

SUMMARY
Adrenergic stimulation of brown adipose tissue (BAT) induces acute and long-term responses. The acute adrenergic response activates thermogenesis by uncoupling oxidative phosphorylation and enabling increased substrate oxidation. Long-term, adrenergic signaling remodels BAT, inducing adaptive transcriptional changes that expand thermogenic capacity. Here, we show that the estrogen-related receptors alpha and gamma (ERRα, ERRγ) are collectively critical effectors of adrenergically stimulated transcriptional reprogramming of BAT. Mice lacking adipose ERRs (ERRαγAd−/−) have reduced oxidative and thermogenic capacity and rapidly become hypothermic when exposed to cold. ERRαγAd−/− mice treated long term with a β3-adrenergic agonist fail to expand oxidative or thermogenic capacity and do not increase energy expenditure in response to norepinephrine (NE). Furthermore, ERRαγAd−/− mice fed a high-fat diet do not lose weight or show improved glucose tolerance when dosed with β3-adrenergic agonists. The molecular basis of these defects is the finding that ERRs mediate the bulk of the transcriptional response to adrenergic stimulation.
Graphical abstract
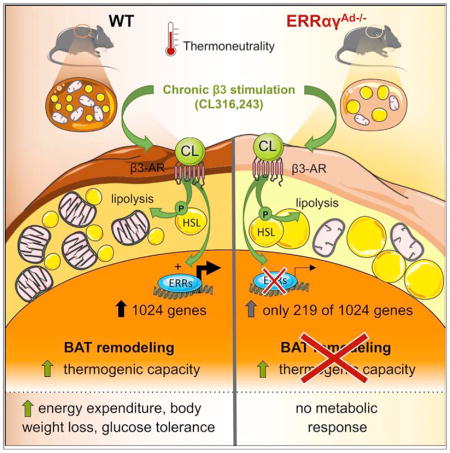
INTRODUCTION
Brown adipose tissue (BAT) specializes in the regulated production of heat, also known as “adaptive” or “non-shivering” thermogenesis (Cannon and Nedergaard, 2004). To fulfill this function, BAT is rich in mitochondria and expresses the mitochondrial uncoupling protein 1 (Ucp1). Upon exposure to cold, local release of the adrenergic agonist norepinephrine (NE) induces thermogenesis by activating Ucp1, which generates heat by dissipating the mitochondrial proton gradient and allowing high rates of substrate oxidation. The capacity of BAT to convert stored chemical energy to heat is important for the defense of body temperature in cold environments and may also serve to expend energy at times of caloric excess, as in “diet-induced thermogenesis,” and thus counteract obesity (Cannon and Nedergaard, 2004). Importantly, the thermogenic capacity of BAT shows plasticity: it can be expanded in response to repeated or prolonged exposure to cold or attenuated and even lost in response to thermoneutral environments, aging, obesity, or denervation of the tissue (Dulloo and Miller, 1984; Ouellet et al., 2011; Saito et al., 2009; Yoneshiro et al., 2011). Although it is well recognized that adrenergic stimulation is the main signal that drives the expansion of thermogenic capacity, the transcription factors that transform this signal into long-term adaptive changes in energy expenditure remain poorly defined.
The estrogen-related receptors (ERRα, ERRβ, and ERRγ) are closely related members of the nuclear receptor family. They have no known endogenous ligands and, despite their name, do not bind estrogen (Giguere et al., 1988). Several features of ERRs suggest that these receptors could play important roles in the adrenergic-driven remodeling of BAT. First, they are highly and preferentially expressed in BAT vs. white adipose tissue (WAT), with ERRα being the most abundant, followed by ERRγ and then ERRβ (Bookout et al., 2006; Gantner et al., 2014). Second, ERRs are strongly activated by PGC-1α and Gadd45γ, two regulators that are induced by cold in BAT (Gantner et al., 2014; Puigserver et al., 1998). Finally, ERRs directly regulate the transcription of genes important for oxidative and thermogenic capacity. ERRα controls genes encoding factors of mitochondrial biogenesis, tricarboxylic acid (TCA) cycle, lipid oxidation, and angiogenesis (Arany et al., 2008; Huss et al., 2004, 2015; Mootha et al., 2004; Schreiber et al., 2004). ERRγ shares many ERRα targets and when overexpressed can induce Ucp1 expression and oxidative capacity in brown adipocytes (Dixen et al., 2013; Dufour et al., 2007; Gantner et al., 2014). However, whole-body ERRα KO mice show no defects in the transcriptional response to adrenergic stimulation, even though they have decreased BAT mitochondrial content and are sensitive to cold (Luo et al., 2003; Villena et al., 2007). The physiological roles of ERRγ or ERRβ in BAT are not known.
To address the role of adipose ERRs in BAT physiology and specifically the transcriptional remodeling that takes place in response to adrenergic stimulation, we have generated adipose tissue-specific knockout mice lacking different combinations of ERRs. Because young mice grown at regular vivarium temperatures need BAT function to effectively defend their body temperature, we have bred and raised WT and ERRAd−/− at thermoneutrality. This approach minimizes confounding factors such as increased adrenergic tone in mice that have BAT defects (Schulz et al., 2013). Here, we show that mice lacking either ERRα alone or both ERRβ and ERRγ in adipose tissue have minor changes in their BAT, whereas mice lacking ERRα and ERRγ (the two main isoforms in BAT) have severe defects in oxidative and thermogenic capacity. Remarkably, when mice are treated with a β3-adrenergic agonist, the transcriptional response of BAT is virtually entirely dependent on ERRs. Mice lacking adipose ERRα and ERRγ also fail to enhance their oxidative and thermogenic capacity, even though adrenergic cytoplasmic signaling is preserved. Our findings show that ERRs are essential for BAT remodeling in response to adrenergic stimulation and suggest that ERR activation may sensitize BAT to adrenergic signals and lead to increased energy expenditure.
RESULTS
ERRs Act Collectively and in a Redundant Manner to Protect BAT Oxidative Capacity In Vivo
We recently showed that ERRs act redundantly to preserve mitochondrial function in primary brown adipocytes (Gantner et al., 2016), suggesting that ERRs may collectively control brown adipocyte oxidative and thermogenic function in vivo. To test this notion, we generated three lines of mice lacking different combinations of ERRs specifically in adipose tissue: mice lacking just ERRα, the most abundant ERR in adipose tissue (ERRαAd−/−); mice lacking ERRβ and ERRγ, the two ERRs that are highly enriched in BAT compared with WAT (ERRβγAd−/−); and mice that lack ERRα and ERRγ, the two most abundant ERRs in BAT and WAT (ERRαγAd−/−) (Dixen et al., 2013; Gantner et al., 2014). To minimize the effect of thermal stress at standard vivarium temperatures, all mice were born and raised at thermoneutrality (30°C). Loss of ERRs in BAT was confirmed at the mRNA level for all three isoforms (Figure 1A) and at the protein level for ERRα and ERRγ (Figure 1B); ERRβ protein could not be detected reliably in WT BAT with currently available antibodies. Decreases in ERR mRNA were also observed in inguinal WAT, but not skeletal muscle, confirming that the adiponectin promoter-driven knockout was tissue specific (Figures S1A and S1B). Notably, ERRβ mRNA was nearly undetectable in the BAT of ERRαγAd−/− mice, suggesting that double knockout of ERRα and ERRγ results in the practical loss of all three ERRs (Figure 1A). We next measured the mRNA levels in BAT of known ERR target genes involved in the TCA cycle and oxidative phosphorylation (OxPhos; Dufour et al., 2007; Schreiber et al., 2004; Villena et al., 2007). Loss of ERRα alone (ERRαAd−/−) showed decreases in many but not all of these genes, whereas loss of ERRβ and ERRγ (ERRβγAd−/−) had no significant effect (Figure 1C). However, the parallel loss of ERRα and ERRγ (ERRαγAd−/−) resulted in larger decreases and in all tested ERR targets, consistent with ERRs complementing each other in regulating gene expression in BAT (Figure 1C). Similarly, ERRαAd−/− and ERRβγAd−/− mice showed minor defects in BAT OxPhos protein and mtDNA content, but ERRαγAd−/− mice displayed a dramatic loss of BAT OxPhos complexes I–IV and a ~60% reduction in mtDNA content (Figures 1D–1F). These data show that ERRs display robust redundancy in protecting BAT oxidative capacity in vivo. Thus, to test the collective role of ERRs in adipose tissue function, we focused on studying BAT function in the ERRαγAd−/− mice, which showed pronounced defects in oxidative capacity.
Figure 1. ERRs Act Collectively and in a Redundant Manner to Promote BAT Mitochondrial Oxidative Capacity.

(A) Relative ERR mRNA levels in BAT from WT or littermate mice with adipose-specific deletions of ERRα (ERRαAd−/−), ERRβ and ERRγ (ERRβγAd−/−), or ERRα and ERRγ (ERRαγAd−/−), expressed relative to the levels of each gene in WT BAT and normalized to Ppia. Numbers above bars represent Ct (threshold cycle) values for each ERR isoform in WT BAT.
(B) Western blot of ERRα, ERRγ, and RAN (ras-related nuclear protein; loading control).
(C) Relative mRNA levels of genes acting in the tricarboxylic acid (TCA) cycle and/or oxidative phosphorylation (OxPhos) and normalized to Ppia.
(D) Representative western blot of OxPhos complexes I–V and RAN (loading control) protein.
(E) Quantification of OxPhos complexes, based on blots of panel D (expressed relative to levels in WT BAT and normalized to RAN protein). (F) Relative mitochondrial DNA (mtDNA) copy number, normalized to gDNA in BAT of the same mice.
(A–F) Mice were born and raised at thermoneutrality (30°C) and euthanized at 12 weeks of age. Data are mean ± SD of 14 WT, 4 ERRαAd−/−, 6 ERRβγAd−/−, and 5 ERRαγAd−/− mice. *p < 0.05, **p < 0.01, ***p < 0.001 vs. WT BAT. WT data in panels A, C, E, and F are the accrued data of all WT littermates of ERRα floxed, ERRβγ floxed, and ERRαγ floxed mice. There were no differences between WT mice of the different cohorts. The same loading control (RAN) is presented in panels B and D. See also Figure S1, Table S3.
