

Abstract
The Melastatin-related Transient Receptor Potential member 7 (TRPM7) is a unique fusion protein with both ion channel function and enzymatic α-kinase activity. TRPM7 is essential for cellular systemic magnesium homeostasis and early embryogenesis, it promotes calcium transport during global brain ischemia and emerges as a key player in cancer growth. TRPM7 channels are negatively regulated through G-protein-coupled receptor-stimulation, either by reducing cellular cyclic adenosine monophosphate (cAMP) or depleting phosphatidylinositol bisphosphate (PIP2) levels in the plasma membrane. We here identify that heterologous overexpression of human TRPM7-K1648R mutant will lead to disruption of protease or purinergic receptor-induced calcium release. The disruption occurs at the level of Gq, which requires intact TRPM7 kinase phosphorylation activity for orderly downstream signal transduction to activate phospholipase (PLC)β and cause calcium release. We propose that this mechanism may support limiting GPCR-mediated calcium signaling in times of insufficient cellular ATP supply.
Keywords: TRPM7, α-kinase, heterotrimeric G-protein
INTRODUCTION
The Transient receptor potential melastatin 7 (TRPM7) is a member of the melastatin-related subfamily of TRP channels. The protein is unique in that it has both ion channel function and enzymatic α-kinase activity, with the ion transport domain in the N-terminal, and serine/threonine protein kinase domain in the C-terminal. TRPM7 is ubiquitously expressed and is essential for a variety of biological and physiological processes as well as early embryonic development (reviewed in [1]). TPRM7 protein promotes magnesium (Mg2+) transport into cells via its channel domain and is essential for cellular Mg2+ homeostasis. Importantly, TRPM7 is reported as a regulator of cell proliferation, apoptosis, and differentiation, and therefore TRPM7 is considered as a key player in various diseases that include brain ischemia and cancer.
TRPM7 is negatively regulated by intracellular Mg2+ and physiological amounts of adenosine triphosphate (Mg·ATP) [2–4]. A loss of TRPM7 kinase function leads to desensitization of TRPM7 to its intracellular inhibitors [5]. Enzymatic activity of the TRPM7 kinase is not required for the TRPM7 channel, rather, the binding of Mg·ATP to the kinase domain is required for phosphorylation of other protein targets. At the same time, the intrinsic kinase is a regulatory domain affecting TRPM7 ion conductance [5–7]. For example, TRPM7 activity is modulated by its kinase via protein kinase A (PKA) signaling, responding to changes in intracellular cyclic adenosine monophosphate (cAMP) levels through Gi and Gs-coupled receptors [8]. In addition, muscarinic type 1 (M1) G-protein-coupled receptor (GPCR) stimulation regulates TRPM7 through activating phospholipase (PLC)β via Gq. This leads to depletion of phosphatidylinositol 4,5-bisphosphate (PIP2) in the plasma membrane and inhibition of TRPM7 [9, 10]. Few phosphorylation substrates of the TRPM7 kinase are known, amongst them are annexin A1, eukaryotic elongation factor, myosin IIA, phospholipase C (PLC)β and PLCγ, as well as histones [1, 11].
Thrombin, a serine protease coagulation factor, binds to protease-activated receptor-1 (PAR-1), which is a member of a family of 4 GPCRs (reviewed in [12, 13]). This induces a conformational change in PAR-1 leading to the separation of the Gα and the Gβγ subunits of the G-protein and further downstream signaling. PAR-1 preferentially couples to Gq, leading to PLC-mediated inositol trisphosphate (InsP3) production and intracellular calcium (Ca2+) release. Thrombin can also bind to PAR-4. While PAR-1 causes fast and transient Ca2+ release, PAR-4 induces slowly increasing intracellular Ca2+ levels (reviewed in [14]). PARs are ubiquitously expressed throughout mammalian tissue, and the physiological role of particularly PAR-1 includes platelet aggregation, endothelial barrier disruption and smooth muscle cell proliferation [12]. PAR-1 is an emerging therapeutic target in cancer [13]. Trypsin, an alternate serine protease, primarily activates GPCRs through PAR-2 binding [15].
We previously reported that HEK293-TREx cells heterologously overexpressing mouse TRPM7 (mTRPM7) did not respond with a Ca2+ release transient to thrombin stimulation, although control cells did [8]. Here, we set out to investigate how human TRPM7 (hTRPM7) interferes with thrombin-induced receptor stimulation. We find that the phosphorylation activity of the hTRPM7 kinase regulates Gq, thereby controlling downstream signaling events of this heterotrimeric G-protein.
MATERIALS AND METHODS
Cell culture
Tetracycline (Tet)-inducible HEK293-TREx cells stably transfected with HA-tagged human TRPM7 wild type (hTRPM7) and the phosphotransferase activity-deficient point mutation in the hTRPM7 kinase domain, K1648R [5], were cultured in DMEM medium (Sigma, USA) containing 10% fetal bovine serum (FBS) (Corning, USA), blasticidin (5 μg/ml) (Gibco, USA), and zeocin (0.4 mg/ml) (Gibco). Overexpression was induced adding 1 μg/ml tetracycline (Gibco) to the culture medium. Patch-clamp current measurements were performed 0-20 hours post- tetracycline induction. Cells were maintained at 37°C under 95% air and 5% CO2 conditions.
Electrophysiology
For whole-cell patch clamp technique assessing overexpressed hTRPM7 in Figure 1 the extracellular solution contained (in mM): 140 Na-Cl, 2.8 KCl, 2 MgCl2, 1 CaCl2, 10 HEPES-NaOH, 10 Glucose (pH 7.2, 300 mOsm). External solutions for Figures 3&4 contained (in mM): 140 Na-Gluconate, 2.8 KCl, 2 MgSO4, 1 CaCl2, 10 HEPES-NaOH, 10 Glucose (pH 7.2, 300 mOsm). Intracellular pipette-filling solutions for Figure 1 contained (in mM): 120 K-glutamate, 8 NaCl, 0.3 Na·GTP, 200 μM K5-Fura-2 (Invitrogen, USA), 10 HEPES-KOH (pH 7.2, 300 mOsm). For Figures 3&4 intracellular solution contained (in mM): 120 K-glutamate, 8 NaCl, 2 MgCl2 (700 μM free Mg2+), 1.5 Mg·ATP, 0.3 Na·GTP, 200 μM K5-Fura-2, 10 HEPES-KOH (pH 7.2, 300mOsm). Additionally, for Figure 4, Na·GTP was replaced with 0.3 mM GTPγS. Free intracellular Mg2+ concentration was calculated with WebMaxC Standard. For thrombin (Sigma) application, we made stock solutions of 500 Units in Milli-Q-filtered water, diluted to 10, 20 or 30 Units in extracellular solution containing (in mM): 140 Na-Gluconate, 2.8 KCl, 2 MgSO4, 10 HEPES-NaOH (pH 7.2, 300mOsm). All salts were from Sigma, USA.
