

Abstract
Caenorhabditis elegans is an exceptional model organism in which to study lipid metabolism and energy homeostasis. Many of its lipid genes are conserved in humans and are associated with metabolic syndrome or other diseases. Examination of lipid accumulation in this organism can be carried out by fixative dyes or label-free methods. Fixative stains like Nile red and oil red O are inexpensive, reliable ways to quantitatively measure lipid levels and to qualitatively observe lipid distribution across tissues, respectively. Moreover, these stains allow for high-throughput screening of various lipid metabolism genes and pathways. Additionally, their hydrophobic nature facilitates lipid solubility, reduces interaction with surrounding tissues, and prevents dissociation into the solvent. Though these methods are effective at examining general lipid content, they do not provide detailed information about the chemical composition and diversity of lipid deposits. For these purposes, label-free methods such as GC-MS and CARS microscopy are better suited, their costs notwithstanding.
Keywords: Basic Protocol, Issue 133, Lipid, Nile red, oil red O, nematode, Caenorhabditis elegans, metabolic syndrome, fat, type II diabetes, obesity, somatic, germline
Introduction
Lipids are essential for life. They are integral components of membranes, act as secondary messengers and signal transducers, and have crucial functions in energy storage. When lipid metabolism is dysregulated, it leads to diseases like obesity and type II diabetes, which are pressing public health concerns9. Caenorhabditis elegans (C. elegans) is an excellent model organism in which to study lipid metabolism because it has a relatively short life cycle, a transparent body, a known cell lineage, and a fully sequenced genome. Primarily a hermaphrodite, C. elegans allows researchers to raise large numbers of isogenic animals in short periods of time to carryout high-throughput forward genetic screens to study a wide array of metabolic genes and pathways4. This approach has revealed a high degree of conservation in 273 C. elegans lipid metabolism genes among humans, mice, rats and drosophila. Furthermore, over 300 lipid genes in C. elegans have human orthologues that are associated with diseases unrelated to metabolic syndrome11. Traditionally, examination of lipid storage in C. elegans has mostly relied on dye-labeled assays, which provide robust information about lipid accumulation. Less common is a description of where lipids localize and measured differences in lipid abundance across tissues. However, recent work has revealed that lipid distribution can be as important as lipid accumulation6.
Lately, studies have begun integrating methods such as high performance liquid chromatography-mass spectrometry (HPLC-MS), gas chromatography-mass spectrometry (GC-MS), and coherent anti-stokes Raman scattering (CARS) microscopy to address the shortcomings of stain-based approaches by directly analyzing the contents of lipid extracts, specific lipid fractions, and lipid deposits, respectively10,11. Moreover, CARS microscopy has revealed that Nile red can only serve as a proxy for fat accumulation when used as a fixative dye, for its use as a vital stain leads to off-target staining of auto-fluorescent organelles10. However, the required technical expertise and costs associated with these chromatography and microscopy methods make their use untenable for many research questions. In this article, we discuss a convenient and reliable method to fixate and stain neutral lipid deposits in C. elegans using Nile red and oil red O to distinguish lipid abundance in whole animals and in specific tissues.
Nile red, 9-diethylamino-5H-benzo[α]phenoxazine-5-one, is a benzophenoxazone dye that readily dissolves in various organic solvents, but is mostly insoluble in water. It is an excellent lysochrome dye used to stain neutral lipids such as triglycerides or cholesterol esters because it features a strong color, solubilizes well in lipids, has negligible interaction with surrounding tissues, and is less soluble in the solvent than in lipids. It has an excitation and emission maxima of 450-500 and 520 nm, respectively1. When Nile red-stained C. elegans is viewed for green fluorescence, discrete lipid bodies can be observed throughout the intestine and other tissues either in clusters or evenly dispersed, depending on the animal’s genotype or experimental treatment7.
Oil red O is a lysochrome, fat-soluble dye used to stain triglycerides and lipoproteins. It is called an azo dye because its chemical structure contains two azo groups attached to three aromatic rings. It is difficult to ionize, which renders it highly soluble in lipids. Its stain color is red and its light absorption maximum is 518 nm3. C. elegans stained with oil red O show red lipid droplets that stand out against the animal’s transparent body, which facilitates qualitative assessment of lipid distribution among different tissues6.
