years).
Among the 111 CTE cases with standardized informant reports on clinical symptoms, a reported progressive clinical course was common in participants with both mild and severe CTE pathology, occurring in 23 (85%) mild cases and 84 (100%) severe cases (Table 3). Behavioral or mood symptoms were common in participants with both mild and severe CTE pathology, with symptoms occurring in 26 (96%) mild cases and 75 (89%) severe cases. Impulsivity, depressive symptoms, apathy, and anxiety occurred in 23 (89%), 18 (67%), 13 (50%), and 14 (52%) mild cases and 65 (80%), 46 (56%), 43 (52%), and 41 (50%) severe cases, respectively. Additionally, hopelessness, explosivity, being verbally violent, being physically violent, and suicidality (including ideation, attempts, or completions) occurred in 18 (69%), 18 (67%), 17 (63%), 14 (52%), and 15 (56%) mild cases, respectively. Substance use disorders were also common in participants with mild CTE, occurring in 18 (67%) mild cases. Symptoms of posttraumatic stress disorder were uncommon in both groups, occurring in 3 (11%) mild cases and 9 (11%) severe cases.
Table 3.
Clinical Features Reported in 111 American Football Players Diagnosed as Having CTE, Stratified by Neuropathological Severitya
| Clinical Features | No. (%) of Brain Donors | ||
|---|---|---|---|
| Mild CTE | Severe CTE | Total | |
| Progressive course | 23 (85) | 84 (100) | 107 (96) |
| Cognitive symptomsb | 23 (85) | 80 (95) | 103 (93) |
| Memory | 19 (73) | 76 (92) | 95 (86) |
| Executive function | 19 (73) | 67 (81) | 86 (79) |
| Attention | 18 (69) | 67 (81) | 85 (78) |
| Language | 10 (39) | 54 (66) | 64 (59) |
| Visuospatial | 7 (27) | 44 (54) | 51 (47) |
| Fluctuating cognition | 2 (8) | 17 (21) | 19 (18) |
| Dementiab | 9 (33) | 71 (85) | 80 (72) |
| Behavioral or mood symptomsb | 26 (96) | 75 (89) | 101 (91) |
| Impulsivity | 23 (89) | 65 (80) | 88 (82) |
| Depressive symptoms | 18 (67) | 46 (56) | 64 (59) |
| Explosivity | 18 (67) | 38 (45) | 56 (51) |
| Apathy | 13 (50) | 43 (52) | 56 (51) |
| Anxiety | 14 (52) | 41 (50) | 55 (51) |
| Hopelessness | 18 (69) | 36 (46) | 54 (52) |
| Verbal violence | 17 (63) | 28 (34) | 45 (41) |
| Social inappropriateness | 13 (48) | 26 (32) | 39 (36) |
| Physical violence | 14 (52) | 23 (28) | 37 (34) |
| Paranoia | 11 (41) | 26 (31) | 37 (34) |
| Suicidality (ideation, attempts, or completions) | 15 (56) | 21 (25) | 36 (33) |
| Visual hallucinations | 6 (23) | 22 (27) | 28 (26) |
| Mania | 6 (22) | 3 (4) | 9 (8) |
| Posttraumatic stress disorder (exposure and symptoms consistent with) | 3 (11) | 9 (11) | 12 (11) |
| Substance use disorder | 18 (67) | 41 (49) | 59 (53) |
| Alcohol | 13 (50) | 31 (37) | 44 (41) |
| Anabolic steroid | 0 | 4 (5) | 4 (4) |
| Other | 14 (54) | 23 (28) | 37 (34) |
| Motor symptomsb | 13 (48) | 63 (75) | 76 (68) |
| Gait instability | 7 (26) | 55 (66) | 62 (56) |
| Slowness | 5 (19) | 42 (50) | 47 (42) |
| Coordination difficulties | 7 (26) | 38 (45) | 45 (41) |
| Falls | 4 (15) | 39 (46) | 43 (39) |
| Tremor | 5 (19) | 33 (39) | 38 (34) |
| Dysphagia | 3 (11) | 14 (18) | 17 (16) |
| Dysarthria | 5 (19) | 10 (13) | 15 (14) |
| Headache | 8 (30) | 11 (14) | 19 (18) |
| Diagnoses in life | |||
| Motor neuron disease | 1 (4) | 3 (4) | 4 (4) |
| Parkinson disease | 1 (4) | 5 (6) | 6 (6) |
| Alzheimer disease | 1 (4) | 21 (25) | 22 (20) |
| Obstructive sleep apnea (diagnosis or symptoms) | 7 (27) | 36 (46) | 43 (41) |
| Rapid eye movement sleep behavior disorder (diagnosis or symptoms) | 7 (27) | 23 (29) | 30 (29) |
Abbreviation: CTE, chronic traumatic encephalopathy.
