

Abstract
Enoyl-acyl carrier protein (ACP) reductase II, FabK, is a critical rate-limiting enzyme in the bacterial type II fatty acid synthesis (FAS-II) pathway. FAS-II pathway enzymes are markedly disparate from their mammalian analogs in the FAS-I pathway in both structure and mechanism. Enzymes involved in bacterial fatty acid synthesis represent viable drug targets for Gram-negative pathogens and historical precedent exists for targeting them in the treatment of diseases of the oral cavity. The Gram-negative organism Porphyromonas gingivalis represents a key causative agent of the costly and highly prevalent disease known as chronic periodontitis and exclusively expresses FabK as its enoyl reductase enzyme in the FAS-II pathway. Together, these characteristics distinguish P. gingivalis FabK (PgFabK) as an attractive and novel narrow-spectrum antibacterial target candidate. PgFabK is a flavoenzyme, dependent on FMN and NADPH as cofactors for the enzymatic reaction, which reduces the enoyl substrate via a ‘Ping-Pong’ mechanism. Herein we report the structure of the PgFabK enzyme, determined using x-ray crystallography to 1.9Å resolution with endogenous FMN fully resolved and the NADPH cofactor partially resolved. PgFabK possesses a TIM-barrel motif, and all flexible loops are visible. The determined structure has allowed insight into the structural basis for the NADPH dependence seen in PgFabK and the role of a monovalent cation that has been observed in previous studies to be stringently required for FabK activity. The PgFabK structure and the insights gleaned from its analysis will facilitate structure-based drug discovery efforts toward the prevention and treatment of P. gingivalis infection.
Keywords: Porphyromonas gingivalis, periodontal disease, enoyl-ACP reductase II, FabK, crystal structure
1. Introduction
Periodontitis is a polymicrobial-associated inflammatory disease-causing destruction to the bone and tooth-supported tissues. It is the major cause of tooth loss in the world and is associated with a high global burden in terms of treatment and preventative costs (Schaudinn et al., 2009, Snowden et al., 2003). The bacterial species Porphyromonas gingivalis is associated with a particularly aggressive, chronic disease that is often resistant to standard treatments (Lovegrove, 2004, Chavarry et al., 2009, Fitzpatrick & Katz, 2010, Joshipura et al., 2009). Additionally, recent studies have revealed P. gingivalis to be a ‘keystone’ species in periodontal disease, able to modulate the host immune response, allowing other pathogenic species to thrive (Hajishengallis et al., 2011). These studies have suggested that specific antimicrobial therapy directed toward P. gingivalis may have a strong clinical impact on community-wide oral health. Because P. gingivalis is frequently resistant to standard treatments, the development of novel treatments targeting previously unexploited pathways is critically needed.
One attractive, yet largely undeveloped area of antibacterial development is the bacterial fatty acid synthesis pathway, FAS-II. This pathway is distinct from that in mammals and has been shown to be essential in Gram-negative bacteria and some Gram-positive bacteria, which makes it an appealing and potentially selective antibacterial target (Gerusz, 2010, Kingry et al., 2013). Although novel, the use of agents targeting the FAS-II pathway in the prevention and treatment of periodontal disease is not entirely without precedent. The antibacterial triclosan, a known inhibitor of FabI (enoyl-ACP reductase I), has been used as a component of oral hygiene products in the past (Blinkhorn et al., 2009, Vered et al., 2009). P. gingivalis expresses an isozyme of FabI, FabK (enoyl-ACP reductase II) which is responsible for a key, rate-limiting reduction step in the synthesis of bacterial fatty acids (Figure 1). Additional enoyl-reductase isozymes which are known to be selectively expressed in bacteria are FabL and FabV, enoyl-ACP reductase III and IV, respectively. Some bacteria are known to co-express multiple enoyl reductase isozymes, including E. faecalis which expresses FabI and FabK, and P. aeruginosa which expresses FabI, FabL and FabV. However, in P. gingivalis, the FabK enzyme is the sole isozyme and is completely resistant to triclosan and other FabI active agents (Marrakchi et al., 2003). Although the existence of multiple isozymes at this step in the pathway precludes the possibility of developing broad spectrum antimicrobials, a possibility exists that relatively selective antibacterial agents may be developed that can target harmful bacterial species, such as P. gingivalis, while avoiding activity against so-called ‘beneficial bacteria’.
Figure 1.