Protocol
1. Nile Red (NR) Staining of Lipids
- Preparation of 5 mg/mL NR stock solution
- In a 500 mL bottle, add 100 mg of NR powder to 200 mL of 100% acetone.
- Cover the bottle with aluminum foil to avoid any exposure to light.
- Before use, stir the solution for 2 h in the dark.
- For long term use, store the NR stock solution in a tightly sealed bottle without any light exposure. Scale NR stock solution according to research needs. Ensure that the same stock solution is used across NR-staining experiments to obtain consistent staining and imaging results.
- Preparation of NR working solution
- For every 1 mL of 40% isopropanol (v/v), add 6 μL of NR stock solution.
-
Prepare 600 μL of NR working solution for each sample.NOTE: Make fresh NR working solution right before staining. Depending on NR stock solution needs, use either a 15 mL or 50 mL graduated conical tube.
- Preparation of worms for NR lipid staining
- Grow worms to early L4 stage at 20 °C on nematode growth medium (NGM) seeded with late-log OP50 E. coli.
- Wash the worms off the plate with 1 mL of 1x phosphate-buffered saline + 0.01% Triton X-100 (PBST) solution and put the worm suspension in a 1.5 mL microfuge tube.
- Centrifuge the worms at 560 × g for 1 min. Remove the supernatant and repeat this step until E. coli is cleared from suspension.
- Add 100 μL of 40% isopropanol to the worm pellet and incubate it at room temperature for 3 min.
-
Centrifuge the worms at 560 × g for 1 min and remove the supernatant without disrupting the worm pellet.NOTE: Use higher volumes of PBST to wash the worms off the plates if using larger plates or staining more worms per plate. Do not wash the worms in PBST more than 15 min before the sample fixation. Perform additional washes if the supernatant is not cleared of bacteria. Carry out the incubation in 40% isopropanol using a nutator or rocker. Always monitor the tubes to make sure full sample agitation is occurring.
- Lipid staining with NR
- In the dark, add 600 μL of NR working solution to each sample. Invert the tubes three times and fully mix the worms in NR solution.
- Rotate the sample in the dark at room temperature for 2 h.
- Following the incubation, centrifuge the worms at 560 × g for 1 min and remove the supernatant.
- Add 600 μL of PBST and incubate the samples in the dark for 30 min to remove excess NR stain.
-
Centrifuge the samples at 560 × g for 1 min and remove all but approximately 50 μL of supernatant.NOTE: NR incubation does not require agitation. Settling of worms is common during this step.
- Preparation of slides for microscope imaging
- Resuspend the worm pellet in remaining supernatant.
- Place 5 μL of worm suspension on a microscope slide and put a coverslip on carefully to avoid trapping any air bubbles.
- Seal the coverslip with nail polish before imaging worms.
- Prepare only a couple slides at a time. This will ensure NR imaging remains constant across samples. The quality of NR-stained images diminishes after 6 h. Discrete lipid droplets are difficult to observe and background fluorescence increases, which interferes with NR signal detection.
- Imaging of NR-stained worms
- Image the worms at 5× magnification to capture several animals per field of view.
- Switch to 10× magnification for better quantification of individual worms.
- Use FITC/GFP channel to image NR-stained worms and try different exposure times to determine the optimal conditions for quantification.
-
Save files in TIF format to avoid losing data due to compression.NOTE: Typical exposure times range from 100-1000 ms. Having an optimal exposure time, use it for all samples to maintain imaging consistency.
- NR staining image quantification
- Upload micrographs to ImageJ. In the Plugins pull-down menu, use the Bio-Formats function if the image file is not recognized.
- In the Image pull-down menu, under the Stacks function, use Images-to-Stack to create an image stack when several micrographs are compared.
- In the same pull-down menu, adjust Brightness/Contrast of the image stack and propagate changes to all images by clicking Apply.
- Employ the Polygon selection tool to delineate each imaged worm and use the Measure function under the Analysis pull-down menu to quantify fluorescence intensity emitted by NR.
- For each image, measure five background locations and calculate the average background fluorescence intensity.
- Subtract the background fluorescence intensity from each imaged worm using the formula N = G − (A × B), where N stands for net fluorescence, G for gross fluorescence, A for total worm area and B for average background fluorescence.