There were 111 participants with standardized informant reports, including 27 participants with mild CTE and 84 participants with severe CTE. Sample sizes differed across clinical features because features marked as unknown by the clinician were excluded. For participants with mild CTE, sample sizes ranged from 25 to 27 and for participants with severe CTE, sample sizes ranged from 78 to 84. Mild CTE (CTE neuropathological stages I and II) is characterized by sparse to frequent perivascular CTE lesions at the sulcal depths of the cerebral cortex. Severe CTE (CTE neuropathological stages III and IV) consists of multiple CTE lesions in the cerebral cortex and moderate to severe neurofibrillary degeneration of medial temporal lobe, diencephalon, and brain stem.
Symptoms were present in the last year of life
Cognitive symptoms were common in participants with both mild and severe CTE pathology, with symptoms occurring in 23 (85%) mild cases and 80 (95%) severe cases. Memory, executive function, and attention symptoms occurred in 19 (73%), 19 (73%), and 18 (69%) mild cases and 76 (92%), 67 (81%), and 67 (81%) severe cases, respectively.
Additionally, language and visuospatial symptoms occurred in 54 (66%) and 44 (54%) severe cases, respectively. A premortem diagnosis of AD and a postmortem (but blinded to pathology) consensus diagnosis of dementia were common in severe cases, occurring in 21 (25%) and 71 (85%), respectively. There were no asymptomatic (ie, no mood/behavior or cognitive symptoms) CTE cases. Motor symptoms were common in severe cases, occurring in 63 (75%). Gait instability and slowness of movement occurred in 55 (66%) and 42 (50%) severe cases, respectively. Symptom frequencies remained similar when only pure CTE cases (ie, those with no neuropathological evidence of comorbid neurodegenerative disease) were considered (eTable in the Supplement).
Among the 111 CTE cases with standardized informant reports on clinical symptoms, 47 (42.3%; median age at death, 76 years [IQR, 63–81 years]) initially presented with cognitive symptoms, 48 (43.2%; median age at death, 66 years [IQR, 54–73 years]) initially presented with behavior or mood symptoms, and 16 (14.4%; median age at death, 65.5 years [IQR, 39–78]) initially presented with both cognitive symptoms and behavior or mood symptoms. Forty (85%) of those initially presenting with only cognitive symptoms were reported to have behavior or mood symptoms at the time of death and 43 (90%) of those initially presenting with only behavior or mood symptoms were reported to have cognitive symptoms at the time of death. Dementia was present at the time of death in 36 (77%) of those initially presenting with cognitive symptoms, 33 (69%) of those initially presenting with behavior or mood symptoms, and 11 (69%) of those initially presenting with both cognitive and behavior or mood symptoms.
The most common primary cause of death was neurodegenerative for all 3 groups (cognitive, 26 [55%]; behavior or mood, 16 [33%]; both cognitive and behavior or mood, 6 [38%]). Substance use disorders, suicidality, and family history of psychiatric illness were common among those who initially presented with behavior or mood symptoms, occurring in 32 (67%), 22 (47%), and 23 (49%) cases, respectively.
