

Abstract
Trichomonas vaginalis is a sexually transmitted, eukaryotic parasite that causes trichomoniasis, the most common non-viral sexually-transmitted disease in the U.S. and worldwide. Little is known about the molecular mechanisms involved in the host immune response to this widespread parasite. Here we report that T. vaginalis induces NLRP3 inflammasome activation in human macrophages, leading to caspase-1 activation and the processing of pro-IL-1β to the mature and bioactive form of the cytokine. Using inhibitor-based approaches, we show that NLRP3 activation by T. vaginalis involves host cell detection of extracellular ATP via P2X7 receptors and potassium efflux. In addition, our data reveal that T. vaginalis inflammasome activation induces macrophage inflammatory cell death by pyroptosis, known to occur via caspase-1 cleavage of the gasdermin D protein which assembles to form pores in the host cell membrane. We found that T. vaginalis-induced cytolysis of macrophages is attenuated in gasdermin D knockout cells. Lastly, in a murine challenge model, we detected IL-1β production in vaginal fluids in response to T. vaginalis infection in vivo. Together, our findings mechanistically dissect how T. vaginalis contributes to the production of the pro-inflammatory IL-1β cytokine and uncovers pyroptosis as a mechanism by which the parasite can trigger host macrophage cell death.
Keywords: Trichomonas vaginalis, inflammasomes, pyroptosis, cytokines, macrophages, parasite-host interactions
Introduction
Trichomonas vaginalis is a eukaryotic parasite that infects the urogenital tract of women and men. The parasite is transmitted through sexual intercourse, and after colonizing its human host causes the disease trichomoniasis, which is the most common non-viral sexually transmitted infection (STI) in the United States [1] and worldwide [2]. While T. vaginalis STI can be asymptomatic in many individuals [3], others experience inflammation of the vagina, cervix, or prostate [4]. The inflammatory response mounted against the parasite is predicted to fuel multiple adverse health effects associated with T. vaginalis infection. These include a higher incidence of premature births [5], increased risk of cervical [6, 7] or aggressive prostate cancer [8, 9], and increased risk of acquiring [10] and potentially transmitting the human immunodeficiency virus (HIV) to a sexual partner [11].
Despite the serious public health threat posed by T. vaginalis, limited knowledge exists regarding the molecular mechanisms by which T. vaginalis elicits inflammation. As an extracellular parasite, T. vaginalis survives in the human body via uptake of nutrients from host cells to which it can adhere and/or phagocytose and lyse, including vaginal or prostate epithelial cells [12], and red or white blood cells [13]. Currently, human clinical data regarding the specific innate immune response to T. vaginalis infections are scarce, and existing knowledge largely stems from human co-infection studies or from in vitro experiments. Cauci and Culhane reported a significant increase in vaginal IL-1β levels in pregnant women who had bacterial vaginosis and a T. vaginalis co-infection compared to pregnant women with only bacterial vaginosis [14]. Indeed, inflammatory profiling of human monocytes and monocyte-derived macrophages in response to T. vaginalis infection identified IL-1β as an upregulated cytokine [15–17]. However, the cellular mechanisms and requirements for production of this important immune response factor during macrophage and T. vaginalis encounters are unknown. Here we delineate the cellular pathway that is activated in macrophages by T. vaginalis leading to the maturation and release of IL-1β.
Inflammasomes are multimeric cytosolic complexes that sense the presence of pathogens or changes in cellular homeostasis and activate commensurate innate immune responses. The inflammasome complex is defined by the responding sensor protein, which has characteristic structural domains and includes the nucleotide-binding oligomerization domain-like receptors (NLRs) NLRP1, NLRP3, NLRC4, as well as the AIM2 and Pyrin proteins [18]. Inflammasome assembly is initiated by NLR recognition of pathogen-associated molecular patterns (PAMPs) or endogenous danger-associated molecular patterns (DAMPs) [18]. Upon detecting these stimuli, the sensor proteins oligomerize and associate with the adaptor protein ASC in a multi-protein complex that recruits pro-caspase-1 and promotes its autoproteolytic processing [19, 20]. Mature protease caspase-1 then cleaves the pro-forms of inflammatory cytokines IL-1β and IL-18 to generate the mature bioactive forms capable of binding their cognate receptors and initiating cell signaling [21–27], Two major outcomes of inflammasome activation are processing and release of mature IL-1β and induction of a rapid cell death termed pyroptosis [28, 29].