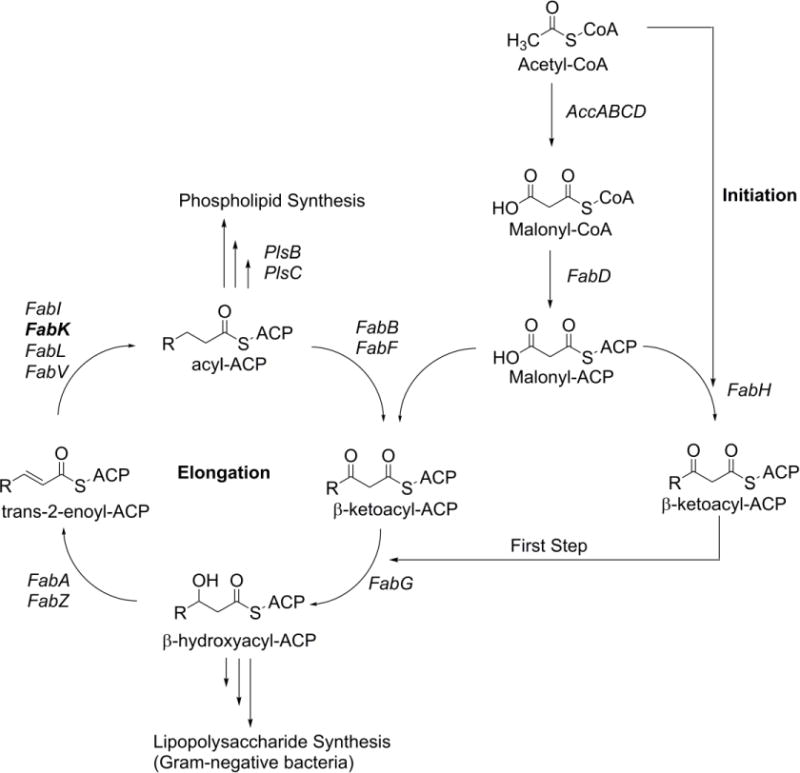
Bacterial FAS-II Pathway. Adapted with permission from Gerusz, V., et al. (2012). Journal of Medicinal Chemistry, 55, 9914-9928. Copyright 2012 American Chemical Society.
The FabK enzyme from Streptococcus pneumoniae (SpFabK) has been previously characterized (Marrakchi et al., 2003) and structures, both apo and with a known inhibitor bound, have been determined for this organism, leading to several key observations (Saito et al., 2008, Saito et al., 2006). SpFabK is a flavoenzyme, dependent upon an FMN prosthetic group and NADH cofactor, and uses a ‘Ping-Pong’, bi-bi enzyme mechanism to reduce the enoyl substrate (Hevener et al., 2012, Saito et al., 2008). SpFabK is structurally distinct from FabI (as well as FabV and FabL) and possesses an overall triose-phosphate isomerase (TIM) barrel structural fold, whereas FabI, FabV and FabL fall within the short-chain dehydrogenase/reductase (SDR) superfamily of reductases and possess a classical Rossmann fold for binding to their NAD(P)H cofactor (White et al., 2005, Oppermann et al., 2003). Interestingly, an intrinsic NADH oxidation activity, in the absence of enoyl substrate, was reported for SpFabK (Marrakchi et al., 2003). The significance of this activity is unclear, but it was hypothesized that FabK was related to bacterial NADH oxidases involved in anaerobic catabolism of glucose (Marrakchi et al., 2003). Lastly, the activity of the SpFabK enzyme was shown to be critically dependent on monovalent cation salts, and a preference for NH4+ salts was seen with respect to the potentiation of activity. No activity was seen with the use of bivalent cation salts and removal of the monovalent salts resulted in an apparent loss of structural integrity of the enzyme (Marrakchi et al., 2003).
We have previously reported preliminary kinetic characterization and crystallization studies of P. gingivalis FabK (Hevener et al., 2012). PgFabK has a 41% overall sequence identity compared to SpFabK. Unlike NADH for S. pneumoniae FabK, P. gingivalis FabK showed a strong preference for NADPH. Km values for both NADPH and the enoyl substrate, crotonyl-CoA, were determined and balanced assay conditions reported. We also noted a slight, intrinsic NADPH oxidation with PgFabK in the absence of substrate, and interestingly, the enzyme kinetics suggested a pattern of substrate inhibition at higher concentrations of NADPH (Hevener et al., 2012). However, several critical issues still impede successful structure-based design efforts toward the identification of PgFabK inhibitors: First, the binding mode of the NADPH/NADP+ cofactor has not been established, nor has the structural basis for the dependence of PgFabK on NADPH over NADH been established. Second, missing loop elements from the SpFabK crystal structures only allow partial visualization of the active site. And lastly, the structural basis for the cation dependence and the intrinsic activity that has been noted has not been established. Herein, we report the structure of P. gingivalis FabK determined to 1.9 Å resolution by x-ray crystallography. In this structure, all flexible loops are present surrounding the active site. Additionally, the endogenous FMN prosthetic group is fully visible and the NADPH cofactor is partially resolved in two distinct binding sites. A metal binding pocket in the active site is clearly occupied by a sodium ion and provides insights, discussed here, as to the structural basis for the cation dependence noted.
2. Materials and methods
2.1. Reagents, chemicals, biologicals, and equipment
The protein expression cell line, BL21-Gold (DE3), catalog #230132, was obtained from Agilent Technologies (Santa Clara, CA). The pET-15b plasmid vector was obtained from Novagen/Millipore (Madison, WI). Genomic DNA (Porphyromonas gingivalis strain W83, BAA-308D-5) was obtained from ATCC (Manassas, VA). Oligonucleotide primers were obtained from Integrated DNA Technologies (IDT, Coralville, IA). The IMAC-Ni column (HisTrap HP, 5 ml) and gel filtration columns (HiLoad 26/600 and 16/600, Superdex 200 PG) were obtained from GE Healthcare (Piscataway, NJ). Ampicillin, isopropyl β-D-thiogalactopyranoside (IPTG), β-nicotinamide adenine dinucleotide 2′-phosphate reduced tetrasodium salt (NADPH) were obtained from Sigma-Aldrich Chemicals (St. Louis, MO). Crystallization screening kits (SaltRx, Index, Crystal Screen, PEG/Ion) and supplies were obtained from Hampton Research (Aliso Viejo, CA). Expression media LB and TB broth (BD Difco™), MOPS buffer and NaCl (Acros Organics), and glycerol (Fisher Chemical) were obtained from Fisher Scientific (Pittsburgh, PA).