Discussion
In a convenience sample of 202 deceased former players of American football who were part of a brain donation program, a high proportion were diagnosed neuropathologically with CTE. The severity of CTE pathology was distributed across the highest level of play, with all former high school players having mild pathology and the majority of former college, semiprofessional, and professional players having severe pathology. Behavior, mood, and cognitive symptoms were common among those with mild and severe CTE pathology and signs of dementia were common among those with severe CTE pathology.
Nearly all of the former NFL players in this study had CTE pathology, and this pathology was frequently severe. These findings suggest that CTE may be related to prior participation in football and that a high level of play may be related to substantial disease burden. Several other football-related factors may influence CTE risk and disease severity, including but not limited to age at first exposure to football, duration of play, player position, cumulative hits, and linear and rotational acceleration of hits. Recent work in living former football players has shown that age at first exposure may be related to impaired cognitive performance29 and altered corpus callosum white matter30 and that cumulative hits may be related to impairment on self-report and objective measures of cognition, mood, and behavior,31 although it is unclear if any of these outcomes are related to CTE pathology. Furthermore, it is unclear if symptomatic hits (concussions) are more important than asymptomatic hits resulting in subconcussive injury. As with other neurodegenerative diseases, age may be related to risk and pathological severity in CTE. It will be important for future studies to resolve how different measures of exposure to football and age influence the outcome.
In cases with severe CTE pathology, accumulations of amyloid-β, α-synuclein, and TDP-43 were common. These findings are consistent with previous studies that show deposition of multiple neurodegenerative proteins after exposure to TBI32 and with work showing that neuritic amyloid-β plaques are associated with increased CTE neuropathological stage.33 Diagnoses of comorbid neurodegenerative diseases, including AD, Lewy body disease, motor neuron disease, and frontotemporal lobar degeneration, were also common in cases with severe CTE pathology. Overall, 19% of participants with CTE had comorbid Lewy body disease, which aligns with a recent observation by Crane et al34 regarding the increased prevalence of Lewy body pathology after single TBI. Chronic traumatic encephalopathy was not assessed in the analysis by Crane et al; to investigate the possibility of CTE after single TBI would require more extensive sampling of the depths of the cortical sulci with ptau immunostaining, as silver stains typically do not detect CTE pathology.
Behavioral, mood, and cognitive symptoms were common among participants with either mild or severe CTE pathology. In participants with severe CTE pathology, there was marked ptau pathology in brain regions that have been associated with symptoms frequently reported: impulsivity, depressive symptoms, apathy, anxiety, and explosivity (prefrontal cortex, amygdala, locus coeruleus); episodic memory symptoms (hippocampus and entorhinal and perirhinal cortices); and attention and executive function symptoms (prefrontal cortex). Participants with mild CTE pathology often had these symptoms despite having relatively circumscribed cortical pathology and absence of ptau pathology in the hippocampus, entorhinal cortex, or amygdala. This may suggest that other pathologies not captured by the pathological data set, such as neuroinflammation, axonal injury, or astrocytosis, or pathologies in neuroanatomical regions not evaluated contribute to these clinical symptoms. Microglial neuroinflammation appears to precede tau accumulation in CTE,35 suggesting it may play a role in early symptoms.
Informants reported that 43% of participants had behavior or mood symptoms as their initial presentation. Many of these participants had a substance use disorder, demonstrated suicidality, or had a family history of psychiatric illness. Behavior or mood symptoms may be the initial presentation for a subset of individuals with CTE, or alternatively, CTE ptau pathology may lower the threshold for psychiatric manifestations in susceptible individuals. These clinical observations confirm and expand on previous reports of 2 primary clinical presentations of CTE.9
There is substantial evidence that CTE is a progressive, neurodegenerative disease. In this study, 107 participants (96%) had a progressive clinical course based on informant report. In addition, pathological severity of CTE was correlated with age at death (Table 3). However, a postmortem study evaluates brain pathology at only 1 time point and is by definition cross-sectional. In addition, the participants were not observed longitudinally during life. Although associations with age in cross-sectional samples can result from age-related progression within individuals, they can also arise from birth cohort effects, differential survival, or age- related differences in how individuals were selected into the study. Population-based prospective studies are needed to address the issue of progression of CTE pathology and age at symptom onset.