Loss of ERRα and ERRγ Impairs BAT Thermogenic Capacity without Affecting Adipogenesis or BAT Identity
BAT is characterized by its brown color, the presence of small multilocular lipid droplets, a high density of mitochondria, and the expression of Ucp1 (Cannon and Nedergaard, 2004). In mice lacking adipose ERRα and ERRγ, the interscapular BAT depot was paler than WT BAT (Figure 2A) and showed notably increased lipid accumulation (Figure 2B). Electron microscopy imaging revealed sparse BAT mitochondria that were often malformed and with few visible cristae (Figure 2C). The abnormal mitochondrial structure was coupled with decreased expression of genes important for mitochondrial morphology and cristae structure (Figure 2D). Importantly, loss of adipose ERRs did not result in decreased expression of markers of BAT differentiation (Prdm16, Zic1, Lhx8, Ebf2) or adipogenesis (Pparg, Fabp4), indicating that the lipid accumulation and changes in mitochondria were not secondary to defects in the program of development or differentiation of BAT (Figure 2E). In support of the dramatic decrease in expression of TCA genes and OxPhos complexes I–IV shown in Figure 1, other genes of these pathways were also significantly reduced (Cox7a1 and Cs) in ERRαγAd−/− BAT (Figure 2F). ERRαγAd−/− BAT also displayed decreased expression of genes important for fatty acid oxidation (Figure 2G) and increased expression of genes that regulate fatty acid biosynthesis and storage (Figure 2H), which may contribute to the increase in lipid accumulation. Additional lipid metabolism genes and other pathways, such as creatine metabolism, were also altered in ERRαγAd−/− mice (Figures S2A and S2B). Finally, the expression of two hallmark genes essential for thermogenesis, Ucp1 and Dio2, was severely defective in BAT of ERRαγAd−/− mice (Figure 2I). Ucp1 protein levels were also dramatically decreased (Figure 2J), in a manner consistent again with largely redundant roles of ERRs (Figure S2C). The decreased Ucp1 expression suggested that thermogenic activity would be greatly reduced. Indeed, when exposed to 4°C, ERRαγAd−/− mice were very cold intolerant and became hypothermic in less than 2.5 hr (Figure 2K). These findings show that adipose ERRs are required to maintain proper oxidative and thermogenic capacity in BAT and to defend body temperature upon acute cold exposure.
Figure 2. Loss of ERRα and ERRγ Impairs BAT Thermogenic Capacity without Affecting Adipogenesis or BAT Identity.

(A) Gross morphology of interscapular BAT depots from WT and ERRαγAd−/− mice.
(B) H&E staining of BAT at 400× magnification.
(C) Electron microscopy of mitochondria and surrounding lipid droplets in BAT. L, lipid droplet.
(D–I, L, and M) Relative mRNA levels of genes involved in mitochondrial morphology and cristae structure (D), adipogenesis and BAT identity (E), OxPhos and TCA cycle (F), fatty acid (FA) oxidation (G), FA biosynthesis and storage (H), thermogenesis (I), nuclear regulation of the mitochondrial oxidative metabolism program (L), and mitochondrial regulation of mitochondrial biogenesis (M).
(J) Western blot of Ucp1 protein and RAN (loading control).
(K) Body temperature of WT and ERRαγAd−/− female mice either at 30°C or moved from 30°C to 4°C for 2.5 hr (n = 5). **p < 0.01, ***p < 0.001 vs. WT 4°C.
Mice were born and raised at thermoneutrality (30°C) and euthanized at 12 weeks of age. mRNA data are from male and female mice combined and normalized to Ppia (n = 7–18). Protein data shown here are from male mice. Data are mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 vs. WT BAT. See also Figure S2 and Table S3.
To gain insights into the level at which ERRs control mitochondrial content and thermogenic function, we next assessed the expression levels of other transcriptional regulators of oxidative metabolism and thermogenesis genes. The two nuclear transcriptional co-regulators that drive mitochondrial biogenesis and Ucp1 expression in BAT, Ppargc1a and Ppargc1b (Puigserver et al., 1998; Uldry et al., 2006), were moderately up- and down-regulated, respectively, at the mRNA level (Figure 2L); assessment of their protein expression showed a trend for both co-regulators to be at higher levels in ERRαγAd−/− than WT BAT (Figure S2D). The mRNA levels of other regulators of BAT oxidative capacity and Ucp1 were not altered (Nrf1 and Gadd45γ), moderately (320%) decreased (Gabpa), or increased (Irf4) (Figure 2L). The small changes in the expression of these regulators are thus unlikely to explain the dramatic loss of mitochondrial structure and proteins. On the other hand, we observed decreases in the expression of nuclear genes that encode regulators of mtDNA expression, such as Tfam, Tfb2m, Polrmt, and most notably the ERR targets Sirt3 and Endog (Giralt et al., 2011; McDermott-Roe et al., 2011; Figure 2M), suggesting that the loss of ERRs is sufficient to compromise expression of genes important for mitochondrial biogenesis and thermogenesis even when other known nuclear transcriptional regulators of these programs are largely unaffected.
Adipose ERRs Are Essential for the Remodeling of BAT Induced by β3-Adrenergic Agonists
Adrenergic stimulation of BAT promotes tissue remodeling by enhancing mitochondrial biogenesis and Ucp1 expression, thereby increasing long-term oxidative and thermogenic capacity (Cannon and Nedergaard, 2004). To determine if ERRs are required for BAT remodeling in response to adrenergic stimulation, mice were injected with the β3-specific adrenergic agonist CL316,243 or PBS as a control, for 10 days. As expected, WT mice treated with CL316,243 had darker BAT depots (Figure 3A; lower left panel) and decreased lipid accumulation, relative to WT mice treated with PBS (Figure 3B; lower left panel). These responses to CL316,243 treatment were completely dependent on ERRs, as PBS- and CL316,243-treated ERRαγAd−/− mice had similar pale color and high lipid content in their BAT depots (Figures 3A and 3B). Moreover, CL316,243 treatment resulted in mitochondrial changes, such as increased cristae density, in WT but not in ERRαγAd−/− mice (Figure 3C). Instead, mitochondria from ERRαγAd−/− mice treated with CL316,243 appeared to form abnormal circle-shaped cristae structures (Figure 3C; lower right panel). mtDNA content increased upon CL316,243 treatment in both WT and ERRαγAd−/− mice but remained very low in ERRαγAd−/− mice (Figure 3D). In WT BAT, CL316,243 treatment also elicited greater than 2-fold increases in protein expression of OxPhos complexes I and II and smaller but significant increases in complexes III and IV. These increases were considerably blunted in the BAT of ERRαγAd−/− mice (Figures 3E and 3F). Finally, the ability of CL316,243 to induce Ucp1 protein (Figure 3G) and Ucp1 mRNA (Figure 3H) in WT BAT was largely lost in the BAT of ERRαγAd−/− mice. Notably, the defects in the response to CL316,243 were not due to impaired adrenergic signaling. Primary brown adipocytes isolated from ERRαγAd−/− mice displayed full induction of hormone-sensitive lipase (HSL) phosphorylation and of lipolysis upon NE or CL316,243 stimulation (Figures S3A and S3B). Moreover, acute treatment of mice with CL316,243 induced the phosphorylation of HSL, the MAPK p38, and the transcription factor ATF2 in ERRαγAd−/− mice (Figures S3C and S3D). Finally, CL316,243 induced lipolysis in ERRαγAd−/− BAT explants as efficiently, if not better than in WT BAT explants (Figure S3E).
Figure 3. Effect of 10 Days’ Treatment with CL316,243 on BAT of WT and ERRαγAd−/− Mice.

(A) Gross morphology of interscapular BAT from WT and ERRαγAd−/− mice. Upper panel: PBS (before and after dissection from surrounding WAT). Lower panel: CL316,243 (before and after dissection from surrounding WAT).
(B) H&E staining of BAT at 400× magnification.
(C) Electron microscopy of mitochondria and surrounding lipid droplets in BAT. L, lipid droplet; arrows point to abnormal cristae formations.
(D) Relative mtDNA copy number, normalized to gDNA (n = 9–19).
(E) Representative western blot of OxPhos complexes I–V, ERRα, ERRγ, and RAN (loading control).
(F) Quantification of OxPhos western blots, normalized to RAN protein levels (n = 6–9).
(G) Representative western blot of Ucp1 protein and RAN (loading control).
(H) Ucp1 mRNA, normalized to Ppia mRNA (n = 6–7).
Male mice were born and raised at thermoneutrality (30°C), treated with PBS or CL316,243 for 10 days, and euthanized at 12 weeks of age. Data are mean ± SEM **p < 0.01, ***p < 0.001 vs. WT in the same treatment; #p < 0.05, ##p < 0.01, ###p < 0.001 vs. PBS in the same genotype. The same loading control (RAN) is presented in panels E and G. See also Figure S3 and Table S3.
To obtain an unbiased view of the contribution of ERRs to the β3-adrenergic-induced remodeling of BAT, we next used RNA sequencing to compare the global transcriptional changes induced by 10 days of CL316,243 treatment in WT and ERRαγAd−/− BAT depots. CL316,243 enhanced the expression of 1,024 genes in WT mice but only 352 genes in ERRαγAd−/− mice (applying cut-offs of 1.5-fold increase and p < 0.05; Figures 4A and 4B). Strikingly, only 219 of the 1,024 genes induced by CL316,243 in WT mice were significantly induced in ERRαγAd−/− BAT depots, showing that ERRs are required for the vast majority (380%) of the transcriptional adaptation to chronic β3-adrenergic stimulation (Figures 4A and 4B). Moreover, the absolute levels of 109 of the 219 genes induced in both WT and KO mice were significantly lower in ERRαγAd−/− BAT relative to WT BAT (Figures 4Aii and 4Bii). Overall, 389% of the genes induced by CL316,243 in WT BAT were dependent on ERRs for their full induction. Pathway analyses of the differentially expressed genes identified “mitochondrial dysfunction” and “oxidative phosphorylation” as the top two canonical pathways regulated by CL316,243 in WT BAT; neither of these pathways was significantly regulated by CL316,243 in ERRαγAd−/− BAT (Table 1). Of the top pathways regulated by CL316,243 in WT BAT, “Fatty-acid β-oxidation” remained preserved and became the top regulated pathway in ERRαγAd−/− BAT (Tables 1 and S1). Thus the loss of ERRs selectively altered the nature of the CL316,243-induced response by affecting specific and highly regulated pathways, rather than just attenuating CL316,243-induced responses across all pathways. Notably, the role of ERRs in the CL316,243-induced transcriptional remodeling extended beyond the predicted regulation of mitochondrial, OxPhos, and TCA cycle genes. Among the 20 genes highest induced by CL316,243 in WT BAT, 14 genes rely on ERRs for their induction and these include genes of lipid, phospholipid, and glycosphingolipid synthesis, as well as signaling proteins, a K+ channel, a carbonic anhydrase, and proteins of unknown function (Figure S4A).