2.2. Macromolecule production
We have previously reported the cloning, expression and purification of FabK from Porphyromonas gingivalis (Hevener et al., 2012). In short, a standard prokaryotic protein expression protocol was used to express the PgFabK enzyme. The fabK gene was cloned by PCR amplification using genomic DNA from the W83 P. gingivalis strain. Primers were designed in-house and ordered from a commercial source (Table 1). The gene was cloned into the BamHI and NdeI restriction sites of pET-15b. After sequence confirmation, the E. coli strain BL21 (DE3) was used for expression of PgFabK. The expressed protein is purified using two chromatographic steps: an immobilized metal affinity (IMAC) step using nickel chelating resin followed by size exclusion chromatography. FMN was co-purified along with the target protein and was not supplemented during expression or purification stages. Typical yield is approximately 20mg of protein per liter of culture media at greater than 95% purity. The His-tag is not removed, and all studies reported here involved the use of the affinity-tagged protein.
Table 1.
Macromolecule production information
| Source organism | Porphyromonas gingivalis (strain: W83 BAA-308D-5) |
|---|---|
| DNA source | gDNA |
| Forward primer | 5′-GAA TAA GCA TAT GAA TAG AAT TTG CGA ATT ATT GGG T-3′ |
| Reverse primer | 5′-AGA TGG ATC CTC ATA TCT CAG TGG G-3′ |
| Cloning vector | pET-15b |
| Expression vector | pET-15b |
| Expression host | E. coli BL21 (DE3) |
| Complete amino acid sequence of the construct produced | MGSSHHHHHHSSGLVPRGSHMNRICELLGIEHPIISGGMVWCSGWKLASAVSNCGGLGLIGAGSMHPDNLEHHIRSCKAATDKPFGVNVPLLYPEMDKIMEIIMREHVPVVVTSAGSPKVWTAKLKAAGSKVIHVVSSATFARKSEAAGVDAIVAEGFEAGGHNGREETTTLCLIPEVVDAVNIPVVAAGGIASGRAVAAALALGADAVQVGTRFALSEESSAHEDFKAHCRRSVEGDTMLSLKAVSPTRLLKNKFYQDVFAAEQRGASVEELRELLGRGRAKQGIFEGDLHEGELEIGQAVSQISHAETVAEIMVDLVD GYKRSLAGMPTEI |
2.3. Crystallization
Protein crystals were grown using the hanging drop vapor diffusion method. A coarse matrix screen was employed using commercially available kits to identify preliminary crystallization conditions. The protein was incubated for 15 minutes at room temperature with a 10-fold molar excess of NADPH prior to crystallization setup. Initial crystal growth conditions were 0.15 M Na Citrate, pH 5.6, 10% PEG 3350, 5mg/ml protein in drops 1:1 with well buffer (Table 2). Crystals were visible within one week.
Table 2.
Crystallization
| Method | Vapor diffusion, hanging drop |
|---|---|
| Plate type | 24-well VDX™ plate with sealant (Hampton Research) |
| Temperature (K) | 291 |
| Protein concentration (mg ml−1) | 5 mg |
| Buffer composition of protein solution | Initial: 10 mM MOPS, pH 7.5 with 100 mM
NH4Cl, 1.4 mM NADPH, and 5 mM DTT Micro-seeding: 0.15 M sodium citrate, pH 5.6, 10% w/v PEG3350 |
| Composition of reservoir solution | Initial crystal: 0.15 M Na citrate, pH 5.6,
10% PEG 3350 Micro-seeding: 0.2 M Na formate, pH 7.0, 20% PEG 3350 |
| Volume and ratio of drop | 3μL (protein stock):3μL (Index 90):1μL Seed stock (Seed Stock From: 10% PEG 3350, 0.15 M Na Citrate pH 5.6) |
| Volume of reservoir (μL) | 500 |
Microseeding
Crystals were harvested from the initial hit condition, looped through 50% glycerol (with well buffer) for cryoprotection, and screened for their ability to diffract x-rays using an in-house x-ray diffractometer. Because they diffracted poorly, a series of microseeding trials were undertaken using the initial protein crystals to generate seed stocks and rescreen. This technique resulted in the identification of a novel crystallization condition, 0.2 M Na formate, pH 7.0, 20% PEG 3350 that resulted in crystals with significantly better diffraction over the initial conditions.
2.4. Data collection and processing
PgFabK crystals were harvested, looped through 50% glycerol for cryoprotection, and cooled to 100 K for data collection. The complete data set was collected at 21-ID-G, LS-CAT, sector 21, at the Advanced Photon Source, Argonne National Laboratory, Lemont, IL, USA (Table 3). The data were processed using the program XDS and scaled with XSCALE (Kabsch, 2010a, Kabsch, 2010b).
Table 3.