The strengths of this study are that this is the largest CTE case series ever described to our knowledge, more than doubling the size of the 2013 report,6 and that all participants were exposed to a relatively similar type of repetitive head trauma while playing the same sport. In addition, the comprehensive neuropathological evaluation and retrospective clinical data collection were independently performed while blinded to the findings of the other investigators.
This study had several limitations. First, a major limitation is ascertainment bias associated with participation in this brain donation program. Although the criteria for participation were based on exposure to repetitive head trauma rather than on clinical signs of brain trauma, public awareness of a possible link between repetitive head trauma and CTE may have motivated players and their families with symptoms and signs of brain injury to participate in this research. Therefore, caution must be used in interpreting the high frequency of CTE in this sample, and estimates of prevalence cannot be concluded or implied from this sample. Second, the VA-BU-CLF brain bank is not representative of the overall population of former players of American football; most players of American football have played only on youth or high school teams, but the majority of the brain bank donors in this study played at the college or professional level. Additionally, selection into brain banks is associated with dementia status, depression status, marital status, age, sex, race, and education.36 Third, this study lacked a comparison group that is representative of all individuals exposed to American football at the college or professional level, precluding estimation of the risk of participation in football and neuropathological outcomes.
Conclusions
In a convenience sample of deceased football players who donated their brains for research, a high proportion had neuropathological evidence of CTE, suggesting that CTE may be related to prior participation in football.
Supplementary Material
Key Points.
Question
What are the neuropathological and clinical features of a case series of deceased players of American football neuropathologically diagnosed as having chronic traumatic encephalopathy (CTE)?
Findings
In a convenience sample of 202 deceased players of American football from a brain donation program, CTE was neuropathologically diagnosed in 177 players across all levels of play (87%), including 110 of 111 former National Football League players (99%).
Meaning
In a convenience sample of deceased players of American football, a high proportion showed pathological evidence of CTE, suggesting that CTE may be related to prior participation in football.
Acknowledgments
Funding/Support: This study received support from NINDS (grants U01 NS086659, R01 NS078337, R56 NS078337, U01 NS093334, and F32 NS096803), the National Institute on Aging (grants K23 AG046377, P30AG13846 and supplement 0572063345-5, R01 AG1649), the US Department of Defense (grant W81XWH-13-2-0064), the US Department of Veterans Affairs (I01 CX001038), the Veterans Affairs Biorepository (CSP 501), the Veterans Affairs Rehabilitation Research and Development Traumatic Brain Injury Center of Excellence (grant B6796-C), the Department of Defense Peer Reviewed Alzheimer’s Research Program (grant 13267017), the National Operating Committee on Standards for Athletic Equipment, the Alzheimer’s Association (grants NIRG-15-362697 and NIRG-305779), the Concussion Legacy Foundation, the Andlinger Family Foundation, the WWE, and the NFL.
Footnotes
Author Contributions: Drs Mez and McKee had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. Drs Mez, Daneshvar, and Mr Kiernan are co–first authors.
Concept and design: Mez, Daneshvar, Abdolmohammadi, Murphy, Montenigro, Kowall, Cantu, Stern, McKee.
Acquisition, analysis, or interpretation of data: Mez, Daneshvar, Kiernan, Abdolmohammadi, Alvarez, Huber, Alosco, Solomon, Nowinski, McHale, Cormier, Kubilus, Martin, Murphy, Baugh, Montenigro, Chaisson, Tripodis, Weuve, McClean, Goldstein, Katz, Stern, Stein, McKee.
Drafting of the manuscript: Mez, Daneshvar, Abdolmohammadi, Alosco, Martin, Murphy, Montenigro, McKee.
Critical revision of the manuscript for important intellectual content: Mez, Daneshvar, Kiernan, Abdolmohammadi, Alvarez, Huber, Alosco, Solomon, Nowinski, McHale, Cormier, Kubilus, Baugh, Chaisson, Tripodis, Kowall, Weuve, McClean, Cantu, Goldstein, Katz, Stern, Stein, McKee.