In this study, we show that T. vaginalis activates NLRP3 inflammasomes in human macrophages leading to bioactive IL-1β production and pyroptotic cell death. The parasite-driven NLRP3 inflammasome activation involves sensing of ATP as a danger associated molecular pattern (DAMP) and potassium efflux, and IL-1β release in response to T. vaginalis can be detected in vivo in a mouse vaginal challenge study. Thus T. vaginalis activation of NLRP3 inflammasomes leads to a strong macrophage proinflammatory response against the parasite, consistent with recent findings in prostate epithelial cells [30], but also leads to elimination of the macrophages in the process. The balance of innate immune detection and inflammatory response is likely to influence T. vaginalis colonization and pathogenesis differentially depending on the magnitude and stage of infection.
Materials and Methods
Growth of cells and culture
T. vaginalis strains RU393 (New York, USA, ATCC 50142, Ref. [31]) and MSA1132 (Mt. Dora, FL, USA, Ref. [12]), were both obtained from Patricia Johnson’s Laboratory at UCLA. Parasites were treated with 50 μg/ml chloramphenicol and 5 μg/ml tetracycline for 6 days and then frozen down as stocks. Parasites were thawed from these stocks and passaged daily for less than two weeks as previously described [32], in the continued presence of chloramphenicol and tetracycline. Human THP-1 monocytes (ATCC TIB-202) were cultured in RPMI medium supplemented with 10% fetal bovine serum (FBS), 0.05 mM 2-mercaptoethanol, 0.2% D-glucose, 10 mM HEPES, and 1 mM sodium pyruvate. THP-1 monocytes were differentiated to macrophages by treating cells with 25 nM PMA (Fisher) for 24 h.
THP-1 Cas9 cell line
A Cas9 stable cell line was generated by transducing THP-1 cells with Lenti-Cas9-Blast lentivirus (Addgene 52962-LV). Spinfection was performed by spinning down THP-1 cells with viral particles supplemented with 10 μg/ml polybrene at 1,000 × g, 32°C, for 1 h. After 48 h, cells were selected for stable expression of Streptococcus pyogenes Cas9 using blasticidin (10 μg/ml). Ten days days after selection, cells were harvested and Cas9 expression confirmed by immunoblotting and qPCR.
Cloning of GSDMD sgRNAs
Two sgRNA sequences previously shown to successfully target the Gasdermin D gene were cloned into the lentiGuide-Puro vector (Addgene, Plasmid# 52963) as described previously [33]. The sgRNA sequences were as follows: GSDMD KO1 5’-TGAGTGTGGACCCTAACACC-3’ (from Ref [34]) and GSDMD KO2 5’-CTTGCTTTAGACGTGCAGCG-3’ (from Ref [33], GeCKOv2 Library, identifier # HGLibA_20413).
GSDMD knockout cell lines
Lentivirus was packaged in HEK 293T cells (ATCC CRL-3216) using the Fugene HD transfection reagent (Promega) and 4 μg of the Gasdermin D sgRNA-lentiGuide-Puro construct, 4 μg of the packaging plasmid psPAX2 (Addgene, Plasmid# 12260), and 4 μg of the VSV-G envelope expressing plasmid pMD2-G (Addgene, Plasmid# 12259). THP-1 Cas9 cells (described above) were spinfected with 10 μg/ml polybrene and viral particles for 1 h at 1,000 × g, 32°C. After 48 h, cells were selected for stable expression of the sgRNAs using puromycin (1 μg/ml). After 10 days of selection, cells were cloned by limiting dilution. Two clonal cell lines (KO1 and KO2), one from each sgRNA, were harvested and Gasdermin D knockout confirmed by immunoblotting assays and PCR analysis.
T. vaginalis co-culture experiments:
1 × 105 THP-1 cells were seeded and differentiated in 96-well plates. T. vaginalis RU393 parasites were spun down at 2,061 × g, washed once, then resuspended in RPMI+2% heat-inactivated FBS. Serial dilutions were performed to attain the desired T. vaginalis to macrophage ratios. Media was aspirated from the THP-1 macrophages and 100 μl of T. vaginalis cell suspensions added to each well. For inhibitor or chemical treatment experiments, parasites were pre-mixed with the vehicle or test compound then added to the macrophages.