Figure 1. Heterologous overexpression of human TRPM7 inhibits thrombin-induced Ca2+ release.
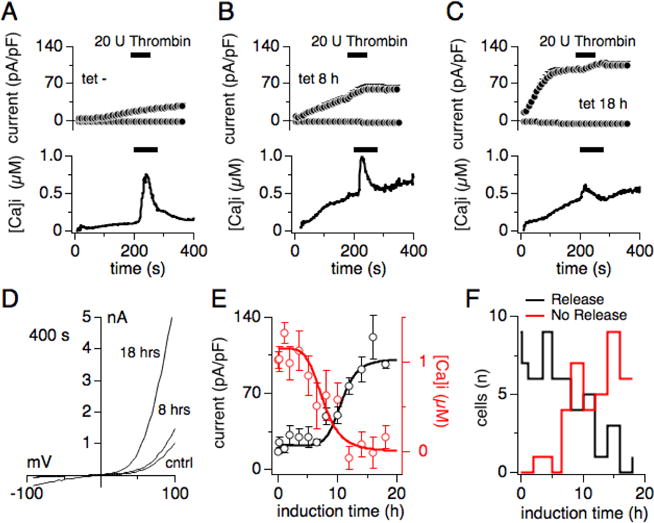
HEK293-TREx cells stably transfected with tetracycline-inducible human TRPM7 (hTRPM7; [5]) were subjected to combined voltage-clamp and Fura-2 measurements to assess whole-cell currents and intracellular Ca2+ changes. Voltage ramps were given from −100 mV to +100 mV over 50 ms. Current amplitudes were extracted at −80 mV and +80 mV, normalized to cell size in pF, averaged and plotted versus time of the experiment. As indicated by the black bars, 20 U thrombin, added to an external standard solution in 0 mM Ca2+, was applied to individual cells to evoke receptor-stimulated Ca2+ release. Ca2+ traces were averaged and plotted over time of the experiment. Internal solution was supplemented with 0.3 mM Na·GTP and 200 μM Fura-2, in the absence of any ATP supplementation. Error bars are S.E.M. (A) The graph depicts current development of endogenous TRPM7 in non-induced hTRPM7-HEK293-TREx cells (upper panel) and intracellular Ca2+ changes (lower panel) (n = 10). (B) Shown is the overexpressed hTRPM7 current development in hTRPM7-HEK293-TREx cells induced for 8 hours with tetracycline (upper panel) and intracellular Ca2+ changes (lower panel) (n = 11). (C) Same experimental setup as in (A) and (B), but here cells were tetracycline-induced for 18 hours (n = 7). (D) Current-voltage traces of TRPM7 currents extracted from representative cells that were either non-induced (cntrl), tetracycline-induced for 8 hrs and 18 hrs, and at 400 s into the experiment. (E) Dose-response curve of hTRPM7 expression and thrombin-induced Ca2+-release in dependence of induction time. Averaged peak currents (black circles) assessed at 400 s into the experiment are plotted against tetracycline induction time (n = 4-12). Red circles represent the maximal change in Ca2+ signal induced by extracellular thrombin application, and measured in the same cells (n = 4-12). (F) TRPM7 overexpression reduces the likelihood of thrombin-induced Ca2+ release. The graph plots the number of cells that either release (in black) or do not release (in red) Ca2+ in response to extracellular thrombin.
Figure 3. Thrombin-induced Ca2+ release requires intracellular Mg·ATP and a functional TRPM7 kinase, and is rescued by constitutively active heterotrimeric G-proteins.
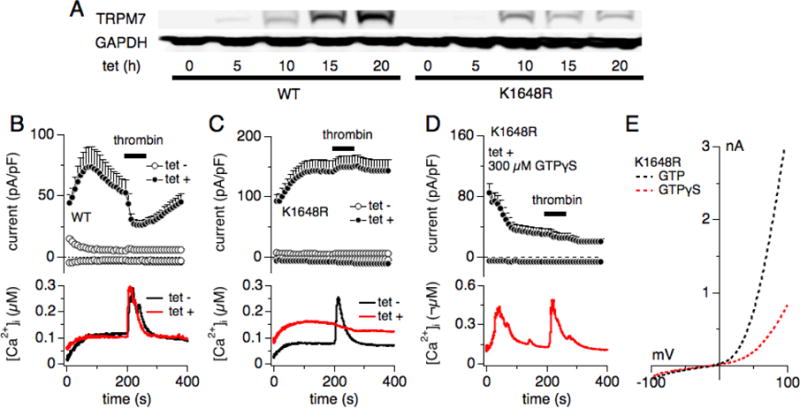
(A) Western blot of wild-type (WT) hTRPM7 expression levels and a TRPM7 mutant deficient in phosphotransferase activity (hTRPM7-K1648R; [5]) in HEK293-TREX cells non-induced or induced with 1μg/ml tetracycline at 5 h, 10 h, 15 h and 20 h exposure time. (B–E) Combined whole-cell patch clamp and Fura-2 experiments were performed on tetracycline-inducible HEK293-TREx cells overexpressing either WT hTRPM7 or hTRPM7-K1648R. Cells were either non-induced (tet−; control) or induced for 20 hrs with tetracycline (tet+). Application of 30 U thrombin in an external solution without Ca2+ was as indicated by the black bar. Error bars are S.E.M. (B) Upper panel: Normalized and averaged current development of overexpressed hTRPM7. Internal pipette solution was supplemented with 0.3 mM Na·GTP, 1.5 mM Mg·ATP, 2 mM MgCl2 (700 μM free Mg2+) and 200 μM Fura-2. These conditions suppressed endogenous TRPM7 current development (open circles, n = 12) and allowed inhibition of the overexpressed WT currents by 30 U thrombin (filled circles; n = 21). Lower panel: Averaged concomitant changes in intracellular Ca2+ concentration in response to thrombin application in non-induced hTRPM7-HEK293-TREx cells (black line) and induced for 20 hrs (red line). (C) Same experimental conditions as in (B), assessed for the hTRPM7-K1648R mutant (non-induced control n = 11, induced n = 33). Note both the absence of thrombin-induced current inhibition and Ca2+ release. (D) Combined whole-cell patch clamp and Fura-2 experiments were performed on tetracycline-inducible HEK293-TREx cells overexpressing hTRPM7-K1648R. Cells were induced for 20 hrs with tetracycline (tet +). Application of 30U thrombin in an external solution without Ca2+ as indicated by the black bar. Error bars are S.E.M. Upper panel: Normalized and averaged current development of overexpressed hTRPM7-K1648R. Internal pipette solution was supplemented with 0.3 mM Na·GTPγS, 1.5 mM Mg·ATP, 2 mM MgCl2 (700 μM free Mg2+) and 200 μM Fura-2. These conditions suppressed TRPM7-K1648R current development (filled circles, n = 23). Lower panel: Averaged concomitant changes in intracellular Ca2+ concentration in response to thrombin application. Note the recovery of thrombin-induced Ca2+ release. (E) Dotted traces represent averaged I/V curves of hTRPM7-K1648R mutant cells perfused with control internal solution (300 μM Na·GTP, 1.5 mM Mg·ATP, 700 μM free Mg2+) and extracted at 200 s (in black, n = 33), or perfused with 300 μM GPTγS in the internal solution, and at 200 s (in red; n = 23).