Statistical analysis: Mez, Daneshvar, Abdolmohammadi, Huber, Montenigro, Tripodis, Weuve.
Obtained funding: Mez, Nowinski, McKee.
Administrative, technical, or material support: Daneshvar, Kiernan, Abdolmohammadi, Alvarez, Huber, Alosco, McHale, Cormier, Kubilus, Murphy, Baugh, Montenigro, Chaisson, Kowall, McClean, Stein, McKee.
Supervision: Mez, Daneshvar, Abdolmohammadi, Solomon, Cantu, Stern, McKee.
Conflict of Interest Disclosures: All authors have completed and submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Dr Nowinski reported that he receives travel reimbursements for various unpaid advisory roles from the NFL Players’ Association, Major League Lacrosse, World Wrestling Entertainment (WWE), National Collegiate Athletic Association (NCAA), and the Ivy League; receives royalties from the publication of his book Head Games: The Global Concussion Crisis, published by Head Games The Film; served as a consultant for MC10 Inc as recently as 2013; serves as chief executive officer of the Concussion Legacy Foundation; and receives speaking honoraria and travel reimbursements for educational lectures. Ms Baugh reported that she receives research funding through the NCAA and the Harvard Football Players Health Study, which is funded by the NFL Players’ Association. Dr Cantu reported that he receives compensation from the NFL as senior advisor to its Head, Neck and Spine Committee, from the National Operating Committee on Standards for Athletic Equipment as chair of its Scientific Advisory Committee and from the Concussion Legacy Foundation as cofounder and medical director for some talks given and receives royalties from Houghton Mifflin Harcourt and compensation from expert legal opinion. Dr Stern reported that he has received research funding from the NFL, the NFL Players’ Association, and Avid Radiopharmaceuticals Inc; is a member of the Mackey-White Committee of the NFL Players’ Association; is a paid consultant to Amarantus BioScience Holdings Inc, Avanir Pharmaceuticals Inc, and Biogen; and receives royalties for published neuropsychological tests from Psychological Assessment Resources Inc and compensation from expert legal opinion. Dr McKee reported that she has received funding from the NFL and WWE and is a member of the Mackey-White Committee of the NFL Players’ Association.
Role of the Funder/Sponsor: The funders of the study had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; or decision to submit for publication.
Additional Contributions: We acknowledge the use of the resources and facilities at VA Boston Healthcare System and the Edith Nourse Rogers Memorial Veterans Hospital (Bedford, Massachusetts). We also acknowledge the help of all members of the CTE Center at Boston University School of Medicine, Concussion Legacy Foundation, and the individuals and families whose participation and contributions made this work possible.
