

Abstract
Background & Aims
MicroRNA (miRNA) is highly stable in biospecimens and provides tissue-specific profiles, making it a useful biomarker of carcinogenesis. We aimed to discover a set of miRNAs that could accurately discriminate Barrett’s esophagus (BE) from normal esophageal tissue and to test its diagnostic accuracy when applied to samples collected by a non-invasive esophageal cell sampling device.
Methods
We analyzed miRNA expression profiles of 2 independent sets of esophageal biopsy tissues collected during endoscopy from 38 patients with BE tissues and 90 patients with non-BE esophagus (controls) using Agilent microarray and Nanostring counter assays. Consistently upregulated miRNAs were quantified by real-time PCR in esophageal tissues collected by Cytosponge from patients with BE or without BE. miRNAs from plasmids and anti-sense oligonucleotides were expressed in NES normal esophageal squamous cells and effects on proliferation and gene expression patterns were analyzed.
Results
We identified 15 miRNAs that were significantly upregulated in BE vs control tissues. Of these, 11 (MIR215, MIR194, MIR 192, MIR196a, MIR199b, MIR10a, MIR145, MIR181a, MIR30a, MIR7, MIR199a) were validated in Cytosponge samples. The miRNAs with the greatest increases in BE tissues (7.9-fold increase inexpression or more, P<0.0001: MIR196a, MIR192, MIR194, and MIR215) each identified BE vs control tissues with area under the curve (AUC) values of 0.82 or more. We developed an optimized multivariable logistic regression model based on expression levels of 6 miRNAs (MIR7, MIR30a, MIR181a, MIR192, MIR196a, and MIR199a) that identified patients with BE with an AUC value of 0.89, 86.2% sensitivity, and 91.6% specificity. Expression level of MIR192, MIR196a, MIR199a, combined with Trefoil Factor 3 (TFF3), identified patients with BE with an AUC of 0.93, 93.1% sensitivity, and 93.7% specificity. Hypo-methylation was observed in the promoter region of the highly upregulated cluster MIR192-194. Overexpression of these miRNAs in NES cells increased their proliferation, via GRHL3 and PTEN signaling.
Conclusions
In analyses of miRNA expression patterns of BE vs non-BE tissues, we identified a profile that can identify Cytosponge samples from patients with BE with an AUC of 0.93. Expression of MIR194 is increased in BE samples via epigenetic mechanisms that might be involved in BE pathogenesis.
Keywords: gene regulation, biomarker, esophageal adenocarcinoma, diagnosis
Introduction
Esophageal adenocarcinoma is a highly lethal malignancy with a five-year survival of less than 20%1. Although the precursor metaplasia, Barrett’s esophagus (BE), provides an opportunity for surveillance and early detection, 95% of esophageal adenocarcinoma patients are diagnosed in individuals without a prior diagnosis of BE2, 3. This conundrum necessitates the development of new strategies and tools to identify a larger proportion of individuals who have BE.
Any potentially useful screening tool for BE needs to be highly sensitive (to avoid harm caused by false-negatives), highly specific (to avoid financial costs of conducting unnecessary secondary investigations), and logistically feasible and affordable in order to be suitable on a large scale. A minimally-invasive, pan-esophageal cell sampling device, the Cytosponge, coupled with immunohistochemical staining for Trefoil Factor 3 (TFF3), has been shown to have cost-effective utility in diagnosing BE4 with applicability to the primary care setting5. In the BEST2 case-control study, there was encouraging sensitivity and specificity (sensitivity 79.9% for all segment lengths, using an “intention-to-treat” analysis whereby samples lacking columnar cells- indicating that the Cytosponge did not reach the stomach- are included; specificity 92.4%)6. As most patients with BE will not progress to cancer, we have investigated additional nucleic acid biomarkers for the Cytosponge, including methylation and p53 mutations in order to stratify patients according to their risk of progression to cancer7, 8. Ideally, a single automated platform using nucleic acids (DNA and RNA) extracted from the Cytosponge could be used to diagnose and risk stratify patients in parallel rather than relying on a two-step process involving an immunohistochemical biomarker.
MicroRNAs (miRNAs) are a type of small (18-22 nucleotides) non-protein coding RNA that bind to messenger RNAs (mRNA) via their seed sequence to repress gene expression post-transcriptionally9, 10. miRNAs are found across the genome, sometimes within introns of genes, and clusters of miRNA loci are commonly observed. Regulatory elements that control transcription are usually shared within a miRNA cluster or with neighbouring genes, although the latter is under a more complex modulation11. MicroRNAs have central roles in endogenous processes including metabolism, inflammation, and carcinogenesis, and each microRNA has the potential to regulate a diverse array of gene transcripts12. miRNA profiles have been shown to be tissue and disease specific13 and are minimally affected by processes used to generate formalin-fixed, paraffin-embedded (FFPE) samples14, 15. These features of miRNAs make them appealing biomarkers. Previous studies have identified several candidate miRNA biomarkers specific for BE—however, these were limited by relatively small sample sizes, lack of any functional validation, and reliance on a single profiling platform which constrained the diversity of miRNAs that are quantified16-21. Furthermore, we were particularly interested to test the application of an accurate miRNA classifier to Cytosponge samples for the purposes of diagnosing BE.
The aims of the study were (i) to discover a miRNA signature that could distinguish BE from normal esophagus (NE) across two distinct profiling platforms; (ii) to validate this miRNA signature using a Cytosponge case-control sample set; and (iii) to examine the functional consequences of the most upregulated miRNAs in vitro.
Methods
Sample selection
The samples for the different parts of the study are summarised in Figure 1. Patients in sample set A were from an ongoing prospective Barrett’s biobank (Ethics No. LREC 01/149) and sample set B from endoscopic samples collected as part of BEST1 (Ethics No. 06/Q0108/272) and BEST2 (Ethics No. 10/H0308/71). There was no overlap between the two sample sets. All samples were obtained following ethical approval and individual informed consent.
Figure 1. Schematic illustration of the study design and strategy.