Data collection and processing
| Values for the outer shell are given in parentheses. | |
|---|---|
| Diffraction source | Synchrotron (APS beamline 21-ID-F) |
| Wavelength (Å) | 0.97 |
| Temperature (K) | 100 |
| Detector | Rayonix MX-225 CCD |
| Crystal-detector distance (mm) | 200.5 |
| Rotation range per image (°) | 1 |
| Total rotation range (°) | 360 |
| Exposure time per image (s) | 1 |
| Space group | P212121 |
| a, b, c (Å) | 48.65, 86.65, 150.51 |
| α, β, γ (°) | 90, 90, 90 |
| Mosaicity (°) | 0.1 |
| Resolution range (Å) | 20–1.938 (2.05–1.938) |
| Total No. of reflections | 648327 (97141) |
| No. of unique reflections | 90851 (14235) |
| Completeness (%) | 99.5 (97.5) |
| Redundancy | 7.1 (6.8) |
| 〈 I/σ(I)〉 | 15.2 (4.2) |
| Rr.i.m. | 0.145 (0.583) |
| Overall B factor from Wilson plot (Å2) | 21 |
| CC1/2 | 0.997 (0.892) |
2.5. Structure determination and refinement
The PgFabK structure was determined via molecular replacement with Phaser from the CCP4 package using the SpFabK structure previously reported (Winn et al., 2011, Saito et al., 2008, Saito et al., 2006). Model refinement was performed using refmac and model building by Coot (Murshudov et al., 1997, Emsley et al., 2010, McCoy et al., 2007). Restraint files for the FMN, NDP, and GOL ligands were obtained from the CCP4 monomer library. The structure was validated using MolProbity (Davis et al., 2007). Figures 2, 4 and 5 were produced using the UCSF Chimera package from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIH P41 RR001081), and rendered using POV-ray, Persistence of Vision Pty. Ltd. (Pettersen et al., 2004).
Figure 2.
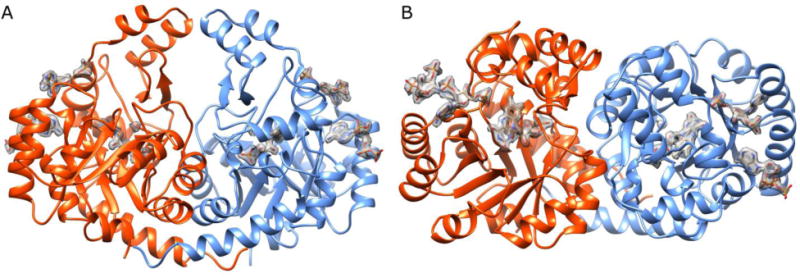
Overall PgFabK Structure. A. PgFabK enzyme depicted as the dimer (chain A orange, chain B blue). FMN prosthetic group and NADPH cofactors are shown with density contour map. B. Rotation of A by 90° about X-axis.
Figure 4.
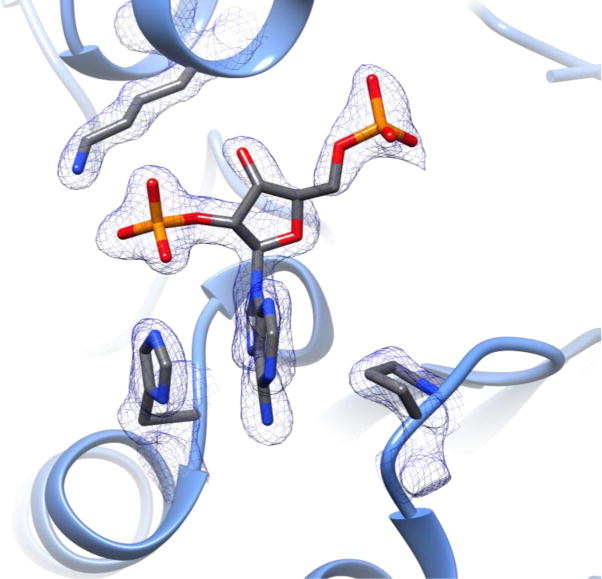
PgFabK NADPH Interactions. 2Fo-Fc density map of the active site NADPH fragment and interacting residues (shown at 1.25 r.m.s.d.).
Figure 5.
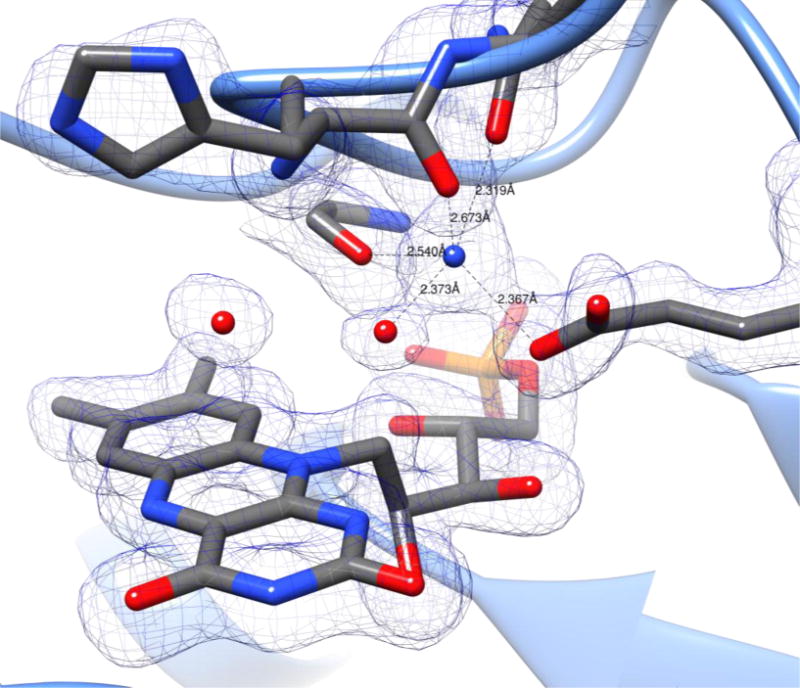
Sodium Ion Coordination in Active Site. Electron density contour map shown at 1.25 r.m.s.d. shows an approximate trigonal bipyramidal coordination of the sodium ion.