References
- 1.Corsellis JA, Bruton CJ, Freeman-Browne D. The aftermath of boxing. Psychol Med. 1973;3(3):270–303. doi: 10.1017/s0033291700049588. [DOI] [PubMed] [Google Scholar]
- 2.Hof PR, Knabe R, Bovier P, Bouras C. Neuropathological observations in a case of autism presenting with self-injury behavior. Acta Neuropathol. 1991;82(4):321–326. doi: 10.1007/BF00308819. [DOI] [PubMed] [Google Scholar]
- 3.Geddes JF, Vowles GH, Nicoll JA, Révész T. Neuronal cytoskeletal changes are an early consequence of repetitive head injury. Acta Neuropathol. 1999;98(2):171–178. doi: 10.1007/s004010051066. [DOI] [PubMed] [Google Scholar]
- 4.Omalu BI, DeKosky ST, Minster RL, Kamboh MI, Hamilton RL, Wecht CH. Chronic traumatic encephalopathy in a National Football League player. Neurosurgery. 2005;57(1):128–134. doi: 10.1227/01.neu.0000163407.92769.ed. [DOI] [PubMed] [Google Scholar]
- 5.Omalu BI, DeKosky ST, Hamilton RL, et al. Chronic traumatic encephalopathy in a National Football League player: part II. Neurosurgery. 2006;59(5):1086–1092. doi: 10.1227/01.NEU.0000245601.69451.27. [DOI] [PubMed] [Google Scholar]
- 6.McKee AC, Stern RA, Nowinski CJ, et al. The spectrum of disease in chronic traumatic encephalopathy. Brain. 2013;136(Pt 1):43–64. doi: 10.1093/brain/aws307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bieniek KF, Ross OA, Cormier KA, et al. Chronic traumatic encephalopathy pathology in a neurodegenerative disorders brain bank. Acta Neuropathol. 2015;130(6):877–889. doi: 10.1007/s00401-015-1502-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McKee AC, Cairns NJ, Dickson DW, et al. TBI/CTE group The first NINDS/NIBIB consensus meeting to define neuropathological criteria for the diagnosis of chronic traumatic encephalopathy. Acta Neuropathol. 2016;131(1):75–86. doi: 10.1007/s00401-015-1515-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stern RA, Daneshvar DH, Baugh CM, et al. Clinical presentation of chronic traumatic encephalopathy. Neurology. 2013;81(13):1122–1129. doi: 10.1212/WNL.0b013e3182a55f7f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mez J, Solomon TM, Daneshvar DH, et al. Assessing clinicopathological correlation in chronic traumatic encephalopathy: rationale and methods for the UNITE study. Alzheimers Res Ther. 2015;7(1):62. doi: 10.1186/s13195-015-0148-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Corrigan JD, Bogner J. Initial reliability and validity of the Ohio State University TBI identification method. J Head Trauma Rehabil. 2007;22(6):318–329. doi: 10.1097/01.HTR.0000300227.67748.77. [DOI] [PubMed] [Google Scholar]
- 12.Seichepine DR, Stamm JM, Daneshvar DH, et al. Profile of self-reported problems with executive functioning in college and professional football players. J Neurotrauma. 2013;30(14):1299–1304. doi: 10.1089/neu.2012.2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Robbins CA, Daneshvar DH, Picano JD, et al. Self-reported concussion history: impact of providing a definition of concussion. Open Access J Sports Med. 2014;5:99–103. doi: 10.2147/OAJSM.S58005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vonsattel JPG, Aizawa H, Ge P, et al. An improved approach to prepare human brains for research. J Neuropathol Exp Neurol. 1995;54(1):42–56. doi: 10.1097/00005072-199501000-00006. [DOI] [PubMed] [Google Scholar]
- 15.Vonsattel JPG, Del Amaya MP, Keller CE. Twenty-first century brain banking: processing brains for research: the Columbia University methods. Acta Neuropathol. 2008;115(5):509–532. doi: 10.1007/s00401-007-0311-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McKee AC, Cantu RC, Nowinski CJ, et al. Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J Neuropathol Exp Neurol. 2009;68(7):709–735. doi: 10.1097/NEN.0b013e3181a9d503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McKee AC, Gavett BE, Stern RA, et al. TDP-43 proteinopathy and motor neuron disease in chronic traumatic encephalopathy. J Neuropathol Exp Neurol. 2010;69(9):918–929. doi: 10.1097/NEN.0b013e3181ee7d85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Newell KL, Hyman BT, Growdon JH, Hedley-Whyte ET. Application of the National Institute on Aging (NIA)–Reagan Institute criteria for the neuropathological diagnosis of Alzheimer disease. J Neuropathol Exp Neurol. 1999;58(11):1147–1155. doi: 10.1097/00005072-199911000-00004. [DOI] [PubMed] [Google Scholar]