Figure 4. Analyses of RNA Sequencing Data of BAT of WT and ERRαγAd−/− Mice Treated with CL316,243.

(A) Heat maps of genes significantly induced (p < 0.05) by CL316,243 in WT and ERRαγAd−/− BAT, using a 1.5-fold cut-off. Minimum and maximum log transformed values are 39 and 6, respectively, with visual contrasts set to 2. (i) Genes induced significantly by CL316,243 in WT BAT only. (ii) Genes induced by CL316,243 in both WT and ERRαγAd−/− BAT, but expression is significantly higher in WT CL316,243 vs. ERRαγAd−/− CL316,243. (iii) Genes induced by CL316,243 in both WT and ERRαγAd−/− mice. (iv) Genes induced by CL316,243 in ERRαγAd−/− BAT only.
(B) Venn diagram of genes significantly induced (p < 0.05) by CL316,243 in BAT. Sections i–iv correspond with genes displayed in heat maps (A).
(C) Heat maps of genes significantly reduced (p < 0.05) in ERRαγAd−/− vs. WT BAT, in PBS- and/or CL316,243-treated mice, using a 1.5-fold cut-off. Minimum and maximum log transformed values were 35 and 6, respectively, with visual contrasts set to 2. (i) Genes reduced in PBS-treated mice only. (ii) Genes reduced in both PBS- and CL316,243-treated ERRαγAd−/− mice. (iii) Genes reduced in CL316,243-treated ERRαγAd−/− mice only.
(D) Venn diagrams of genes significantly reduced (p < 0.05) in ERRαγAd−/− BAT. Sections i–iii correspond with genes displayed in heat maps (C).
Male mice were born and raised at thermoneutrality (30°C), treated with PBS or CL316,243 for 10 days, and euthanized at 12 weeks of age (n = 3). See also Figure S4 and Tables 1, S1, and S2.
Table 1.
Top Canonical Pathways Regulated by CL316,243 in WT BAT and Their Ranking in ERRαγAd−/− BAT
| Canonical Pathway | WT Mice | ERRαγAd−/− Mice | ||||||
|---|---|---|---|---|---|---|---|---|
| Rank | Downregulated | Upregulated | −log(p Value) | Rank | Downregulated | Upregulated | −log(p Value) | |
| Mitochondrial dysfunction | 1 | 8/142 (6%) | 79/142 (56%) | 9.29E+00 | NA | |||
| Oxidative phosphorylation | 2 | 2/85 (2%) | 62/85 (73%) | 8.92E+00 | NA | |||
| TCA cycle II (eukaryotic) | 3 | 0/19 (0%) | 17/19 (89%) | 7.22E+00 | 27 | 0/19 (0%) | 13/19 (68%) | 2.03E+00 |
| Fatty acid β-oxidation I | 4 | 0/26 (0%) | 16/26 (62%) | 6.80E+00 | 1 | 0/26 (0%) | 21/26 (81%) | 8.28E+00 |
| Glutaryl-CoA degradation | 5 | 1/15 (7%) | 11/15 (73%) | 6.66E+00 | 39 | 1/15 (7%) | 11/15 (73%) | 1.72E+00 |
| Tryptophan degradation III (eukaryotic) | 6 | 1/15 (7%) | 11/15 (73%) | 6.66E+00 | 40 | 1/15 (7%) | 11/15 (73%) | 1.72E+00 |
| Valine degradation I | 7 | 0/17 (0%) | 13/17 (76%) | 5.81E+00 | 8 | 0/17 (0%) | 15/17 (88%) | 4.01E+00 |
| Acetyl-CoA biosynthesis I (pyruvate dehydrogenase complex) | 8 | 0/5 (0%) | 5/5 (100%) | 4.34E+00 | 215 | 0/5 (0%) | 3/5 (60%) | 5.08E−01 |
| Allograft rejection signaling | 9 | 11/19 (58%) | 0/19 (0%) | 4.23E+00 | 5 | 12/19 (63%) | 0/19 (0%) | 4.60E+00 |
| LXR/RXR activation | 10 | 13/64 (20%) | 16/64 (25%) | 4.20E+00 | 15 | 17/64 (27%) | 8/64 (13%) | 2.76E+00 |
| Autoimmune thyroid disease signaling | 11 | 9/13 (69%) | 0/13 (0%) | 4.12E+00 | 30 | 8/13 (62%) | 1/13 (8%) | 1.95E+00 |
| Isoleucine degradation I | 12 | 1/13 (8%) | 9/13 (69%) | 4.12E+00 | 69 | 1/13 (8%) | 11/13 (85%) | 1.21E+00 |
| Complement system | 13 | 13/23 (57%) | 1/23 (4%) | 4.11E+00 | 3 | 14/23 (61%) | 2/23 (9%) | 6.83E+00 |
| Antigen presentation pathway | 14 | 11/24 (46%) | 1/24 (4%) | 3.90E+00 | 4 | 17/24 (71%) | 1/24 (4%) | 5.58E+00 |
| Gluconeogenesis I | 15 | 2/17 (12%) | 10/17 (59%) | 3.88E+00 | 104 | 6/17 (35%) | 3/17 (18%) | 9.28E−01 |
NA, non-applicable; LXR, liver X receptor; RXR, retinoid X receptor; these pathways were not detected as regulated by CL316,243 in ERRαγAd−/− BAT. See also Figures 4, S4 and Tables S1 and S2.
To gain insights into the global roles of BAT ERRs, we next analyzed the RNA-sequencing data to define genes and pathways whose expression is affected by ERRs, by comparing WT and ERRαγAd−/− BAT of PBS- or CL316,243-treated mice (Figures 4C and 4D). The top ERR-regulated pathways were “oxidative phosphorylation,” “mitochondrial dysfunction,” and “TCA cycle,” i.e., the same pathways that were induced by CL316,243 in WT BAT (Tables S2 and 1). Notably, a heatmap representation of ERR-dependent gene expression shows that the levels of many of these genes were enhanced by CL316,243 in WT BAT (Figure 4C). Furthermore, ERRs affected the expression of a broader set of genes in CL316,243-treated than in PBS-treated mice, suggesting that ERR signaling was enhanced by the β3-adrenergic agonist (Figure 4D). In summary, there was a high correlation between ERR dependency and CL316,243 induction of gene expression, consistent with the notions that (1) the majority of the transcriptional changes induced by CL316,243 require ERRs and (2) ERR signaling is enhanced by CL316,243.
ERRs Contribute to the CL316,243-Induced Browning of Inguinal WAT
In addition to its effects on BAT, β3-adrenergic stimulation leads to the appearance of brown-like adipocytes in inguinal WAT (ingWAT), a process known as browning or beiging (Granneman et al., 2005; Guerra et al., 1998). Therefore we assessed the ability of CL316,243 to induce browning of ingWAT in WT and ERRαγAd−/− mice. CL316,243 treatment led to the prominent appearance of multilocular adipocytes in the ingWAT of WT mice but had minimal effects on the adipocytes of the ingWAT of ERRαγAd−/− mice (Figure 5A). CL316,243 also significantly increased the protein levels of all five OxPhos complexes in WT ingWAT (Figures 5B and 5C), a response that was blunted in ERRαγAd−/− mice. Similarly, CL316,243 treatment dramatically induced Ucp1 protein and mRNA in WT ingWAT (nearly 100-fold) but had notably more modest effects (310-fold) on Ucp1 expression in ERRαγAd−/− ingWAT (Figures 5D and 5E). Similarly blunted responses to CL316,243 were seen for several other genes involved in the TCA and OxPhos (Figure 5F). The reduced response to CL316,243 was not due to impaired β3-adrenergic signaling, as primary inguinal white adipocytes isolated from these mice displayed full induction of HSL phosphorylation and lipolysis upon NE or CL316,243 stimulation (Figure S5). In summary, the ingWAT of ERRαγAd−/− mice showed impaired “browning,” as assessed by histological analysis and measurements of mitochondrial protein and Ucp1 expression, suggesting that ERRs also contribute to the remodeling of ingWAT driven by β3-adrenergic signaling.
Figure 5. Effect of 10 Days’ Treatment with CL316,243 on ingWAT of WT and ERRαγAd−/− Mice.

(A) H&E staining of inguinal white adipose tissue (ingWAT) at 400× magnification.
(B) Representative western blot of OxPhos complexes I–V, ERRα, and RAN (loading control).
(C) Quantification of OxPhos complex levels, normalized to RAN protein levels.
(D) Western blot of Ucp1 protein and RAN (loading control).
(E and F) mRNA levels of Ucp1 and TCA/OxPhos genes, normalized to Ppia and expressed relative to levels in WT mice treated with PBS (n = 8–14).
Mice are from the same experiment as in Figure 3. Data are mean ± SEM *p < 0.05, **p < 0.01, ***p < 0.001 vs. WT in the same treatment; #p < 0.05, ##p < 0.01, ###p < 0.001 vs. PBS in the same genotype. The same loading control (RAN) is presented in panels B and D. See also Figure S5 and Table S3.