2.6. Analytical Gel Filtration
A HiLoad 16/600 Superdex 75 PG (120 ml, 16 × 600 mm) size exclusion column was equilibrated at 277 K with one and a half column volumes (180 ml) of Buffer A, containing 10 mM MOPS (pH 7.0), 50mM NH4Cl, and 2 mM DTT, using a 1 mL per minute flow rate. Three milliliters of concentrated PgFabK protein (approximately 6 mg/ml) in Buffer A was loaded onto the equilibrated column at 1 ml/min flow rate. Elution was done for the duration of one CV (120 ml), and the FabK protein was eluted around 60 ml of elution volume. Three reference proteins with known molecular weights, 94 kDa, 44 kDa, 17 kDa, were also tested with the same column and the flow rate.
3. Results and discussion
3.1. Overall Structure of PgFabK
The PgFabK crystal structure was determined to 1.94 Å resolution and refined to Rwork/Rfree of 0.161/0.207. The structure contains two domains per 33 kDa monomer and includes the endogenous FMN prosthetic group fully resolved, the NADPH cofactor partially resolved, with all flexible loops fully visible. The bulk of each monomer is comprised of residues 1-198 of the N-terminal domain, which form 8 alternating α-helix-β-strand repeats with the β-strands forming an internal core encompassed by the α-helices, making up a classic TIM barrel motif (Figure 2). The single FMN cofactor binds non-covalently atop the barrel near the C-terminal ends of the β-strands. Residues 198-289 comprise much of the C-terminal domain, forming a putative lid above the TIM barrel and containing a β-hairpin that resides proximally to the His143 catalytic residue (see below). Residues 290-308 form an additional α-helix adjacent to the TIM barrel. Six sodium ions, three associated with each chain, were modelled into the final structure (Table 4), with an average overall B factor of 20 Å2. Sodium ions were selected as initial attempts to place water molecules at these positions resulted in higher B factors and the coordinating geometry was consistent with metal ions versus water molecules (Figure S1).
Table 4.
Structure determination and refinement
| Values for the outer shell are given in parentheses. | |
|---|---|
| Resolution range (Å) | 20.00–1.94 (1.988–1.938) |
| Completeness (%) | 99.7 |
| σ cutoff | None |
| No. of reflections, working set | 45527 (3152) |
| No. of reflections, test set | 2424 (175) |
| Final Rcryst | 0.161 (0.210) |
| Final Rfree | 0.207 (0.272) |
| Cruickshank DPI | 0.151 |
| No. of non-H atoms | |
| Protein | 4638 |
| Ion (Na+) | 6 |
| Ligand (FMN) | 62 |
| Ligand (NDP) | 116 |
| Ligand (GOL) | 6 |
| Water | 319 |
| Total | 5147 |
| R.m.s. deviations | |
| Bonds (Å) | 0.017 |
| Angles (°) | 1.6 |
| Average B factors (Å2) | |
| Protein | 13 |
| Ion (Na+) | 20 |
| Ligand (FMN) | 9.5 |
| Ligand (NDP) | 25 |
| Ligand (GOL) | 22 |
| Water | 18 |
| Ramachandran plot | |
| Favored (%) | 97.6 |
| Allowed (%) | 100.0 |
Several interactions exist between the N-terminal TIM barrel β-strands and the FMN cofactor. The isoalloxazine ring system of FMN forms hydrogen bonds with Asn68 and Ser94, a backbone hydrogen bond with Val20, as well as interactions with several water molecules. The ribityl moiety of the FMN molecule forms hydrogen bonds with Glu136 and Gln190, in addition to forming a backbone hydrogen interaction with Met19 and multiple water molecule interactions. The C-terminal “lid” domain appears to form a pocket between itself and the N-terminal domain in which the phosphate from the FMN cofactor binds. Here, the phosphate moiety forms a hydrogen bond with Thr193, backbone interactions with Gly171, Gly192, and Thr193, as well as interactions of its own with several water molecules.
Previously determined structures of SpFabK and TmFabK were consistent with a dimeric enzyme, but the functionality of the dimer was not confirmed. The PgFabK homodimer interface buries an extensive surface area of about 2,200 Å2 involving 33 hydrogen bonds and 9 salt bridges as calculated by the PDBePISA server (Krissinel & Henrick., 2007). Additionally, a favorable solvation energy gain upon interface formation, estimated at −28.1 kcal/mol., indicates a high probability that the enzyme is a functional dimer. This was further confirmed by investigating the oligomeric state of this enzyme in solution by size-exclusion chromatography. PgFabK eluted between 94 kDa and 44 kDa (Figure 3), confirming of a functional 66 kDa dimer. All of this is conclusively corroborated by the electron density and overall determined crystal structure showcasing the dimer.