- 19.Montine TJ, Phelps CH, Beach TG, et al. National Institute on Aging; Alzheimer’s Association National Institute on Aging–Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol. 2012;123(1):1–11. doi: 10.1007/s00401-011-0910-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McKeith IG. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the Consortium on DLB International Workshop. J Alzheimers Dis. 2006;9(3 (suppl)):417–423. doi: 10.3233/jad-2006-9s347. [DOI] [PubMed] [Google Scholar]
- 21.Cairns NJ, Bigio EH, Mackenzie IR, et al. Consortium for Frontotemporal Lobar Degeneration Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol. 2007;114(1):5–22. doi: 10.1007/s00401-007-0237-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Litvan I, Hauw JJ, Bartko JJ, et al. Validity and reliability of the preliminary NINDS neuropathologic criteria for progressive supranuclear palsy and related disorders. J Neuropathol Exp Neurol. 1996;55(1):97–105. doi: 10.1097/00005072-199601000-00010. [DOI] [PubMed] [Google Scholar]
- 23.Dickson DW. Neuropathology of non-Alzheimer degenerative disorders. Int J Clin Exp Pathol. 2009;3(1):1–23. [PMC free article] [PubMed] [Google Scholar]
- 24.Bigio EH. Update on recent molecular and genetic advances in frontotemporal lobar degeneration. J Neuropathol Exp Neurol. 2008;67(7):635–648. doi: 10.1097/NEN.0b013e31817d751c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mackenzie IR, Neumann M, Bigio EH, et al. Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol. 2010;119(1):1–4. doi: 10.1007/s00401-009-0612-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brownell B, Oppenheimer DR, Hughes JT. The central nervous system in motor neurone disease. J Neurol Neurosurg Psychiatry. 1970;33(3):338–357. doi: 10.1136/jnnp.33.3.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Love S, Louis D, Ellison DW. Greenfield’s Neuropathology, 2-Volume Set. 8th. Boca Raton, FL: CRC Press; 2008. [Google Scholar]
- 28.Su JH, Cummings BJ, Cotman CW. Early phosphorylation of tau in Alzheimer’s disease occurs at Ser-202 and is preferentially located within neurites. Neuroreport. 1994;5(17):2358–2362. doi: 10.1097/00001756-199411000-00037. [DOI] [PubMed] [Google Scholar]
- 29.Stamm JM, Bourlas AP, Baugh CM, et al. Age of first exposure to football and later-life cognitive impairment in former NFL players. Neurology. 2015;84(11):1114–1120. doi: 10.1212/WNL.0000000000001358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stamm JM, Koerte IK, Muehlmann M, et al. Age at first exposure to football is associated with altered corpus callosum white matter microstructure in former professional football players. J Neurotrauma. 2015;32(22):1768–1776. doi: 10.1089/neu.2014.3822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Montenigro PH, Alosco ML, Martin BM, et al. Cumulative head impact exposure predicts later-life depression, apathy, executive dysfunction, and cognitive impairment in former high school and college football players. J Neurotrauma. 2017;34(2):328–340. doi: 10.1089/neu.2016.4413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Uryu K, Chen X-H, Martinez D, et al. Multiple proteins implicated in neurodegenerative diseases accumulate in axons after brain trauma in humans. Exp Neurol. 2007;208(2):185–192. doi: 10.1016/j.expneurol.2007.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stein TD, Montenigro PH, Alvarez VE, et al. Beta-amyloid deposition in chronic traumatic encephalopathy. Acta Neuropathol. 2015;130(1):21–34. doi: 10.1007/s00401-015-1435-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Crane PK, Gibbons LE, Dams-O’Connor K, et al. Association of traumatic brain injury with late-life neurodegenerative conditions and neuropathologic findings. JAMA Neurol. 2016;73(9):1062–1069. doi: 10.1001/jamaneurol.2016.1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cherry JD, Tripodis Y, Alvarez VE, et al. Microglial neuroinflammation contributes to tau accumulation in chronic traumatic encephalopathy. Acta Neuropathol Commun. 2016;4(1):112. doi: 10.1186/s40478-016-0382-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Haneuse S, Schildcrout J, Crane P, Sonnen J, Breitner J, Larson E. Adjustment for selection bias in observational studies with application to the analysis of autopsy data. Neuroepidemiology. 2009;32(3):229–239. doi: 10.1159/000197389. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.