Adipose ERRs Are Important for the Acute Enhancement of Whole-Body Metabolism by β-Adrenergic Stimulation
Adrenergic activation of BAT enhances whole-body energy expenditure (Xiao et al., 2015). To determine the relative significance of adipose ERRs to whole-body metabolism, we tested the acute metabolic response of WT and ERRαγAd−/− mice to adrenergic agonists, using indirect calorimetry. Notably, ERRαγAd−/− and WT littermates treated with PBS showed no differences in oxygen consumption, body temperature, or respiratory exchange ratio (RER), suggesting that the decreased oxidative and thermogenic capacity of ERRαγAd−/− mice is not important for whole-body metabolism in a thermoneutral, basal state (Figures 6A–6C and S6A). An injection of CL316,243 led to a dramatic increase in oxygen consumption in WT mice but had a minimal effect, similar to injection of PBS, in ERRαγAd−/− mice (Figure 6D), demonstrating that ERRs are required for the metabolic response to CL316,243. There were no differences in physical activity between the groups (Figures S6B and S6D). Parallel to the increase in oxygen consumption, CL316,243 caused a small but significant and sustained (over 36 hr) increase in basal body temperature of WT mice that was not seen in ERRαγAd−/− mice (Figure 6E). CL316,243 also led to a significant decrease in the RER of WT mice, consistent with increased lipid metabolism (Figures 6C and S6C). The RER of CL316,243-treated ERRαγAd−/− mice was not significantly different from that of WT and ERRαγAd−/− mice injected with PBS (Figure 6C, S6A, and S6C), although it is possible that a CL316,243-induced increase in adipose tissue lipolysis may have increased lipid utilization in other tissues, such as skeletal muscle. Interestingly, WT mice treated with CL316,243 for 10 days displayed a mild, but significant, increase in oxygen consumption even on the day after the injection (during the active phase, 32–44 hr after an injection [Figure 6F]), suggesting a prolonged effect of chronic β3-adrenergic stimulation on whole-body energy expenditure. Again, this was not seen in ERRαγAd−/− mice. Overall, our data show that ERRs are required for the metabolic response to β3-adrenergic stimulation.
Figure 6. Metabolic Responses to Adrenergic Stimulation in WT and ERRαγAd−/− Mice.

(A) Oxygen consumption (VO2) in WT and ERRαγAd−/− mice injected with PBS for 10 days, on the last day of injections.
(B) Core body temperature of WT and ERRαγAd−/− mice injected with PBS for 10 days, on the last day of injections.
(C) Average respiratory exchange ratio (RER) over 6 hr following a PBS or CL316,243 injection.
(D) VO2 in WT and ERRαγAd−/− mice injected with CL316,243 for 10 days, on the last day of injections.
(E) Core body temperature of WT and ERRαγAd−/− mice injected with CL316,243 for 10 days, on the last day of injections.
(F) Average VO2 over the day and night on the day after the last PBS or CL316,243 injection (day, 20–32 hr after last injection; night, 32–44 hr after the last injection).
(G) Acute VO2 response to norepinephrine (NE) in WT and ERRαγAd−/− mice that had been treated with PBS for 10 days.
(H) Acute VO2 response to NE in WT and ERRαγAd−/− mice that had been treated with CL316,243 for 10 days.
Male mice were born and raised at thermoneutrality (30°C), treated with PBS or CL316,243 for 10 days, and euthanized at 12–14 weeks of age. NE responses were tested 2 days after the last PBS or CL316,243 injection. Data are mean ± SEM (n = 5–10). *p < 0.05, **p < 0.01, ***p < 0.001, WT vs. ERRαγAd−/− for each condition; #p < 0.05, ##p < 0.01 vs. WT PBS. ZT = zeitgeber time. Different mice were used for calorimetry and telemetry experiments. See also Figure S6.
The β3-adrenergic agonist CL316,243 is a pharmacologic agent that acts selectively and potently on adipose tissue. To test the relative significance of ERRs in a more physiological context, we next compared the metabolic response of WT and ERRαγAd−/− mice to the native agonist, NE, which activates a wider range of adrenergic receptors, including β1 and α1 types (Nedergaard and Cannon, 2014). NE was administered to naive mice (treated with PBS), as well as to mice with “trained” BAT (treated with CL316,243 for 10 days). NE injection evoked significantly higher oxygen consumption rates in WT mice than in ERRαγAd−/− mice, in both PBS- and CL316,243-treated groups (Figures 6G and 6H), showing that adipose ERRs are important for whole-body oxidative and thermogenic capacity in a physiological context. Furthermore, the difference in maximal oxygen consumption rates between WT and ERRαγAd−/− mice treated with NE was more pronounced in mice that had received CL316,243 for 10 days relative to mice treated with PBS (Figure S6E), supporting the notion that CL316,243-dependent remodeling of adipose tissues increased the thermogenic capacity in WT but not in ERRαγAd−/− mice.
ERRs Are Required for CL316,243-Induced Weight Loss and Improved Glucose Tolerance in HFD Mice
The mice studied earlier were fed a chow diet and were fairly lean. CL316,243 treatment induced a decrease in the adiposity of WT mice that was not seen in ERRαγAd−/− mice, but it did not result in significant changes in body weight (Figures S6F and S6G). CL316,243 has been shown to confer greater metabolic improvements in mice fed a high-fat diet (HFD) compared with chow diet (Xiao et al., 2015). Hence, we next put WT and ERRαγAd−/− mice on an HFD before evaluating the effects of CL316,243 treatment. WT and ERRαγAd−/− mice had a similar food intake (Figure S7A), and after 18 weeks on the HFD, similar body weights and glucose tolerance (Figures 7A and 7B). Subsequent treatment of the mice with CL316,243 (14 injections over 4 weeks, given every other day, since we observed sustained effects of CL316,243 on energy expenditure [Figure 6F]) led to significantly higher body weight loss in WT mice than in ERRαγAd−/− mice (Figures 7C and 7D), with WT mice losing on average 320% and ERRαγAd−/− mice losing just 3.7% of their body weight. CL316,243 treatment also improved glucose tolerance in WT mice (Figure 7E) but had no effect in ERRαγAd−/− mice (Figure 7F), revealing that adipose ERRs are required for the beneficial metabolic effects of β3-agonists.
Figure 7. Effect of Long-term Adrenergic Stimulation on Body Weight and Glucose Tolerance of HFD-fed WT and ERRαγAd−/− Mice.

(A) Body weights of WT and ERRαγAd−/− mice fed a 60% high fat diet (HFD) for 18 weeks (n = 12–18).
(B) Glucose tolerance test (GTT) of WT and ERRαγAd−/− mice fed a 60% HFD and before treatment. Values are presented as percentage of starting blood glucose (WT, 111.7 ±6.1 mg/dL; ERRαγAd−/−, 107.1 ±3.7 mg/dL; p = 0.52), (n = 12–18).
(C) Body weights of WT and ERRαγAd−/− mice before and after treatment with PBS or CL316,243 (n = 6–9). ***p < 0.001.
(D) Body weight loss of WT and ERRαγAd−/− mice, expressed as percentage of body weight before treatment (n = 6–9). *p < 0.01; ***p < 0.001.
(E) GTT of WT mice treated with PBS or CL316,243. Values are presented as percentage of starting blood glucose (WT PBS, 74.3 ± 9.9 mg/dL; WT CL316,243, 99.4 ± 8.2 mg/dL; p = 0.074). (n = 6–9). *p < 0.05, **p < 0.01, ***p < 0.001 vs. WT CL316,243.
(F) GTT of ERRαγAd−/− mice treated with PBS or CL316,243. Values are presented as percentage of starting blood glucose (ERRαγAd−/− PBS, 79.9 ± 4.9 mg/dL; ERRαγAd−/− CL316,243, 75.2 ± 6.2 mg/dL; p = 0.56) (n = 6–9).
Female mice were born and raised at thermoneutrality (30°C) and fed a 60% HFD starting at weaning. At 22 weeks of age, mice were given an intraperitoneal injection of PBS or CL316,243 every other day for 4 weeks. Mice were euthanized at 26 weeks of age. Data are presented as mean ± SEM.
DISCUSSION
Deciphering the transcriptional networks that regulate BAT development and function is important for developing therapeutic strategies to increase energy expenditure via non-shivering thermogenesis for the treatment of obesity. In the last several years, there has been much progress in understanding transcriptional mechanisms that control the commitment of stem cells to the brown/beige preadipocyte lineage and/or the differentiation of preadipocytes to mature brown or beige adipocytes (Lynes and Tseng, 2015; Wang and Seale, 2016). Once differentiated, however, brown adipocytes require additional signals and factors to maintain and expand their thermogenic capacity (Cannon and Nedergaard, 2004; Peirce et al., 2014). Notably, exposure to cold and pharmacologic administration of β3-adrenergic agonists can enhance the thermogenic capacity and/or energy expenditure in both rodents and humans (Cannon and Nedergaard, 2004; Cypess et al., 2015; van der Lans et al., 2013; Yoneshiro et al., 2013). We show here that ERRs perform a central and essential role in mature brown adipocytes in determining both the basal thermogenic capacity and the ability to expand this capacity. Although past studies revealed a role for ERRα in building the high basal mitochondrial content of BAT (Villena et al., 2007), the current study provides multiple lines of evidence that highlight that ERRs, collectively, play a substantially more important role than previously appreciated. First, double deletion of ERRα and ERRγ in mature adipocytes resulted in an extreme loss of mitochondrial content, with the remaining mitochondria having abnormal cristae and overall morphology and almost no Ucp1 protein; in comparison, mice lacking just ERRα (the most abundant BAT ERR isoform) had a milder reduction in mitochondrial content, and their mitochondria had normal morphology, oxidative function, and Ucp1 expression (Villena et al., 2007). Second, mice lacking adipose ERRα and ERRγ show remarkable defects in their adaptive response to adrenergic stimulation, failing to remodel their transcriptome, expand their thermogenic capacity, and attain the metabolic benefits of β3-adrenergic agonists.