Figure 4. Protease- and purinergic receptor-induced Ca2+ release and TRPM7 kinase activity.
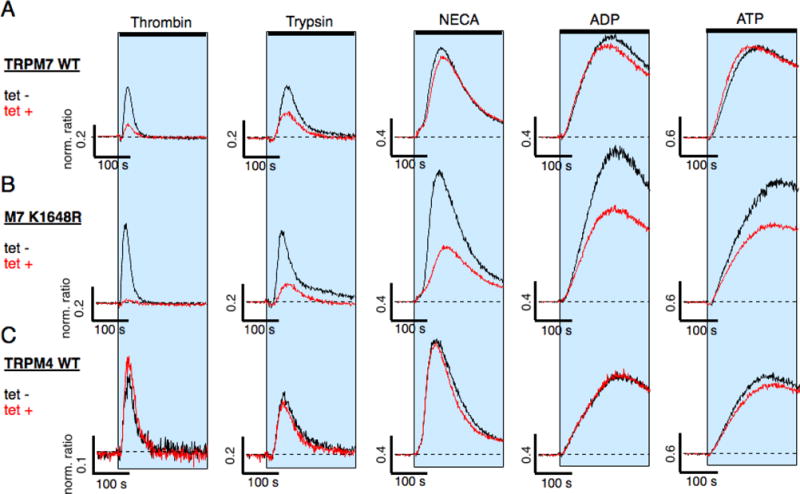
HEK293-TREx cells overexpressing either human wild-type TRPM7 (TRPM7 WT), a TRPM7 mutant deficient in phosphotransferase activity (M7-K1648R), or human wild-type TRPM4 (TRPM4 WT) were investigated for Ca2+ signals through protease-activated receptor stimulation using thrombin or trypsin, and metabotrophic purinergic-receptor stimulation using NECA, ADP or ATP. Ca2+ release was assessed using a fluorescent kinetic plate reader in the 384 well format (see methods). The 340 nm/380 nm ratio was normalized to one to the average of the 10 data points acquired before compound application in each of the 384 wells. The normalized ratio was then plotted against time of the experiment. The shaded areas indicate exposure to the respective agonists in extracellular solution absent of Ca2+: either thrombin (30 U), trypsin (100 nM), NECA (3 mM), ADP (3 mM) or ATP (3 mM) on (A) hTRPM7-WT (n = 4-8), p(B) hTRPM7-K1648R (n = 4-8), (C) hTRPM4 (n = 4). Cells were either non-induced (tet−; black line) or induced for 20 hrs with tetracycline (tet+, red line).
Patch clamp experiments were performed in the whole-cell configuration and combined with concomitant Fura-2 single cell Ca2+ imaging [16]. TRPM7 currents were elicited by a ramp protocol from −100 mV to +100 mV from a holding potential of 0 mV over 50 ms and acquired at 0.5 kHz. Inward current amplitudes over the course of the experiment were extracted at −80 mV, outward currents at +80 mV and plotted versus time. Data were normalized to cell capacitance measured immediately after whole-cell break-in as pA/pF. Capacitance was measured using the automated capacitance cancellation function of the EPC-9 (HEKA, Lambrecht, Germany). All values are given as mean ± standard error of mean (S.E.M). Patch pipettes (Sutter Instrument, USA) were pulled and polished, and had a tip resistance of 2-3 MΩ with the solutions used. For fluorescence measurements, the ratiometric Ca2+ dye Fura-2 was excited at 360 nm and 380 nm using an ITC-16 analog-digital board (Instrutech, USA), and X-Chart software (HEKA, Germany) controlling a monochromatic mirror grid (TILL Photonics, Germany). Fura-2 emission was captured using a custom-build photomultiplier tube from Hamamatsu Photonics, KK. Data were acquired at 5 Hz.
Western blot
To induce expression of HA-tagged human TRPM7 wild-type (hTRPM7) or K1648R mutant (hTRPM7-K1648R), 1 μg/mL tetracycline was added to the media for 5, 10, 15, 20 hrs. The cells were harvested and dissolved in RIPA buffer (25 mM Tris-HCl pH 7.4, 150 mM NaCl, 0.1% sodium dodecyl sulfate, 0.5% Triton X-100 and 0.5% sodium deoxycholate) containing a protease inhibitor cocktail (104 mM AEBSF, 80 μM Aprotinin, 4 mM Bestatin, 1.4 mM E-64, 2 mM Leupeptin and 1.5 mM Pepstatin A: Sigma-Aldrich, USA) for 30 min at 4 °C. Non-induced cells represent 0 hrs induction. After incubation for 30 mins, the lysates were centrifuged at 13,000 rpm for 15 min at 4 °C and the protein concentrations of the lysates were measured using a protein assay reagent (Bio-Rad Laboratories, USA). Soluble lysates were boiled with NuPAGE LDS sample buffer (Invitrogen, USA) at 98 °C for 5 min. Equal amounts of protein (100 μg) was loaded and separated by NuPAGE 3-8% gel (Invitrogen) and transferred to a PVDF membrane. Protein was detected by immunoblotting using the antibodies of rat monoclonal anti-HA (3F10, Roche, Switzerland), mouse monoclonal anti- GAPDH (6C5, Abcam, UK) and followed by treatment with IRDye infrared fluorescence-conjugated anti-rat and anti-mouse secondary antibodies (LI-COR, USA). GAPDH was evaluated as an internal control. Antibody-bound protein was visualized using Odyssey CLx Imaging System (LI-COR).