Figure 3.
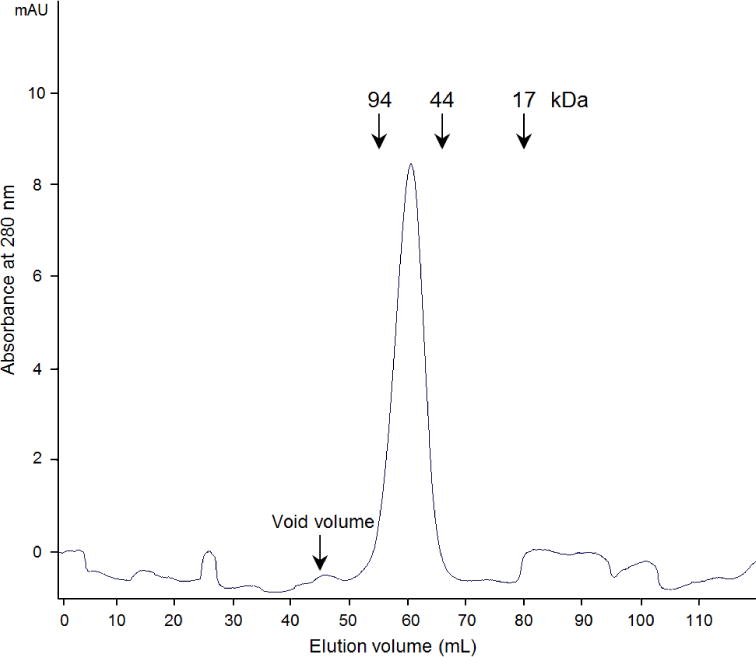
UV Profile of Gel Filtration of PgFabK. The protein elution peak relative to control proteins suggests a functional PgFabK dimer (monomer is 33 kDa).
3.2. Structural Basis of NADPH Dependence
While the FabK isozyme from Streptococcus pneumoniae was reported to be highly selective for NADH over NADPH as the nucleotide cofactor (Marrakchi et al., 2003), we have observed the Porphyromonas gingivalis FabK enzyme to show greater selectivity for NADPH (Hevener et al., 2012). To elucidate the structural basis for these observations, the NADPH cofactor was co-crystallized with the FabK protein (see Methods above) using a 10-fold molar excess of NADPH. The refined model shows the presence of two partially resolved NADPH molecules bound to each FabK monomer, one near the active site and one located distant to the active site. The second NADPH molecule is approximately 8.5 Å distant from the active site NADPH molecule, with the adenine ring system occupying a hydrophobic pocket created by Phe267, Trp21, His53, Met45, and Asn49. The presence of the second (adventitious) NADPH molecule is presumed to be a co-crystallographic artefact arising from the use of a 10-fold molar excess of NADPH in the crystallization buffer. However, we note that the average B-factor for the second NADPH molecule is slightly lower than that calculated for the NADPH molecule residing in the active site, 21 and 29 Å2, respectively.
Both NADPH molecules are only partially resolved, with no visible electron density for the nicotinamide moiety past the diphosphate linker at an r.m.s.d. of 1 (Figures S2 & S3). This may be due to hydrolytic cleavage of the nicotinamide group at the diphosphate moiety (Wu et al., 1986), or structural disorder. A small deviation from the optimal value for the O1X-P2B-O3X angle (119.9° with standard deviation of 3.0 from the CCP4 monomer library versus 111.9° here) is noted, though based upon the density maps (Figures S2 and S3) the ligand does not appear to be misfit. The NADPH nicotinamide group is responsible for the hydride transfer to the FMN prosthetic group during the first part of the double-displacement enzymatic reaction (Saito et al., 2008); due to the absence of density for this moiety, the complete structural detail of the PgFabK catalytic mechanism cannot be fully elucidated. However, the adenine ring system, ribose ring, and both the 2′- and 5′ phosphates have clear electron density and this portion of the NADPH molecule is believed to be bound to the enzyme in its true binding conformation. The 2′-ribosyl phosphate group of NADPH is observed to make favorable electrostatic interactions with the positively charged Lys263 as well as the His46 residue, which is likely either positively charged or protonated on the epsilon nitrogen (Figure 4). The corresponding residues in the NADH-binding S. pneumoniae FabK enzyme are Ala267 and Pro47, which may explain the difference in cofactor preference between these two enzymes. The adenine ring system of the bound NADPH is observed to stack between His46 and Pro74 and to make favorable electrostatic binding interactions with both Glu75 and Tyr73. The distance of the NADPH adenosyl moiety to the FMN prosthetic group in the FabK active site would appear to allow for correct placement of the missing nicotinamide moiety near the FMN isoalloxazine ring system (Figure S4), lending further credence to the possibility that, though only partially resolved, the binding conformation of the NADPH fragment reflects the true binding conformation of the full NADPH molecule. Further studies are underway to confirm this hypothesis.