ERRα and ERRγ have been shown to bind similar sites on the genome and to regulate overlapping sets of genes (Dufour et al., 2007; Huss et al., 2015), suggesting that a certain degree of redundancy in their action is to be expected. Recent studies have also highlighted the complimentary roles of ERRs in determining the oxidative capacity in primary brown adipocytes and in the heart (Gantner et al., 2016; Wang et al., 2015). Yet, the extent to which mice lacking either just ERRα (ERRαAd−/−) or ERRβ and ERRγ together (ERRβγAd−/−) maintain their mitochondrial content and OxPhos levels, despite a substantial loss of total ERR protein, is surprising. This functional compensation is seen without an upregulation of the remaining ERR isoforms, suggesting that BAT, at least in young mice, expresses ERRs at much higher levels than necessary for maintaining basal oxidative function. It will be interesting to assess ERR expression in the BAT depots of mice and humans at different ages, to establish if this high ERR reserve diminishes with age, as BAT function declines.
A most striking finding in our study is the key role of ERRs in the adrenergic response of BAT. This was unexpected, based on studies of the ERRα KO mouse, which showed normal induction of gene expression in response to adrenergic stimulation (Villena et al., 2007). Importantly, adrenergic signaling, judged by phosphorylation of HSL, p38, and ATF2 and induction of lipolysis, is intact in BAT lacking ERRs, suggesting that ERRs act downstream of such signals to effect the transcriptional changes elicited by adrenergic stimulation. ERRs likely integrate adrenergic signaling in more than one way. PGC-1α and PGC-1β, whose expression and/or activity are increased via cAMP, protein kinase A (PKA), and p38 signaling, are potent co-activators of all three ERRs (Gantner et al., 2014; Huss et al., 2002; Kamei et al., 2003; Schreiber et al., 2003). Moreover, Gadd45γ, whose expression is also induced by adrenergic signaling in a cAMP- and PKA-dependent manner, activates p38 and ERRβ/γ-dependent transcription in a PGC-1 independent manner (Gantner et al., 2014). Notably, although the absence of ERRs does not block adrenergic signaling, it may lead to a partial activation of the pathway in the basal state. We observed higher levels of phos-pho-p38, phospho-ATF2, and PGC-1α in the absence of adrenergic stimulation. The underlying mechanisms are not clear.
Once activated, ERRs are likely direct positive regulators of many adrenergically induced genes. Although some of the pathways induced by β3-adrenergic agonism (e.g., mitochondrial function, oxidative phosphorylation, and TCA cycle) were known to be regulated by ERRs (Hock and Kralli, 2009; Villena and Kralli, 2008), our RNA sequencing data show a much broader set of adrenergic responses, extending beyond the control of mitochondrial biogenesis, that rely on ERRs. Some of the highly induced genes, such as Ucp1, Dio2, and Fndc5 (Figure S4), have ERR response elements and are regulated by ERRs (Debevec et al., 2007; Dixen et al., 2013; Gantner et al., 2016; Wrann et al., 2013), suggesting that others are also direct targets. Notably, there are adrenergically induced genes that are not dependent on ERRs, consistent with adrenergic signaling being intact, and ERRs not required for all components of the adrenergic response. In particular, the induction of the FAO pathway is retained. Ingenuity Pathway Analysis for regulators of the genes in the FAO pathway suggests that this may be due to Pparα, whose expression is increased by CL316,243 in the BAT of ERRαγAd−/− mice, and Klf15, which is modestly increased in the BAT of ERRαγAd−/− (Figure S4). Even though the induction of FAO genes is retained, FAO is likely defective in ERRαγAd−/− BAT, owing to the mitochondrial impairment. Finally and importantly, we do not expect that all genes and proteins whose expression is reduced in the BAT of ERRαγAd−/− mice are direct ERR targets. Loss of mitochondria and mitochondria-derived signals, as well as of the further actions of direct ERR targets (e.g., a potential paracrine function of Fndc5), may contribute to the loss of adaptive responses to adrenergic stimulation. In addition, the decrease in mitochondrial content likely contributes to the post-translational loss of expression of many mitochondrial proteins whose stability requires proper localization, including Ucp1.
The thermogenic capacity of BAT can be harnessed to increase energy expenditure and counteract obesity and obesity-related metabolic dysfunction (Lee et al., 2014). Consistent with this notion, when treated with the β3-adrenergic agonist CL316,243, WT mice increased their energy expenditure, lost weight, and showed improvements in glucose homeostasis. ERRαγAd−/− littermates were defective in all three responses, demonstrating the importance of ERR-controlled pathways in adipose tissue for the effects of β3-adrenergic stimulation on energy balance. Nonetheless, ERRαγAd−/− mice were not prone to obesity in the absence of CL316,243 administration, whether on chow or HFD diet. This was irrespective of gender, as similar findings were seen in female and male mice fed a HFD (Figure S7). On the one hand, this finding is not surprising, because our studies were performed at thermoneutrality, when BAT receives minimal adrenergic stimulation (Xiao et al., 2015). Other mutant mice with BAT thermogenic defects are also not prone to obesity (Emmett et al., 2017; Lee et al., 2016). On the other hand, BAT thermogenic capacity has been proposed to be important for diet-induced thermogenesis, i.e., the ability to increase energy expenditure in response to caloric excess, as in HFD (Feldmann et al., 2009; Kazak et al., 2017). There can be many explanations as to why ERRαγAd−/− mice do not show increased obesity. First, diet-induced thermogenesis, the basis of which is less well understood than cold-induced thermogenesis, may involve signals and pathways that are sufficiently retained in ERRαγAd−/− mice. Second, minor differences in vivarium temperatures or in the length of time spent at thermoneutrality may explain the different outcomes seen with different models. In our study, mice were born, raised, and studied at thermoneutrality, to minimize the impact that standard vivarium temperatures have on mice with defective BAT (Schulz et al., 2013). Finally, the adrenergic system can be activated by stress, so additional differences in vivarium conditions housing may affect findings.
Even though loss of BAT may not always give rise to obesity, it is clear that increased BAT capacity, coupled to activation, as seen at standard vivarium temperatures for mice, can lead to protection from obesity (Cohen and Spiegelman, 2015). Indeed, mice with increased adipose PGC-1α/ERRα signaling, as seen in mice lacking the tumor suppressor folliculin, show elevated oxidative metabolism, resistance to hypothermia, and protection from HFD-induced obesity (Yan et al., 2016). Although ERRs are nuclear receptors with no known endogenous ligand, crystallography studies show that they are amenable to drug design, with hydrophobic pockets that can accommodate synthetic ligands (Coward et al., 2001; Kallen et al., 2007; Wang et al., 2006). Synthetic agonists, such as GSK4716, can enhance the activity of ERRβ and ERRγ (Wang et al., 2006; Zuercher et al., 2005). Newer GSK4716-derived molecules can activate ERRγ and promote WAT browning (Xu et al., 2015), although the specificity of these ligands has not been tested yet in vivo. Our findings suggest that pharmacological activation of ERRs may be beneficial by increasing brown (and beige) adipose tissue thermogenic capacity. Moreover, increased ERR activity may sensitize adipose tissues to the action of adrenergic agonists. The concept of using β3-adrenergic agonists to pharmacologically activate BAT in humans was floated long ago but has been difficult to apply (Lee and Greenfield, 2015). Recently, mirabregon, a β3-adrenergic agonist used in the treatment of overactive bladder, was shown to activate BAT in humans, albeit at doses four times higher than that of the current US Food and Drug Administration–approved dose and with adverse effects on the cardiovascular system (Cypess et al., 2015). Administration of ERR agonists could prime the adipose tissue for a more effective response to β3-adrenergic agonism, thereby enabling the use of lower doses. Future studies will be needed to evaluate the potential of ERR agonists to enhance BAT thermogenesis and energy expenditure, alone or in combination with β3-adrenergic stimulation.
In conclusion, we have demonstrated that ERRs are critical transcriptional regulators that control adipose tissue plasticity and orchestrate the adaptive remodeling induced by adrenergic stimuli. Our findings underscore the potential of targeting ERRs to expand energy expenditure capacity, which may be harnessed for the treatment of metabolic disease.
METHODS
All methods can be found in the accompanying Transparent Methods supplemental file.
Supplementary Material
Figure S1. Related to Figure 1. ERR mRNA expression in inguinal WAT and skeletal muscle. (A) Relative Esrra, Esrrb and Esrrg mRNA levels in inguinal WAT (IngWAT) of WT and ERRαγAd−/− mice, normalized to Ppia. Data are mean ± SEM (n = 7–14). ***p < 0.001.
(B) Relative Esrra, Esrrb and Esrrg mRNA levels in skeletal muscle (plantaris) of WT and ERRαγAd−/− mice, normalized to Ppia. Data are mean ± SEM (n = 6).
Figure S2. Related to Figure 2. Gene and protein expression levels in BAT of WT and ERRAd−/− mice. (A,B) Relative mRNA levels of energy handling (A) and creatine metabolism (B) genes in BAT of WT and ERRαγAd−/− mice, normalized to Ppia. Data are mean ± SEM (n = 9–18). ***p < 0.001.
(C,D) Representative western blots of UCP1 and RAN (loading control) and their quantification, in BAT from ERRαAd−/− (C) or ERRβγAd−/− (D), relative to WT of each group. Data are mean ± SD (n = 3).
(E) Representative western blots of PGC-1α and PGC-1β in BAT of WT and ERRαγAd−/− mice (n=3). Ponceau stain is shown as loading control.
Figure S3. Related to Figure 3. Adrenergic signaling in brown adipocytes and BAT of WT and ERRαγAd−/− mice. (A) Representative western blots of phosphorylated hormone-sensitive lipase (HSL), total HSL, ERRα, and α-tubulin (loading control) in primary brown adipocytes of WT and ERRαγAd−/− mice, treated with vehicle, 100 nM norepinephrine (NE) or 50 nM CL316,243 for 10 min.
(B) Lipolysis in primary brown adipocytes of WT and ERRαγAd−/− mice, treated as in panel A for 2 hrs. Data are mean ± SEM (n = 4). ***p < 0.001 vs. vehicle in the same genotype.