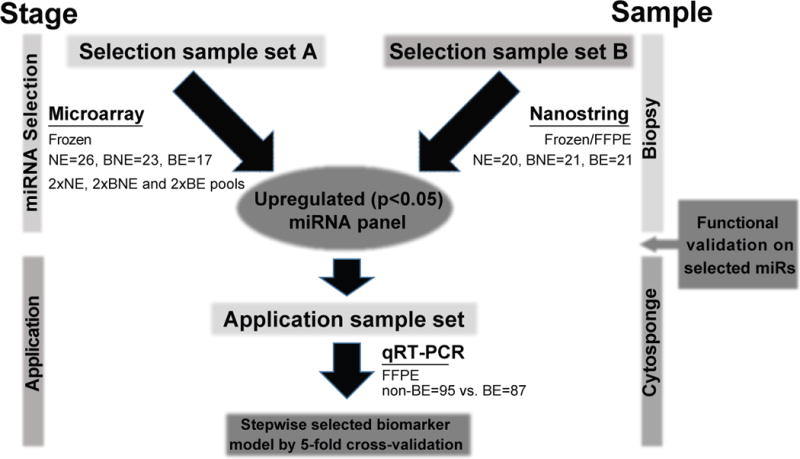
Summary of methods divided into two stages of Selection (A and B) and application using both biopsy-derived and Cytosponge-derived samples.
Sample sets A and B comprised cases and controls and for sample set A pools were created. Cases comprised patients with a known diagnosis of BE attending for surveillance and controls were individuals referred to endoscopy because of dyspepsia and/or reflux symptoms. All biopsy samples were subject to an expert histopathological review prior to inclusion. NE biopsy samples contained stratified squamous epithelium and an absence of columnar cells. BE biopsy samples contained intestinal metaplasia without dysplasia or neoplasia.
The upregulated miRNAs were then tested in Cytosponge samples. These samples were randomly selected from the BEST2 comprising cases (BE) and controls (individuals referred for endoscopy because of dyspepsia or reflux symptoms with BE). The Cytosponge sample had to have sufficient material remaining for miRNA analysis in order to be included.
miRNA extraction
RNA from frozen regular forceps biopsy samples was extracted using the miRNeasy Mini Kit (Qiagen) according to the manufacturer’s protocol. For FFPE blocks, 2-4 scrolls of 10mm were cut and extracted using the miRNeasy FFPE Kit (Qiagen) according to the manufacturer’s protocol. Total RNA concentrations were measured by ND-1000 spectrophotometer (NanoDrop Technologies) and 2100 Bioanalyzer (Agilent Technologies).
Microarray expression analysis
miRNA microarray was performed with the Human miRNA Microarray kit v1.0 (8 × 15K, consists of 534 probes of 470 human and 64 virus mature miRNAs) (Agilent Technologies) with 100ng total RNA per sample per the manufacturer’s protocol. The hybridised chip was scanned using the G2565BA Microarray Scanner (Agilent Technologies) and analysed using GenePix Pro software v4.1 (Molecular Devices Corporation). Platform annotations were re-annotated to miRBase 21.0 using miRiadne22. Raw intensities were then log2-transformed and normalised by quantile with differentially expressed miRNAs identified using linear models implemented in the limma package (version 3.32.2) for R (version 3.4.0)23. We filtered any miRNAs with fewer than 2/6 pools expressing at above 6.5 (corresponding to intensity above 90) and if multiple probes mapped to the same miRNA, the probe with the highest average expression was selected24.
Nanostring nCounter analysis
Samples were sent to Johns Hopkins Medical Institute (JHMI) Deep Sequencing and Core Facility for NanoString nCounter analysis. RNA samples were processed according to the manufacturer’s protocol for the nCounter Human miRNA Expression Assay v2 kit which profiled 800 human miRNAs (NanoString, Seattle, WA). We used 100ng of each total RNA sample as input into the nCounter Human miRNA sample preparation. Hybridization with the capture probe set was incubated for 16 hours. Counts were collated for each sample by the nCounter Digital Analyzer and raw counts were imported into nSolver version 3.0.
Internal negative control probes included in each assay were used to determine a background threshold (2 standard deviations above the mean negative control probe count value) for each sample. Background was subtracted from raw count values for each probe and counts set to 0 for all probes at or below the background threshold. Background-adjusted counts were then normalised using the functions ‘calcNormFactors’ (‘method’ set to “TMM”) and ‘estimateDisp’ (‘robust’ set to “TRUE”) with differentially expressed miRNAs identified using a generalised linear model-likelihood ratio test implemented in the edgeR package (version 3.18.1) for R25. We filtered any miRNAs with fewer than 25% of samples expressing at 1 count per million or higher and accounted for samples from the same patient with a term in the model.
TCGA data analysis
The miRNA expression values (miRNA Expression Quantification data files) for esophageal cancer samples were obtained from https://gdc.cancer.gov/. Tumour samples from esophageal adenocarcinoma patients were selected based on histological diagnosis. Case TCGA-L5-A4OI was excluded because of missing annotation for why two miRNA quantification files were available for this case. The R package biomaRt26 in combination with the Feb 2014-Ensembl archive was used to map the gene annotations from miRBase identifiers to Ensembl gene identifiers and HGNC symbols. Only mappings with HGNC symbols related to miRNAs were retained. miRBase identifiers mapping to multiple Ensembl or HGNC symbols were removed.
Expression-based correlation between miRNAs
The data were transformed using variance stabilization from the DESeq227 R package. Pre-selected miRNAs were investigated for correlation among their expression values in the tumours. Expression values were averaged in case multiple Ensembl identifiers mapped to the same HGNC symbol. miRNAs with no variation across the samples were excluded. The matrix of Pearson’s correlation coefficients was clustered using hierarchical clustering with Euclidean distance and complete agglomeration as implemented in heatmap.2-function from the gplots R package.
Methylation analysis
Methylation data from 10 squamous and 20 Barrett’s cases were generated using Illumina EPIC Array platform. All samples were processed through ChAMP1 program in R platform and data were normalized using BMIQ2 algorithm. The Mann-Whitney test was used for observing any differences in the methylation levels at MIR192 and MIR194-2 between squamous and Barrett’s groups. The median beta of all probes annotated to MIR192 and MIR194-2 were considered for this.
Quantative Real-time PCR (qRT-PCR)
RNA was reverse transcribed to cDNA using the miScript II RT Kit (Qiagen) using the manufacturer’s protocol. qRT-PCR reactions were performed in triplicate in a 384-well plate using LightCycler 480 Instrument II (Roche) according to the manufacturer’s protocol. Primer sequences are detailed in SupTable 5.
The threshold cycle (Ct) was determined by the Second Derivative Maximum method. The expression of each target was normalised relative to the geometric mean of endogenous controls. Endogenous controls for miRNA (MIR103, MIR191, MIR21) and mRNA (GAPDH, ACTB, RPS18) targets were selected by a literature review. Consistent miRNA and mRNA endogenous control expression was validated using internal and published microarray datasets28, 29.