Cytokine and Caspase-1 protein analysis
IL 1-β cytokine levels and caspase-1 protein levels present in cell culture supernatants were quantified using the DuoSet IL-1β ELISA kit and the Quantikine Human Caspase-1/ICE ELISA Kit (both from R&D Systems).
IL-1β Signaling Assay
HEK-Blue™ IL-1β Cells (InvivoGen) were purchased and grown per the company’s specifications. 50,000 IL-1β Sensor Cells were seeded per well of a 96-well plate. 50 μl of cell supernatant from each experimental sample or a recombinant human IL-1β (InvivoGen) dilution was added to the IL-1β Sensor Cells and incubated at 37 °C, 5% CO2, for 18 h. Secreted alkaline phosphatase activity was assessed by adding 50 μl of the HEK-Blue IL-1β reporter cell supernatants onto 150 μl of QUANTI-Blue™ reagent (InvivoGen) and measuring the optical density at 620 nm using an EnSpire Plate Reader (PerkinElmer). Background levels from THP-1 only or THP-1 cells treated with vehicle were substracted from all values to report IL-1β signalling levels above background.
Caspase-1 Activity Assay
Caspase-1 activity was assessed using the Caspase-1/ICE Colorimetric Assay Kit (Biovision). 2 × 106 THP-1 macrophages were seeded and differentiated in 6-well plates. T. vaginalis RU393 parasites were spun down at 2,061 × g, washed once, and resuspended in RPMI+ 2% heat-inactivated FBS; 1.5 ml of parasite suspensions were added to each well. After a 2.5 h co-incubation, the cell supernatants were removed and cells lysed with cell lysis buffer. Caspase-1 activity was assessed per manufacturer’s specifications in whole cell lysates normalized by equal protein amounts.
Reagents and inhibitors
Escherichia coli LPS, ATP, KCI, NaCl, and Adenosine 5’-triphosphate, periodate oxidized sodium salt (oxATP) were purchased from SIGMA. The Ac-YVAD-CMK caspase-1 peptide inhibitor was purchased from Enzo, and the NLRP3/AIM2 inhibitor CRID3 was purchased from Tocris.
Filter Experiment Assays
Co-incubation experiments were performed in the presence of a Millicell® 96 Cell Culture Insert Plate PCF with pore size of 0.4 μm (Millipore). For contact conditions T. vaginalis cell suspensions were placed under the filter to allow direct interaction between T. vaginalis and THP-1 cells. For filter conditions that prevented physical contact, T. vaginalis was placed on top of the filter.
LDH Cytolysis Assays:
Cytotolysis was assessed by assaying for LDH release from cells using the CytoTox-ONE™ Homogeneous Membrane Integrity Assay Kit (Promega). Manufacturer’s specifications were used to perform the assays and calculate the percent cytolysis compared to 100% lysis controls. For cytolysis analysis of Gasdermin D knockout cell lines, cell lysis was compared to THP-1 Cas9 parental cells (THP-1 wildtype).
Immunoblot Analysis
2 × 106 THP-1 macrophages were seeded and differentiated per well of a 6-well plate. After co-incubation with T. vaginalis, cells were washed two times with PBS and lysed with RIPA Buffer (Thermo Scientific) containing HALT™ protease inhibitor cocktail. Samples were separated by SDS-PAGE, immunoblotted, and visualized with SuperSignal® WestPico Chemiluminescent Substrate (Thermo Scientific) and CL-XPosure™ Film (Thermo Scientific). The following antibodies were used for immunoblotting: anti-IL-1β (AF-201-NA, Cell Signaling), anti β-actin (A5316, SIGMA), anti-Gasdermin D (20770-1-AP, Proteintech), anti-goat IgG HRP-conjugated (HAF017, R&D), ECL Amersham anti-mouse IgG HRP-Linked (NA931, GE Healthcare).
Animal Experiments
Six-week-old female C57BI/6 mice were purchased from The Jackson Laboratory. Mice were pretreated by daily intraperitoneal (i.p.) injections with 100 μl of 2 mg/ml dexamethasone sodium phosphate Injection, USP (WEST-WARD Pharmaceuticals) for 4 d prior to infection. The day before infection mice were also injected i.p. with 100 μl of 5 mg/mL β-estradiol resuspended in sesame oil (both from SIGMA). For animal infections, T. vaginalis MSA1132 strain was washed twice and resuspended in sterile Dulbecco’s phosphate buffered saline (DPBS, Corning). 6×106 T. vaginalis in a final volume of 10-15 μl were used to infect mice vaginally. Three days after infection, vaginal lavages were collected by washing the vaginal lumen four times with 50 μl of DPBS (200 μl total) using Gel-Loading pipet tips (Fisher Scientific).