(C,D) Western blots of phosphorylated and total HSL, p38 MAPK and ATF2 in BAT of WT and ERRαγAd−/− mice treated with PBS or CL316,243 for 45 min. Quantification data are mean ± SEM (n = 3). For p-p38, quantification is of the upper band. *p < 0.05 vs. vehicle in the same genotype. ND, not detected, NS, not significant.
(E) Lipolysis in BAT explants of WT and ERRαγAd−/− mice, treated ex vivo with vehicle or 1 μM CL316,243 for 2 hrs. Data are mean ± SEM (n = 3). ***p < 0.001 vs. vehicle in the same genotype.
Figure S4. Related to Figure 4. Gene expression levels in BAT of WT and ERRαγAd−/− mice treated with CL316,243. (A) Expression levels of genes that are in the top 20 induced by the ten days of CL316,243 in WT BAT and dependent on ERRs for their induction. Fourteen of the top 20 CL316,243-induced genes relied on ERRs and are shown here. Several of these genes have been reported as highly induced also by cold in BAT (Marcher et al., 2015). Data are mean ± SD of CPM (counts per million reads), measured by RNA-sequencing (n = 3).
(B,C) Expression levels of thermogenesis genes Ucp1, Dio2 (B), and regulators of lipid metabolism Ppara and Klf15 (C) in BAT of WT and ERRαγAd−/− mice, treated with PBS or CL316,243 for 10 days. Data are mean ± SD of CPM (counts per million reads), measured by RNA-sequencing (n = 3).
(A–C) **p < 0.01, ***p < 0.001 vs. WT in the same treatment; #p < 0.05, ###p < 0.001 vs. PBS in the same genotype.
Figure S5. Related to Figure 5. Adrenergic signaling in inguinal white adipocytes from WT and ERRαγAd−/− mice. (A) Representative western blots of phosphorylated hormone-sensitive lipase (HSL)S563, total HSL, ERRα, and α-tubulin (loading control) in primary inguinal adipocytes from WT and ERRαγAd−/− mice. Differentiated adipocytes were treated with either DMSO (vehicle), 100 nM norepinephrine (NE) or 50 nM CL316,243 for 10 minutes.
(B) Lipolysis (glycerol release in media) in primary inguinal adipocytes isolated from WT and ERRαγAd−/− mice. Differentiated adipocytes were treated with either DMSO (vehicle), 100 nM NE or 50 nM CL316,243 for 2 hrs. Data are mean ± SEM (n = 4). ***p < 0.001 vs. vehicle in the same genotype.
Figure S6. Related to Figure 6. Metabolic responses to adrenergic stimulation in WT and ERRαγAd−/− mice. (A,C) Average respiratory exchange ratio (RER) at 1 hour intervals of WT and ERRαγAd−/− mice injected with PBS (A) or CL316,243 (C) for ten days, on the last day of injections.
(B,D) Physical activity levels of WT and ERRαγAd−/− mice injected with PBS (B) or CL316,243 (D) for ten days, on the last day of injections.
(E) Maximum oxygen consumption (VO2) reached following an acute norepinephrine (NE) injection in WT and ERRαγAd−/− mice, previously treated with PBS or CL316,243 for ten days. **p < 0.01, ***p < 0.001 vs. WT in the same treatment; #p < 0.05 vs. PBS in the same genotype.
(F) Body weights of WT and ERRαγAd−/− mice treated with PBS or CL316,243 for ten days.
(G) Body fat percentage of WT and ERRαγAd−/− mice treated with PBS or CL316,243 for ten days. ##p < 0.01 vs. WT PBS. (A–G) Male mice were born and raised at thermoneutrality (30 °C), treated with PBS or CL316,243 for ten days, and euthanized at 12–14 weeks of age. NE responses were determined 2 days after the last PBS or CL316,243 injection. Data are presented as mean ± SEM (n = 5–10). ZT, Zeitgeber time. A.U., Arbitrary units.
Figure S7. Related to Figure 7. Food intake and body weights of HFD-fed WT and ERRαγAd−/− mice. (A) Total food intake (kCal) of female WT and ERRαγAd−/− mice shown in Figure 7 during 10 weeks on the 60% high fat diet (HFD). Data are the mean ± SEM consumption per mouse in group-housed mice (n = 8–25).
(B–D) Body weights (B), total food intake (C) and metabolic efficiency (D) of male WT and ERRαγAd−/− mice fed a control chow or 60% HFD for 10 weeks. Data are the mean ± SEM of singly-housed mice (n = 8–9).
Table S1. The top canonical pathways regulated (p < 0.05) by CL316,243 in ERRαγAd−/− BAT, and their ranking in WT BAT, using a 1.5-fold cut-off. Related to Figure 4.
Table S2. The top canonical pathways regulated (p < 0.05) by ERRs in PBS or CL316,243-treated mice, using a 1.5-fold cut-off. Related to Figure 4.
Table S3. Primer sequences used for Q-PCR. Related to Figures 1, 2, 3 and 5.
HIGHLIGHTS.
Adipose ERRs collectively control brown fat oxidative and thermogenic capacity
Adipose ERRs are essential for BAT remodeling induced by β-adrenergic agonism
ERRs control the bulk of the transcriptional response to adrenergic stimulation
Mice that lack adipose ERRs show no metabolic benefits of β-adrenergic agonism
Acknowledgments
We thank Dr. Johan Auwerx for providing the ERR floxed mouse strains; Dr. Malcolm Parker at the TSRI Microscopy core for the EM analyses; Yoshitake Cho, Enrique Saez, Bernard Kok, and other Saez laboratory members for discussions and advice. The study was supported by the National Institute of Health grant R01DK095686 (to A.K.), GM113894 (to B.C.), and the AHA post-doctoral award 16POST31350029 (to E.L.B.).
Footnotes
AUTHOR CONTRIBUTIONS
Conceptualization, E.L.B. and A.K.; Methodology, E.L.B and A.K.; Investigation, E.L.B., B.C.H., E.E., J.-S.W., M.L.G., S.C., M.S.-A., and V.A.; Writing – Original draft, E.L.B. and A.K.; Writing – Review and Editing, E.L.B., B.C.H., E.E., J.-S.W., M.L.G., V.A., M.S.-A., B.C., and A.K.; Supervision, E.L.B. and A.K.; Funding Acquisition, E.L.B. and A.K.; Resources, B.C. and M.S.-A.
DATA AND SOFTWARE AVAILABILITY
The RNA sequencing gene expression data can be found at GEO: GSE104285.
Supplemental Information includes Transparent Methods, seven figures, and three tables and can be found with this article online at https://doi.org/10.1016/j.isci.2018.03.005.
DECLARATION OF INTERESTS
The authors declare no competing interests.
References
- Arany Z, Foo SY, Ma Y, Ruas JL, Bommi-Reddy A, Girnun G, Cooper M, Laznik D, Chinsomboon J, Rangwala SM, et al. HIF-independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC-1alpha. Nature. 2008;451:1008–1012. doi: 10.1038/nature06613. [DOI] [PubMed] [Google Scholar]
- Bookout AL, Jeong Y, Downes M, Yu RT, Evans RM, Mangelsdorf DJ. Anatomical profiling of nuclear receptor expression reveals a hierarchical transcriptional network. Cell. 2006;126:789–799. doi: 10.1016/j.cell.2006.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon B, Nedergaard J. Brown adipose tissue: function and physiological significance. Physiol Rev. 2004;84:277–359. doi: 10.1152/physrev.00015.2003. [DOI] [PubMed] [Google Scholar]
- Cohen P, Spiegelman BM. Brown and beige fat: molecular parts of a thermogenic machine. Diabetes. 2015;64:2346–2351. doi: 10.2337/db15-0318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coward P, Lee D, Hull MV, Lehmann JM. 4-Hydroxytamoxifen binds to and deactivates the estrogen-related receptor gamma. Proc Natl Acad Sci USA. 2001;98:8880–8884. doi: 10.1073/pnas.151244398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cypess AM, Weiner LS, Roberts-Toler C, Franquet Elia E, Kessler SH, Kahn PA, English J, Chatman K, Trauger SA, Doria A, et al. Activation of human brown adipose tissue by a beta3-adrenergic receptor agonist. Cell Metab. 2015;21:33–38. doi: 10.1016/j.cmet.2014.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debevec D, Christian M, Morganstein D, Seth A, Herzog B, Parker M, White R. Receptor interacting protein 140 regulates expression of uncoupling protein 1 in adipocytes through specific peroxisome proliferator activated receptor isoforms and estrogen-related receptor alpha. Mol Endocrinol. 2007;21:1581–1592. doi: 10.1210/me.2007-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixen K, Basse AL, Murholm M, Isidor MS, Hansen LH, Petersen MC, Madsen L, Petrovic N, Nedergaard J, Quistorff B, et al. ERRgamma enhances UCP1 expression and fatty acid oxidation in brown adipocytes. Obesity (Silver Spring) 2013;21:516–524. doi: 10.1002/oby.20067. [DOI] [PubMed] [Google Scholar]
- Dufour CR, Wilson BJ, Huss JM, Kelly DP, Alaynick WA, Downes M, Evans RM, Blanchette M, Giguere V. Genome-wide orchestration of cardiac functions by the orphan nuclear receptors ERRalpha and gamma. Cell Metab. 2007;5:345–356. doi: 10.1016/j.cmet.2007.03.007. [DOI] [PubMed] [Google Scholar]
- Dulloo AG, Miller DS. Energy balance following sympathetic denervation of brown adipose tissue. Can J Physiol Pharmacol. 1984;62:235–240. doi: 10.1139/y84-035. [DOI] [PubMed] [Google Scholar]
- Emmett MJ, Lim HW, Jager J, Richter HJ, Adlanmerini M, Peed LC, Briggs ER, Steger DJ, Ma T, Sims CA, et al. Histone deacetylase 3 prepares brown adipose tissue for acute thermogenic challenge. Nature. 2017;546:544–548. doi: 10.1038/nature22819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmann HM, Golozoubova V, Cannon B, Nedergaard J. UCP1 ablation induces obesity and abolishes diet-induced thermogenesis in mice exempt from thermal stress by living at thermoneutrality. Cell Metab. 2009;9:203–209. doi: 10.1016/j.cmet.2008.12.014. [DOI] [PubMed] [Google Scholar]