Application of selected miRNAs to Cytosponge samples
For validation, miRNAs were selected based on having a log2 fold change > 1 and adjusted p-value < 0.05 (Benjamini-Hochberg correction) in both biopsy miRNA profiling sets. In the Cytosponge set, we calculated mean fold-change differences between BE and NE Cytosponge samples as well as p values based on the Mann-Whitney test. For all multivariable miRNA models, we used five-fold cross-validation using Stata version 14.0 (StataCorp LP) to obtain estimates of performance criteria. Subjects were randomly assigned to five mutually-exclusive groups with approximately equal numbers of BE cases and NE controls in each group. For a given fold, we used the four retained groups to model the selected miRNAs as continuous variables using logistic regression from which we estimated prediction probabilities for the group that was omitted from the fold. We repeated this prediction procedure five times, each time sequentially omitting a single distinct group of subjects to estimate prediction probabilities of case-control status. Prediction probabilities of ≥0.478 (87 BE/182 total) were interpreted to indicate BE case status which was then used to estimate sensitivity, specificity, and the area under the receiver operating characteristic curve (AUC). The first multivariable miRNA model for which we assessed performance criteria included all miRNAs that replicated (were positively associated with BE) in univariate analyses in this Cytosponge sample set. Next, we assessed a reduced multivariable model selected from a stepwise logistic regression model (significance level for removal from the model=0.1) using the total validation case-control sample set. We next assessed addition of TFF3 to this reduced model. Last, we assessed the performance criteria of a multivariable model selected using the same stepwise model to select from all miRNAs that replicated as well as TFF3. Performance criteria for all multivariable models was assessed used five-fold cross-validation as previously described.
In vitro cell culture, transfection and viability assay
NES cells (gift from R. Souza, University of Texas Southwestern Medical Center) were cultured in a supplemented 3:1 mixture of DMEM/Ham’s F12 medium (Invitrogen) as previously described30. Cell numbers were determined by trypan blue cell exclusion method after 48-hour transfection.
Plasmid sequence (SupFig 4) and transfection miRNA expression plasmids were cloned by replacing the insert from a pcDNA3.1 plasmid (Addgene plasmid #21114) with inserts cloned by PCR (primers detailed in SupTable 5). Plasmid cloning was validated by Sanger sequencing using a CMV-F primer (SupFig 4). Transfection was performed by using Lipofectamine 3000 with expression plasmid or anti-miR (Dharmacon miRIDIAN microRNA Hairpin Inhibitor) according to the manufacturer’s protocol. All data reflects at least two biological replicates and refers to fold change versus empty vector, with each experiment normalised to untransfected controls, 48 hours following transfection unless otherwise stated.
Results
Patient characteristics
This study examined samples derived from three sets of BE patients and NE controls as summarised in Table 1 and Fig 1. The number of samples for each tissue type in sample sets A and B were similar: samples were selected from 26 NE and 40 BE patients (17 for BE tissues, 23 for BNE [squamous epithelium from above the Barrett’s segment]). The extracted material was compiled to form 2 NE, 2 BNE and 2 BE pools of miRNAs for Agilent Technologies Human miRNA Microarray v1.0; 20 NE and 21 BE (BE and BNE) for Nanostring Human miRNA Expression Assay v2. To maximise the power of detection and accuracy of any findings, fresh frozen preserved endoscopically collected biopsy samples were prioritised for the profiling assays, where possible. Furthermore, matched patient tissue samples were used in Nanostring nCounter assay, which offered insights into the miRNA expression profile of BE and BNE within the same BE patient.
Table 1.
Summary of patient characteristics
| Variables | Selection sample set A | Selection sample set B | Application sample set | ||||
|---|---|---|---|---|---|---|---|
| NE | (B)NE | BE | NE | BE | Controls | BE Cases | |
| Numbers | 26 | 23 | 17 | 20 | 21 | 95 | 87 |
| Age (years) - median (IQR) | 56 (44-72) | 66 (62-73) | 63 (57-72) | 60.5 (55-64) | 63 (54-70) | 55 (43-68) | 66 (54-72) |
| Male:female ratio | 0.73 | 3.6 | 4.67 | 1.86 | 9.5 | 0.98 | 4.8 |
| Ethnicity: White | NA | NA | NA | 20 | 19 | 89 | 84 |
| Ethnicity: Other | NA | NA | NA | 0 | 2 | 6 | 3 |
| BMI - median (IQR) | NA | 26.8 (25.0-28.7) | 25.6 (22.9-27.5) | 28.2 (25.0-33.2) | 28.0 (26.2-30.4) | 27.3 (24.1-31.6) | 28.4 (26.2-31.3) |
| Wait-to-hip ratio - median (IQR) | NA | NA | NA | 0.89 (0.82-0.93) | 0.95 (0.88-0.99) | 0.88 (0.83-0.95) | 0.95 (0.91-0.99) |
| Maximum M length of BE (cm) -median (IQR) | NA | 2 (1-2) | 4 (4-6) | NA | 6 (4-9) | NA | 4 (3-7.5) |
| Smoking (no) | 17 | 10 | 9 | 8 | 6 | 54 | 46 |
| Smoking (ex) | 1 | 4 | 1 | 7 | 13 | 25 | 28 |
| Smoking (active) | 0 | 2 | 0 | 5 | 2 | 14 | 13 |
| Smoking (NA) | 8 | 7 | 7 | 0 | 0 | 2 | 0 |
For the application of the miRNA panel Cytosponge samples were randomly selected from cases and controls who had participated in the BEST2 study (details in methods). Cases comprised individuals with histopathologically-verified BE biopsies and Cytosponge samples. For all parts of the study BE patients were older, more likely to be male and have higher wait-hip ratio than controls, which is consistent with known risk factor for BE31 (Table 1).
Upregulated miRNAs detect BE in esophageal biopsy samples
Previous studies have indicated suboptimal correlations between different miRNA profiling platforms17-21. To maximise the robustness of a miRNA signature differentially expressed in BE versus NE, we therefore performed two parallel high-throughput approaches (Agilent Technologies microarray and Nanostring nCounter) on two independent sample sets (A and B, Fig 1). Furthermore, given the high tissue specificity of miRNA profiles and the possibility of a field effect from adjacent Barrett’s, we used two control NE tissues. These comprised squamous tissues from healthy controls without Barrett’s (NE) as well as squamous epithelium from above the Barrett’s segment in cases (BNE), (see study design Fig 1 and Table 1). The miRNA profile of BNE and NE samples clustered together (SupFig 1) and there were 4 miRNAs (MIR451, MIR144, MIR191, MIR375) that showed differential expression between NE and BNE samples (SupTable 1). However, the fold changes were modest. In view of the similarities between BNE and NE miRNA expression profiles, these groups were combined for subsequent analyses.