Fluorescent 96-well or 384-well kinetic plate reader assay
To detect the optimal thrombin concentration for receptor-stimulated calcium release, a state-of-the-art 96-well fluorescent kinetic plate reader was used (Hamamatsu FDSS-7000EX, Hamamatsu Photonics KK, Japan). Our TRPM7 screening assay developed for the FLEX station (Molecular Devices, USA; [17]) was adapted to the FDSS-7000EX the following way: hTRPM7-HEK293-TREx cells (100,000 cells/well) were plated in poly-L-lysine-coated black 96 well plates with clear bottom (Greiner, Monroe, NC) and induced by 1 μg/mL tetracycline after 5 hours of cell plating. After 18 hours of tetracycline post-induction, the culture medium was removed, replaced with Krebs-Ringer-HEPES (KRH) buffer supplemented with 2 μM Fura-2 AM (Invitrogen), 0.4% pluronic F-127 (Sigma-Aldrich) and incubated at 37°C for 1 hour. The plates were washed 3 times with KRH buffer after Fura-2 AM incubation, fresh KRH buffer was added, and plates were transferred to the FDSS-7000EX. Fura-2 was excited using 340 nm and 380 nm wavelengths and their ratio plotted versus time to observe changes in intracellular Ca2+ levels. Baseline fluorescence was measured for 1 min before application of thrombin (10, 20, 30 U, respectively), diluted in KRH buffer without CaCl2, and data acquisition continued for another 4 min. The emission signal was captured at 540 nm. KRH buffer bath solution contained (in mM): 135 NaCl, 5 KCl, 1 CaCl2, 1.5 MgCl2, 20 HEPES-NaOH, 5.6 Glucose, 2 probenecid (Sigma-Aldrich).
For the 384-well plate assay, hTRPM7-HEK293-TREx cells and hTRPM4 HEK293-TREx cells (20,000 cells/well) were plated in poly-L-lysine-coated black 384 well plates with clear bottom (Greiner) and induced by 1 μg/mL tetracycline after 5 hours of cell plating. After 20 hours of tetracycline induction, the culture medium was removed, replaced with DMEM containing 10% FBS supplemented with 2 μM Fura-2 AM, 0.4% pluronic F-127, 2 mM probenecid and incubated at 37°C for 1 hour. The plates were washed 3 times with standard external solution after Fura-2 AM incubation, fresh buffer added, and transferred to the FDSS-7000EX. Intracellular Ca2+ levels were measured by the same protocol of 96 well. Trypsin, NECA, ATP and ADP were diluted in standard external solution without Ca2+ and applied after 1 min of starting to measure intracellular Ca2+ by the FDSS-7000EX. Standard external solution contained (in mM): 140 NaCl, 2.8 KCl, 2 MgCl2, 20 HEPES-NaOH, 5.6 Glucose, 2 probenecid.
Statistical analysis
Patch-clamp data were acquired with PatchMaster software (HEKA, Germany) and exported to IGOR Pro (Wavemetrics, USA). Current amplitudes were extracted from IGOR Pro where all values for mean, standard deviation and standard error of the mean were calculated. In some cases p-values based on 2-tail, paired Student’s T-tests were calculated (IGOR Pro, Wavemetrics, USA).
RESULTS
Heterologous overexpression of human TRPM7 inhibits G-protein receptor-coupled Ca2+ release
Although the kinase domain of TRPM7 has been reported to bind phospholipase C (PLC), this interaction does not seem to influence TRPM7 channel activity per se. Rather, TRPM7 channel conductance is inhibited by depletion of phosphatidylinositol bisphosphate (PIP2) in the plasma membrane upon activation of PLCβ [9, 10, 18]. Since PLC does not seem to directly influence TRPM7 channel activity, and receptor-induced calcium (Ca2+) release appears altered in TRPM7-overexpressing cells [8], we wondered whether the PLC-kinase interaction might cause changes in receptor-stimulated Ca2+ signaling. We performed combined whole-cell patch-clamp and Fura-2 fluorescence experiments in HEK293-TREx cells overexpressing human TRPM7 (hTRPM7-HEK293) to assess changes in intracellular calcium [16]. hTRPM7-HEK293 were either left untreated or exposed to tetracycline for 10 min up to 18 hours before whole-cell patch clamp experiments (Fig. 1). The intracellular solution was supplemented with 0.3 mM guanosine triphosphate (Na·GTP) and 200 μM Fura-2, but did not contain any supplemented adenosine triphosphate (Mg·ATP) to maximize TRPM7 current amplitudes (see methods). TRPM7 currents were allowed to develop fully before application of 20 U thrombin (in the absence of extracellular Ca2+) between 200 s and 280 s. Non-induced hTRPM7-HEK293 cells developed endogenous TRPM7-like currents that were not inhibited by thrombin application, and responded to thrombin stimulation with a fast and large Ca2+ release transient from a flat baseline (Fig. 1A). Induction of hTRPM7 overexpression after 8 hrs (Fig. 1B) and 18 hrs (Fig. 1C) showed a steady increase in the intracellular Ca2+ baseline in parallel with increased hTRPM7 current amplitudes (Fig. 1D), and a clear, although diminished thrombin-induced Ca2+ transient. Plotting averaged peak TRPM7 currents (assessed at 400 s) and averaged peak Ca2+ release by thrombin over tetracycline induction time revealed an inverse relationship between hTRPM7 current magnitude and the ability of thrombin to cause significant Ca2+ release (Fig. 1E). Note that the difference of peak release amplitude (Fig. 1A lower panel; Fig. 1B lower panel) and first Ca2+ release data point (Fig. 1E) is because the peak release does not occur at exactly the same time in each cell. This broadens the release transient and reduces the apparent peak release. Maximal currents were obtained after 14 hrs induction time, whereas receptor-induced Ca2+ release was almost completely suppressed after 12 hrs of tetracycline exposure. In addition, the number of cells responding with Ca2+ release to thrombin stimulation also decreased with longer induction times (Fig. 1F). We conclude that overexpression of hTRPM7 perturbs thrombin-induced Ca2+ signaling.
Thrombin-induced Ca2+ release is dose-dependent
We next performed a dose-response experiment for thrombin using a fluorescent kinetic plate reader in the 96 well plate format allowing the measurement of fluorescence and see the real-time kinetic reactions of cells. Using the kinetic plate reader, we assessed thrombin-induced intracellular Ca2+ changes in intact hTRPM7-HEK293 cells (see methods; Fig. 2A). hTRPM7-HEK293 cells were either induced with tetracycline for 20 hrs (Fig. 2C) or not (Fig. 2B), and exposed to various concentrations of thrombin diluted in extracellular solution without Ca2+. Analyzing the normalized 340/380 nm ratio over time, Ca2+ release was impaired in hTRPM7 overexpressing cells compared to non-induced cells regardless of thrombin concentration (Fig. 2D) in analogy to our results obtained from combined whole-cell and Fura-2 patch-clamp experiments (Fig. 1). A quantitative peak amplitude analysis revealed a concentration of 30 U thrombin as optimal for the induction of Ca2+ release (Fig. 2D). These data also confirmed that overexpression of hTRPM7 leads to a statistically significant suppression of thrombin-induced changes in cellular Ca2+ regardless of agonist concentration (p < 0.05; Fig. 2D) suggesting that this effect is not mediated by changes in receptor expression or agonist sensitivity.