Statistical analysis
For in vitro experiments, statistical significance was calculated using a Student’s t-test. For IL-1β detection in animal studies, a Grubb’s test was performed to test for the presence of experimental outliers. One cytokine value from the uninfected mice group (777.768 pg/ml) and one value from the T. vaginalis-infected group (266.183 pg/ml) were identified as outliers. A Mann–Whitney test was used to test for statistical significance across the remaining samples. p<0.05 was considered as statistically significant.
Results
T. vaginalis stimulates bioactive IL-1β secretion in a caspase-1-dependent manner
The NLRP3 inflammasome is highly expressed and best characterized in cells of the myeloid cell lineage including macrophages and dendritic cells [35, 36]. As macrophages are one of the most abundant immune effector cells in the female reproductive tract [37], we investigated whether T. vaginalis activates NLRP3 inflammasomes in human macrophages. For our studies, we used the human THP-1 monocyte-like cell line, which are differentiated to macrophages using phorbol 12-myristate acetate (PMA) treatment. NLRP3 inflammasome activation involves two signals. Signal 1, known as the priming step, occurs after receptor engagement and signal transduction leads to NF-κB activation, which in turn stimulates increased transcription of the genes encoding pro-IL-1β and NLRP3 [38–40]. Signal 2, the activation step, is initiated by diverse microbial PAMPs and host cell DAMPs which lead to the physical assembly of the NLPR3 inflammasome complex. Examples of NLRP3 Signal 2 activators include bacterial surface proteins, microbial pore-forming toxins, and viral RNA, as well as host-cell DAMPs such as extracellular ATP, amyloid-β, and monosodium urate crystals [41, 42].
We found a dose-dependent increase in IL-1β secretion from THP-1 macrophages at increasing multiplicities of infection (MOI) of T. vaginalis (Fig.1A); LPS and ATP, two well described NLRP3 inflammasome inducers that provide Signal 1 and Signal 2, respectively [43], served as positive controls. While IL-1β protein has been detected by ELISA in the monocyte/macrophage cytokine response to T. vaginalis exposure [15–17], it was unclear if this represented the pro-form of IL-1β released secondary to parasite-induced cell lysis or whether it was the processed and bioactive form of the inflammatory cytokine. To test the bioactivity of IL-1β generated upon THP-1 macrophage exposure to T. vaginalis, we added cell supernatants from the co-culture experiments to HEK-Blue IL-1β sensor cells (InvivoGen). These reporter cells express the IL-1 receptor (IL-1R) to initiate cell signaling and trigger production of a SEAP reporter enzyme that can convert a colorimetric substrate to provide a readout for IL-1β signaling. With recombinant bioactive IL-1β serving as a positive control for IL-1β signalling activity, Fig. 1B shows T. vaginalis co-incubation with THP-1 macrophages led to processing of pro-IL-1β and generation of bioactive IL-1β capable of IL-1 R-mediated cell signaling. LPS and ATP served as a positive control for NLRP3 inflamamsome activation in the THP-1 cells. Immunoblot analysis in Fig. 1C confirms detection of the 17 kDa bioactive IL-1β cleavage product in whole cell lysates from THP-1 macrophages exposed to T. vaginalis, which was not detected in the uninfected control. Conversely, the full length pro-IL-1β protein which has a molecular weight of 31 kDa but migrates with an apparent molecular weight ~36 kDa, was detected in both uninfected THP-1 macrophages and macrophages exposed to T. vaginalis. Therefore, macrophage co-incubation with T. vaginalis leads to active production of processed and bioactive IL-1β.
Fig. 1: THP-1 exposure to T. vaginalis leads to bioactive IL-1β production via caspase-1 activity.