When comparing squamous (BNE + NE) and Barrett’s (BE) samples miRNA Microarray (Agilent Technologies) expression analysis in sample set A identified 28 upregulated miRNAs of 470 total measured (SupTable 2) while Nanostring nCounter Human v2 microRNA Expression Assay analysis in sample set B identified 46 upregulated miRNAs out of 800 (SupTable 3). Cross-referencing of upregulated miRNAs (defined as log fold change >1 and adjusted p-value < 0.05) identified 15 miRNAs significantly upregulated in both sample sets (Fig 2).
Figure 2. Upregulated miRNAs from cross-platform analysis.
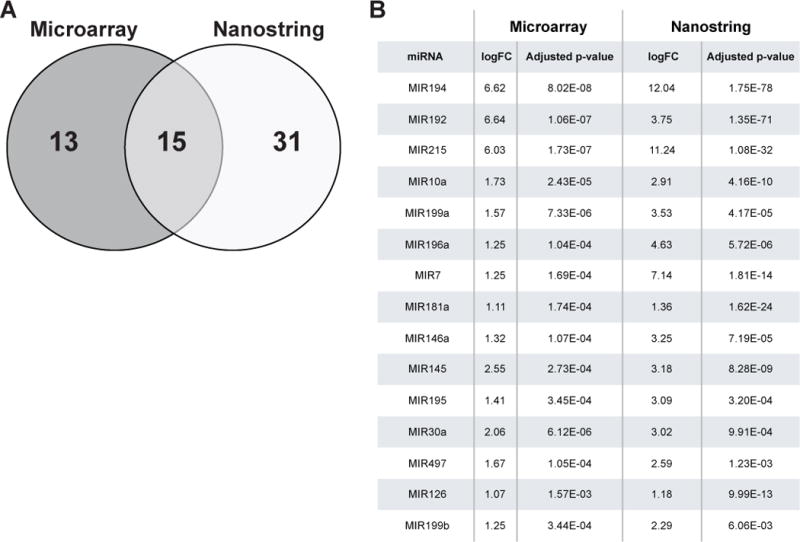
(A) Venn diagram showing upregulated miRNAs detected by Agilent microarray and Nanostring profiling in Selection sample sets A and B respectively. Detailed fold changes for each upregulated miRNA listed in SupTable 2 and 3. 15 consensus miRNAs were determined by cross-referencing of miRNAs detected by each platform. Log of fold changes (logFC) and adjusted p-values of these 15 miRNAs are listed in (B), ranked by mean of adjusted p-value.
Novel miRNA panel as effective biomarkers for non-invasive BE diagnosis
To assess the diagnostic performance of these 15 miRNAs using Cytosponge samples, their expression was examined by qRT-PCR in a distinct set of 95 control and 87 BE samples randomly selected from the BEST2 Cytosponge study6. Upon univariate analysis, MIR215, 194, 192 and 196a were significantly (p<0.0005) and highly (fold changes of relative expression of 13.0, 9.7, 8.5 and 7.9, respectively) upregulated in case versus control Cytosponge samples (Fig 3A i-iv) with areas under the receiver operating characteristic curve (AUC) of 0.82 (95% confidence interval (CI): 0.75-0.88), 0.88 (95% CI: 0.83-0.93), 0.89 (95% CI: 0.84-0.94), and 0.90 (95% CI: 0.85-0.94), respectively (Fig 3B). This increased expression pattern for BE vs NE was also replicated for a further 7 of the 15 miRNAs upon univariate analysis: 199b, 10a, 145, 181a, 30a, 7 and 199a (fold changes >1 between BE and control patients, Fig 3A v-xi). However, miRNAs 195, 126, 497 and 146a failed to replicate in this Cytosponge set of cases and controls (fold change <1 between BE and control patients, Fig 3A xii-xv). These four miRNAs were, therefore, not considered for multivariable models.
Figure 3. Validation of upregulated miRNAs in case-control Cytosponge sample set.

(A i-xv) Relative miRNA expression determined by qRT-PCR. Fold changes (FC) and mean (colored line) are presented for each miRNA. Significance determined by Mann-Whitney test: *, p<0.05; **, p<0.01; ***, p<0.001; ****, p<0.0001. (B) AUC (Area under the ROC Curve) and 95% CI (Confidence Interval) for each miRNA and TFF3 were calculated using validation qRT-PCR results.
The AUC of a model including age, sex, race/ethnicity, body mass index, waist-to-hip ratio and smoking status was 0.71 (95%CI:0.64, 0.77) which dramatically improves when the predictive tissue biomarkers are added (Table 2). Five-fold cross-validation of a multivariable biomarker model that included the 11 validated miRNAs provided an AUC of 0.87 (95% CI: 0.82-0.92) with a sensitivity 83.9% and specificity 90.5% (Table 2) using a predicted probability threshold of ≥0.478 (87 BE/182 total) to assign BE case status (see Methods). Stepwise selection in the total set followed by five-fold cross validation suggested that a subset of miRNAs (MIR7, 30a, 181a, 192, 196, 199a) slightly improved the AUC to 0.89 (95% CI: 0.84-0.93) with 86.2% sensitivity and 91.6% specificity (Table 2). Inclusion of TFF3 improved the AUC to 0.92 (95% CI: 0.88-0.96) although statistically there is no significant difference with or without TFF3 (Table 2). Stepwise selection of the 11 validated miRNAs and TFF3 retained just three miRNAs (MIR192, 196a and 199a) as well as TFF3 and five-fold cross validation of this model provided an AUC of 0.93 (95% CI: 0.90-0.97) with sensitivity 93.1% and specificity 93.7% (Table 2).
Table 2.
AUCs and 95%CIs of stepwise selected biomarker models using five-fold Cross-validation
| Model Details | Predictors | AUC (95%CI) | AUC (95%CI) with risk factors for BE |
|---|---|---|---|
| Risk factors for Barrett’s esophagus | Age, sex, ethnicity, smoking, BMI, waist-hip ratio | 0.71 (0.64, 0.77) | - |
| All miRNAs that univariately positively predicted BE in Cytosponge application | MIR7, 10a, 30a, 145, 181a, 192, 194, 196, 199a, 199b, 215 | 0.87 (0.82, 0.92) | 0.84 (0.79, 0.89) |
| Stepwise selection of miRNAs from initial model | MIR7, 30a, 181a, 192, 196, 199a | 0.89 (0.84, 0.93) | 0.88 (0.83, 0.93) |
| Above model plus TFF3 | MIR7, 30a, 181a, 192, 196, 199a plus TFF3 | 0.92 (0.88, 0.96) | 0.92 (0.88, 0.96) * |
| Stepwise selection of miRNAs from initial model and TFF3 (3 miRNAs and TFF3) | MIR192, 196, 199a, plus TFF3 | 0.93 (0.90, 0.97) | 0.91 (0.87, 0.95) |
Model did not include smoking due to failure to converge
Epigenetic alteration in BE could contribute to aberrant co-expression of cluster miRNAs
Interestingly, 3 of the most significantly unregulated miRNAs are from two miRNA clusters, MIR192-194-2 (11q13.1) and MIR215-194-1 (1q41, intron of RNU5F-1 and IARS2) which have been reported to respond to p53 activation32. While MIR194-1 and MIR194-2 are located on different chromosomes, they share an identical mature sequence and target the same type of mRNAs. Clustered miRNAs usually share a similar expression pattern33 and the correlation matrix based on the case-control sample set revealed the co-expression of MIR192 and MIR194 (r=0.787, Fig 4Ai). This observation can be replicated using TCGA esophageal adenocarcinoma miRNAseq data (n= 88) as independent data set (r=0.945, Fig 4Aii).