- Gantner ML, Hazen BC, Conkright J, Kralli A. GADD45gamma regulates the thermogenic capacity of brown adipose tissue. Proc Natl Acad Sci USA. 2014;111:11870–11875. doi: 10.1073/pnas.1406638111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gantner ML, Hazen BC, Eury E, Brown EL, Kralli A. Complementary roles of estrogen-related receptors in brown adipocyte thermogenic function. Endocrinology. 2016;157:4770–4781. doi: 10.1210/en.2016-1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giguere V, Yang N, Segui P, Evans RM. Identification of a new class of steroid hormone receptors. Nature. 1988;331:91–94. doi: 10.1038/331091a0. [DOI] [PubMed] [Google Scholar]
- Giralt A, Hondares E, Villena JA, Ribas F, Diaz-Delfin J, Giralt M, Iglesias R, Villarroya F. Peroxisome proliferator-activated receptor-gamma coactivator-1alpha controls transcription of the Sirt3 gene, an essential component of the thermogenic brown adipocyte phenotype. J Biol Chem. 2011;286:16958–16966. doi: 10.1074/jbc.M110.202390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granneman JG, Li P, Zhu Z, Lu Y. Metabolic and cellular plasticity in white adipose tissue I: effects of beta3-adrenergic receptor activation. Am J Physiol Endocrinol Metab. 2005;289:E608–E616. doi: 10.1152/ajpendo.00009.2005. [DOI] [PubMed] [Google Scholar]
- Guerra C, Koza RA, Yamashita H, Walsh K, Kozak LP. Emergence of brown adipocytes in white fat in mice is under genetic control. Effects on body weight and adiposity. J Clin Invest. 1998;102:412–420. doi: 10.1172/JCI3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hock MB, Kralli A. Transcriptional control of mitochondrial biogenesis and function. Annu Rev Physiol. 2009;71:177–203. doi: 10.1146/annurev.physiol.010908.163119. [DOI] [PubMed] [Google Scholar]
- Huss JM, Garbacz WG, Xie W. Constitutive activities of estrogen-related receptors: transcriptional regulation of metabolism by the ERR pathways in health and disease. Biochim Biophys Acta. 2015;1852:1912–1927. doi: 10.1016/j.bbadis.2015.06.016. [DOI] [PubMed] [Google Scholar]
- Huss JM, Kopp RP, Kelly DP. Peroxisome proliferator-activated receptor coactivator-1alpha (PGC-1alpha) coactivates the cardiac-enriched nuclear receptors estrogen-related receptor-alpha and -gamma. Identification of novel leucine-rich interaction motif within PGC-1alpha. J Biol Chem. 2002;277:40265–40274. doi: 10.1074/jbc.M206324200. [DOI] [PubMed] [Google Scholar]
- Huss JM, Torra IP, Staels B, Giguere V, Kelly DP. Estrogen-related receptor alpha directs peroxisome proliferator-activated receptor alpha signaling in the transcriptional control of energy metabolism in cardiac and skeletal muscle. Mol Cell Biol. 2004;24:9079–9091. doi: 10.1128/MCB.24.20.9079-9091.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallen J, Lattmann R, Beerli R, Blechschmidt A, Blommers MJ, Geiser M, Ottl J, Schlaeppi JM, Strauss A, Fournier B. Crystal structure of human estrogen-related receptor alpha in complex with a synthetic inverse agonist reveals its novel molecular mechanism. J Biol Chem. 2007;282:23231–23239. doi: 10.1074/jbc.M703337200. [DOI] [PubMed] [Google Scholar]
- Kamei Y, Ohizumi H, Fujitani Y, Nemoto T, Tanaka T, Takahashi N, Kawada T, Miyoshi M, Ezaki O, Kakizuka A. PPARgamma coactivator 1beta/ERR ligand 1 is an ERR protein ligand, whose expression induces a high-energy expenditure and antagonizes obesity. Proc Natl Acad Sci USA. 2003;100:12378–12383. doi: 10.1073/pnas.2135217100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazak L, Chouchani ET, Lu GZ, Jedrychowski MP, Bare CJ, Mina AI, Kumari M, Zhang S, Vuckovic I, Laznik-Bogoslavski D, et al. Genetic depletion of adipocyte creatine metabolism inhibits diet-induced thermogenesis and drives obesity. Cell Metab. 2017;26:660–671. doi: 10.1016/j.cmet.2017.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Choi J, Aja S, Scafidi S, Wolfgang MJ. Loss of adipose fatty acid oxidation does not potentiate obesity at thermoneutrality. Cell Rep. 2016;14:1308–1316. doi: 10.1016/j.celrep.2016.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee P, Greenfield JR. Non-pharmacological and pharmacological strategies of brown adipose tissue recruitment in humans. Mol Cell Endocrinol. 2015;418(Pt 2):184–190. doi: 10.1016/j.mce.2015.05.025. [DOI] [PubMed] [Google Scholar]
- Lee YH, Jung YS, Choi D. Recent advance in brown adipose physiology and its therapeutic potential. Exp Mol Med. 2014;46:e78. doi: 10.1038/emm.2013.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Sladek R, Carrier J, Bader JA, Richard D, Giguere V. Reduced fat mass in mice lacking orphan nuclear receptor estrogen-related receptor alpha. Mol Cell Biol. 2003;23:7947–7956. doi: 10.1128/MCB.23.22.7947-7956.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynes MD, Tseng YH. The thermogenic circuit: regulators of thermogenic competency and differentiation. Genes Dis. 2015;2:164–172. doi: 10.1016/j.gendis.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott-Roe C, Ye J, Ahmed R, Sun XM, Serafin A, Ware J, Bottolo L, Muckett P, Canas X, Zhang J, et al. Endonuclease G is a novel determinant of cardiac hypertrophy and mitochondrial function. Nature. 2011;478:114–118. doi: 10.1038/nature10490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha VK, Handschin C, Arlow D, Xie X, St Pierre J, Sihag S, Yang W, Altshuler D, Puigserver P, Patterson N, et al. Erralpha and Gabpa/b specify PGC-1alpha-dependent oxidative phosphorylation gene expression that is altered in diabetic muscle. Proc Natl Acad Sci USA. 2004;101:6570–6575. doi: 10.1073/pnas.0401401101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedergaard J, Cannon B. The browning of white adipose tissue: some burning issues. Cell Metab. 2014;20:396–407. doi: 10.1016/j.cmet.2014.07.005. [DOI] [PubMed] [Google Scholar]
- Ouellet V, Routhier-Labadie A, Bellemare W, Lakhal-Chaieb L, Turcotte E, Carpentier AC, Richard D. Outdoor temperature, age, sex, body mass index, and diabetic status determine the prevalence, mass, and glucose-uptake activity of 18F-FDG-detected BAT in humans. J Clin Endocrinol Metab. 2011;96:192–199. doi: 10.1210/jc.2010-0989. [DOI] [PubMed] [Google Scholar]
- Peirce V, Carobbio S, Vidal-Puig A. The different shades of fat. Nature. 2014;510:76–83. doi: 10.1038/nature13477. [DOI] [PubMed] [Google Scholar]
- Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92:829–839. doi: 10.1016/s0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- Saito M, Okamatsu-Ogura Y, Matsushita M, Watanabe K, Yoneshiro T, Nio-Kobayashi J, Iwanaga T, Miyagawa M, Kameya T, Nakada K, et al. High incidence of metabolically active brown adipose tissue in healthy adult humans: effects of cold exposure and adiposity. Diabetes. 2009;58:1526–1531. doi: 10.2337/db09-0530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber SN, Emter R, Hock MB, Knutti D, Cardenas J, Podvinec M, Oakeley EJ, Kralli A. The estrogen-related receptor alpha (ERRalpha) functions in PPARgamma coactivator 1alpha (PGC-1alpha)-induced mitochondrial biogenesis. Proc Natl Acad Sci USA. 2004;101:6472–6477. doi: 10.1073/pnas.0308686101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber SN, Knutti D, Brogli K, Uhlmann T, Kralli A. The transcriptional coactivator PGC-1 regulates the expression and activity of the orphan nuclear receptor estrogen-related receptor alpha (ERRalpha) J Biol Chem. 2003;278:9013–9018. doi: 10.1074/jbc.M212923200. [DOI] [PubMed] [Google Scholar]
- Schulz TJ, Huang P, Huang TL, Xue R, McDougall LE, Townsend KL, Cypess AM, Mishina Y, Gussoni E, Tseng YH. Brown-fat paucity due to impaired BMP signalling induces compensatory browning of white fat. Nature. 2013;495:379–383. doi: 10.1038/nature11943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uldry M, Yang W, St-Pierre J, Lin J, Seale P, Spiegelman BM. Complementary action of the PGC-1 coactivators in mitochondrial biogenesis and brown fat differentiation. Cell Metab. 2006;3:333–341. doi: 10.1016/j.cmet.2006.04.002. [DOI] [PubMed] [Google Scholar]
- van der Lans AA, Hoeks J, Brans B, Vijgen GH, Visser MG, Vosselman MJ, Hansen J, Jorgensen JA, Wu J, Mottaghy FM, et al. Cold acclimation recruits human brown fat and increases nonshivering thermogenesis. J Clin Invest. 2013;123:3395–3403. doi: 10.1172/JCI68993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villena JA, Hock MB, Chang WY, Barcas JE, Giguere V, Kralli A. Orphan nuclear receptor estrogen-related receptor alpha is essential for adaptive thermogenesis. Proc Natl Acad Sci USA. 2007;104:1418–1423. doi: 10.1073/pnas.0607696104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villena JA, Kralli A. ERRalpha: a metabolic function for the oldest orphan. Trends Endocrinol Metab. 2008;19:269–276. doi: 10.1016/j.tem.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Zuercher WJ, Consler TG, Lambert MH, Miller AB, Orband-Miller LA, McKee DD, Willson TM, Nolte RT. X-ray crystal structures of the estrogen-related receptor-gamma ligand binding domain in three functional states reveal the molecular basis of small molecule regulation. J Biol Chem. 2006;281:37773–37781. doi: 10.1074/jbc.M608410200. [DOI] [PubMed] [Google Scholar]
- Wang T, McDonald C, Petrenko NB, Leblanc M, Wang T, Giguere V, Evans RM, Patel VV, Pei L. Estrogen-related receptor alpha (ERRalpha) and ERRgamma are essential coordinators of cardiac metabolism and function. Mol Cell Biol. 2015;35:1281–1298. doi: 10.1128/MCB.01156-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Seale P. Control of brown and beige fat development. Nat Rev Mol Cell Biol. 2016;17:691–702. doi: 10.1038/nrm.2016.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrann CD, White JP, Salogiannnis J, Laznik-Bogoslavski D, Wu J, Ma D, Lin JD, Greenberg ME, Spiegelman BM. Exercise induces hippocampal BDNF through a PGC-1alpha/FNDC5 pathway. Cell Metab. 2013;18:649–659. doi: 10.1016/j.cmet.2013.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao C, Goldgof M, Gavrilova O, Reitman ML. Anti-obesity and metabolic efficacy of the beta3-adrenergic agonist, CL316243, in mice at thermoneutrality compared to 22 degrees C. Obesity (Silver Spring) 2015;23:1450–1459. doi: 10.1002/oby.21124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S, Mao L, Ding P, Zhuang X, Zhou Y, Yu L, Liu Y, Nie T, Xu T, Xu Y, et al. 1-Benzyl-4-phenyl-1H-1,2,3-triazoles improve the transcriptional functions of estrogen-related receptor gamma and promote the browning of white adipose. Bioorg Med Chem. 2015;23:3751–3760. doi: 10.1016/j.bmc.2015.03.082. [DOI] [PubMed] [Google Scholar]
- Yan M, Audet-Walsh E, Manteghi S, Dufour CR, Walker B, Baba M, St-Pierre J, Giguere V, Pause A. Chronic AMPK activation via loss of FLCN induces functional beige adipose tissue through PGC-1alpha/ERRalpha. Genes Dev. 2016;30:1034–1046. doi: 10.1101/gad.281410.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneshiro T, Aita S, Matsushita M, Kayahara T, Kameya T, Kawai Y, Iwanaga T, Saito M. Recruited brown adipose tissue as an antiobesity agent in humans. J Clin Invest. 2013;123:3404–3408. doi: 10.1172/JCI67803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneshiro T, Aita S, Matsushita M, Okamatsu-Ogura Y, Kameya T, Kawai Y, Miyagawa M, Tsujisaki M, Saito M. Age-related decrease in cold-activated brown adipose tissue and accumulation of body fat in healthy humans. Obesity. 2011;19:1755–1760. doi: 10.1038/oby.2011.125. [DOI] [PubMed] [Google Scholar]
- Zuercher WJ, Gaillard S, Orband-Miller LA, Chao EY, Shearer BG, Jones DG, Miller AB, Collins JL, McDonnell DP, Willson TM. Identification and structure-activity relationship of phenolic acyl hydrazones as selective agonists for the estrogen-related orphan nuclear receptors ERRbeta and ERRgamma. J Med Chem. 2005;48:3107–3109. doi: 10.1021/jm050161j. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Related to Figure 1. ERR mRNA expression in inguinal WAT and skeletal muscle. (A) Relative Esrra, Esrrb and Esrrg mRNA levels in inguinal WAT (IngWAT) of WT and ERRαγAd−/− mice, normalized to Ppia. Data are mean ± SEM (n = 7–14). ***p < 0.001.
(B) Relative Esrra, Esrrb and Esrrg mRNA levels in skeletal muscle (plantaris) of WT and ERRαγAd−/− mice, normalized to Ppia. Data are mean ± SEM (n = 6).
Figure S2. Related to Figure 2. Gene and protein expression levels in BAT of WT and ERRAd−/− mice. (A,B) Relative mRNA levels of energy handling (A) and creatine metabolism (B) genes in BAT of WT and ERRαγAd−/− mice, normalized to Ppia. Data are mean ± SEM (n = 9–18). ***p < 0.001.
(C,D) Representative western blots of UCP1 and RAN (loading control) and their quantification, in BAT from ERRαAd−/− (C) or ERRβγAd−/− (D), relative to WT of each group. Data are mean ± SD (n = 3).
(E) Representative western blots of PGC-1α and PGC-1β in BAT of WT and ERRαγAd−/− mice (n=3). Ponceau stain is shown as loading control.
Figure S3. Related to Figure 3. Adrenergic signaling in brown adipocytes and BAT of WT and ERRαγAd−/− mice. (A) Representative western blots of phosphorylated hormone-sensitive lipase (HSL), total HSL, ERRα, and α-tubulin (loading control) in primary brown adipocytes of WT and ERRαγAd−/− mice, treated with vehicle, 100 nM norepinephrine (NE) or 50 nM CL316,243 for 10 min.
(B) Lipolysis in primary brown adipocytes of WT and ERRαγAd−/− mice, treated as in panel A for 2 hrs. Data are mean ± SEM (n = 4). ***p < 0.001 vs. vehicle in the same genotype.
(C,D) Western blots of phosphorylated and total HSL, p38 MAPK and ATF2 in BAT of WT and ERRαγAd−/− mice treated with PBS or CL316,243 for 45 min. Quantification data are mean ± SEM (n = 3). For p-p38, quantification is of the upper band. *p < 0.05 vs. vehicle in the same genotype. ND, not detected, NS, not significant.
(E) Lipolysis in BAT explants of WT and ERRαγAd−/− mice, treated ex vivo with vehicle or 1 μM CL316,243 for 2 hrs. Data are mean ± SEM (n = 3). ***p < 0.001 vs. vehicle in the same genotype.
Figure S4. Related to Figure 4. Gene expression levels in BAT of WT and ERRαγAd−/− mice treated with CL316,243. (A) Expression levels of genes that are in the top 20 induced by the ten days of CL316,243 in WT BAT and dependent on ERRs for their induction. Fourteen of the top 20 CL316,243-induced genes relied on ERRs and are shown here. Several of these genes have been reported as highly induced also by cold in BAT (Marcher et al., 2015). Data are mean ± SD of CPM (counts per million reads), measured by RNA-sequencing (n = 3).
(B,C) Expression levels of thermogenesis genes Ucp1, Dio2 (B), and regulators of lipid metabolism Ppara and Klf15 (C) in BAT of WT and ERRαγAd−/− mice, treated with PBS or CL316,243 for 10 days. Data are mean ± SD of CPM (counts per million reads), measured by RNA-sequencing (n = 3).
(A–C) **p < 0.01, ***p < 0.001 vs. WT in the same treatment; #p < 0.05, ###p < 0.001 vs. PBS in the same genotype.
Figure S5. Related to Figure 5. Adrenergic signaling in inguinal white adipocytes from WT and ERRαγAd−/− mice. (A) Representative western blots of phosphorylated hormone-sensitive lipase (HSL)S563, total HSL, ERRα, and α-tubulin (loading control) in primary inguinal adipocytes from WT and ERRαγAd−/− mice. Differentiated adipocytes were treated with either DMSO (vehicle), 100 nM norepinephrine (NE) or 50 nM CL316,243 for 10 minutes.
(B) Lipolysis (glycerol release in media) in primary inguinal adipocytes isolated from WT and ERRαγAd−/− mice. Differentiated adipocytes were treated with either DMSO (vehicle), 100 nM NE or 50 nM CL316,243 for 2 hrs. Data are mean ± SEM (n = 4). ***p < 0.001 vs. vehicle in the same genotype.
Figure S6. Related to Figure 6. Metabolic responses to adrenergic stimulation in WT and ERRαγAd−/− mice. (A,C) Average respiratory exchange ratio (RER) at 1 hour intervals of WT and ERRαγAd−/− mice injected with PBS (A) or CL316,243 (C) for ten days, on the last day of injections.
(B,D) Physical activity levels of WT and ERRαγAd−/− mice injected with PBS (B) or CL316,243 (D) for ten days, on the last day of injections.
(E) Maximum oxygen consumption (VO2) reached following an acute norepinephrine (NE) injection in WT and ERRαγAd−/− mice, previously treated with PBS or CL316,243 for ten days. **p < 0.01, ***p < 0.001 vs. WT in the same treatment; #p < 0.05 vs. PBS in the same genotype.
(F) Body weights of WT and ERRαγAd−/− mice treated with PBS or CL316,243 for ten days.
(G) Body fat percentage of WT and ERRαγAd−/− mice treated with PBS or CL316,243 for ten days. ##p < 0.01 vs. WT PBS. (A–G) Male mice were born and raised at thermoneutrality (30 °C), treated with PBS or CL316,243 for ten days, and euthanized at 12–14 weeks of age. NE responses were determined 2 days after the last PBS or CL316,243 injection. Data are presented as mean ± SEM (n = 5–10). ZT, Zeitgeber time. A.U., Arbitrary units.
Figure S7. Related to Figure 7. Food intake and body weights of HFD-fed WT and ERRαγAd−/− mice. (A) Total food intake (kCal) of female WT and ERRαγAd−/− mice shown in Figure 7 during 10 weeks on the 60% high fat diet (HFD). Data are the mean ± SEM consumption per mouse in group-housed mice (n = 8–25).
(B–D) Body weights (B), total food intake (C) and metabolic efficiency (D) of male WT and ERRαγAd−/− mice fed a control chow or 60% HFD for 10 weeks. Data are the mean ± SEM of singly-housed mice (n = 8–9).
Table S1. The top canonical pathways regulated (p < 0.05) by CL316,243 in ERRαγAd−/− BAT, and their ranking in WT BAT, using a 1.5-fold cut-off. Related to Figure 4.
Table S2. The top canonical pathways regulated (p < 0.05) by ERRs in PBS or CL316,243-treated mice, using a 1.5-fold cut-off. Related to Figure 4.
Table S3. Primer sequences used for Q-PCR. Related to Figures 1, 2, 3 and 5.