3.3. Insights into the Enzyme Mechanism
A comparison of aligned sequences of the FabK enzyme from three species for which the structure has been determined, S. pneumoniae, T. maritima, and P. gingivalis, show a high degree of similarity between the structures, with 43% overall identity between the P. gingivalis and T. maritima sequences and 41% overall identity between P. gingivalis and S. pneumoniae (Figure S5). The T. maritima and S. pneumoniae sequences have 49% overall identity. Across all three species, 34% of residues are identical, overall, with the active site similarity being significantly higher. Within a 4 Å region defined by the phenylimidazole derivative FabK inhibitor in the S. pneumoniae co-crystal structure (Saito et al., 2008), 42% of residues are identical across all three species, increasing to 58% if similarity (Val/Ala, Leu/Ile & Asp/Glu) is considered. Interestingly, most of the active site differences come from the P. gingivalis protein, as the T. maritima and S. pneumonia active sites are 90% similar. Notable differences in the P. gingivalis active site are Asn144, and Glu277 which differ significantly from the corresponding residues in the other two species’ enzymes. A highly conserved region from Ala188 to Phe195 corresponds to the FMN binding region, which differs amongst all three species only by a methionine substitution in TmFabK at the position corresponding to Val191.
An analysis of highly conserved residues in the FabK active site can provide some insight into residues involved in the catalytic mechanism. A protein family analysis based upon globally conserved residues reveals that the FabK enzyme falls in the nitronate monooxygenase (NMO, formerly 2-nitropropane dioxygenase) family of flavoproteins. Previous reports on the mechanism of NMO enzymes have suggested a role for an active site histidine residue acting as a catalytic base in the oxidative reaction (Gadda & Francis, 2010, Francis & Gadda, 2008). Consistent with this, a globally conserved histidine residue (His143 in P. gingivalis) is located near the active site of the FabK enzyme as part of a structurally conserved G-G-H motif on loop 6 of the TIM barrel structure. We suggest a possible role of the His residue as a proton donor/acceptor (catalytic acid/base) in the FabK reduction reaction, facilitating reduction of FMN by the NADPH substrate, and the subsequent reduction of the enoyl substrate by reduced FMN. Additionally, globally conserved residues Trp21 located in loop 1 and Pro74 in loop 3 are adjacent to the NADPH binding site discussed above and may play a functional role in facilitating the binding of the NADPH cofactor.
3.4. Role of the Stringently Required Monovalent Cations
The activity of the FabK enzyme has previously been reported to depend on the presence of a monovalent cation salt in the enzyme conditions, with NH4Cl having the most potent effect (Marrakchi et al., 2003). This effect has been observed and reported for the PgFabK enzyme as well (Hevener et al., 2012). Further, studies with the S. pneumoniae FabK enzyme, including high-throughput screening, reported the use of NH4Cl as the salt in the assay buffer (Ozawa et al., 2007). This suggests a possible structural or catalytic role for the monovalent cation, though it is interesting to note that the order reported by Marrakchi and coworkers for the salt activation of enzymatic activity closely follows the Hofmeister series (Hofmeister, 1888). This may suggest an additional role of the monovalent cation salt in facilitating the solubility and stability of the protein.
In the structure reported herein, a sodium ion was identified in the active site of the PgFabK enzyme, adjacent to the FMN binding site (Figure 5). This density was modelled as a sodium ion due to the presence of a relatively high concentration of sodium in the storage and crystallization buffers, as well as the types and distance of the coordinating atoms from the metal. The sodium ion is coordinated by an active site water (2.37 Å), the Glu136 OE2 atom (2.37 Å), the Asn144 OD1 atom (2.32 Å) and the Gly141 backbone carbonyl oxygen (2.54 Å). The His143 backbone carbonyl oxygen also appears to play a coordinating role, though the distance is further at 2.67 Å. The usual coordination number of monovalent cations such as sodium or potassium is five or six (trigonal bipyramidal or octahedral) while the divalent cations prefer a coordination number of six (Harding et al., 2010). Additionally, the normal cation to donor atom distance for sodium is 2.38 – 2.41 Å. The observed coordination of the sodium ion in this structure may explain, in part, the activity activation of this enzyme by monovalent cation salts, particularly NH4Cl. The distances and geometries between the sodium cation and the coordinating atoms in the active site residues suggest a 5-point coordination (trigonal bipyramidal) of the sodium ion, however the coordination is slightly distorted from the ideal trigonal bipyramidal geometry and the coordinating His134 oxygen falls outside the typical cation to donor range. A 4-point coordination, as would be seen with the NH4+ cation may be more favorable as the tetragonal geometry would be less distorted. Further, the structure suggests that the 6-point coordination (octahedral) that would be preferred by divalent cations could not be achieved, which may explain the negative effect on activity of the divalent cation salts previously reported (Marrakchi et al., 2003). Whether the active site cation plays a catalytic role or simply a structural role remains to be determined.
4. Conclusions
FabK (enoyl-ACP reductase II) is a novel antimicrobial target in the bacterial fatty acid synthesis pathway. As the sole enoyl reductase enzyme in P. gingivalis, a major causative organism in chronic periodontitis, FabK has strong potential for development as a drug target. Using x-ray crystallography methods, we have identified conditions leading to crystals which diffracted to high resolution and determined the PgFabK crystal structure to 1.9 Å. This FabK structure is the first in which all flexible loops are fully resolved and the binding site, with the FMN prosthetic group and a partially resolved NADPH cofactor, is visible. The results of these studies have improved our understanding of the FabK target with respect to the role of the cofactor and monovalent cation in the catalytic mechanism and will facilitate future rational drug discovery efforts targeting the enzyme.
Supplementary Material
Synopsis.
The structure of the PgFabK enzyme was determined via x-ray crystallography to 1.9Å resolution with all flexible loops visible, FMN fully resolved, and the NADPH cofactor partially resolved. Characterization includes the structural basis of NADPH dependence and the role of a required monovalent cation.
Acknowledgments
Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor for the support of this research program (Grant 085P1000817). Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357.
Funding information Funding was provided by NIAID R21-AI26755, UTHSC Hevener (PI), and NIDCR 5T32-DE018381, UIC College of Dentistry, MOST Program.
References
- Blinkhorn A, Bartold PM, Cullinan MP, Madden TE, Marshall RI, Raphael SL, Seymour GJ. Br Dent J. 2009;207:117–125. doi: 10.1038/sj.bdj.2009.669. [DOI] [PubMed] [Google Scholar]
- Chavarry NG, Vettore MV, Sansone C, Sheiham A. Oral Health Prev Dent. 2009;7:107–127. [PubMed] [Google Scholar]
- Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, 3rd, Snoeyink J, Richardson JS, Richardson DC. Nucleic Acids Res. 2007;35:W375–383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Acta Cryst. 2010;D66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzpatrick SG, Katz J. J Dent. 2010;38:83–95. doi: 10.1016/j.jdent.2009.10.007. [DOI] [PubMed] [Google Scholar]
- Francis K, Gadda G. Biochemistry. 2008;47:9136–9144. doi: 10.1021/bi801013e. [DOI] [PubMed] [Google Scholar]
- Gadda G, Francis K. Arch Biochem Biophys. 2010;493:53–61. doi: 10.1016/j.abb.2009.06.018. [DOI] [PubMed] [Google Scholar]
- Gerusz V. Annu Rep Med Chem. San Diego, CA: Elsevier; 2010. pp. 295–311. [Google Scholar]
- Hajishengallis G, Liang S, Payne MA, Hashim A, Jotwani R, Eskan MA, McIntosh ML, Alsam A, Kirkwood KL, Lambris JD, Darveau RP, Curtis MA. Cell Host Microbe. 2011;10:497–506. doi: 10.1016/j.chom.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding MM, Nowicki MW, Walkinshaw MD. Crystallography Reviews. 2010;16:247–302. [Google Scholar]
- Hevener KE, Mehboob S, Boci T, Truong K, Santarsiero BD, Johnson ME. Protein Expr Purif. 2012;85:100–108. doi: 10.1016/j.pep.2012.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmeister F. Arch Exp Pathol Pharmacol. 1888;24:247–260. [Google Scholar]
- Joshipura K, Zevallos JC, Ritchie CS. Compend Contin Educ Dent. 2009;30:430–439. [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. Acta Cryst. 2010a;D66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. Acta Cryst. 2010b;D66:133–144. doi: 10.1107/S0907444909047374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingry LC, Cummings JE, Brookman KW, Bommineni GR, Tonge PJ, Slayden RA. J Bacteriol. 2013;195:351–358. doi: 10.1128/JB.01957-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E, Henrick K. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- Lovegrove JM. J N Z Soc Periodontol. 2004:7–21. [PubMed] [Google Scholar]
- Marrakchi H, Dewolf WE, Jr, Quinn C, West J, Polizzi BJ, So CY, Holmes DJ, Reed SL, Heath RJ, Payne DJ, Rock CO, Wallis NG. Biochem J. 2003;370:1055–1062. doi: 10.1042/BJ20021699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Acta Cryst. 1997;D53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Oppermann U, Filling C, Hult M, Shafqat N, Wu X, Lindh M, Shafqat J, Nordling E, Kallberg Y, Persson B, Jornvall H. Chem Biol Interact. 2003;143–144:247–253. doi: 10.1016/s0009-2797(02)00164-3. [DOI] [PubMed] [Google Scholar]
- Ozawa T, Kitagawa H, Yamamoto Y, Takahata S, Iida M, Osaki Y, Yamada K. Bioorg Med Chem. 2007;15:7325–7336. doi: 10.1016/j.bmc.2007.08.050. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Saito J, Yamada M, Watanabe T, Iida M, Kitagawa H, Takahata S, Ozawa T, Takeuchi Y, Ohsawa F. Protein Sci. 2008;17:691–699. doi: 10.1110/ps.073288808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito J, Yamada M, Watanabe T, Kitagawa H, Takeuchi Y. Acta Cryst. 2006;F62:576–578. doi: 10.1107/S1744309106017039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaudinn C, Gorur A, Keller D, Sedghizadeh PP, Costerton JW. J Am Dent Assoc. 2009;140:978–986. doi: 10.14219/jada.archive.2009.0307. [DOI] [PubMed] [Google Scholar]
- Snowden CB, Miller TR, Jensen AF, Lawrence BA. Public Health Rep. 2003;118:10–17. doi: 10.1016/S0033-3549(04)50212-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vered Y, Zini A, Mann J, DeVizio W, Stewart B, Zhang YP, Garcia L. J Clin Dent. 2009;20:62–65. [PubMed] [Google Scholar]
- White SW, Zheng J, Zhang YM, Rock Annu Rev Biochem. 2005;74:791–831. doi: 10.1146/annurev.biochem.74.082803.133524. [DOI] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS. Acta Cryst. 2011;D67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JT, Wu LH, Knight JA. Clin Chem. 1986;32:314–319. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.