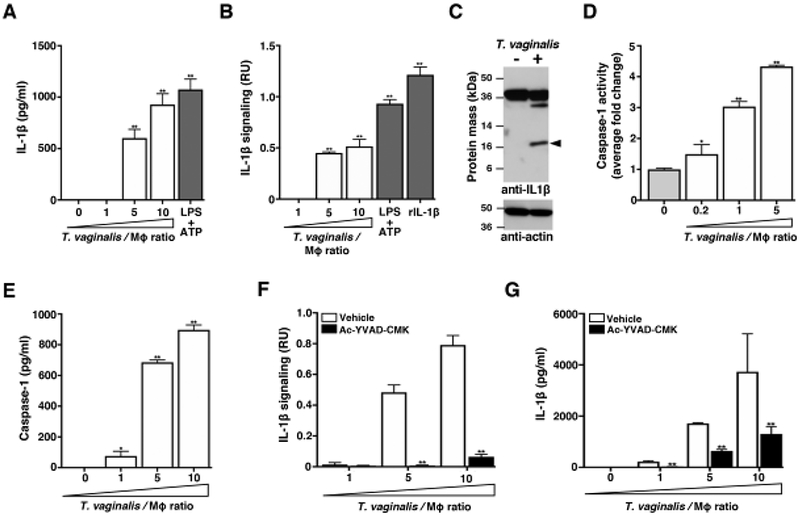
THP-1 macrophages were incubated with T. vaginalis using different multiplicities of infection (MOIs, T. vaginalis: host cell) for 4 h unless otherwise stated. As a positive control for inflammasome activation, THP-1 cells were stimulated with 10 ng/ml LPS+ 5 mM ATP. To test for the presence of processed IL-1β, experiment supernatants were placed on HEK-Blue IL-1β reporter cells. Bioactive IL-1β binds to the IL-1 receptor on IL-1β reporter cells, activating production of a reporter enzyme. Reporter enzyme activity was quantified spectrophotometrically and is proportional to IL-1β signalling activity shown as relative units (RU) after subtracting background readings from supernatants of THP-1 cells alone or THP-1 cells treated with vehicle control. As a positive control, recombinant bioactive IL-1β (rIL-1β) was added to the HEK-Blue IL-1β reporter cells. (A) IL-1β in cell supernatants was measured by ELISA. (B) Bioactive IL-1β was detected using the HEK-Blue IL-1β reporter cells. (C) Immunoblot detection of the IL-1β cleavage product (17 kDa, arrow) in whole cell lysates of THP-1 macrophages exposed to T. vaginalis for 4 h at an MOI of 1. Full length pro-IL-1β (31 kDa) could be detected in both samples. Actin loading control and molecular weight markers in kDa are also shown. (D) THP-1 macrophages were exposed to T. vaginalis for 2.5 h. The amount of caspase-1 activity was assayed by measuring cleavage of the p-nitroanilide-labeled YVAD peptide (YVAD-pNA) spectrophotometrically. The fold-change relative to THP-1 cells alone is shown. (E) Amounts of caspase-1 protein released into cell supernatants was quantified by ELISA. (F-G) THP-1 cells were co-incubated with T. vaginalis in the presence of the caspase-1 peptide inhibitor Ac-YCAD-CMK or vehicle control. Levels of bioactive IL-1β were detected using the IL-1β reporter cells (F) and IL-1β protein levels detected by ELISA (G). For each experiment, samples were assayed at least in triplicate. Representative results from two (D, E) or three (A-C and F-G) independent experiments is shown. Graphs show the mean with standard deviations, *p-value<0.05, **p-value<0.01 compared to THP-1 cells (A, B, D, E) or vehicle treatment at that multiplicity of infection (F and G).
Canonical inflammasome assembly and activation leads to the autoproteolytic processing of the proenzyme form of caspase-1 (45 kDa) to its active form composed of a heterodimeric tetramer of the p20 and p10 subunits [44]. To quantify caspase-1 activity, we assayed cleavage of the chromogenic peptide YVAD-pNA. After 2.5 h co-incubation with T. vaginalis, we detected a ~4-fold increase in caspase-1 activity from THP1 cells at MOI = 5 and a ~3-fold-increase at MOI = 1 (Fig. 1D). Caspase-1 can be released into cell supernatants after inflammasome activation [45], and we found a dose-dependent release of high amounts of caspase-1 in THP-1 supernatants following exposure to increasing MOIs of T. vaginalis (Fig. 1E). Treatment with the specific Ac-YVAD-CMK peptide inhibitor of caspase-1 markedly ablated bioactive IL-1β production (Fig. 1F), a result corroborated when total IL-1β was quantified by ELISA (Fig. 1G). Together, these results indicate that upon exposure to T. vaginalis, caspase-1 activity is increased in THP-1 cells and leads to the processing of pro-IL-1β to bioactive IL-1β.