-
To normalize the fluorescence intensity by worm size, divide each worm’s net fluorescence by its total area. The results are reported as fluorescence intensity, in arbitrary units (a.u.), per pixel.NOTE: Avoid exporting images in JPEG format. While compression makes files more manageable to store, data integrity is compromised by this format. Make sure that all images are modified and analyzed identically. Remember to include a scale bar to properly evaluate size at different magnifications. To better visualize differences in fluorescence intensity using ImageJ, switch image type from RGB to 8-bit and select Fire from the image lookup tables (LUT) menu. This creates a heat-map image in which increased color brightness correlates with increased fluorescence intensity.
2. Oil Red O Staining (ORO) of Lipids
- Preparation of ORO stock solution
- In a 250 mL bottle, add 500 mg of ORO powder to 100 mL of 100% isopropanol and mix it well.
- Once prepared, store the solution tightly sealed with no light exposure.
- Preparation of ORO working solution
- Dilute ORO stock solution in water (3:2) to 60% isopropanol.
-
Before use, filter the working ORO solution through a 0.2 μm cellulose acetate sterile syringe filter.NOTE: For best solution quality, prepare working ORO solution the day before, and let it mix overnight. However, if solution is needed same day, dilute and mix ORO solution on a rocker for at least 2 h before use. Before rocking, wrap conical tube in paraffin tape to avoid leakage of ORO solution.
- Preparation of worms for ORO lipid staining
- Grow the worms at 20 °C on nematode growth medium (NGM) seeded with late-log OP50 E. coli to desired life stage.
- Add 1 mL of PBST solution to the plate and swirl until all worms are off the plate. Tilt the plate and wash it with 1 mL of PBST. Transfer the worm suspension to a 1.5 mL microfuge tube.
- Centrifuge the worms at 560 × g for 1 min. Remove the supernatant without disturbing the pellet and repeat the washing step with 1 mL of PBST three times. Remove all supernatant but 100 μL.
- Add 600 μL of 40% isopropanol to the worm pellet and rock it at room temperature for 3 min.
-
Centrifuge the worms at 560 × g for 30 s and remove all supernatant but 100 μL without disrupting the worm pellet.NOTE: Use higher volumes of PBST to wash worms off plates if doing high-throughput staining. Do not wash worms in PBST more than 15 min prior to sample fixation. Do additional washes if supernatant is not cleared of bacteria. Carry out incubation in 40% isopropanol using a nutator or rocker. Always monitor tubes to make sure full sample agitation is occurring.
- Lipid staining with ORO
- Add 600 μL ORO working solution to each sample. Invert the tube three times and mix the worms well in ORO.
- Rotate the samples at 30 rpm for 2 h at room temperature.
- Centrifuge the samples at 560 × g for 1 min and eliminate all supernatant but 100 μL.
- Resuspend the samples in 600 μL of PBST and rotate the tubes at 30 rpm for 30 min to remove excess ORO stain.
- Centrifuge samples at 560 × g for 1 min and eliminate all but 50 μL of supernatant.
-
To better distinguish the location of the animal’s germline and intestinal cells, stain nuclei by adding 1 μL of (2-(4-amidinophenyl)-1 H-indole-6-carboxamidine) (DAPI) for every 1 mL of ORO working solution. Carry out ORO staining for 2 h with adding DAPI prior to mounting slides for imaging. Intestinal nuclei are larger in size and the gonad is distinguished by the developing germ cells.NOTE: After ORO staining, worms may adhere to the sides of the microfuge tube and fail to form a noticeable pellet. If this happens, centrifuge the worms again or allow worms to settle by gravity for at least 10 min.
- Preparation of the slides for worm imaging
- Resuspend the worms in the remaining supernatant and mix the solution well.
- Put 5 μL of worm suspension on a microscope slide and place a coverslip carefully, ensuring that no air bubbles form.
-
Seal the coverslip with nail polish and image worms.NOTE: After fixing and staining, worms can be very rigid and difficult to pipette. To circumvent this, make wide-bore pipettes by cutting off their tips.
- Store the slides in a micro-centrifuge rack at 4 °C for up to 24 h if not imaging immediately after staining.
- Imaging of ORO-stained worms
- Use a color-capable camera to image ORO-stained worms.
- Use 5× magnification to image several worms in one field of view.
- Switch to 10× magnification for better examination of individual worms.
-
Export images in TIF format to avoid data loss due to compression.NOTE: If staining with ORO + DAPI, switch the color-capable camera to one that can capture fluorescence.