Figure 4. Co-expression of MIR192/194 and hypo-methylation found in the promotor region of miRNA cluster MIR192-194-2.
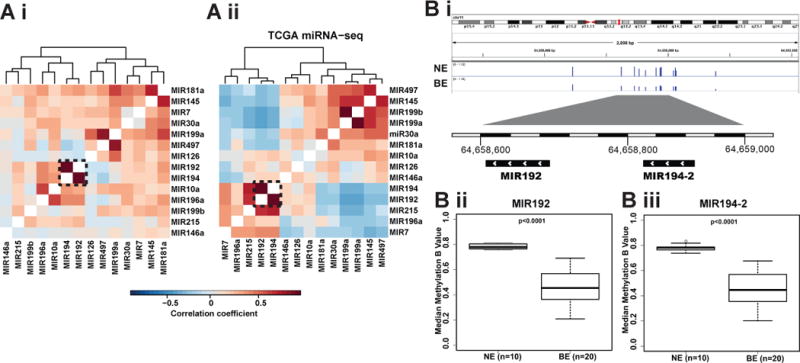
(A) Heatmap showing the Pearson correlation coefficient (color key) between 15 consensus upregulated miRNA expressions based on validation data from this study (A i) and TCGA esophageal adenocarcinoma miRNA sequencing data (A ii). Dendrograms show the hierarchical clustering based on the complete linkage method and Euclidean pair-wise distance. Genomic region of cluster MIR192-194-2 shows DNA methylation probe peaks in the promoter region (<1 kb) in NE and BE samples (B i). Median methylation beta values were plotted for MIR192 and MIR194-2 (B ii). Significance was determined by Mann-Whitney test.
To understand the increased co-expression of MIR192 and MIR194-2 in BE, we searched for genomic and epigenetic alterations. By using whole genomic sequencing data from our previous genomic studies34-36, no recurrent somatic mutations were found in the known regulatory regions of MIR192-194-2. Interestingly, the promoter regions of MIR192-194-2 (<1kb from transcription starting site) were highly methylated in the NE samples (n=10), whereas a significant hypo-methylation (p<0.0001) was found in the distribution of methylation intensity in the BE samples (n=20), which is well-known to be correlated with target gene overexpression37 (Fig 4B i-iii).
MIR194 dependent signalling prompts esophageal cell growth in vitro
Next we set out to examine the role of upregulated MIR192/194 through their target mRNAs in BE. We predicted targets of miRNAs 192 and 194 using the TargetScan 7.1 algorithm which searches for conserved 3′UTR sites that match the seed region (nucleotides 2-7) of each miRNA38. We also incorporated mRNAs downregulated >20% following MIR192 transfection using data from a published microarray dataset to populate our list of predicted targets39. We hypothesised that true targets of these miRNAs would be downregulated in BE versus NE biopsy samples. Using recent microarray datasets28, 40, 53 such putative targets were identified and following a literature review to prioritise targets with known tumour suppressor roles in cancer, 6 were selected for further validation (SupTable 4). qRT-PCR confirmed downregulation of all of these targets and the increased expression of MIR192 and MIR194 (SupFig 2).
To demonstrate repression of putative miRNA targets in vitro, cell line NES derived from NE30 was transfected with MIR192 or MIR194 expression plasmids. GRHL3, one putative target mRNA of MIR194 (Fig 5A) was significantly downregulated on MIR194 overexpression (fold change >3, Fig 5B i-ii) while the other 5 miRNA targets examined were not significantly repressed upon transfection (SupFig 3). To further characterise this relationship, NES cells were transfected with antisense oligonucleotides against MIR194 (anti-MIR194). This was associated with concurrent downregulation of MIR194 and upregulation of GRHL3 compared to control anti-miRNA (Fig 5Ci-ii). Comparative analysis of the GRHL3 3′UTR across vertebrates using TargetScan 7.1 showed that it contains two conserved 7mer-m8 binding sites for MIR194 (Fig 5A)41. In summary, MIR194 negatively regulated GRHL3 expression both in silico and in vitro.
Figure 5. Dysregulated MIR194 expression drove cell proliferation in vitro through MIR194-GRHL3-PTEN axis.
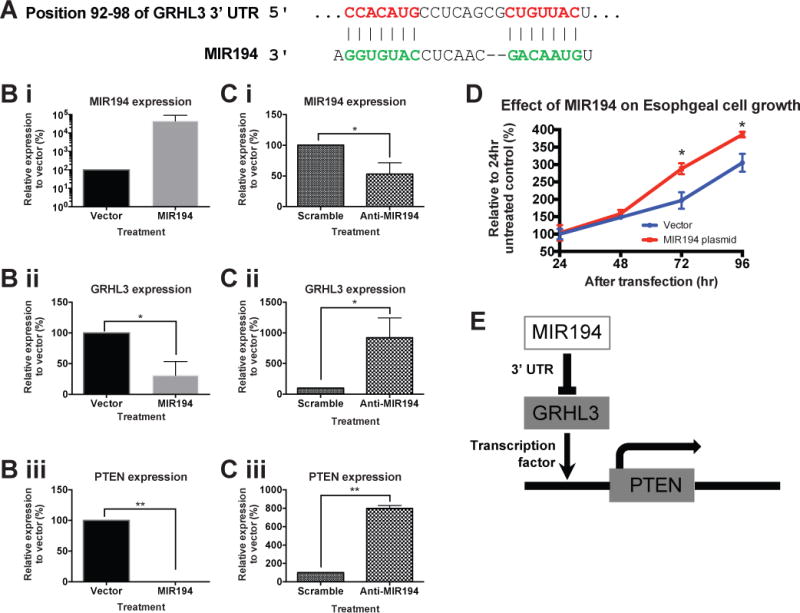
(A) Genomic alignment of GRHL3 mRNA 3′UTR with MIR194. Normal esophageal cell line NES were transfected with either MIR194 plasmid (B) or anti-MIR194 (C). Relative expression of MIR194 (i), GRHL3 (ii) and PTEN (iii) in NES cell line was examined by qRT-PCR. Error bars show standard deviation of the mean: *, p<0.05; **, p<0.01. (D) Cell growth was determined by trypan blue exclusion assays every 24 hours for a period of 96 hours after MIR194 transfection relative to vector control. Error bars show standard deviation of the mean: *, p<0.05. (E) Diagram shows MIR194-GRHL3-PTEN cascade network.