T. vaginalis activates NLRP3 Inflammasomes
Multiple innate immune sensor proteins can be assembled to form different types of inflammasome complexes [18]. Many inflammasomes are activated by bacterial components such as LPS or flagellin [46] that are not found in the eukaryotic T. vaginalis parasite. NLRP3 inflammasomes were strong molecular candidates for the observed generation of bioactive IL-1β, since they can also be activated in response to host cell perturbations and by sensing DAMPs potentially released during cellular injury inflicted by T. vaginalis. To investigate the role of NLRP3 as the sensor protein driving IL-1β processing, we co-incubated T. vaginalis with THP-1 cells in the presence or absence of CRID3, which inhibits ASC oligomerization to block both NLRP3 and AIM2 inflammasomes [47]. CRID3 treatment completely ablated bioactive IL-1β signaling from THP-1 macrophages exposed to T. vaginalis at MOI = 5 or 10 (Fig. 2A); similar reductions in total IL-1β detected by ELISA were observed at each MOI (Fig. 2B). A variety of ligands or cell perturbations can serve as upstream activators of NLRP3 inflammasome activation; however, the precise molecular mechanism shared by these various pathways to result in NLRP3 inflammasome activation is still a subject of active investigation. K+ efflux has been identified as a common signaling event in many cases of NLRP3 inflammasome activation [41, 48, 49]. Therefore, we compared the levels of IL-1β signaling in THP-1 cells co-incubated with T. vaginalis in the presence or absence of added extracellular KCI to counteract the effects of K+ efflux [49] and observed a dose-dependent inhibition on IL-1β signaling (Fig. 2C) and total IL-1β protein levels (Fig. 2D). As a control for addition of extracellular ions, exogenous addition of NaCI only slightly modified IL-1β signaling (Fig. 2C) and had no effect on IL-1β protein levels (Fig. 2D). Together these results indicated that T. vaginalis-induction of IL-1β secretion in THP-1 macrophages is mediated through NLRP3 inflammasome activation in a scenario where K+efflux is required.
Fig. 2: T. vaginalis activates NLRP3 Inflammasomes in THP-1 macrophages.
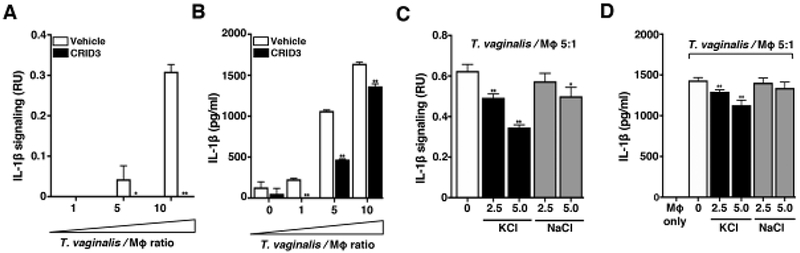
THP-1 macrophages were exposed to T. vaginalis at an MOI of 5 (T. vaginalis: host) for 4 h in the presence of (A-B) CRID3 an NLRP3/AIM2 inflammasome inhibitor or (C-D) exogenous KCl and NaCl. Levels of bioactive IL-1β were detected using the IL-1β reporter cells (A and C) and total IL-1β protein levels were detected by ELISA (B and D). A representative result from 3 independent experiments is shown. Graphs show the mean with standard deviations, *p-value<0.05, **p-value<0.01 compared to THP-1 cells co-incubated with T. vaginalis vehicle treatment.
ATP-P2X7 receptor signaling and macrophage contact contributes to T. vaginalis-induced inflammasome activation
Extracellular ATP serves as a DAMP by binding to cell surface P2X7 receptors that provoke K+ efflux from the cell and subsequent NLRP3 inflammasome activation [50–55]. To test whether ATP plays a role in inflammasome activation by T. vaginalis, we coincubated the parasites with THP-1 macrophages in the presence of oxidized ATP (oxATP), which covalently and irreversibly binds to P2 family receptors (P2XR) inhibiting their ATP-mediated ion transport and ATP-dependent IL-1β release [54, 56], OxATP treatment led to a dose-dependent reduction in IL-1β signaling and IL-1β protein levels in THP-1 macrophages exposed to T. vaginalis (Fig. 3A and 3B). To further probe P2X7 receptor involvement, we used Glyburide (also known as Glybenclamide), which inhibits P2X7 receptors and is also a broad-spectrum inhibitor of ATP-binding cassette transporters. As a result of blocking K+efflux, Glyburide inhibits NLRP3 inflammasomes [57]. Glyburide treatment completely ablated the production of bioactive IL-1β in T. vaginalis-infected THP-1 macrophages (Fig. 3C), and more than halved the amount of IL-1β protein detected by ELISA (Fig. 3D). Together, these results indicate that exposure of macrophages to T. vaginalis leads to inflammasome activation via P2X7 receptor-mediated K+ efflux in response to extracellular ATP.