- Image analysis of ORO-stained worms
- Upload micrograph(s) to ImageJ. In the Plugins pull-down menu, use the Bio-Formats function if the image file is not recognized.
- In the Image pull-down menu, under the Stacks function, use Images-to-Stack to create an image stack when several micrographs are compared.
- Adjust Brightness/Contrast under the same pull-down menu to improve the visibility of lipid droplets.
- Categorize the images according to lipid accumulation throughout the animal’s body or in specific tissues.
- For better evaluation of lipid localization and for the identification of non-specific image artifacts using ImageJ, select the Color function under the Image pull-down menu and click on Stack to RGB to show the signals of each RGB component.
Representative Results
SKN-1 is a bZip, cytoprotective transcription factor that shares homology with mammalian NRF2 and has been shown to mediate fatty acid oxidation. Depending on the glucose concentration in their diet, worms with a constitutively activated skn-1 allele show different lipid levels when stained with Nile red7. Figure 1A-C shows activated skn-1 animals exposed to conditions that lead to increasing lipid levels. NR fluorescence captured using a FITC/GFP channel is prominent along the intestine, but is dimmer in the head, tail, and intestinal lumen. As lipid levels increase, discrete NR-stained particles are more difficult to discern, which may necessitate lower exposure times. Though this method uses fluorescence intensity as a quantitative proxy for lipid accumulation, it is inadequate at distinguishing unspecific staining of cellular structures that are not fat stores and is prone to signal interference from intestinal auto fluorescence. Therefore, alternative label and non-label methods must be used in parallel to unambiguously quantify neutral lipids in the animal10.
Figure 1. Staining of lipids by Nile red.
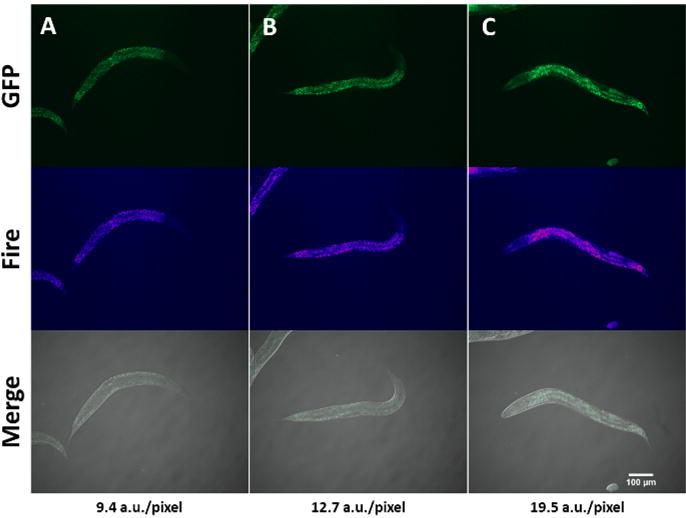
Nile red staining of activated skn-1 mutants under conditions that elicit increased lipid accumulation (AC). The gain of function skn-1 strain (lax188) harbors an E237K amino acid substitution that renders SKN-1 constitutively active. L4-stage worms were exposed to 0, 15 and 30 J/m2 UV-light followed by a 12-h recovery period on unseeded NGM plates before NR-staining and imaging. Images shown are representative of the phenotype. Lipid quantification was performed as mentioned in section 1.7. Fire is an ImageJ look up table (LUT) used to create a thermal image that shows fluorescence intensity differences between images. Merge shows FITC/GFP and DIC images combined. Quantification of fluorescence intensity for each image is shown at the bottom of the image montage. Please click here to view a larger version of this figure.
Age-dependent Somatic Depletion of Fat (Asdf), is a phenotype that occurs in aged worms whereby animals display decreased somatic cell lipids, while germ cell lipids remain unchanged6. Oil red O staining is not a method that reliably quantifies lipid levels, but is excellent for visualizing lipid localization and is useful for determining fat depletion phenotypes like Asdf in the worm. While it is easy to categorize the worms according to the presence or absence of Asdf, it may be difficult to identify animals with intermediate fat loss (Figure 2A). Compared to non-Asdf animals, which show bright red staining throughout the body with few translucent areas (Figure 2B), Asdf worms exhibit noticeable fat depletion in intestinal cells (Figure 2C). Animals in the process of developing Asdf (Figure 2D) often show translucent spots where most fat loss eventually occurs. Moreover, Asdf worms may still feature bright red staining in the head and tail regions because the phenotype is not defined by complete somatic fat loss, but by extensive fat depletion relative to non-aged (non-Asdf) animals. The use of oil red O staining, thus, allows for qualitative determination of lipid localization and the identification of phenotypes that substantially change fat deposits in the worm.