GRHL3 is known to activate PTEN transcription by binding to a conserved site in the PTEN promoter42. Transfection of MIR194 in NES cell line was associated with significant repression of PTEN expression (Fig 5Biii). In contrast, suppression of MIR194 by anti-MIR194 leads to upregulation of PTEN (>7 folds) (Fig 5Ciii). Furthermore, consistent with PTEN’s function as a negative regulator of growth signalling43, PTEN downregulation on MIR194 overexpression was associated with significantly enhanced cell growth in vitro at 72 hours following transfection (Fig 5D). Taken together, our findings highlight the regulation of MIR194 on esophageal cell growth through the MIR194-GRHL3-PTEN axis (Fig 5E).
Discussion
Using patient biopsy and Cytosponge samples, this study identified a panel of miRNAs that are differentially expressed in BE versus NE and accurately diagnosed BE using Cytosponge samples. We demonstrated that these miRNAs may have a functional role in BE etiology, whereby increased expression of MIR194 drives proliferation in an in vitro NE model through the MIR194-GRHL3-PTEN regulatory network.
To improve the profiling signals and reproducibility of miRNA discovery platforms17-21, we used two profiling methods in pathologically-verified biopsy samples. A panel of 15 upregulated miRNAs in BE biopsy samples were identified, as well as some lesser known and novel candidates (MIR196a, 199a/b, 7, 181a). It is reassuring that this study identified some miRNAs, including MIR192, MIR194 and MIR215, that have been shown to be upregulated previously44-46. Stepwise selection was used to identify the minimum panel with the maximum AUC. A subset of miRNAs (MIR7, MIR30a, MIR181a, MIR192, MIR196, MIR199a) provided an AUC of 0.89 with 86.2% sensitivity and 91.6% specificity. A logistic regression model with stepwise selection that included TFF3, provided an optimal panel comprising MIR192, MIR196, and MIR199a and TFF3 with an AUC of 0.93, the greatest sensitivity of 93.1% and greatest specificity of 93.7% (Table 2). It was interesting to note that MIR199a was retained in the final panel despite an individual AUC of 0.50 (Fig3). There was no effect modification between MIR199a and MIR192, MIR196 or TFF3. According to our a priori rules, MIR199a was retained in the stepwise multivariable model based on a low p value. However, excluding it from the five-fold cross validation has no material effect on the AUC (0.93, 95%CI:0.89-0.97). It should be noted that the previously reported TFF3 accuracy data were ascertained from a prospective trial with larger sample numbers than the current study6.
From the perspective of clinical translation, a miRNA assay could be readily adapted to a high throughput setting amenable for large volume screening, whereas TFF3 relies on the preparation of a cell block and histopathological and immunohistochemical assessment by an expert. Here we demonstrate that a miRNA panel (MIR7, MIR30a, MIR181a, MIR192, MIR196, MIR199a) can provide a very similar accuracy with an AUC of 0.89 (86.2% sensitivity and 91.6% specificity) compared with TFF3 alone with an AUC of 0.89 (83.9% sensitivity and 93.7% specificity) when applied to the same sample set (Fig 3). Furthermore, the AUC achieved for a combination of TFF3 and miRNAs is not statistically improved and the laboratory processing for a combination would be more complex. miRNA expression analysis could be performed using an automated pipeline with objective quantitation. Diagnostic miRNAs could also be quantitated in parallel with other nucleic acid biomarkers for risk-stratification7, 8.
However, this study does have limitations. Although development of the Cytosponge predictive algorithm was based on selection of the most promising miRNA candidates from independent sample sets by two profiling platforms, some miRNAs were exclusively assessed on one of the profiling platforms thus precluding their selection for the subsequent Cytosponge samples. In addition, the model performance may be somewhat over-optimistic given the lack of an external independent Cytosponge sample set. Given the complexity of the miRNA-mRNA network and its epigenetic regulation, further study is required to elucidate the precise role of these miRNAs in the pathogenesis of BE.
The correlation matrix of the top 15 miRNAs in the BE and NE dataset suggested that MIR192 and 194 may be co-regulated. This high correlation (r=0.78) was also evident in TCGA esophageal adenocarcinoma data (r=0.945). MIR192 and MIR194 are located within a single 300bp miRNA cluster at 11q13.1. Unlike cluster MIR215-194-1, the MIR192-194-2 cluster is not within an intron of a host gene, suggesting a less complex transcription regulation11. Some initial efforts were made to understand the cause of these upregulated miRNAs by examining genomic and epigenetic alterations and we identified hypo-methylation distribution in the promotor region of the cluster MIR192-194 that could explain the upregulation of clustered miRNAs.
We hypothesised that some or all of these miRNAs may be relevant in the pathogenesis of BE by repression of other genes. Using a combination of in silico and in vitro approaches to find and validate putative targets, we found that MIR194 could repress GRHL3 likely through its conserved binding site in GRHL3′s 3′UTR. GRHL3 positively regulates the tumour suppressor PTEN while GRHL3 knockout results in squamous cell carcinoma development in vivo associated with activation of PI3K signalling42. PTEN functions to negatively regulate signalling in the phosphatidylinositol-3-kinase (PI3K) pathway by dephosphorylating PIP3 to prevent activation of AKT and mTOR thus inhibiting cell survival and proliferation47. In line with evidence that BE is associated with increased proliferation48, we demonstrated that MIR194-mediated GRHL3 repression associated with reduced PTEN expression and increased proliferation in vitro. Previous studies revealed that the loss of PTEN expression is an independent negative prognostic factor in esophageal adenocarcinoma49.
In conclusion, we have identified a panel of upregulated miRNAs which can diagnose BE in biopsy and non-endoscopic Cytosponge samples. Hypo-methylation found in the promoter regions of these biomarker miRNAs could contribute to the dysregulation of miRNAs with phenotypic consequences through their target mRNA network. Further work is required to apply this miRNA strategy to a prospective Cytosponge trial in the primary care setting, and explore the feasibility of a high-throughput, automated platform which could potentially be combined with other nucleic acid biomarkers for risk stratification.