Figure 2. Thrombin-induced Ca2+ release is dose-dependent.
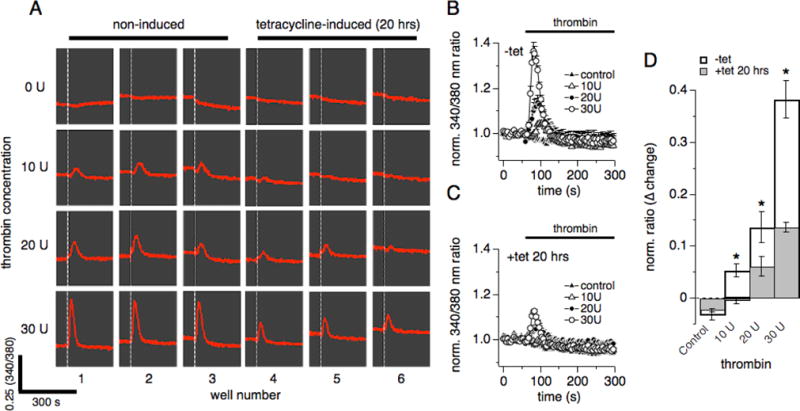
hTRPM7-HEK293-TREx cells were investigated for thrombin-induced Ca2+ release using a fluorescent kinetic plate reader in a 96 well format (see methods). Data capture proceeded at 1 Hz over 300 s. (A) Ratiometric raw data traces of Fura-2 emission signals (in red) measured in 24 individual wells on a 96 well plate over 300 s. Excitation wavelengths were 340 nm and 380 nm. The white dashed vertical bar in each well indicates time of application of either control (0 U), 10 U, 20 U or 30 U of thrombin, repeated in three wells each (1-3 for non-induced cells, 4-6 in tetracycline-induced cells for 20 hrs). (B) The 340 nm/380 nm ratio in non-induced hTRPM7-HEK293-TREx cells was normalized to one using the data points acquired before compound application in each of the 96 wells. Data were averaged and plotted against time of the experiment. Wells were exposed to either 30 U, 20 U, or 10 U thrombin in the absence of extracellular Ca2+, or superfused with external solution without thrombin (control) (n = 3, each condition). (C) Same experimental setup as in (B), however, cells had been induced with tetracycline for 20 hrs to overexpress hTRPM7 (n = 3, each concentration). (D) The graph plots the maximal change in the 340 nm/380 nm ratio induced by thrombin exposure. Data taken from panel (B) and (C) (*p < 0.05).
Thrombin-induced Ca2+ release requires both Mg·ATP and functional TRPM7 kinase activity
One alternative possible reason for the decreased Ca2+ release could be due to low intracellular Mg·ATP levels. In patch-clamped cells, the perfusion of Mg·ATP -free solutions will cause the defusional escape of ATP into the pipette (Fig. 1). In intact cells, the constitutive activity of TRPM7 could increase ATP consumption due to enhanced Ca2+ pump activity to remove the TRPM7-mediated Ca2+ influx (Fig. 2). We therefore repeated the experiment shown in Fig. 1 while adding 1.5 mM Mg·ATP to the intracellular pipette solution (see methods). Interestingly, addition of Mg·ATP to the pipette solution during whole-cell patch clamp and combined Fura-2 experiments had three effects on cells overexpressing hTRPM7: i) it restored the thrombin-mediated Ca2+ release responses in 15/21 cells (71%), similar to non-induced cells (83%, or 10/12 cells; Table 1); ii) it completely restored the magnitude of thrombin-induced Ca2+ release signals, even at 20 hrs of tetracycline induction (Fig. 3B lower panel); iii) thrombin application now also inhibited hTRPM7 currents (Fig. 3B upper panel).
Table 1.
Thrombin-induced Ca2+ release in HEK T-REx overexpressing hTRPM7 or hTRPM7-K1648R
| TRPM7 | Tetracycline Induction time | Supplementation | No. of cells performed patch-clamp | No. of cells with Ca2+ release (%) | No. of cells without Ca2+ release (%) | Average of Ca2+·release (nM) |
|---|---|---|---|---|---|---|
| WT | 0 h | Na• GTP | 12 | 10 (83.3) | 2 (16.7) | 161.7 |
| 20 h | Na• GTP | 21 | 15 (71.4) | 6 (28.6) | 177.3 | |
| K1648R | 0 h | Na• GTP | 11 | 11 (100) | 0 (0) | 176.4 |
| 20 h | Na• GTP | 34 | 1 (2.9) | 33 (97.1) | 0 | |
| 20 h | GTPγS | 23 | 16 (70.0) | 7 (30.0) | 316.2 |
Since the TRPM7 atypical kinase domain requires Mg·ATP for its phosphorylation activity [7, 19], we asked whether a functional TRPM7 kinase domain is required for thrombin-induced Ca2+ release. To this end we investigated HEK293-TREx cells overexpressing hTRPM7-K1648R, which is a single-point mutation affecting the catalytic site of the kinase domain [5]. As shown by Western blot in Figure 3A, tetracycline exposure induced overexpression of both hTRPM7 wild-type and hTRPM7-K1648R protein in a time-dependent manner (Fig. 3A). Both protein species of TRPM7 increased in expression levels after 5 hours of induction. In a next step, we investigated hTRPM7-K1648R with combined whole-cell patch-clamp and Fura-2 techniques using the same conditions as for hTRPM7 in Figure 3B. Here, both TRPM7 current-inhibition and thrombin-induced Ca2+ release were abolished in overexpressed hTRPM7-K1648R cells [5] (Fig. 3C). Only one out of the 34 cells investigated retained thrombin-induced Ca2+ release and concomitant current inhibition (Table 1). In addition, intracellular basal Ca2+ levels were higher in the hTRPM7-K1648R mutant (Fig. 3C lower panel), possibly because these channels are less sensitive to intracellular Mg2+ and Mg·ATP and therefore this mutant is constitutively more active than wild type channels [5]. We conclude that thrombin-induced Ca2+ release requires intact TRPM7 kinase activity.