Figure 2. Staining of lipids by oil red O.
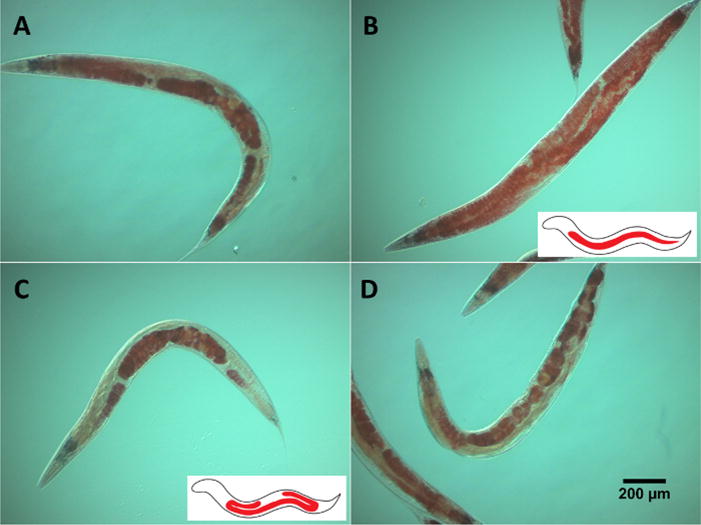
Oil red O staining of activated skn-1 mutants (lax188) showing the presence or absence of the Asdf phenotype (A-D). A & D show animals with intermediate Asdf phenotypes. B & C display opposite ORO-staining. While B is non-Asdf, C shows substantial somatic fat depletion with concomitant increase in germline fat accumulation, which defines the Asdf phenotype. The worms were stained with ORO followed by imaging 144 hafter L1-synchronized animals were placed on NGM media seeded with OP50 bacteria. Images shown are representative of animals with or without the Asdf phenotype. Inset cartoons are modified from Lynn, et al.6 and represent the absence (B) or presence (C) of somatic fat depletion. Please click here to view a larger version of this figure.
Discussion
The rise in obesity and metabolic disease rates makes C. elegans a suitable model to study the mechanisms that regulate fat accumulation in cells and tissues. Recent evidence suggests that the changes in lipid levels are correlated with cellular processes ranging from insulin signaling8, the activation of hormone receptors2, to reproductive output5. Compared to label-free microscopy and chromatography methods, Nile red and oil red O are relatively inexpensive dyes used to stain neutral lipids in the worm consistently and reproducibly10, 12, 13. The first allows for the quantification of total lipid levels, while the second facilitates the evaluation of lipid distribution among tissues such as the hypodermis, intestine, and the germ line. When used in conjunction, NR and ORO enable the researchers to determine how genotype and environmental changes affect lipid accumulation and where in the worm these changes are occurring.
These dyes, nevertheless, do not directly assess lipid accumulation in the same non-invasive manner as does CARS microscopy12. Therefore, they are prone to errors during the fixation and staining, which may compromise measurement accuracy13. Additionally, these dyes may inadvertently interact with the lipofuscin and intestinal granules, resulting in fluorescence unrelated to intracellular neutral fat stores10,14. Additionally, in cases where lipid levels appear low, NR and ORO staining cannot distinguish if the outcome results from permeability issues or reduced fat accumulation. Moreover, the sensitivity of these stains to light requires special measures during storage and active handling that limit the degradation and photo bleaching. However, if caution is exercised when performing and drawing conclusions from label-based assays, NR and ORO staining are excellent means of forward genetic screens to study lipid metabolism pathways and examine their interactions with other physiological functions. The use of daf-2 and fat-6 worms is recommended to make relative comparisons of increased and decreased lipid accumulation, respectively. The washing and fixating steps for NR and ORO staining are similar. Therefore, both can be performed on the same day. However, only the slides with ORO-stained worms can be stored for imaging later because NR staining is inconsistent after 6 h. During the washing, it is critical to include Triton 100-X not only to enhance the permeability to NR and ORO, but also to avoid the worm loss due to excessive adherence to plasticware. Additionally, the worms should be washed less than 30 min before staining to ensure maximum permeability to each stain. While NR and ORO incubation times may be reduced to 1 h and 30 min, respectively, it is encouraged to follow the time suggested in this article for more consistent staining and imaging results. Prolonged exposure of NR solution to light and failure to filter ORO solution before use will increase background fluorescence during imaging. Carrying out these staining protocols requires 3-4 h in addition to imaging time. Though stain incubation offers at most 2 h of pause between steps, it is strongly recommended that the protocols are carried out with as few interruptions as possible to reduce the many sources of error inherent to all dye-based methods of lipid quantification and examination.