Supplementary Material
Acknowledgments
We thank the Human Research Tissue Bank, which is supported by the National Institute for Health Research (NIHR) Cambridge Biomedical Research Centre, from Addenbrooke’s Hospital, Cambridge.
Grant support: The BEST2 study was funded by Cancer Research UK (Grant ref; C14478/A12088, http://www.cancerresearchuk.org/). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The study received infrastructure support from the Cambridge Human Research Tissue Bank, which is supported by the National Institute for Health Research (NIHR) Cambridge Biomedical Research Centre, from Addenbrooke’s Hospital. R.C.F. is funded by an NIHR Professorship and receives core funding from the Medical Research Council and infrastructure support from the Biomedical Research Centre and the Experimental Cancer Medicine Centre. This study was also supported by the Intramural Research Program of the National Institutes of Health, Bethesda, MD, USA; the National Cancer Institute, USA; and the Division of Cancer Epidemiology and Genetics, USA.
Abbreviations
- miRNA
microRNA
- NE
normal esophagus
- BE
Barrett’s esophagus
Footnotes
Disclosures: Since this study was conducted the Cytosponge™-TFF3 technology has been licensed to Covidien GI solutions (now owned by Medtronic) by the Medical Research Council. Rebecca Fitzgerald and Pierre Lao-Sirieix are named inventors on patents pertaining to the Cytosponge™. Covidien Solutions and Medtronic have not been privy to this manuscript or the data therein. All other authors declare no conflicts of interest.
Author Contributions: X.L and S.K designed and performed experiments, analysed data and wrote the manuscript. S.B.C and M.B.C analysed data and R.M.P gave statistical support. J.P and S.J performed genomic data analysis. C.F, M.T, N.GD and S.Mac assisted in technical support and sample management. A.M contributed expertise in pathology for the study. L.O and H.H performed Nanostring nCounter assay. P.L helped conduct the microarray screening. P.R.T and R.M.P contributed and assisted in study design. R.C.F and M.B.C conceived and designed the project, provided funding, reviewed the data and wrote the manuscript.
References
- 1.Vaughan TL, Fitzgerald RC. Precision prevention of oesophageal adenocarcinoma. Nat Rev Gastroenterol Hepatol. 2015;12 doi: 10.1038/nrgastro.2015.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Corley DA, Levin TR, Habel LA, et al. Surveillance and survival in Barrett’s adenocarcinomas: a population-based study. Gastroenterology. 2002;122:633–640. doi: 10.1053/gast.2002.31879. [DOI] [PubMed] [Google Scholar]
- 3.Dulai GS, Guha S, Kahn KL, et al. Preoperative prevalence of Barrett’s esophagus in esophageal adenocarcinoma: A systematic review. Gastroenterology. 2002;122:26–33. doi: 10.1053/gast.2002.30297. [DOI] [PubMed] [Google Scholar]
- 4.Benaglia T, Sharples LD, Fitzgerald RC, et al. Health benefits and cost effectiveness of endoscopic and nonendoscopic cytosponge screening for Barrett’s esophagus. Gastroenterology. 2013;144:62–73.e6. doi: 10.1053/j.gastro.2012.09.060. [DOI] [PubMed] [Google Scholar]
- 5.Kadri SR, Lao-Sirieix P, O’Donovan M, et al. Acceptability and accuracy of a non-endoscopic screening test for Barrett’s oesophagus in primary care: cohort study. BMJ. 2010;341:c4372. doi: 10.1136/bmj.c4372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ross-Innes CS, Debiram-Beecham I, O’Donovan M, et al. Evaluation of a minimally invasive cell sampling device coupled with assessment of trefoil factor 3 expression for diagnosing Barrett’s esophagus: a multi-center case-control study. PLoS Med. 2015;12:e1001780. doi: 10.1371/journal.pmed.1001780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chettouh H, Mowforth O, Galeano-Dalmau N, et al. Methylation panel is a diagnostic biomarker for Barrett’s oesophagus in endoscopic biopsies and non-endoscopic cytology specimens. Gut. 2017 doi: 10.1136/gutjnl-2017-314026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weaver JMJ, Ross-Innes CS, Shannon N, et al. Ordering of mutations in preinvasive disease stages of esophageal carcinogenesis. Nat Genet. 2014;46:837–843. doi: 10.1038/ng.3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ryan BM, Robles AI, Harris CC. Genetic variation in microRNA networks: the implications for cancer research. Nat Rev Cancer. 2010;10:389–402. doi: 10.1038/nrc2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin S, Gregory RI. MicroRNA biogenesis pathways in cancer. Nat Rev Cancer. 2015;15:321–333. doi: 10.1038/nrc3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rodriguez A, Griffiths-Jones S, Ashurst JL, et al. Identification of mammalian microRNA host genes and transcription units. Genome Res. 2004;14:1902–10. doi: 10.1101/gr.2722704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zen K, Zhang C-Y. Circulating microRNAs: a novel class of biomarkers to diagnose and monitor human cancers. Med Res Rev. 2012;32:326–348. doi: 10.1002/med.20215. [DOI] [PubMed] [Google Scholar]
- 13.Lu J, Getz G, Miska EA, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–838. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- 14.de Biase D, Visani M, Morandi L, et al. miRNAs Expression Analysis in Paired Fresh/Frozen and Dissected Formalin Fixed and Paraffin Embedded Glioblastoma Using Real-Time PCR. PLoS One. 2012;7 doi: 10.1371/journal.pone.0035596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li J, Smyth P, Flavin R, et al. Comparison of miRNA expression patterns using total RNA extracted from matched samples of formalin-fixed paraffin-embedded (FFPE) cells and snap frozen cells. BMC Biotechnol. 2007;7:36. doi: 10.1186/1472-6750-7-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mallick R, Patnaik SK, Wani S, et al. A Systematic Review of Esophageal MicroRNA Markers for Diagnosis and Monitoring of Barrett’s Esophagus. Dig Dis Sci. 2016;61:1039–1050. doi: 10.1007/s10620-015-3959-3. [DOI] [PubMed] [Google Scholar]
- 17.Koshiol J, Wang E, Zhao Y, et al. Strengths and limitations of laboratory procedures for microRNA detection. Cancer Epidemiol Biomarkers Prev. 2010;19:907–11. doi: 10.1158/1055-9965.EPI-10-0071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Callari M, Dugo M, Musella V, et al. Comparison of microarray platforms for measuring differential microRNA expression in paired normal/cancer colon tissues. PLoS One. 2012;7:e45105. doi: 10.1371/journal.pone.0045105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chatterjee A, Leichter AL, Fan V, et al. A cross comparison of technologies for the detection of microRNAs in clinical FFPE samples of hepatoblastoma patients. Sci Rep. 2015;5:10438. doi: 10.1038/srep10438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kolbert CP, Feddersen RM, Rakhshan F, et al. Multi-platform analysis of microRNA expression measurements in RNA from fresh frozen and FFPE tissues. PLoS One. 2013;8:e52517. doi: 10.1371/journal.pone.0052517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aldridge S, Hadfield J. Introduction to miRNA profiling technologies and cross-platform comparison. Methods Mol Biol. 2012;822:19–31. doi: 10.1007/978-1-61779-427-8_2. [DOI] [PubMed] [Google Scholar]
- 22.Bonnal RJP, Rossi RL, Carpi D, et al. miRiadne: a web tool for consistent integration of miRNA nomenclature. Nucleic Acids Res. 2015;43:W487–92. doi: 10.1093/nar/gkv381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ritchie ME, Phipson B, Wu D, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miller JA, Cai C, Langfelder P, et al. Strategies for aggregating gene expression data: the collapseRows R function. BMC Bioinformatics. 2011;12:322. doi: 10.1186/1471-2105-12-322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Durinck S, Spellman PT, Birney E, et al. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat Protoc. 2009;4:1184–91. doi: 10.1038/nprot.2009.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hyland PL, Hu N, Rotunno M, et al. Global changes in gene expression of Barrett’s esophagus compared to normal squamous esophagus and gastric cardia tissues. PLoS One. 2014;9:e93219. doi: 10.1371/journal.pone.0093219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fassan M, Volinia S, Palatini J, et al. MicroRNA expression profiling in human Barrett’s carcinogenesis. International Journal of Cancer. 2011;129:1661–1670. doi: 10.1002/ijc.25823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Morales CP, Gandia KG, Ramirez RD, et al. Characterisation of telomerase immortalised normal human oesophageal squamous cells. Gut. 2003;52:327–333. doi: 10.1136/gut.52.3.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shaheen NJ, Richter JE. Barrett’s oesophagus. Lancet. 2009;373:850–861. doi: 10.1016/S0140-6736(09)60487-6. [DOI] [PubMed] [Google Scholar]
- 32.Pichiorri F, Suh SS, Rocci A, et al. Downregulation of p53-inducible microRNAs 192, 194, and 215 impairs the p53/MDM2 autoregulatory loop in multiple myeloma development. Cancer Cell. 2010;18:367–81. doi: 10.1016/j.ccr.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 33.Ventura A, Young AG, Winslow MM, et al. Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell. 2008;132:875–86. doi: 10.1016/j.cell.2008.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Secrier M, Li X, de Silva N, et al. Mutational signatures in esophageal adenocarcinoma define etiologically distinct subgroups with therapeutic relevance. Nat Genet. 2016 doi: 10.1038/ng.3659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ross-Innes CS, Becq J, Warren A, et al. Whole-genome sequencing provides new insights into the clonal architecture of Barrett’s esophagus and esophageal adenocarcinoma. Nat Genet. 2015;47:1038–1046. doi: 10.1038/ng.3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weaver JMJ, Ross-Innes CS, Shannon N, et al. Ordering of mutations in preinvasive disease stages of esophageal carcinogenesis. Nature Genetics. 2014;46:837–843. doi: 10.1038/ng.3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ehrlich M. DNA hypomethylation in cancer cells. Epigenomics. 2009;1:239–59. doi: 10.2217/epi.09.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 39.Georges SA, Biery MC, Kim S-Y, et al. Coordinated Regulation of Cell Cycle Transcripts by p53-Inducible microRNAs, miR-192 and miR-215. Cancer Res. 2008 doi: 10.1158/0008-5472.CAN-08-1846. [DOI] [PubMed] [Google Scholar]
- 40.di Pietro M, Lao-Sirieix P, Boyle S, et al. Evidence for a functional role of epigenetically regulated midcluster HOXB genes in the development of Barrett esophagus. Proceedings of the National Academy of Sciences. 2012;109:9077–9082. doi: 10.1073/pnas.1116933109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Agarwal V, Bell GW, Nam JW, et al. Predicting effective microRNA target sites in mammalian mRNAs. Elife. 2015;4 doi: 10.7554/eLife.05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Darido C, Georgy SR, Wilanowski T, et al. Targeting of the tumor suppressor GRHL3 by a miR-21-dependent proto-oncogenic network results in PTEN loss and tumorigenesis. Cancer Cell. 2011;20:635–648. doi: 10.1016/j.ccr.2011.10.014. [DOI] [PubMed] [Google Scholar]
- 43.Yamada KM, Araki M. Tumor suppressor PTEN: modulator of cell signaling, growth, migration and apoptosis. J Cell Sci. 2001;114:2375–2382. doi: 10.1242/jcs.114.13.2375. [DOI] [PubMed] [Google Scholar]
- 44.Bansal A, Lee IH, Hong X, et al. Discovery and validation of Barrett’s esophagus microRNA transcriptome by next generation sequencing. PLoS One. 2013;8:e54240. doi: 10.1371/journal.pone.0054240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mathe EA, Nguyen GH, Bowman ED, et al. MicroRNA expression in squamous cell carcinoma and adenocarcinoma of the esophagus: associations with survival. Clin Cancer Res. 2009;15:6192–200. doi: 10.1158/1078-0432.CCR-09-1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Revilla-Nuin B, Parrilla P, Lozano JJ, et al. Predictive value of MicroRNAs in the progression of barrett esophagus to adenocarcinoma in a long-term follow-up study. Ann Surg. 2013;257:886–93. doi: 10.1097/SLA.0b013e31826ddba6. [DOI] [PubMed] [Google Scholar]
- 47.Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol. 2012;13:283–96. doi: 10.1038/nrm3330. [DOI] [PubMed] [Google Scholar]
- 48.Çoban Ş, Örmeci N, Savaş B, et al. Evaluation of Barrett’s esophagus with CK7, CK20, p53, Ki67, and COX2 expressions using chromoendoscopical examination. Dis Esophagus. 2013;26:189–196. doi: 10.1111/j.1442-2050.2012.01352.x. [DOI] [PubMed] [Google Scholar]
- 49.Bettstetter M, Berezowska S, Keller G, et al. Epidermal growth factor receptor, phosphatidylinositol-3-kinase catalytic subunit/PTEN, and KRAS/NRAS/BRAF in primary resected esophageal adenocarcinomas: loss of PTEN is associated with worse clinical outcome. Hum Pathol. 2013;44:829–836. doi: 10.1016/j.humpath.2012.08.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.