GTPγS rescues thrombin-induced Ca2+ release independent of TRPM7 kinase activity
Thrombin stimulates protease-activated receptors (PAR1, PAR3 and PAR4), which belong to the seven-transmembrane G protein-coupled family of receptors (GPCRs; reviewed in [12]). Thrombin-stimulation activates the phosphatidylinositol-signaling pathway through coupling to Gq. Therefore, the absence of thrombin-induced Ca2+ release in overexpressed TRPM7-K1648R mutants could be due malfunction of any step in this signaling cascade, including perturbed refilling of intracellular Ca2+ stores. To probe whether the signaling defect seen in hTRPM7 overexpressing cells was due to dysfunctional execution within the GPCR signaling cascade or interference with intracellular Ca2+ store content, we took advantage of GTPγS (guanosine 5′-O-[gamma-thio] triphosphate), a non-hydrolysable heterotrimeric G protein-activating analog of guanosine triphosphate (GTP) [20]. Replacing 300 μM Na·GTP with 300 μM GTPγS in combined whole-cell patch clamp and Fura-2 experiments using hTRPM7-K1648R overexpressing HEK293-TREx cells (20 hrs induction time) caused both inhibition of hTRPM7 mutant currents (Fig. 3D upper panel, 3E) as well as early Ca2+ oscillations (Fig. 3D lower panel). Furthermore, additional stimulation of these cells with 30 U thrombin in Ca2+-free extracellular solution (see methods) was now able to cause significant Ca2+ release (Fig. 3D). Thus, GTPγS is able to fully overcome the Ca2+ release deficiency observed with kinase-mute hTRPM7-K1648R cells.
Protease- and purinergic receptor-induced Ca2+ release and TRPM7 kinase activity
Heterotrimeric G protein complexes are activated by a variety of GPCRs, including both PARs through e.g. thrombin and trypsin, and metabotrophic purinergic receptors through ATP, adenosine diphosphate (ADP) or Adenosine (reviewed in [21]). We wished to know whether the TRPM7 kinase specifically regulates thrombin-induced Ca2+ release, or whether other GPCRs known to interact with Gq would be affected as well. To this end we resorted to our fluorescent kinetic plate reader using the 384-well plate format to assess Ca2+ signals in intact HEK293 cells that were either non-induced or induced with tetracycline for 20 hrs to overexpress either wild-type hTRPM7 (hTRPM7-HEK293) or the hTRPM7-K1648R mutant. Overexpression of human TRPM4 (hTRPM4) [16], another melastatin family member, in HEK293-TRex induced by tetracycline for 20 hours served as control to account for possible overexpression artifacts. To prevent unwanted artifacts due to Ca2+ influx, extracellular Ca2+ was absent from both the standard extracellular solution and the application solution containing the appropriate receptor agonist (30 U thrombin, 100 nM trypsin, 3 mM N-ethylcarboxamido adenosine (NECA), an adenosine receptor agonist, ATP or ADP; see methods; Fig. 4). Analysis of the normalized 340/380 nm ratio under these conditions showed that of the additionally tested agonists only the protease receptor agonist trypsin had diminished Ca2+ release when overexpressing hTRPM7 (Fig. 4A, 4C). Importantly, hTRPM4 overexpression did not interfere with Ca2+ release by any of the tested agonists (Fig. 4C). However, Ca2+ was strongly reduced in overexpressing hTRPM7-K1648R cells when stimulating either purinergic GPCRs or protease-sensitive PARs. We conclude that an intact catalytic site within the hTRPM7 kinase domain is needed to provide feedback regulation for protease- and purinergic GPCR signaling.
DISCUSSION
We here set out to identify how TRPM7 regulates heterotrimeric G protein-induced Ca2+ release. We find that heterologous overexpression of human TRPM7 will lead to disruption of thrombin-induced Ca2+ release unless an adequate intracellular Mg·ATP supply is guaranteed. The disruption occurs at the level of the heterotrimeric G-protein Gq, which requires intact TRPM7 kinase phosphorylation activity for orderly downstream signal transduction resulting in Ca2+ release induced by both protease- and purinergic agonists. We propose that this mechanism may contribute to the limitation of GPCR-mediated Ca2+ signaling in times of insufficient cellular ATP supply.
TRPM7 is a constitutively active divalent cation channel with an enzymatically functional serine/threonine kinase domain. It is well known that free Mg2+ and Mg·nucleotides, in particular Mg·ATP, suppress TRPM7 channel activity at physiologically relevant levels. The Mg·ATP binding site of the kinase domain has been well characterized (reviewed in [1]). Because of the inhibitory action of Mg·ATP, most whole-cell patch clamp experiments are conducted in the absence of both Mg2+ and nucleotides to assure maximal current activation and reduce the signal-to-noise ratio. Both Mg·ATP and Mg·GTP are similarly effective in suppressing TRPM7 currents, however, physiologically significant Mg·GTP levels required for functional G-protein activation are in the lower millimolar range (~0.4 mM; [22]), and therefore unlikely to significantly interfere with TRPM7 currents at those concentrations. However, overexpression of TRPM7 may lead to significant constitutive Ca2+ entry triggering Ca2+ pump activity and ATP consumption under the plasma membrane domain in intact cells.
Store-operated calcium entry is an important signaling mechanism in most immune and other electrically non-excitable cells (SOCE, reviewed in [23]). Although TRPM7 is not a store-operated protein itself, it is synergistically interlaced with SOCE in the regulation of intracellular calcium homeostasis [24]. Experiments in DT40 B lymphocytes carrying various TRPM7 or Ca2+-release activated Ca2+ (CRAC) channel mutations show that the TRPM7 kinase domain negatively regulates SOCE, possibly through stromal interaction molecule 2 (STIM2). In addition, the TRPM7 knock-out experiments show that the channel domain participates in the maintenance of endoplasmic reticulum Ca2+ concentrations in resting cells [24]. While the former mechanism contributes to receptor-mediated Ca2+ signaling, the latter is a homeostatic mechanism independent of receptor stimulation. In primary mouse mast cells, the TRPM7 kinase domain, but not the channel domain, appears to be involved in the regulation of GPCR-mediated stimulation in that it sets the Ca2+ sensitivity of G protein-triggered mast cell degranulation and is essential for GPCR-activated histamine release [25].
The need for an active TRPM7 kinase domain has also been reported for Gi/o, suppressing TRPM7 channel activity through modulating intracellular cyclic adenosine monophosphate (cAMP) and protein kinase A (PKA) [8]. In addition, we observed that overexpressing heterologous mouse TRPM7 (mTRPM7) in HEK293-TREx cells (16-22 hours tetracycline induction) suppressed thrombin-induced Ca2+ release without significantly affecting the filling status of intracellular Ca2+ stores [8]. We therefore assessed the time course of this effect in overexpressed human TRPM7 (hTRPM7) using combined whole-cell patch-clamp and fluorimetric Ca2+ measurements and made three important observations (Figure 1): (1) the proportion of cells responding to thrombin-stimulation decreased as a function of tetracycline-induced expression of hTRPM7; (2) the Ca2+ release transients in responding cells and the increases in hTRPM7 currents were also temporally correlated and proceeded in parallel; and (3) basal Ca2+ levels increased as hTRPM7 currents developed over induction time. Similar observations were made when stimulating intact cells using a 96-well kinetic plate reader to assess Ca2+ signals induced by increasing concentrations of receptor agonist (Figure 2). Here, however, Ca2+ basal levels did not increase over time, presumably because of the cell’s capacity to produce and maintain a certain level of intracellular ATP assuring working plasma membrane (PM) and endoplasmic reticulum (ER) Ca2+-ATPases and better-controlled TRPM7 channel activity.
Clearly, in the case of thrombin stimulation, ATP availability is critical, as was confirmed in combined whole-cell patch clamp and Fura-2 experiments in overexpressing hTRPM7-HEK293 cells: Adding both GTP and ATP to the internal pipette solution even after 20 hrs of tetracycline induction not only restored thrombin-induced Ca2+ release to control levels, but also inhibited hTRPM7 currents upon agonist exposure (Figure 3). hTRPM7 inhibition only occurred in cells that were still able to generate a Ca2+ transient (15/21 cells; Table 1). hTRPM7 currents in cells that did not respond to thrombin were not inhibited (6/21 cells, Table 1), likely because of the break in signal transduction prior to activation of PLCβ and thereby preventing the reduction in PIP2 levels in the vicinity of TRPM7 channels [9, 10]. We previously reported that thrombin had no inhibitory effect on native TRPM7-like currents [8], however, these data were performed in the absence of intracellular Mg·ATP to allow full development of the small native currents. Adding Mg·ATP (1.5 mM) to the intracellular solution completely suppressed native hTRPM7 currents in non-induced HEK293 cells, and therefore thrombin-induced inhibition could not be assessed (Figure 3).
The requirement of ATP to compensate for a loss of Ca2+ release when overexpressing hTRPM7 indicates involvement of the TRPM7 kinase domain in the process. Indeed, cells overexpressing a TRPM7 mutant with a point mutation that eliminates phosphorylation activity (hTRPM7-K1648R; [5]), completely lacked intracellular Ca2+ release (33/34 cells; Table 1; Figure 3C). Ca2+ signaling can be inhibited at several steps of the GPCR signaling cascade. For example, desensitization of GPCRs can occur through phosphorylation by protein kinase C (PKC) or G protein-coupled receptor kinases (GRK), as well as allosteric binding of β-arrestin [26, 27]. However, suppression of thrombin receptor PAR-1 function by TRPM7 kinase phosphorylation seems unlikely, since hTRPM7-K1648R cells responded with suppressed Ca2+ release regardless of which metabotropic GPCR was activated (Fig. 4A & 4B). In addition, the reduction in GPCR-induced Ca2+ release does not seem to be an tetracycline-induced overexpression artifact as heterologous overexpression of human TRPM4, another protein of the TRPM family [16], did not alter its Ca2+ release signal to either of the agonist applied (Fig. 4C). Finally, irreversibly activating G-proteins with the non-hydrolysable GTP-analog GTPγS (reviewed in [28]) not only caused an initial Ca2+ response in whole-cell patch clamp experiments, but also produced significant Ca2+ transients by a subsequent thrombin-receptor stimulation (Fig. 3D). The latter also indicates that Ca2+ stores properly refill through sarco-endoplasmic reticulum ATP (SERCA) pumps. Similarly, PLCβ and inositol triphosphate receptors (IP3R) appear fully functional in generating and responding to InsP3 production (Fig. 3D).
Heterotrimeric G-protein activation rests upon the ability of its Gα subunit to exchange GDP for GTP. This process is limited by Gα‘s intrinsic GTPase function that returns the G protein to its inactive state through GTP hydrolysis. GTPase activity, and with this Gq inactivation, is strongly accelerated by binding to regulating effectors like GTPase-activating proteins (GAPs) and G protein-coupled receptor kinases (GRK) (reviewed in [29]). The binding of these modulators is greatly facilitated by protein kinase A (PKA), which phosphorylates serine residues on regulators of G protein stimulation (RGS), a GAP member protein family, and GRK [30]. Serine phosphorylation of RGS proteins inhibits their ability to turn off GTPase-activity related to Gq [31]. Since both the TRPM7 kinase phosphorylation activity needs to be preserved and, at least for thrombin receptors, sufficient ATP is needed to assure intact Gq signaling, GAPs represent a plausible downstream target of TRPM7. Phospholipase C is a dual-function protein acting as lipase and as GAP [29], and both PLCβ and PLCγ are known interaction partners of the TRPM7 serine/threonine kinase [9, 18]. Although PLCγ is not involved in GPCR signaling, it is known that TRPM7 kinase specifically phosphorylates PLCγ residues S1164 and T1045, which both are relevant in chicken B lymphocyte receptor signaling [18]. However, similar information awaits further study in regards to PLCβ.
Based on these observations, we propose that heterotrimeric G-proteins are targets of TRPM7 kinase phosphorylation activity, either directly, or through modulators of GTP hydrolysis.
Acknowledgments
This work was partially supported by National Institutes of Health NIGMS P01 GM078195 (AF), Hamamatsu/Queen’s PET Imaging, LLC (SS), and DFG LI1750/4-1 (AL). We thank Dr. Clay Wakano from the Queen’s High-Throughput Drug Screening Laboratory for providing access to and support for the 96/384-well kinetic plate reader assay partially supported by Hamamatsu/Queen’s PET Imaging, LLC (AF).
Footnotes
ETHICAL STANDARDS
The experiments comply with the current laws of the United States of America, where they were performed.
CONFLICT OF INTEREST
All authors declare no conflict of interests or competing financial interests.
References
- 1.Fleig A, Chubanov V. TRPM7. Handb Exp Pharmacol. 2014;222:521–546. doi: 10.1007/978-3-642-54215-2_21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kerschbaum HH, Kozak JA, Cahalan MD. Polyvalent cations as permeant probes of MIC and TRPM7 pores. Biophys J. 2003;84:2293–305. doi: 10.1016/S0006-3495(03)75035-8. doi: S0006-3495(03)75035-8 [pii] 10.1016/S0006-3495(03)75035-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nadler MJ, Hermosura MC, Inabe K, et al. LTRPC7 is a Mg.ATP-regulated divalent cation channel required for cell viability. Nature. 2001;411:590–5. doi: 10.1038/35079092. doi: Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S. [DOI] [PubMed] [Google Scholar]
- 4.Demeuse P, Penner R, Fleig A. TRPM7 channel is regulated by magnesium nucleotides via its kinase domain. J Gen Physiol. 2006;127:421–34. doi: 10.1085/jgp.200509410. Research Support, N.I.H., Extramural. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schmitz C, Perraud AL, Johnson CO, et al. Regulation of vertebrate cellular Mg2+ homeostasis by TRPM7. Cell. 2003;114:191–200. doi: 10.1016/s0092-8674(03)00556-7. doi: S0092867403005567 [pii] [DOI] [PubMed] [Google Scholar]
- 6.Matsushita M, Kozak JA, Shimizu Y, et al. Channel function is dissociated from the intrinsic kinase activity and autophosphorylation of TRPM7/ChaK1. J Biol Chem. 2005;280:20793–803. doi: 10.1074/jbc.M413671200. doi: Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S. [DOI] [PubMed] [Google Scholar]
- 7.Yamaguchi H, Matsushita M, Nairn AC, Kuriyan J. Crystal structure of the atypical protein kinase domain of a TRP channel with phosphotransferase activity. Mol Cell. 2001;7:1047–57. doi: 10.1016/s1097-2765(01)00256-8. [DOI] [PubMed] [Google Scholar]
- 8.Takezawa R, Schmitz C, Demeuse P, et al. Receptor-mediated regulation of the TRPM7 channel through its endogenous protein kinase domain. Proc Natl Acad Sci U S A. 2004;101:6009–14. doi: 10.1073/pnas.0307565101. doi: Research Support, U.S. Gov’t, P.H.S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Runnels LW, Yue L, Clapham DE. TRP-PLIK, a bifunctional protein with kinase and ion channel activities. Science. 2001;291:1043–7. doi: 10.1126/science.1058519. [DOI] [PubMed] [Google Scholar]
- 10.Runnels LW, Yue L, Clapham DE. The TRPM7 channel is inactivated by PIP(2) hydrolysis. Nat Cell Biol. 2002;4:329–36. doi: 10.1038/ncb781. doi: Research Support, Non-U.S. Gov’t. [DOI] [PubMed] [Google Scholar]
- 11.Krapivinsky G, Krapivinsky L, Manasian Y, Clapham DE. The TRPM7 chanzyme is cleaved to release a chromatin-modifying kinase. Cell. 2014;157:1061–1072. doi: 10.1016/j.cell.2014.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhao P, Metcalf M, Bunnett NW. Biased signaling of protease-activated receptors. Front Endocrinol. 2014;5:67. doi: 10.3389/fendo.2014.00067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bar-Shavit R, Maoz M, Kancharla A, et al. Protease-activated receptors (PARs) in cancer: Novel biased signaling and targets for therapy. Methods Cell Biol. 2016;132:341–358. doi: 10.1016/bs.mcb.2015.11.006. [DOI] [PubMed] [Google Scholar]
- 14.Sidhu TS, French SL, Hamilton JR. Differential signaling by protease-activated receptors: implications for therapeutic targeting. Int J Mol Sci. 2014;15:6169–6183. doi: 10.3390/ijms15046169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Coughlin SR, Camerer E. PARticipation in inflammation. J Clin Invest. 2003;111:25–27. doi: 10.1172/JCI17564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Launay P, Fleig A, Perraud AL, et al. TRPM4 is a Ca2+-activated nonselective cation channel mediating cell membrane depolarization. Cell. 2002;109:397–407. doi: 10.1016/s0092-8674(02)00719-5. doi: S0092867402007195 [pii] [DOI] [PubMed] [Google Scholar]
- 17.Castillo B, Porzgen P, Penner R, et al. Development and optimization of a high-throughput bioassay for TRPM7 ion channel inhibitors. J Biomol Screen. 2010;15:498–507. doi: 10.1177/1087057110368294. doi: 1087057110368294 [pii] 10.1177/1087057110368294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deason-Towne F, Perraud A-L, Schmitz C. Identification of Ser/Thr phosphorylation sites in the C2-domain of phospholipase C γ2 (PLCγ2) using TRPM7-kinase. Cell Signal. 2012;24:2070–2075. doi: 10.1016/j.cellsig.2012.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ryazanova LV, Dorovkov MV, Ansari A, Ryazanov AG. Characterization of the protein kinase activity of TRPM7/ChaK1, a protein kinase fused to the transient receptor potential ion channel. J Biol Chem. 2004;279:3708–16. doi: 10.1074/jbc.M308820200. [DOI] [PubMed] [Google Scholar]
- 20.Strange PG. Use of the GTPγS ([35S]GTPγS and Eu-GTPγS) binding assay for analysis of ligand potency and efficacy at G protein-coupled receptors. Br J Pharmacol. 2010;161:1238–1249. doi: 10.1111/j.1476-5381.2010.00963.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.D’ Ambrosi N, Volonte C. Metabotropic purinergic receptors in lipid membrane microdomains. Curr Med Chem. 2013;20:56–63. [PubMed] [Google Scholar]
- 22.Traut TW. Physiological concentrations of purines and pyrimidines. Mol Cell Biochem. 1994;140:1–22. doi: 10.1007/BF00928361. [DOI] [PubMed] [Google Scholar]
- 23.Putney JW, Steinckwich-Besançon N, Numaga-Tomita T, et al. The functions of store-operated calcium channels. Biochim Biophys Acta. 2016 doi: 10.1016/j.bbamcr.2016.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Faouzi M, Kilch T, Horgen FD, et al. The TRPM7 channel kinase regulates store-operated calcium entry. J Physiol. doi: 10.1113/JP274006. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zierler S, Sumoza-Toledo A, Suzuki S, et al. TRPM7 kinase activity regulates murine mast cell degranulation. J Physiol. 2016;594:2957–2970. doi: 10.1113/JP271564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jung S-R, Seo JB, Deng Y, et al. Contributions of protein kinases and β-arrestin to termination of protease-activated receptor 2 signaling. J Gen Physiol. 2016;147:255–271. doi: 10.1085/jgp.201511477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Park JY, Lee SY, Kim HR, et al. Structural mechanism of GPCR-arrestin interaction: recent breakthroughs. Arch Pharm Res. 2016;39:293–301. doi: 10.1007/s12272-016-0712-1. [DOI] [PubMed] [Google Scholar]
- 28.Casey PJ, Gilman AG. G protein involvement in receptor-effector coupling. J Biol Chem. 1988;263:2577–2580. [PubMed] [Google Scholar]
- 29.Litosch I. Decoding Gαq signaling. Life Sci. 2016;152:99–106. doi: 10.1016/j.lfs.2016.03.037. [DOI] [PubMed] [Google Scholar]
- 30.Huang J, Zhou H, Mahavadi S, et al. Inhibition of Galphaq-dependent PLC-beta1 activity by PKG and PKA is mediated by phosphorylation of RGS4 and GRK2. Am J Physiol Cell Physiol. 2007;292:C200–208. doi: 10.1152/ajpcell.00103.2006. [DOI] [PubMed] [Google Scholar]
- 31.Brass LF, Ma P. Applying the brakes to platelet activation. Blood. 2012;119:3651–3652. doi: 10.1182/blood-2012-02-406629. [DOI] [PubMed] [Google Scholar]