Fig. 3: Response to extracellular ATP via P2X7 receptor signalling and physical contact contributes to T. vaginalis inflammasome activation.
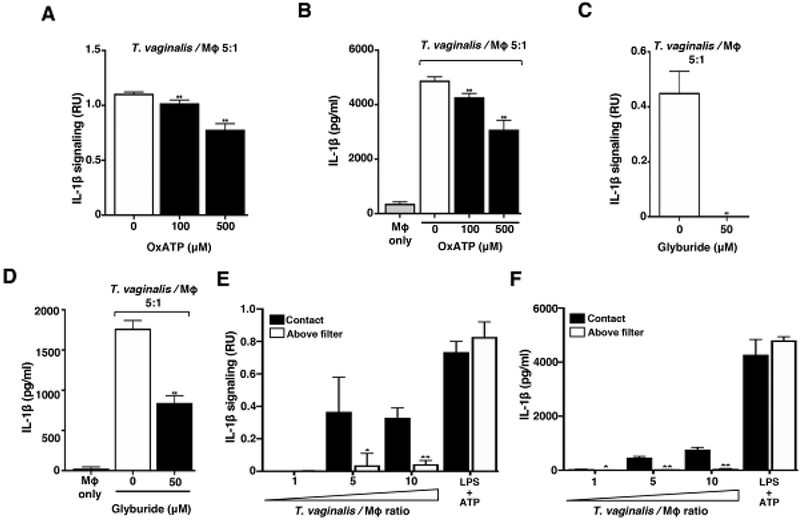
THP-1 macrophages were exposed to T. vaginalis at an MOI of 5 (T. vaginalis: host) for 4 h in the presence of (A-B) oxidized ATP (OxATP) or (C-D) Glyburide, an ATP-gated P2X7 receptor inhibitor. (E-F) A 0.4 μm membrane filter that allowed soluble materials to flow through but prevented contact was placed in between THP-1 macrophages and T. vaginalis in the above filter condition, or parasites were placed below the filter to allow contact between T. vaginalis and macrophages (contact). THP-1 cells were treated with 10 ng/ml LPS+ 5 mM ATP as a positive control for NLRP3 activation. Levels of bioactive IL-1β were detected using the IL-1β reporter cells (A, C, and E) and total IL-1β protein levels were detected by ELISA (B, D, and F). A representative result from 3 independent experiments is shown in A-D, and two independent experiments in E and F. Graphs show the mean with standard deviations. (A-D) **p-value<0.01 compared to THP-1 cells exposed to T. vaginalis in the presence of vehicle control. (E-F) *p-value<0.05, **p-value<0.01 compared to contact with T. vaginalis condition at that MOI.
To determine if direct contact between the parasite and the THP-1 cells was necessary to induce inflammasome activation, we placed a filter in between the macrophages and T. vaginalis, which allows soluble materials to pass through, but prohibits direct contact between the parasites and the THP-1 macrophages. In conditions where T. vaginalis was placed above the filter (no direct contact), the secretion and processing of IL-1β were markedly reduced (Fig. 3E and 3F). Thus, physical contact of the host macrophages with the parasites is critical for sensing, either an unknown PAMP on T. vaginalis or, more likely, a DAMP (e.g. ATP) arising from the host cell generated upon cell lysis that serves as a signal for inflammasome activation.
T. vaginalis inflammasome activation leads to THP-1 macrophage cell death by pyroptosis
In addition to IL-1β secretion, NLRP3 inflammasome activation can lead to an inflammatory cell death called pyroptosis. Cell lysis occurs after the mature caspase-1 protease cleaves the gasdermin D protein [28] which then oligomerizes [58] and forms pores in the plasma membrane [29] prompting cell swelling and membrane rupture. