

Abstract
Huntington disease (HD) is a neurodegenerative disorder caused by an expanded CAG tract in the HD gene. Polyglutamine expansion of huntingtin (htt) results in early, progressive loss of medium spiny striatal neurons as well as cortical neurons that project to the striatum. Excitotoxicity has been postulated to play a key role in the selective vulnerability of striatal neurons in HD. Early excitotoxic neuropathological changes observed in human HD brain include increased quinolinate (QUIN) concurrent with proliferative changes such as increased spine density and dendritic length. In later stages of the disease, degenerative-type changes are apparent, such as loss of dendritic arborization, a reduction in spine density and reduced levels of 3-hydroxykynurenine and QUIN. It is currently unknown whether sensitivity to excitotoxic stress varies between initiation and progression of disease. Here we have assessed the excitotoxic phenotype in the YAC128 mouse model of HD by examining the response to excitotoxic stress at different stages of disease. Our results demonstrate that YAC128 mice display enhanced sensitivity to NMDA (N-methyl-d-aspartate) ex vivo and QUIN in vivo prior to obvious phenotypic changes. By contrast, 10 months-old symptomatic YAC128 mice are resistant to QUIN-induced neurotoxicity. These findings are paralleled by a significant increase in NMDAR-mediated membrane currents in presymptomatic YAC128 dissociated medium spiny neurons (MSNs) progressing to reduced NMDAR-mediated membrane currents with disease progression. These data highlight the dynamic nature of the mutant htt-mediated excitotoxic phenotype and suggests that therapeutic approaches to HD may need to be altered, depending on the stage and development of the disease.
Keywords: Huntington disease, excitotoxicity, mouse models, ischemia, mutant huntingtin, quinolinate
Introduction
HD is an adult onset neurodegenerative disorder characterized by progressive deterioration of cognitive and motor functions (Harper, 2005). Neurodegeneration occurs most severely in the MSNs of the striatum, and at later stages in the cortex and other regions of the brain (Vonsattel et al., 1985). The htt protein is expressed in neurons, including dendrites and axon terminals (Sharp et al., 1995), and is involved in a wide range of cellular activities including mediating glutamate-induced intracellular signaling and vesicle trafficking (DiFiglia et al., 1995; Velier et al., 1998; Metzler et al., 2007).
Several lines of evidence have demonstrated that excitotoxicity is an important mechanism leading to neuronal death in HD (Beal et al., 1991; Graveland et al., 1985; Landwehrmeyer et al., 1995; Ferrante et al., 1991). Early studies demonstrated that intrastriatal injections of glutamate analogs in mice and primates cause lesions similar to those observed in HD patients (McGeer and McGeer, 1976; Beal et al., 1991; Isacson et al., 1985) and behavioral features in primates reminiscent of HD motor dysfunction (Ferrante et al., 1993; Burns et al., 1995).
Golgi impregnation studies, performed on human HD brain tissue, support the role of excitotoxic stress as a critical event in the pathogenesis of the disease. Indeed, there are marked alterations in the neuronal morphology of HD MSNs consistent with excitotoxic damage. (Ferrante et al., 1991; Graveland et al., 1985).
More recently, alterations in components of the glutamate receptor signaling pathway in tissues from patients and mouse models of HD have been observed, including defects in NMDAR activity and disturbances in the kynurenine pathway (Li et al., 2004; Guidetti et al., 2004; Guidetti et al., 2006). Levels of the potent NMDAR agonist QUIN, are elevated in the striatum and cortex of early grade human and murine HD brains. Levels of 3-HK, a metabolic product of the kynurenine pathway known to potentiate QUIN-mediated excitotoxicity, are likewise elevated early in the course of the disease. Increased NMDAR-mediated currents and enhanced excitotoxicity following glutamate/NMDA stimulation have also been demonstrated in several HD models early in the pathogenesis (Cepeda et al., 2001; Levine et al., 1999; Zeron et al., 2004; Graham et al., 2006b). Of note, YAC models which do not exhibit an HD phenotype, such as Shortstop and the C6R mice, are resistant to NMDAR-mediated excitotoxicity (Graham et al., 2006a; Slow et al., 2005). However, it is unknown how susceptibility to excitotoxicity varies depending on the phenotypic stage of the illness. As many compounds that modulate glutamate receptor function are currently being tested for their efficacy in therapeutic trials, it is essential to understand the relationship between susceptibility to excitotoxicity and disease state in order to inform both the timing and mode of therapy.
In the present study, we investigated whether stage of the illness influences sensitivity to excitotoxins using the YAC128 model. The YAC models display an age and CAG dependent phenotype that recapitulates many features of the human disease including, cognitive dysfunction, motor deficits and selective striatal degeneration (Slow et al., 2003; Van Raamsdonk et al., 2005). Our findings demonstrate that YAC128 mice display enhanced sensitivity to multiple excitotoxins in the early phase of the disease, prior to development of cognitive dysfunction and motor abnormalities. However, as the disease manifests and progresses, the mhtt (mutant htt)-induced excitotoxic phenotype evolves to a resistance to excitotoxic stress. We further demonstrate that this biphasic excitotoxic phenotype is paralleled at the electrophysiological level by increased postsynaptic receptor-mediated and synaptically-evoked NMDA currents in presymptomatic YAC128 mice which decrease with advanced disease progression.
Material and Methods
Generation of YAC transgenic mice
A 350kb YAC (353G6), containing the entire HD gene with 128CAG repeat was purified and microinjected into FVB/N pronuclei as described previously (Slow et al., 2003). Founder mice, YAC128 HD53 (heterzyogotes) and HD55 (homozygotes), were identified as formerly described (Hodgson et al., 1996; Slow et al., 2003; Graham et al., 2006b). Mice are maintained on FVB/N (Charles River, Wilmington, MA) background strain. All mice are housed, tested and tissues harvested according to Animal Protocol A07-0106 of the University of British Columbia.
NMDAR-mediated excitotoxicity
Embryos were isolated at E16.5 from time-pregnant YAC128 (HD53) females. Striatal neurons were cultured for 9–10 days and apoptotic cell-death post NMDA stimulation determined as described previously (Metzler et al., 2007). Briefly, striatal neurons were stimulated with 500 μM NMDA and 30 μM glycine in extracellular solution (ECS) in the absence of Mg2+ for 10 min at 37°C. Neurons were washed in ECS and incubated in cell culture media for 24 h. A combination of terminal deoxynucleotidyl transferase-mediated biotinylated dUTP nick end labeling (TUNEL, Roche, Germany) and propidium iodide (PI, Sigma) staining was used to determine NMDA-induced cell-death as previously described (Zeron et al., 2002; Graham et al., 2006a). 200 neurons were counted in a blinded fashion per sample setup in triplicate for each condition and genotype in each of 3 independent experiments.
Quinolinic acid injections
All surgical procedures were performed in accordance with UBC Animal Care Committee guidelines. QUIN (Sigma, St Louis, MO) was dissolved into 0.1M PBS. Mice were anesthetized with 1.5% isofluorane and received intrastriatal injections of 4 or 6nM QUIN. Coordinates of the injection site are as follows: 0.8mm posterior to Bregma, 1.8mm lateral from the midline and 3.5mm below the dorsal surface of the neocortex. Seven days post injection, mice were terminally anesthetized as described below and analysed in a previously described procedure (Zeron et al., 2002; Graham et al., 2006a).
Quantitative Analysis
All quantitative analysis was done blind with respect to genotype. Seven days after intrastriatal QUIN injection mice were terminally anesthetized by intraperitoneal injection of 2.5% avertin and perfused with 3% paraformaldehyde in PBS. The brains were stored in 3% paraformaldehyde for 24 hr at 4°C then removed and stored in PBS. Prior to sectioning, brains were transferred to a 30% sucrose solution containing 0.08% sodium azide in PBS, left overnight then mounted using Tissue-TEK O.C.T. compound (Sakura, Torrance, Ca). Mouse brains were cut using a cryostat (Microm HM 500M, Richard-Allan Scientific, Calamazoo, MI) and coronal sections (25μm) spaced 150μl apart were stained with Flurojade (Histo-Chem, Jefferson, Arkansas). The perimeter of the striatal lesion for each section was traced using StereoInvestigator software (Microbrightfield, Williston, VT, USA). For assessment of the number of Fluoro-Jade-positive cells, a 25 μm × 25 μm counting frame was randomly placed within the delineated lesion area and then systematically moved through the striatum. The number of Fluoro-Jade-positive cells was then determined by StereoInvestigator software (Microbrightfield, Williston, VT).
Permanent middle cerebral artery occlusion
Focal cerebral ischemia was produced using a model of permanent middle cerebral artery occlusion (MCAO) as previously described (Zhang et al., 2003). Anesthesia was induced in YAC128 (HD53) mice and their WT littermates (body weight 16–26 gr.) with 2% (vol/vol) isoflurane (70% N2O/30% O2) and was maintained with a 1-0.5% concentration. Mice were randomly coded and the tester was blind to genotype. Focal cerebral ischemia was induced by an intraluminal 7-0 nylon thread with a silicone tip (180 μm diameter) introduced into the right cervical internal carotid artery. The thread was inserted 9 ± 1.0 mm into the internal carotid artery up to the middle cerebral artery. A laser Doppler perfusion monitor (Perimed AB) was adhered to the right temporal aspect of the animal’s skull and used to confirm middle cerebral artery flow disruption. After 24 hours of MCAO, the mice were sacrificed, each brain rapidly removed and chilled for 2 min. Coronal sections (1-mm thick; n = 6) were cut with a mouse brain matrix. Each slice was immersed in a saline solution containing 2% 2,3,5-triphenyltetrazolium chloride (Sigma, St Louis, MO) at 37°C for 20 min. After staining, each slice was scanned with an HP scanjet 4200C. The stained and unstained areas of the right hemisphere were quantified with imageJ 1.32j (NIH), and these values used to calculate the volume infarct expressed as a percentage of the lesioned hemisphere. Mice were assigned a neurobehavioral score at 2 and 24 h following MCAO by two observers blinded to genotype. A previously reported scale was used (Stavrovskaya et al., 2004, which is a modified version of the original neurologic examination grading system by Bederson et al., 1986): 0, no neurological deficits; 1, failure to extend the left forepaw; 2, circling to contralateral side; 3, loss of ability to walk or loss of righting reflex; and 4, death before 24 h. An average of the two observer’s scores determined the final assigned score.
Acutely Dissociated Neurons
Experiments were conducted in mice at 1.5 months (before overt behavioral symptoms begin) and 7 months (symptomatic). A total of 53 animals in the two age groups (13 and 17 transgenic animals (YAC128 (HD53) at 1.5 and 7 months, respectively, and 9 and 14 WTs, respectively) were used. Experimental procedures were performed in accordance with the US Public Health Service Guide for Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee at UCLA.
Acutely dissociated MSNs were obtained using previously described procedures (Andre et al., 2006; Ariano et al., 2005; Starling et al., 2005; Bargas et al., 1994). Briefly, mice were anesthetized with halothane, transcardiacally perfused with 10 ml cold sucrose solution containing (in mM): 250 sucrose, 11 glucose, 15 HEPES, 1 Na2HPO4, 4 MgSO4, and 2.5 KCl (pH 7.4, 300–310 mOsm), and decapitated. Brains were dissected and sliced (coronal sections, 350 μm) in oxygenated cold sucrose solution and incubated in NaHCO3-buffered Earl’s balanced salt solution. After 1 h dorsal striata were dissected, placed in an oxygenated cell-stir chamber and enzymatically treated for 20 min with papain (0.5 mg/ml, Calbiochem, San Diego, CA) at 35°C in a HEPES-buffered Hank’s balanced salt solution (HBSS, pH 7.4, 300–310 mOsm; Sigma Chemicals, St Louis, MO). The tissue was rinsed with a low Ca2+ HEPES-buffered Na-isethionate solution and mechanically dissociated with a graded series of fire-polished Pasteur pipettes. The cell suspension was plated into a Petri dish containing a HEPES-buffered salt solution.
Recordings were obtained on an upright Zeiss Axioskop Microscope. The internal pipette solution contained (in mM): 175 N-methyl-D-glucamine (NMDG), 40 HEPES, 2 MgCl2, 10 ethylene glycol-bis (-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA), 12 phosphocreatine, 2 Na2ATP, 0.2 Na2GTP, and 0.1 leupeptin (pH 7.2–7.3, 265–270 mOsm, all from Sigma Chemicals, St Louis, MO). The Mg2+-free external solution consisted of (in mM): 135 NaCl, 20 CsCl, 3 BaCl2, 2 CaCl2, 10 glucose, 10 HEPES, 0.02 glycine, and 0.0003 tetrodotoxin (TTX, pH 7.4, 300–310 mOsm, Calbiochem, San Diego, CA). Electrode resistance was typically 5–6 MΩ in the bath. After seal rupture, series resistance was compensated (70–90%) and periodically monitored. Only data from cells for which access resistance values <20 MΩ were included. Signals were detected with an Axoclamp 2B amplifier (Axon Instruments, Foster City, CA). Membrane capacitances and input resistances were measured by applying a 10 mV depolarizing step voltage command and using the membrane test function integrated in pClamp 8.2 (Axon Instruments, Foster City, CA).
Drugs were applied through an array of application capillaries positioned 500–600 μm from the cell using a pressure-driven fast perfusion system. Solution changes were performed by changing the position of the array with a DC drive system controlled by a SF-77B perfusion system (Warner Instruments, Hamden, CT). NMDA was applied for 3 s every 30 s. Responsiveness of cells to 100 μM NMDA in the absence or presence of 50 μM Mg2+ was examined at a holding potential of −70 mV. The NMDAR specific antagonist 2-amino-5-phosphonovalerate was applied to confirm the specificity of NMDA-induced currents (data not shown).
Neurons in Brain Slices
Procedures for sacrificing mice and making slices were similar to those described above except slices were cut in oxygenated ice-cold low-Ca2+ artificial cerebrospinal fluid (ACSF) containing (in mM) 130 NaCl, 1.25 NaH2PO4, 26 NaHCO3, 5 MgCl2, 1 CaCl2, and 10 glucose. After cutting, slices were incubated in a chamber containing oxygenated ACSF and maintained at room temperature (25±2°C). After 1 h slices were placed on the stage of an upright fixed stage Olympus BX51, submerged in continuously flowing ACSF. Whole-cell voltage clamp recordings were performed with a MultiClamp 700A amplifier (Axon Instruments, Foster City, CA) in concert with Clampex acquisition software (v8.1, Axon Instruments). Data were low-pass filtered at 1 kHz and sampled at 10 KHz. The liquid junction potential was 2–3 mV and was not corrected. MSNs were visualized in slices with the aid of infrared videomicroscopy and were identified by somatic size, basic membrane properties (input resistance, membrane capacitance and time constant) (Cepeda et al., 1998). Passive membrane properties of MSNs were determined in voltage clamp mode by applying a depolarizing step voltage command (10 mV) and using the membrane test function integrated in the pClamp8 software (Axon Instruments, Foster City, CA). Series resistance was <25 MΩ and was compensated 70–80% and checked periodically. If the series resistance changed >10% by the end of the experiment the cell was discarded. The patch pipette (4–5 MΩ) contained (in mM): 130 Cs-methanesulfonate, 10 CsCl, 4 NaCl, 1 MgCl2, 5 MgATP, 5 EGTA, 10 HEPES, 0.5 GTP, 10 phosphocreatine, 0.1 leupeptin (pH 7.25–7.3, osmolarity 280–290 mOsm) and 4 QX-314. All drugs used for brain slice experiments were purchased from Sigma (St. Louis, MO).
NMDAR-mediated synaptic currents were evoked by electrical stimulation of afferent cortical fibers using a glass pipette filled with ACSF. The monopolar stimulating electrode was placed on the dorsolateral corpus callosum 150–250 μm from the recording electrode. Stimuli (0.1 ms pulse duration, 0.1 to 1.0 mA intensity) were delivered at intervals >30 sec. Evoked NMDAR-mediated excitatory postsynaptic currents were recorded at a holding potential of +40 mV in ACSF containing 10 μM bicuculline (BIC, a GABAA receptor antagonist) and 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX 10 μM, a NMDAR antagonist).
Statistical analysis
Statistical analysis was done using Student’s t-test, one-way ANOVA (in cases of significant effect of genotype, post-hoc comparisons between genotypes were performed using Turkey post hoc test) or two-way ANOVA (repeat measures for motor function assessment and electrophysiological analyses where appropriate). Linear regression analysis, p values, SEM, means and standard deviations were calculated using Graphpad Prism version 4.0 or SigmaStat, version 2.03 (SPSS, Chicago, IL). Differences between means were considered statistically significant if p<0.05.
Results
Increased sensitivity to excitotoxic stress in YAC128 mice before signs of illness
We previously demonstrated that primary MSNs isolated from YAC72 and YAC128 line HD55+/+ striata at birth are more vulnerable to NR2B-type NMDAR-mediated cell death ex vivo and display enhanced sensitivity to QUIN in vivo compared to WT neurons (Zeron et al., 2002; Zeron et al., 2004; Graham et al., 2006b; Graham et al., 2006a; Shehadeh et al., 2006). We now evaluated whether the most extensively used YAC128 line HD53, which expresses mhtt close to murine endogenous levels and develops a more severe phenotype compared to YAC128 line HD55+/+, also display enhanced susceptibility to excitotoxic stress.
MSNs derived from embryonic WT and HD53 striata were assessed for susceptibility to NMDAR-mediated excitotoxicity as assessed by morphological criteria and TUNEL analysis (Graham et al., 2006a; Shehadeh et al., 2006). We observe a significant increase in apoptotic cell death in HD53 MSNs compared to WT littermates post NMDA treatment (WT=12.5±1.2%, HD53=27.0±2.9%, p=0.002, Fig. 1A and B).
Fig. 1. Increased sensitivity to excitotoxic stress in YAC128 mice before signs of illness.

(A and B) Primary neuronal cultures established from embryonic YAC128 (HD53) and WT striata were assessed for apoptotic cell death 24 hrs post NMDA. MSNs from YAC128 (HD53) striata demonstrate an increase in vulnerability to NMDAR-mediated excitototxicity versus WT with increased cell death observed post NMDA (p<0.01). C) Intrastriatal injection of QUIN in 1.5 month old mice demonstrates an increase in apoptotic Fluoro-Jade positive cells in YAC128 (HD53) striatum compared to WT (p<0.05). D) Quantification of lesion volume demonstrates a trend towards enhanced lesion volume in YAC128 (HD53) compared to WT striata (p<0.07). Lesion volume and mean number of Fluoro-Jade positive cell is ± SEM. Mean percent apoptotic cell death is given ± SD. *p<0.05; **p<0.01.
To determine whether the enhanced susceptibility to NMDAR-mediated excitotoxic stress observed in HD53 MSNs could be replicated in vivo, we next assessed vulnerability to QUIN in young HD53 mice. Assessment of QUIN-induced striatal neurotoxicity in 1.5 month old mice (n=9) reveals both WT and HD53 mice have large numbers of Fluoro-Jade positive neurons, a fluorescent marker of degenerating cells (Schmued et al., 1997; Hansson et al., 1999). However there is a significant increase in the number of degenerating neurons in YAC128 striata compared to WT (WT=45872±14236, HD53=95390±26397; p=0.04, Fig. 1C). Determination of QUIN-induced lesion volumes demonstrates a 95% increase in the YAC128 compared to WT striatum which approaches statistical significance (WT=0.84±0.36mm3, HD53=1.64±0.39 mm3, p=0.07, Fig. 1D).
These data demonstrate that striatal neurons expressing full length mhtt have increased vulnerability to excitotoxins and that sensitivity to excitotoxic stress is an early feature in the YAC models of HD.
Mutant htt exacerbates neuronal dysfunction following stroke injury
As young YAC128 mice demonstrate increased vulnerability to glutamate receptor agonists, we postulated that mhtt may enhance neuronal dysfunction and cell death in response to other excitotoxic stimuli such as ischemia. We therefore sought to validate our previous finding using the stroke-induced model of neurodegeneration. Several studies have suggested that an excitotoxic process, involving excess glutamate release, underlies ischemia-induced injury (Chesselet et al., 1990; Choi and Rothman, 1990; Gonzales et al., 1992; Uemura et al., 1990) and that ischemia produces HD-like striatal damage (Mallard et al., 1995; Uemura et al., 1990; Ferriero et al., 1988; Chesselet et al., 1990). Furthermore, ischemia-induced neuronal injury has been shown to be reduced by glutamate antagonists acting at the level of NMDA receptors (Newell et al., 1995; Morikawa et al., 1998; Zhang et al., 2007). The middle cerebral artery was occluded (MCAO) in FVB/N mice expressing human mhtt (HD53) and littermate controls (n=11) at 1.5 months of age. At 2 and 24 hours post stroke injury, a neurological assessment was performed on all mice to establish acute post-stroke motor function and recovery from ischemia-induced injury, respectively.
Following ischemic injury, YAC128 mice demonstrate a significant delay in the recovery of motor function compared to WT mice (Two-way ANOVA, difference between genotypes p=0.012, F2,20=7.6, difference over time p=0.017, F2,20=6.7, interaction between genotype and time p=0.038, F2,20=4.9, Fig. 2A). Comparison of recovery in WT vs. YAC128 reveals that WT mice recover 23% of motor function 24hrs post stroke. In contrast, YAC128 mice have a trend towards worsening of motor function (WT vs. YAC128, p=0.007, Fig. 2B). In addition, YAC128 transgenic mice demonstrate a 39% increase in stroke-induced lesion volume compared to their WT littermates with a trend towards statistical significance (WT = 27.5±6.1% vs. YAC128 = 38.3±5.4%, p=0.09, Fig. 2C). These observations demonstrate that mhtt increases sensitivity to brain damage in presymptomatic YAC128 mice and parallels our findings in the other excitotoxicity paradigms.
Fig. 2. Mutant huntingtin exacerbates neuronal dysfunction following stroke injury.

A) Assessment of motor function in 1.5 month old YAC128 (HD53) and WT mice 24 hrs post ischemia-induced injury reveals the presence of mhtt delays the recover of motor function in YAC128 (HD53) mice compared to WT (p<0.05). No difference between genotypes is observed for baseline gross sensorimotor behavior 2 hrs post stroke injury. B) Percent recovery from MCAO is significantly reduced in YAC128 (HD53) mice vs. WT (p<0.01). In contrast to WT mice, which demonstrate an improvement in motor function 24 hrs post ischemia (23%), YAC128 (HD53) continue to perform poorly (−4%). C) Representative images of infarct volume in YAC128 (HD53) vs. WT coronal sections reveal the ischemic-induced damage by TTC staining. Quantification of lesion volume demonstrates a 39% increase in YAC128 (HD53) compared to control which approaches statistical significance (p<0.09). Lesion volume is ± SEM. *p<0.05; **p<0.01.
Aging influences vulnerability to excitotoxic stress in vivo
A crucial and initial issue in assessing and interpreting the susceptibility to excitotoxicity at different stages of the illness and at different ages of the phenotype is to determine whether normal aging alone influences susceptibility to excitotoxic stress. We therefore examined sensitivity to QUIN-induced neurotoxicity in FVB WT mice at different ages. Animals received intrastriatal QUIN injections at 1.5, 3, 6, 10, 12 and 18 months of age. Seven days post injection the mice were perfused and the brains processed for Fluoro-Jade staining. A 4nM dose was chosen for the 1.5 and 3 month old mice and a 6nM dose used for the 6 month and older age group. The reason for the different doses reflects the increased sensitivity to QUIN in young mice. Indeed a pilot study revealed that a dose of 6nM led to severe seizures and death in the 1.5 and 3 month old mice.
Overall, striatal lesion volumes in the younger age group (1.5, 3 and 6 months) were larger than those in the older mice (10, 12 and 18 months) despite the lower QUIN dose administered at 1.5 and 3 months (Fig. 3A). No difference in striatal lesion volume is observed between the 1.5 and 3 month old WT mice (p=0.92, n=10). ANOVA analysis of lesion volume in 6, 10, 12 and 18 month old mice reveals a significant difference in susceptibility to QUIN with age (ANOVA: p=0.02; lesion volume in 6 vs 18 month p<0.05, 6m=6.90±2.01 ×108μm3; 18m=0.28±0.09 ×108μm3, n=10). Student t test analysis demonstrates a significant difference between 6 and 18 months, (p<0.01), 10 and 18 months (p<0.05) and a trend towards significance between 6 and 12 months (p=0.056). Linear regression analysis demonstrates a strong inverse correlation between age and QUIN-induced lesion volume in FVB mice (r2=0.9501, p=0.02, Fig. 3B). The characterization of the excitotoxic phenotype in the striata of these WT FVB mice demonstrates a reduced susceptibility to striatal QUIN-induced neurotoxicity with increasing age.
Fig. 3. Aging influences vulnerability to excitotoxic stress in vivo.
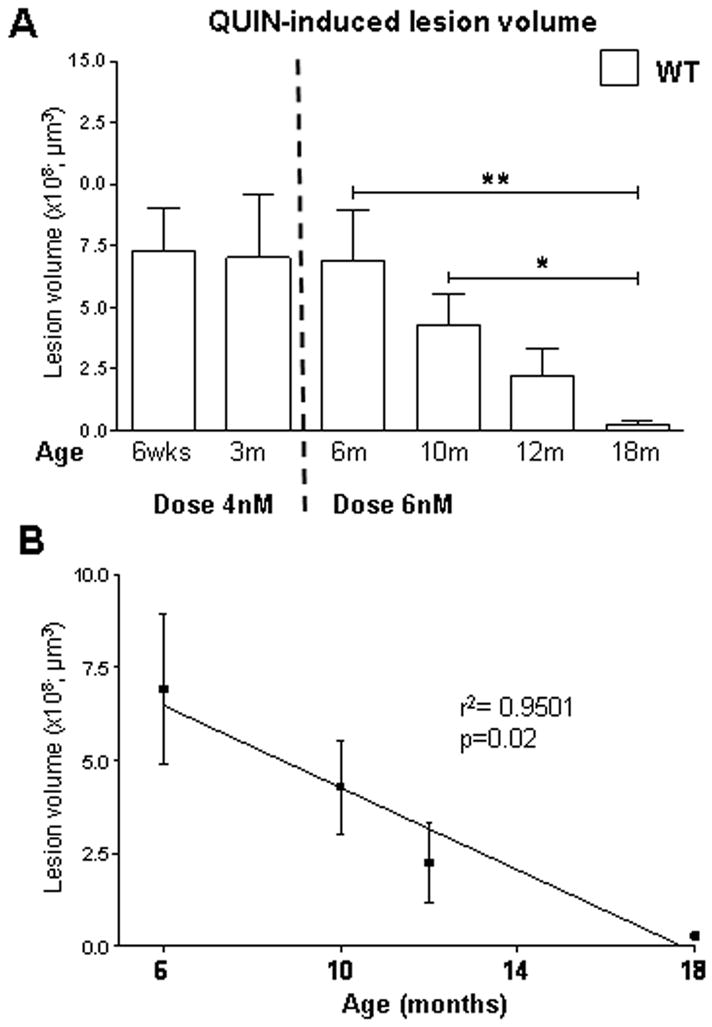
A) Effects of QUIN-induced striatal lesions on aging in FVB mice. An inverse correlation is observed between lesion volume and age of the animal. ANOVA analysis on 6 to 18 month old mice reveals a significant difference in lesion volume with age (p=0.02). B) Linear regression analysis of lesion volume and aging reveal a strong correlation between these outcomes (r2=0.9501, p=0.02). Lesion volume is ± SEM. *p<0.05; **p<0.01.
Susceptibility to excitotoxic stress in YAC128 models of HD is dependent on the stage of the HD phenotype
In order to determine the combined influence of age and disease stage on the excitotoxic phenotype, we next performed intrastriatal injections of QUIN in WT and in two separate YAC128 lines (HD55+/+ and HD53) at different ages. We have previously demonstrated that onset and progression of HD is modulated by levels of mhtt in the YAC128 mouse models (Graham et al., 2006b). HD55+/+ express approximately 20% less mhtt than HD53 and demonstrate a correspondingly less severe phenotype (Fig. 4A). Initially, and prior to obvious and detectable signs of illness in the mice (1.5 and 3 months), the striata of YAC128 mice demonstrate enhanced sensitivity to QUIN-induced neurotoxicity compared to WT (1.5m: WT=0.73±0.17×109μm3, HD53=1.16±0.29×109μm3; p=0.19, n=10; 3m: WT=0.70±0.25×109μm3, HD53=1.04±0.21×109μm3, HD55+/+=1.86±0.28×109μm3, ANOVA p<0.009, WT vs. HD55+/+ p<0.01, n=10) (Fig. 4B). In sharp contrast, HD53 mice with obvious motor, cognitive and neuropathological changes of HD (10 month old), demonstrate resistance to QUIN compared to their WT littermates (WT=0.43±0.12×109μm3, HD53=0.10±0.05×109μm3, p=0.02, n=10, Fig. 4B). Similarly, HD55+/+ mice demonstrate a trend towards resistance at 12 months with a 64% decrease in lesion volume compared to WT control (WT=0.25±0.11×109μm3 HD55+/+=0.09±0.06×109μm3, p=0.24, n=10).
Fig. 4. Susceptibility to excitotoxic stress in YAC128 mouse models of HD is dependent on the stage of the HD phenotype.

A) Levels of mhtt in YAC128 line HD55+/+ and HD53. A more severe phenotype is observed in the higher mhtt expressing line HD53. B) The presence of mhtt influences the response to QUIN-induced neurotoxicity with initial enhanced sensitivity to excitotoxic stress progressing to resistance later in the phenotype. HD55+/+ demonstrate an increase in lesion volume vs. WT at 3 months of age (p<0.01) and a trend towards resistance at 12 month. HD53, which demonstrates enhanced sensitivity at 1.5 months, are resistant to QUIN-induced excitotoxicity at 10 months of age (p<0.05). A significant difference is observed in lesion volume between HD55+/+ and HD53 at 3 months of age (p<0.05). C) Linear regression analysis reveals an increased rate of progression of the excitotoxic phenotype is observed in HD 53 mice as revealed by an increase in the slope of HD53 vs. WT (p=0.01). D) Onset of the resistance phenotype in HD53 occurs earlier than HD55+/+, at 6.9 and 8.6 months respectively. Lesion volume is ± SEM. *p<0.05; **p<0.01.
These data reveal a clear alteration in the excitotoxic phenotype in the HD53 mice from enhanced sensitivity to excitotoxins in presymptomatic mice to resistance at a time when signs of HD are obvious. Comparison of 6 and 10 month lesion volumes in HD53 and WT mice reveal a significant interaction between genotype, age and lesion volume (6m: WT=0.69±0.20×109μm3, HD53=1.11±0.27×109μm3, n=10, two-way ANOVA p<0.004, F2,34=12.6). Linear regression analysis demonstrates that, similar to WT mice, age affects QUIN-induced lesion volume in the YAC128 mice (HD53, r2=0.8661, p=0.06) (Fig. 4C). However, the presence of mhtt accelerates the age-dependent excitotoxic phenotype demonstrated by an increase in the rate of progression in HD53 vs. WT (WT, r2=0.9647, p=0.0005, HD53, r2=0.8661, p=0.06, difference between slopes p=0.01, F1,98=13.3) (Fig. 4C). This is also observed in HD55+/+ mice which demonstrate initial enhanced sensitivity to QUIN-induced lesions at 3 months progressing towards resistance at 12 months. Based on the linear regression data comparing QUIN-induced lesion volume and age, onset of the resistance phenotype would be predicted to occur at 6.9 months in HD53, the YAC HD line with the most severe phenotype and at 8.6 months in HD55+/+ (Fig. 4D).
Disease dependent biphasic changes in NMDAR-mediated currents in YAC128 MSNs
The next step was to examine whether these evolving changes in the excitotoxic phenotype were paralleled by electrophysiological changes. Initially we measured basic membrane properties as an indication of the functional state of neurons. Previously, we have shown that as the phenotype develops in the R6/2 mouse model of HD, input resistance increases while capacitance and time constants decrease (Cepeda et al., 2001; Starling et al., 2005). Input resistance is an index of channel function at rest and is generally considered to reflect functional capacity of specific K+ channels which we have shown are altered in mouse models of HD (Ariano et al., 2005). Capacitance and time constants can be considered measures of somatic and dendritic field size. At 1.5 months of age there are no significant differences in passive membrane properties between neurons obtained from YAC128 (HD53) and WT mice (Table.1). At 7 months however, when obvious motor and cognitive changes are present in the YAC128 line HD53 mice, input resistance increases in neurons from HD53 mice (Table.1, p<0.05). Mean capacitance and time constant are significantly reduced at 7 months in neurons from HD53 mice (Table.1, p<0.05 for both capacitance and time constant). These data demonstrate that electrophysiological changes are occurring in the YAC128 model with development of the phenotype.
Table 1.
Passive Membrane Properties
| Age | Input Resistance (MΩ) | Capacitance (pF) | Time Constant (msec) |
|---|---|---|---|
| 1.5 months | |||
| WT(n=19) | 1.59±0.14 | 13.9±0.5 | 102±9 |
| YAC128 (n=18) | 1.95±0.21 | 13.5±0.56 | 128+14 |
| 7 months | |||
| WT (n=22) | 1.83±0.18 | 13.9±0.52 | 136±12 |
| YAC128 (n=28) | 2.53±0.31* | 12.7±0.36* | 105±6* |
indicates p<0.05 comparison of YAC128 and WT.
We next examined postsynaptic NMDA currents in the acutely isolated neurons as an indication of NMDAR function. Application of 100 μM NMDA produces a characteristic response in MSNs consisting of an initial peak followed by a steady state component at all ages (Fig. 5). We measured peak current and peak current density. Current density was obtained by dividing peak current by cell capacitance to normalize values with respect to the size of the cell (Alzheimer et al., 1993). At 1.5 months peak NMDA currents and current densities are significantly increased in neurons obtained from HD53 mice (p<0.01; p<0.001 for peak current and current density, respectively; Fig. 5). Addition of 50 μM Mg2+ produced an approximately equal percentage decrease in peak currents in both groups (75±2.7 versus 76±1.5% for WT and HD53, respectively). In marked contrast at 7 months peak NMDA currents and current densities are significantly smaller in neurons obtained from HD53 compared to WT mice (p<0.01; p<0.001 for peak current and current density, respectively; Fig. 5). The change is due primarily to a significant increase in peak current and current density in WTs (p<0.001). Although there are decreases in mean peak currents and densities in neurons from HD53 mice between 1.5 and 7 months these are not statistically significant.
Figure 5. Biphasic changes in NMDA receptor-mediated currents and current densities in acutely dissociated MSNs from YAC128 and WT mice.
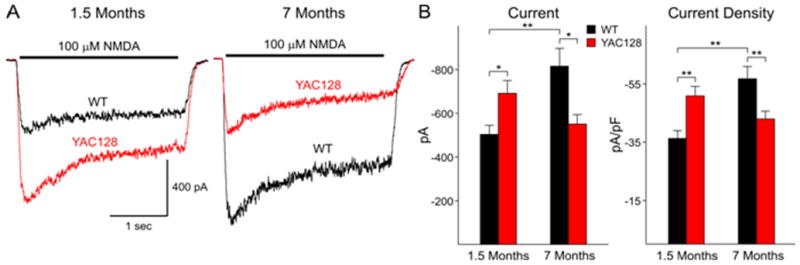
A) Examples of typical traces obtained from MSNs from WT and YAC128 (HD53) mice at 1.5 and 7 months. B) Bar graphs showing increased mean currents and current densities at 1.5 months and decreased mean currents and current densities at 7 months between YAC128 (HD53) and WT mice, respectively. * p<0.05; ** p<0.01.
We next examined NMDA currents evoked synaptically at the two ages to determine both pre- and postsynaptic contributions. At a holding potential of +40 mV and in the presence of BIC and CNQX evoked synaptic responses mediated by activation of NMDARs appear as outward currents with a peak between 15–20 msec after the stimulus followed by a slow decay. At 1.5 months thresholds for evoking NMDAR-mediated synaptic currents are higher in neurons obtained from WT than HD53 mice (0.24±0.02 versus 0.16±0.01 mA for WT versus HD53; p<0.005) indicating greater excitability. Peak currents also are significantly greater in neurons from HD53 than WT mice between 0.2–0.5 mA stimulus intensities (p<0.05) (Fig. 6A, C-left graph). At higher stimulus intensities (0.6–1.0 mA) evoked peak currents are similar. In contrast to the increase in NMDAR mediated current observed at 1.5 months, at 7 months neurons from HD53 mice display smaller mean peak synaptic currents. Peak currents are significantly smaller at the highest stimulus intensities (0.9–1.0 mA; p<0.05)(Fig. 6B, 2C-right graph). Thresholds for evoking synaptic responses are similar at 7 months (0.25±0.02 versus 0.24±0.02 mA in for WT versus HD53, respectively). Comparisons were also made between cells from WT and HD53 mice at the two ages. There are no significant differences in mean currents at each intensity for neurons obtained from WT mice at 1.5 and 7 months. In marked contrast, mean currents at 0.2–0.8 mA stimulus intensities are significantly smaller for neurons obtained from HD53 mice at 7 compared to 1.5 months (p<0.001–p<0.05).
Figure 6. Biphasic changes in NMDA receptor-mediated synaptically-evoked currents in MSNs in slices from YAC128 and WT mice.

(A and B) Examples of typical synaptic responses at increasing stimulus intensities obtained from MSNs from WT and YAC128 (HD53) mice at 1.5 and 7 months, respectively. (C and D) Graphs showing increased mean currents at 1.5 months and decreased mean currents at 7 months between YAC128 (HD53) and WT mice, respectively. * p<0.05.
Discussion
This study demonstrates that susceptibility to excitotoxic stress is different in the YAC128 mouse models of HD depending on the stage of development of the phenotype. At an early age, prior to obvious signs, YAC128 mice display enhanced sensitivity to multiple excitotoxic stressors such as NMDA, QUIN and ischemia demonstrated by an increase in neuronal dysfunction and cell death compared to WT mice. These findings are paralleled at the cellular level by a significant increase in NMDAR-mediated membrane currents in YAC128 neurons compared to WT. Furthermore, thresholds for evoking NMDAR-mediated membrane currents are decreased in dissociated neurons from YAC128, correlating with the enhanced sensitivity to exogenous agonists observed. This increased susceptibility occurs prior to detection of cognitive and motor abnormalities in the HD mice. The excitotoxic phenotype in the YAC128 model gradually progresses to resistance to QUIN-induced neurotoxicity with disease manifestation and progression. Parallel changes are observed in MSNs isolated from YAC128 striata which demonstrate decreased NMDAR membrane currents with disease progression. At this stage we also observe smaller mean peak synaptic currents in the YAC128 neurons compared to WT. These findings suggest that susceptibility to excitotoxic stress is a dynamic process in HD and supports therapeutic strategies aimed at decreasing levels of excitotoxic stress prior to detection of obvious signs of the illness.
In the present study we demonstrate that presymptomatic YAC128 mice have increased sensitivity to NMDA ex vivo and QUIN in vivo, consistent with findings from several mouse models of HD (Table 2, 3). Studies performed on human HD brain tissue also highlight excitotoxic neuropathological changes as an important marker of the disease. Increased levels of glutamate are observed in the striatum of patients with early HD (Taylor-Robinson et al., 1996). As medium spiny neurons of the neostriatum receive glutamatergic input from most cortical regions, the increased levels of glutamate and/or QUIN may cause overactivation of NMDARs leading to excitotoxicity. Furthermore, Golgi impregnation studies performed on human HD brain tissue reveal there are marked alterations in the neuronal morphology of spiny striatal neurons, potentially as a result of excess glutamate receptor stimulation (Graveland et al., 1985; Ferrante et al., 1991). Increased spine density and altered dendritic morphology is observed in other neurological conditions where excitotoxic has been implicated as an underlying mechanism, including epilepsy, fragile X syndrome, schizophrenia and HIV-related dementias (Smart and Halpain, 2000). An increased number of dendritic spines and/or dendritic arbors may promote excitability and exacerbate neuronal cell death.
Table 2. Vulnerability to NMDAR-mediated excitotoxic stress in HD mouse models.
Several HD mouse models demonstrate enhanced sensitivity to NMDAR-mediated excitotoxic stress early in the phenotype.
| Line | CAG | Method | Age (Months) | Result vs. wt | Phenotype | Strain | Reference | |
|---|---|---|---|---|---|---|---|---|
| Truncated HD models | R6/2 | ~150/200 | NMDA/50μM/slice | <2 | similar | presymptomatic | CBA/C57BL6 | Levine et al., 1999 |
| “ | >2 | enhanced | symptomatic | Levine et al., 1999 | ||||
| Shortstop | 120 | NMDA/1ere | P0 | resistant | no phenotype | FVB | Slow et al., 2005 | |
| Full length HD models | knockin | 71 | NMDA/50μM/slice | <3 | similar | presymptomatic | C57BL6 | Levine et al., 1999 |
| 71 | NMDA/50μM/, slice | >3 | similar | presymptomatic | C57BL6 | Levine et al., 1999 | ||
| knockin | 94 | NMDA/50μM/slice | <3 | enhanced | presymptomatic | C57BL6 | Levine et al., 1999 | |
| 94 | NMDA/50μM/slice | >3 | enhanced | presymptomatic | C57BL6 | Levine et al., 1999 | ||
| YAC72 | 72 | NMDA/1ere | P0 | enhanced | presymptomatic | FVB | Zeron et al., 2002 | |
| YAC128/HD55+/− | 120 | NMDA/1ere | P0 | enhanced | presymptomatic | FVB | Graham et al., 2006 | |
| YAC128/HD55+/+ | 120 | NMDA/1ere | P0 | enhanced | presymptomatic | FVB | Graham et al., 2006 | |
| YAC128/HD53 | 120 | NMDA/1ere | P0 | enhanced | presymptomatic | FVB |
Table 3. Vulnerability to QUIN-induced neurotoxicity in HD mouse models.
Vulnerability to QUIN-induced neurotoxicity is dependent on the stage of the HD phenotype in several HD mouse models of disease.
| Line | CAG | Dose | Age (months) |
Result vs. wt | Phenotype | Strain | Reference | |
|---|---|---|---|---|---|---|---|---|
| Truncated HD models | R6/1 | ~115 | 20nM | 0.75 | enhanced | presymptomatic | CBA/C57BL6 | Hansson et al., 2001 |
| 20nM | 2 | partial resistant | presymptomatic | Hansson et al., 2001 | ||||
| 20nM | 4.5 | resistant | symptomatic | Hansson et al., 2001 | ||||
| 20nM | 5.5/9 | resistant | symptomatic | Hansson et al., 1999 | ||||
| R6/2 | ~150/200 | 20nM | 0.75 | partial resistance | presymptomatic | CBA/C57BL6 | Hansson et al., 2001 | |
| 20nM | 1.5 | resistant | presymptomatic | Hansson et al., 2001 | ||||
| 20nM | 2.5/3 | resistant | symptomatic | Mac Gibbon et al., 2002 | ||||
| KA/20mg/kg | 0.75 and 2 | resistant | pre/symptomatic | Morton et al., 2000 | ||||
| tg46/tg100 | 46/100 | 30nM | 4–78 | similar | presymptomatic/symptomatic | S5L/C57BL6 | Petersen et al., 2002 | |
| N171 | 82 | 30nM | 4 | resistant | symptomatic | B6C3F1/J | Jarabek et al., 2004 | |
| Shortstop | 120 | 4nM | 3,6,10 | resistant | no phenotype | FVB | Slow et al., 2005; unpublished | |
| Full length HD modes | YAC72 | 72 | 8nM | 6,10 | enhanced | presymptomatic | FVB | Zeron et al., 2002 |
| YAC128/HD55+/+ | 120 | 4nM | 3 | enhanced | presymptomatic | Graham et al., 2006 | ||
| 6nM | 12 | trend resistant | symptomatic | |||||
| YAC128/HD53 | 120 | 4nM | 1.5 | enhanced | presymptomatic | FVB | ||
| 4nM | 3 | similar (trend↑) | symptomatic | |||||
| 6nM | 6 | similar (trend↑) | symptomatic | |||||
| 6nM | 10 | resistant | symptomatic |
In addition to the early enhanced sensitivity to NMDA and QUIN in the YAC model, we also observe a significant effect of mhtt on recovery from ischemia-induced injury. Transient global ischemia in rats yields selective striatal neuronal loss reminiscent of HD and similar to the QUIN acute model of HD (Meade et al., 2000). However, conflicting results have been published in other mouse models of HD with regards to susceptibility to ischemia. The R6/1 mice are resistant to cerebral global ischemia (Schiefer et al., 2002), while no effect is observed in the HdhQ92 knockin model to mild focal ischemia (Namura et al., 2002). The reason for this apparent discrepancy may simply reflect the stage of disease of the animals at the time of testing. Indeed, a longitudinal study of susceptibility to an ischemic challenge in the R6/2 model revealed an age-dependent biphasic response with initial sensitivity progressing to resistance with disease progression (Klapstein and Levine, 2005). The delay in recovery in YAC128 mice following stroke likely reflects increased neuronal damage that results from enhanced sensitivity to excitotoxicity compared to WT animals and may also be influenced by increased neuroinflammation in YAC128 animals. Indeed, YAC128 mice show enhanced immune activation (Björkqvist et al., 2008) which is known to accelerate cerebral ischemic injury (Lee et al., 2005).
Consistent with studies on rats, our data demonstrate that FVB WT mice have an age-dependent decrease in susceptibility to excitotoxic stress. Several lines of evidence support the hypothesis that age-related effects, such as reduced levels of NMDAR (Magnusson and Cotman, 1993; Peterson and Cotman, 1989), increased kynurenic acid concentrations (Moroni et al., 1988) and diminished striatal excitatory synaptic input, lead to decreased vulnerability to excitotoxins with normal aging (Cepeda et al., 1989; Cepeda et al., 1996; Finn et al., 1991). Furthermore, morphological changes occur including decreases in synaptic density, spine density and loss of distal dendritic segments, correlating with the reduced susceptibility observed (Ingham et al., 1989; Levine et al., 1987).
Indeed, the YAC128 models demonstrate an age-dependent response to neuronal excitability. However, in the YAC models there is an acceleration of the bi-phasic response to excitotoxic stress, suggesting mhtt lowers the threshold for activation of neuronal excitability and sensitizes neurons to excitotoxic damage. Once initiated, these pathways appear to progress inexorably towards resistance to excitotoxicity.
In vivo studies done in the R6/2 model demonstrate that the R6/2 line is resistant to QUIN-induced neurotoxicity (Hansson et al., 2001). However, in the more sensitive brain slice model, it has been shown that the R6/2 model displays early enhanced sensitivity to NMDA (Levine et al., 1999; Starling et al., 2005). Interestingly, and similar to our findings in the YAC mice, studies done on the less severely affected R6/1 line show a biphasic response to QUIN-induced neuronal damage with initial increased lesion volume progressing to a resistance later in the phenotype (Hansson et al., 2001). This raises the possibility that therapeutic approaches to down-regulate excitotoxic stress early may provide benefit in the YAC and R6 models. The critical questions remaining are whether the resistant phenotype observed is due to selective loss of the neuronal types that are highly vulnerable to excitotoxins, cortico-striatal dysfunction with consequent reduction of spine density and/or synapse loss in MSNs or an attempt by the neurons to invoke compensatory mechanisms to down-regulate the response to excitotoxic stress.
The electrophysiological data from both acutely dissociated neurons and striatal slices demonstrate a disease-dependent biphasic change in NMDAR function level and provide physiological data supporting the excitotoxicity studies. In acutely dissociated and MSNs in slices from YAC128 mice before the development of the phenotype, NMDA responses are larger than those from WTs. These early increases are in agreement with data from cultured neurons in other YAC models (Zeron et al., 2002) and R6/2 mice (Cepeda et al., 2001; Starling et al., 2005) as well NMDAR-mediated synaptic responses examined in slices in YAC72 mice (Li et al., 2004). Different mechanisms might underlie this enhancement, including altered receptor subunit composition, changes in Ca2+ signaling and/or transduction systems and altered receptor trafficking (Fan and Raymond, 2007). Measurements of NR1/NR2B mRNA levels in human HD neostriatum have been shown to be increased within specific areas early in the disease (Arzberger et al., 1997). In addition, NR2A and NR2B receptor gene variations have been shown to modify age at onset in HD (Arning et al., 2005).
As the disease phenotype develops and progresses the differences reverse and by 7 months, NMDA responses are smaller in MSNs from YAC128 compared to WTs. In dissociated neurons this reversal is paralleled by increases in peak currents and current densities in MSNs of WT but not YAC128 mice. In the YAC128 model, there is an increased rate of removal/degradation of surface NMDA receptors in MSNs paralleled by enhanced NR2B cleavage at 12 months of age in the striatum (Cowan et al., 2008). This enhanced NR2B degradation with disease progression may underlie the resistance phenotype observed. Additional factors that may contribute include neuronal loss in the YAC animals (Lerch et al., 2008) and/or degenerative changes, such as thinning and loss of dendritic length and spines, as seen in other models of HD (Klapstein et al., 2001). Interestingly, the appearance of striatal inclusions in both the YAC128 and R6/2 models correlate with onset of the resistance phenotype (Slow et al., 2005; Hansson et al., 2001). This suggests that nuclear inclusions may trigger mechanisms that protect neurons against excitotoxic stress. Indeed, it has recently been demonstrated that expression of mhtt is associated with accumulation of Hsp70, (King et al., 2008), over-expression of which provides protection against glutamate, malonate and 3-nitropropionic acid (Morkushin et al., 2005; Dedeoglu et al., 2002)
In brain slices of young mice, the YAC128 cell currents have the same peak amplitude as WTs, however are evoked at lower stimulus intensity. These data might indicate a larger NMDA current per synapse, but fewer synapses. In older mice, the NMDA currents from YAC128 mice have similar characteristics to those of WT animals in terms of stimulus intensity, but are smaller, consistent with the current and current density data. Overall, the results may imply that extrasynaptic receptors mediate increased QUIN sensitivity early, whereas decreased synaptic and extrasynaptic effects mediate decreased QUIN sensitivity as the disease progresses.
We have shown previously in other HD mouse models that the development of symptoms is paralleled by a disconnection between the cortex and striatum and a reduction in spontaneous excitatory synaptic currents of MSNs (Cepeda et al., 2003). Synaptic NMDAR activation would be affected by such a disconnection. This component will not necessarily be evident in the acutely dissociated neurons. Furthermore, it is well known that cortical input influences susceptibility to excitotoxicity within the striatum. Indeed, lesioning of the cortex has been shown to prevent excitotoxic damage from occurring in the striatum (Biziere and Coyle, 1979; McGeer et al., 1978). The decrease in lesion size over age may reflect a natural progressive disconnection between cortex and striatum that is exacerbated in the HD models (Cepeda et al., 2007) leading to even smaller lesions in transgenic mice.
Numerous studies have been done in the R6/2 line in an attempt to explain the resistance phenotype. In this model the resistance phenotype occurs prior to overt cell death of neurons or astrocytes (Hansson et al., 1999). Increased Ca2+buffering capacity is observed in the R6/2 line (Hansson et al., 2001), potentially suggesting that pre-conditioning of the neurons leads to an increase capacitance to handle higher levels of Ca2+. Surprisingly however, no increase in proteins expected to modulate Ca2+ buffering are observed. There is however decreased spine density in striatal neurons from symptomatic R6/2 mice (Klapstein et al., 2001), correlating with the reduced susceptibility to excitotoxic stress.
Human HD brain also shows decreased dendritic length and spine density in later stages of the disease (Graveland et al., 1985; Ferrante et al., 1991). During excessive excitotoxic stimulation, such as using excessive levels of glutamate analogs and/or ischemia-induced damage, neurons undergo rapid alterations in dendritic structures including focal swelling and spine loss (review (Smart and Halpain, 2000). In the case of HD, it may be that a steady accumulation of glutamate and QUIN initially enhances neuronal sprouting and excitotoxic stress. Indeed, in vitro studies have shown that low levels of glutamate cause increased spine formation (Richards et al., 2005). However, the constant mhtt-induced stress may over time lead to a pre-conditioned state in the neurons, causing an increase in threshold levels required to trigger neuronal excitability. In the case of the YAC mice we observe increased levels of QUIN at a time when the neurons are resistant to exogenous glutamate stimulation. Alternatively, it is possible that the alterations observed in spines in end stage human HD brain reflect a response to decreased levels of glutamate and or QUIN. In fact, spines require signals from their afferent neurons to remain intact (Kemp and Powell, 1971). QUIN levels in humans would support this observation with levels initially increased and subsequently down-regulated in the advanced stage of the disease (Guidetti et al., 2004).
The results of this study support a critical role for excitotoxic mechanisms early in the pathogenesis of HD and suggest there may be a window of opportunity for the therapeutic use of compounds which modulate glutamate receptor activity. Our evidence supports the most efficacious use of glutamate antagonists early in the course of the disease, preferably during the presymptomatic stage when excitotoxic mechanisms are predominant.
Acknowledgments
We thank members of our lab, in particular Jeff Carroll, Lisa Bertram, Nagat Bissada and Lili Wang for their support. Funding for this work was provided by the Huntington Disease Society of America, Cure HD Initiative, Canadian Institutes of Health Research, Michael Smith Foundation for Health Research (R.K.G.) and National Institute of Health USPHS NS41574. M.R.H is Killam University Professor and holds a Canada Research Chair in Human Genetics.
Reference List
- Andre VM, Cepeda C, Venegas A, Gomez Y, Levine MS. Altered cortical glutamate receptor function in the R6/2 model of Huntington’s disease. J Neurophysiol. 2006;95:2108–2119. doi: 10.1152/jn.01118.2005. [DOI] [PubMed] [Google Scholar]
- Ariano MA, Cepeda C, Calvert CR, Flores-Hernandez J, Hernandez-Echeagaray E, Klapstein GJ, Chandler SH, Aronin N, DiFiglia M, Levine MS. Striatal potassium channel dysfunction in Huntington’s disease transgenic mice. J Neurophysiol. 2005;93:2565–2574. doi: 10.1152/jn.00791.2004. [DOI] [PubMed] [Google Scholar]
- Arning L, Kraus PH, Valentin S, Saft C, Andrich J, Epplen JT. NR2A and NR2B receptor gene variations modify age at onset in Huntington disease. Neurogenetics. 2005;6:25–28. doi: 10.1007/s10048-004-0198-8. [DOI] [PubMed] [Google Scholar]
- Arzberger T, Krampfl K, Leimgruber S, Weindl A. Changes of NMDA receptor subunit (NR1, NR2B) and glutamate transporter (GLT1) mRNA expression in Huntington’s disease--an in situ hybridization study. J Neuropathol Exp Neurol. 1997;56:440–454. doi: 10.1097/00005072-199704000-00013. [DOI] [PubMed] [Google Scholar]
- Bargas J, Howe A, Eberwine J, Cao Y, Surmeier DJ. Cellular and molecular characterization of Ca2+ currents in acutely isolated, adult rat neostriatal neurons. J Neurosci. 1994;14:6667–6686. doi: 10.1523/JNEUROSCI.14-11-06667.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beal MF, Ferrante RJ, Swartz KJ, Kowall NW. Chronic quinolinic acid lesions in rats closely resemble Huntington’s disease. J Neurosci. 1991;11:1649–59. doi: 10.1523/JNEUROSCI.11-06-01649.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bederson JB, Pitts LH, Tsuji M, Nishimura MC, Davis RL, Bartkowski H. Rat middle cerebral artery occlusion: evaluation of the model and development of a neurologic examination. Stroke. 1986;17(3):472–6. doi: 10.1161/01.str.17.3.472. [DOI] [PubMed] [Google Scholar]
- Biziere K, Coyle JT. Effects of cortical ablation on the neurotoxicity and receptor binding of kainic acid in striatum. J Neurosci Res. 1979;4(5–6):383–98. doi: 10.1002/jnr.490040507. [DOI] [PubMed] [Google Scholar]
- Björkqvist M, Wild EJ, Thiele J, Silvestroni A, Andre R, Lahiri N, Raibon E, Lee RV, Benn CL, Soulet D, et al. A novel pathogenic pathway of immune activation detectable before clinical onset in Huntington’s disease. J Exp Med. 2008;205(8):1869–77. doi: 10.1084/jem.20080178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns LH, Pakzaban P, Deacon TW, Brownell AL, Tatter SB, Jenkins BG, Isacson O. Selective putaminal excitotoxic lesions in non-human primates model the movement disorder of Huntington disease. Neuroscience. 1995;64:1007–1017. doi: 10.1016/0306-4522(94)00431-4. [DOI] [PubMed] [Google Scholar]
- Cepeda C, Wu N, André VM, Cummings DM, Levine MS. The corticostriatal pathway in Huntington’s disease. Prog Neurobiol. 2007;81(5–6):253–71. doi: 10.1016/j.pneurobio.2006.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cepeda C, Hurst RS, Calvert CR, Hernández-Echeagaray E, Nguyen OK, Jocoy E, Christian LJ, Ariano MA, Levine MS. Transient and progressive electrophysiological alterations in the corticostriatal pathway in a mouse model of Huntington’s disease. J Neurosci. 2003;23(3):961–9. doi: 10.1523/JNEUROSCI.23-03-00961.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cepeda C, Ariano MA, Calvert CR, Flores-Hernandez J, Chandler SH, Leavitt BR, Hayden MR, Levine MS. NMDA receptor function in mouse models of Huntington disease. J Neurosci Res. 2001;66:525–539. doi: 10.1002/jnr.1244. [DOI] [PubMed] [Google Scholar]
- Cepeda C, Li Z, Levine MS. Aging reduces neostriatal responsiveness to N-methyl-D-aspartate and dopamine: an in vitro electrophysiological study. Neuroscience. 1996;73:733–750. doi: 10.1016/0306-4522(96)00056-5. [DOI] [PubMed] [Google Scholar]
- Cepeda C, Colwell CS, Itri JN, Chandler SH, Levine MS. Dopaminergic modulation of NMDA-induced whole cell currents in neostriatal neurons in slices: contribution of calcium conductances. J Neurophysiol. 1998;79:82–94. doi: 10.1152/jn.1998.79.1.82. [DOI] [PubMed] [Google Scholar]
- Cepeda C, Walsh JP, Hull CD, Buchwald NA, Levine MS. Intracellular neurophysiological analysis reveals alterations in excitation in striatal neurons in aged rats. Brain Res. 1989;494:215–226. doi: 10.1016/0006-8993(89)90589-1. [DOI] [PubMed] [Google Scholar]
- Chesselet MF, Gonzales C, Lin CS, Polsky K, Jin BK. Ischemic damage in the striatum of adult gerbils: relative sparing of somatostatinergic and cholinergic interneurons contrasts with loss of efferent neurons. Exp Neurol. 1990;110:209–218. doi: 10.1016/0014-4886(90)90032-n. [DOI] [PubMed] [Google Scholar]
- Choi DW, Rothman SM. The role of glutamate neurotoxicity in hypoxic-ischemic neuronal death. Annu Rev Neurosci. 1990;13:171–182. doi: 10.1146/annurev.ne.13.030190.001131. [DOI] [PubMed] [Google Scholar]
- Cowan CM, Mannie MY, Fan J, Shehadeh J, Zhang LY, Graham RK, Hayden MR, Raymond LA. Chronically elevated striatal calpain activity in YAC transgenic Huntington disease mouse model: Impact on NMDA receptor function and toxicity. J Neurosci. 2008;28(48):12735–12725. doi: 10.1523/JNEUROSCI.4619-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedeoglu A, Ferrante RJ, Andreassen OA, Dillmann WH, Beal MF. Mice overexpressing 70-kDa heat shock protein show increased resistance to malonate and 3-nitropropionic acid. Exp Neurol. 2002;176(1):262–5. doi: 10.1006/exnr.2002.7933. [DOI] [PubMed] [Google Scholar]
- DiFiglia M, Sapp E, Chase K, Schwarz C, Meloni A, Young C, Martin E, Vonsattel JP, Carraway R, Reeves SA. Huntingtin is a cytoplasmic protein associated with vesicles in human and rat brain neurons. Neuron. 1995;14:1075–1081. doi: 10.1016/0896-6273(95)90346-1. [DOI] [PubMed] [Google Scholar]
- Fan MM, Raymond LA. N-methyl-D-aspartate (NMDA) receptor function and excitotoxicity in Huntington’s disease. Prog Neurobiol. 2007;81:272–293. doi: 10.1016/j.pneurobio.2006.11.003. [DOI] [PubMed] [Google Scholar]
- Ferrante RJ, Kowall NW, Cipolloni PB, Storey E, Beal MF. Excitotoxin lesions in primates as a model for Huntington’s disease: histopathologic and neurochemical characterization. Exp Neurol. 1993;119:46–71. doi: 10.1006/exnr.1993.1006. [DOI] [PubMed] [Google Scholar]
- Ferrante RJ, Kowall NW, Richardson EP., Jr Proliferative and degenerative changes in striatal spiny neurons in Huntington’s disease: a combined study using the section-Golgi method and calbindin D28k immunocytochemistry. J Neurosci. 1991;11:3877–3887. doi: 10.1523/JNEUROSCI.11-12-03877.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferriero DM, Arcavi LJ, Sagar SM, McIntosh TK, Simon RP. Selective sparing of NADPH-diaphorase neurons in neonatal hypoxia-ischemia. Ann Neurol. 1988;24:670–676. doi: 10.1002/ana.410240512. [DOI] [PubMed] [Google Scholar]
- Finn SF, Hyman BT, Storey E, Miller JM, Beal MF. Effects of aging on quinolinic acid lesions in rat striatum. Brain Res. 1991;562:276–280. doi: 10.1016/0006-8993(91)90631-5. [DOI] [PubMed] [Google Scholar]
- Gonzales C, Lin RC, Chesselet MF. Relative sparing of GABAergic interneurons in the striatum of gerbils with ischemia-induced lesions. Neurosci Lett. 1992;135:53–58. doi: 10.1016/0304-3940(92)90134-s. [DOI] [PubMed] [Google Scholar]
- Graham RK, Deng Y, Slow EJ, Haigh B, Bissada N, Lu G, Pearson J, Shehadeh J, Bertram L, Murphy Z, Warby SC, Doty CN, Roy S, Wellington CL, Leavitt BR, Raymond LA, Nicholson DW, Hayden MR. Cleavage at the caspase-6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell. 2006a;125:1179–1191. doi: 10.1016/j.cell.2006.04.026. [DOI] [PubMed] [Google Scholar]
- Graham RK, Slow EJ, Deng Y, Bissada N, Lu G, Pearson J, Shehadeh J, Leavitt BR, Raymond LA, Hayden MR. Levels of mutant huntingtin influence the phenotypic severity of Huntington disease in YAC128 mouse models. Neurobiol Dis. 2005b;21:444–455. doi: 10.1016/j.nbd.2005.08.007. [DOI] [PubMed] [Google Scholar]
- Graveland GA, Williams RS, DiFiglia M. Evidence for degenerative and regenerative changes in neostriatal spiny neurons in Huntington’s disease. Science. 1985;227:770–773. doi: 10.1126/science.3155875. [DOI] [PubMed] [Google Scholar]
- Guidetti P, Bates GP, Graham RK, Hayden MR, Leavitt BR, Macdonald ME, Slow EJ, Wheeler VC, Woodman B, Schwarcz R. Elevated brain 3-hydroxykynurenine and quinolinate levels in Huntington disease mice. Neurobiol Dis. 2006;23:190–197. doi: 10.1016/j.nbd.2006.02.011. [DOI] [PubMed] [Google Scholar]
- Guidetti P, Luthi-Carter RE, Augood SJ, Schwarcz R. Neostriatal and cortical quinolinate levels are increased in early grade Huntington’s disease. Neurobiol Dis. 2004;17:455–461. doi: 10.1016/j.nbd.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Hansson O, Guatteo E, Mercuri NB, Bernardi G, Li XJ, Castilho RF, Brundin P. Resistance to NMDA toxicity correlates with appearance of nuclear inclusions, behavioural deficits and changes in calcium homeostasis in mice transgenic for exon 1 of the huntington gene. Eur J Neurosci. 2001;14:1492–1504. doi: 10.1046/j.0953-816x.2001.01767.x. [DOI] [PubMed] [Google Scholar]
- Hansson O, Petersen A, Leist M, Nicotera P, Castilho RF, Brundin P. Transgenic mice expressing a Huntington’s disease mutation are resistant to quinolinic acid-induced striatal excitotoxicity. Proc Natl Acad Sci U S A. 1999;96:8727–8732. doi: 10.1073/pnas.96.15.8727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper PS. Huntington disease. J R Soc Med. 2005;98:550. doi: 10.1258/jrsm.98.12.550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgson JG, Smith DJ, McCutcheon K, Koide HB, Nishiyama K, Dinulos MB, Stevens ME, Bissada N, Nasir J, Kanazawa I, Disteche CM, Rubin EM, Hayden MR. Human huntingtin derived from YAC transgenes compensates for loss of murine huntingtin by rescue of the embryonic lethal phenotype. Hum Mol Genet. 1996;5:1875–1885. doi: 10.1093/hmg/5.12.1875. [DOI] [PubMed] [Google Scholar]
- Ingham CA, Hood SH, Arbuthnott GW. Spine density on neostriatal neurons changes with 6-hydroxydopamine lesions and with age. Brain Res. 1989;503:334–338. doi: 10.1016/0006-8993(89)91686-7. [DOI] [PubMed] [Google Scholar]
- Isacson O, Brundin P, Gage FH, Bjorklund A. Neural grafting in a rat model of Huntington’s disease: progressive neurochemical changes after neostriatal ibotenate lesions and striatal tissue grafting. Neuroscience. 1985;16:799–817. doi: 10.1016/0306-4522(85)90095-8. [DOI] [PubMed] [Google Scholar]
- Jarabek BR, Yasuda RP, Wolfe BB. Regulation of proteins affecting NMDA receptor-induced excitotoxicity in a Huntington’s mouse model. Brain. 2004;127:505–516. doi: 10.1093/brain/awh058. [DOI] [PubMed] [Google Scholar]
- Kemp JM, Powell TP. The termination of fibres from the cerebral cortex and thalamus upon dendritic spines in the caudate nucleus: a study with the Golgi method. Philos Trans R Soc Lond B Biol Sci. 1971;262:429–439. doi: 10.1098/rstb.1971.0105. [DOI] [PubMed] [Google Scholar]
- King MA, Goemans CG, Hafiz F, Prehn JHM, Wyttenbach A, Tolkovsky AM. Cytoplasmic inclusions of htt exon 1 containing an expanded polyglutamine tract suppress execution of apoptosis in sympathetic neurons. J Neurosci. 2008;28(53):14401–14415. doi: 10.1523/JNEUROSCI.4751-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klapstein GJ, Levine MS. Age-dependent biphasic changes in ischemic sensitivity in the striatum of Huntington’s disease R6/2 transgenic mice. J Neurophysiol. 2005;93:758–765. doi: 10.1152/jn.00483.2004. [DOI] [PubMed] [Google Scholar]
- Klapstein GJ, Fisher RS, Zanjani H, Cepeda C, Jokel ES, Chesselet MF, Levine MS. Electrophysiological and morphological changes in striatal spiny neurons in R6/2 Huntington’s disease transgenic mice. J Neurophysiol. 2001;86:2667–2677. doi: 10.1152/jn.2001.86.6.2667. [DOI] [PubMed] [Google Scholar]
- Landwehrmeyer GB, Standaert DG, Testa CM, Penney JB, Jr, Young AB. NMDA receptor subunit mRNA expression by projection neurons and interneurons in rat striatum. J Neurosci. 1995;15:5297–307. doi: 10.1523/JNEUROSCI.15-07-05297.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JC, Cho GS, Kim HJ, Lim JH, Oh YK, Nam W, Chung JH, Kim WK. Accelerated cerebral ischemic injury by activated macrophages/microglia after lipopolysaccharide microinjection into rat corpus callosum. Glia. 2005;50(2):168–81. doi: 10.1002/glia.20164. [DOI] [PubMed] [Google Scholar]
- Lerch JP, Carroll JB, Spring S, Bertram LN, Schwab C, Hayden MR, Henkelman RM. Automated deformation analysis in the YAC128 Huntington disease mouse model. Neuroimage. 2008;39:32–39. doi: 10.1016/j.neuroimage.2007.08.033. [DOI] [PubMed] [Google Scholar]
- Levine MS, Klapstein GJ, Koppel A, Gruen E, Cepeda C, Vargas ME, Jokel ES, Carpenter EM, Zanjani H, Hurst RS, Efstratiadis A, Zeitlin S, Chesselet MF. Enhanced sensitivity to N-methyl-D-aspartate receptor activation in transgenic and knockin mouse models of Huntington’s disease. J Neurosci Res. 1999;58:515–532. [PubMed] [Google Scholar]
- Levine MS, Lloyd RL, Hull CD, Fisher RS, Buchwald NA. Neurophysiological alterations in caudate neurons in aged cats. Brain Res. 1987;401:213–230. doi: 10.1016/0006-8993(87)91407-7. [DOI] [PubMed] [Google Scholar]
- Li L, Murphy TH, Hayden MR, Raymond LA. Enhanced striatal NR2B-containing N-methyl-D-aspartate receptor-mediated synaptic currents in a mouse model of Huntington disease. J Neurophysiol. 2004;92:2738–2746. doi: 10.1152/jn.00308.2004. [DOI] [PubMed] [Google Scholar]
- Magnusson KR, Cotman CW. Age-related changes in excitatory amino acid receptors in two mouse strains. Neurobiol Aging. 1993;14:197–206. doi: 10.1016/0197-4580(93)90001-r. [DOI] [PubMed] [Google Scholar]
- Mallard EC, Waldvogel HJ, Williams CE, Faull RL, Gluckman PD. Repeated asphyxia causes loss of striatal projection neurons in the fetal sheep brain. Neuroscience. 1995;65:827–836. doi: 10.1016/0306-4522(94)00504-x. [DOI] [PubMed] [Google Scholar]
- MacGibbon GA, Hamilton LC, Crocker SF, Costain WJ, Murphy KM, Robertson HA, Denovan-Wright EM. Immediate-early gene response to methamphetamine, haloperidol, and quinolinic acid is not impaired in Huntington’s disease transgenic mice. J Neurosci Res. 2002;67:372–378. doi: 10.1002/jnr.10100. [DOI] [PubMed] [Google Scholar]
- McGeer EG, McGeer PL, Singh K. Kainate-induced degeneration of neostriatal neurons: dependency upon corticostriatal tract. Brain Res. 1978;139(2):381–3. doi: 10.1016/0006-8993(78)90941-1. [DOI] [PubMed] [Google Scholar]
- McGeer EG, McGeer PL. Duplication of biochemical changes of Huntington’s chorea by intrastriatal injections of glutamic and kainic acids. Nature. 1976;263:517–9. doi: 10.1038/263517a0. [DOI] [PubMed] [Google Scholar]
- Meade CA, Figueredo-Cardenas G, Fusco F, Nowak TS, Jr, Pulsinelli WA, Reiner A. Transient global ischemia in rats yields striatal projection neuron and interneuron loss resembling that in Huntington’s disease. Exp Neurol. 2000;166:307–323. doi: 10.1006/exnr.2000.7530. [DOI] [PubMed] [Google Scholar]
- Metzler M, Wong TP, Liu L, Helm J, Georgiou J, Wang Y, Bissada N, Cheng K, Roder JC, Wang YT, Hayden MR. NMDAR function and NMDAR-dependent phosphorylation of huntingtin is altered by endocytic protein HIP1. J Neurosci. 2007;27(9):2298–2308. doi: 10.1523/JNEUROSCI.5175-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mokrushin AA, Pavlinova LI, Plekhanov AY. Heat shock protein HSP70 increases the resistance of cortical cells to glutamate excitotoxicity. Bull Exp Biol Med. 2005;140(1):1–5. doi: 10.1007/s10517-005-0396-x. [DOI] [PubMed] [Google Scholar]
- Morikawa E, Mori H, Kiyama Y, Mishina M, Asano T, Kirino T. Attenuation of focal ischemic brain injury in mice deficient in the epsilon1 (NR2A) subunit of NMDA receptor. J Neurosci. 1998;18(23):9727–32. doi: 10.1523/JNEUROSCI.18-23-09727.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moroni F, Russi P, Carla V, Lombardi G. Kynurenic acid is present in the rat brain and its content increases during development and aging processes. Neurosci Lett. 1988;94:145–150. doi: 10.1016/0304-3940(88)90285-6. [DOI] [PubMed] [Google Scholar]
- Morton AJ, Leavens W. Mice transgenic for the human Huntington’s disease mutation have reduced sensitivity to kainic acid toxicity. Brain Res Bull. 2000;52:51–59. doi: 10.1016/s0361-9230(00)00238-0. [DOI] [PubMed] [Google Scholar]
- Namura S, Hirt L, Wheeler VC, McGinnis KM, Hilditch-Maguire P, Moskowitz MA, Macdonald ME, Persichetti F. The HD mutation does not alter neuronal death in the striatum of Hdh(Q92) knock-in mice after mild focal ischemia. Neurobiol Dis. 2002;11:147–154. doi: 10.1006/nbdi.2002.0532. [DOI] [PubMed] [Google Scholar]
- Newell DW, Barth A, Malouf AT. Glycine site NMDA receptor antagonists provide protection against ischemia-induced neuronal damage in hippocampal slice cultures. Brain Research. 1995;675(1–2):38–44. doi: 10.1016/0006-8993(95)00039-s. [DOI] [PubMed] [Google Scholar]
- Petersen A, Chase K, Puschban Z, DiFiglia M, Brundin P, Aronin N. Maintenance of susceptibility to neurodegeneration following intrastriatal injections of quinolinic acid in a new transgenic mouse model of Huntington’s disease. Exp Neurol. 2002;175:297–300. doi: 10.1006/exnr.2002.7885. [DOI] [PubMed] [Google Scholar]
- Peterson C, Cotman CW. Strain-dependent decrease in glutamate binding to the N-methyl-D-aspartic acid receptor during aging. Neurosci Lett. 1989;104:309–313. doi: 10.1016/0304-3940(89)90594-6. [DOI] [PubMed] [Google Scholar]
- Richards DA, Mateos JM, Hugel S, de PV, Caroni P, Gahwiler BH, McKinney RA. Glutamate induces the rapid formation of spine head protrusions in hippocampal slice cultures. Proc Natl Acad Sci U S A. 2005;102:6166–6171. doi: 10.1073/pnas.0501881102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiefer J, Alberty A, Dose T, Oliva S, Noth J, Kosinski CM. Huntington’s disease transgenic mice are resistant to global cerebral ischemia. Neurosci Lett. 2002;334:99–102. doi: 10.1016/s0304-3940(02)01111-4. [DOI] [PubMed] [Google Scholar]
- Schmued LC, Albertson C, Slikker W., Jr Fluoro-Jade: a novel fluorochrome for the sensitive and reliable histochemical localization of neuronal degeneration. Brain Res. 1997;751:37–46. doi: 10.1016/s0006-8993(96)01387-x. [DOI] [PubMed] [Google Scholar]
- Sharp AH, Loev SJ, Schilling G, Li SH, Li XJ, Bao J, Wagster MV, Kotzuk JA, Steiner JP, Lo A. Widespread expression of Huntington’s disease gene (IT15) protein product. Neuron 1995. 1995;14:1065–1074. doi: 10.1016/0896-6273(95)90345-3. [DOI] [PubMed] [Google Scholar]
- Shehadeh J, Fernandes HB, Zeron Mullins MM, Graham RK, Leavitt BR, Hayden MR, Raymond LA. Striatal neuronal apoptosis is preferentially enhanced by NMDA receptor activation in YAC transgenic mouse model of Huntington disease. Neurobiol Dis. 2006;21:392–403. doi: 10.1016/j.nbd.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Slow EJ, Graham RK, Osmand AP, Devon RS, Lu G, Deng Y, Pearson J, Vaid K, Bissada N, Wetzel R, Leavitt BR, Hayden MR. Absence of behavioral abnormalities and neurodegeneration in vivo despite widespread neuronal huntingtin inclusions. Proc Natl Acad Sci U S A. 2005;102:11402–11407. doi: 10.1073/pnas.0503634102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slow EJ, van RJ, Rogers D, Coleman SH, Graham RK, Deng Y, Oh R, Bissada N, Hossain SM, Yang YZ, Li XJ, Simpson EM, Gutekunst CA, Leavitt BR, Hayden MR. Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum Mol Genet. 2003;12:1555–1567. doi: 10.1093/hmg/ddg169. [DOI] [PubMed] [Google Scholar]
- Smart FM, Halpain S. Regulation of dendritic spine stability. Hippocampus. 2000;10:542–554. doi: 10.1002/1098-1063(2000)10:5<542::AID-HIPO4>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Starling AJ, Andre VM, Cepeda C, de Lima M, Chandler SH, Levine MS. Alterations in N-methyl-D-aspartate receptor sensitivity and magnesium blockade occur early in development in the R6/2 mouse model of Huntington’s disease. J Neurosci Res. 2005;82:377–386. doi: 10.1002/jnr.20651. [DOI] [PubMed] [Google Scholar]
- Stavrovskaya IG, Narayanan MV, Zhang W, Krasnikov BF, Heemskerk J, Young SS, Blass JP, Brown AM, Beal MF, Friedlander RM, Kristal BS. Clinically approved heterocyclics act on a mitochondrial target and reduce stroke-induced pathology. J Exp Med. 2004;200(2):211–22. doi: 10.1084/jem.20032053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor-Robinson SD, Weeks RA, Bryant DJ, Sargentoni J, Marcus CD, Harding AE, Brooks DJ. Proton magnetic resonance spectroscopy in Huntington’s disease: evidence in favour of the glutamate excitotoxic theory. Mov Disord. 1996;11:167–173. doi: 10.1002/mds.870110209. [DOI] [PubMed] [Google Scholar]
- Uemura Y, Kowall NW, Beal MF. Selective sparing of NADPH-diaphorase-somatostatin-neuropeptide Y neurons in ischemic gerbil striatum. Ann Neurol. 1990;27:620–625. doi: 10.1002/ana.410270606. [DOI] [PubMed] [Google Scholar]
- Van Raamsdonk JM, Pearson J, Slow EJ, Hossain SM, Leavitt BR, Hayden MR. Cognitive dysfunction precedes neuropathology and motor abnormalities in the YAC128 mouse model of Huntington’s disease. J Neurosci. 2005;25:4169–4180. doi: 10.1523/JNEUROSCI.0590-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velier J, Kim M, Schwarz C, Kim TW, Sapp E, Chase K, Aronin N, DiFiglia M. Wild-type and mutant huntingtins function in vesicle trafficking in the secretory and endocytic pathways. Exp Neurol. 1998;152:34–40. doi: 10.1006/exnr.1998.6832. [DOI] [PubMed] [Google Scholar]
- Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP., Jr Neuropathological classification of Huntington’s disease. J Neuropathol Exp Neurol. 1985;44:559–577. doi: 10.1097/00005072-198511000-00003. [DOI] [PubMed] [Google Scholar]
- Zeron MM, Fernandes HB, Krebs C, Shehadeh J, Wellington CL, Leavitt BR, Baimbridge KG, Hayden MR, Raymond LA. Potentiation of NMDA receptor-mediated excitotoxicity linked with intrinsic apoptotic pathway in YAC transgenic mouse model of Huntington’s disease. Mol Cell Neurosci. 2004;25:469–479. doi: 10.1016/j.mcn.2003.11.014. [DOI] [PubMed] [Google Scholar]
- Zeron MM, Hansson O, Chen N, Wellington CL, Leavitt BR, Brundin P, Hayden MR, Raymond LA. Increased sensitivity to N-methyl-D-aspartate receptor-mediated excitotoxicity in a mouse model of Huntington’s disease. Neuron. 2002;33:849–860. doi: 10.1016/s0896-6273(02)00615-3. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Li M, Drozda M, Chen M, Ren S, Mejia Sanchez RO, Leavitt BR, Cattaneo E, Ferrante RJ, Hayden MR, Friedlander RM. Depletion of wild-type huntingtin in mouse models of neurologic diseases. J Neurochem. 2003;87(1):101–6. doi: 10.1046/j.1471-4159.2003.01980.x. [DOI] [PubMed] [Google Scholar]
- Zhang F, Li C, Wang R, Han D, Zhang QG, Zhou C, Yu HM, Zhang GY. Activation of GABA receptors attenuates neuronal apoptosis through inhibiting the tyrosine phosphorylation of NR2A by Src after cerebral ischemia and reperfusion. Neurosci. 2007;150(4):938–49. doi: 10.1016/j.neuroscience.2007.09.070. [DOI] [PubMed] [Google Scholar]