Acknowledgments
This work was made possible by the NIH grant: R01GM109028 (S.P.C.)
Footnotes
Video Link
The video component of this article can be found at https://www.jove.com/video/57352/
Disclosures
The authors declare no conflicts of interest.
References
- 1.Greenspan P, Mayer EP, Fowler SD. Nile red: a selective fluorescent stain for intracellular lipid droplets. J Cell Biol. 1985;100(3):965–973. doi: 10.1083/jcb.100.3.965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guo Y, Cordes KR, Farese RV, Walther TC. Lipid droplets at a glance. J Cell Sci. 2009;122(6):749–752. doi: 10.1242/jcs.037630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Horobin RW, Kiernan JA. BIOS Scientific Publishers. 2002. Conn’s biological stains: a handbook of dyes, stains and fluorochromes for use in biology and medicine. [Google Scholar]
- 4.Hulme SE, Whitesides GM. Chemistry and the worm: Caenorhabditis elegans as a platform for integrating chemical and biological research. Angew Chem Int Edit. 2011;50(21):4774–4807. doi: 10.1002/anie.201005461. [DOI] [PubMed] [Google Scholar]
- 5.Khanna A, Johnson DL, Curran SP. Physiological roles for mafr-1 in reproduction and lipid homeostasis. Cell Rep. 2014;9(6):2180–2191. doi: 10.1016/j.celrep.2014.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lynn DA, Dalton HM, Sowa JN, Wang MC, Soukas AA, Curran SP. Omega-3 and-6 fatty acids allocate somatic and germline lipids to ensure fitness during nutrient and oxidative stress in Caenorhabditis elegans. P Natl Acad Sci USA. 2015;112(50):15378–15383. doi: 10.1073/pnas.1514012112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pang S, Lynn DA, Lo JY, Paek J, Curran SP. SKN-1 and Nrf2 couples proline catabolism with lipid metabolism during nutrient deprivation. Nat Commun. 2014;5 doi: 10.1038/ncomms6048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saltiel AR, Kahn CR. Insulin signalling and the regulation of glucose and lipid metabolism. Nature. 2001;414(6865):799–806. doi: 10.1038/414799a. [DOI] [PubMed] [Google Scholar]
- 9.Witting M, Schmitt-Kopplin P. The Caenorhabditis elegans lipidome: A primer for lipid analysis in Caenorhabditis elegans. Arch Biochem Biophys. 2016;589:27–37. doi: 10.1016/j.abb.2015.06.003. [DOI] [PubMed] [Google Scholar]
- 10.Yen K, Le TT, Bansal A, Narasimhan SD, Cheng JX, Tissenbaum HA. A comparative study of fat storage quantitation in nematode Caenorhabditis elegans using label and label-free methods. PloS One. 2010;5(9):e12810. doi: 10.1371/journal.pone.0012810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang Y, Zou X, Ding Y, Wang H, Wu X, Liang B. Comparative genomics and functional study of lipid metabolic genes in Caenorhabditis elegans. BMC Genomics. 2013;14(1):164. doi: 10.1186/1471-2164-14-164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Folick A, Min W, Wang MC. Label-free imaging of lipid dynamics using Coherent Anti-stokes Raman Scattering (CARS) and Stimulated Raman Scattering (SRS) microscopy. Curr Opin Genet Dev. 2011;21(5):585–90. doi: 10.1016/j.gde.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pino EC, Webster CM, Carr CE, Soukas AA. Biochemical and high throughput microscopic assessment of fat mass in Caenorhabditis elegans. J Vis Exp. 2013;30(73) doi: 10.3791/50180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O’ Rourke EJ, Soukas AA, Carr CE, Ruvkun G. C. elegans major fats are stored in vesicles distinct from lysosome-related organelles. Cell Metab. 2009;10(5):430–5. doi: 10.1016/j.cmet.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]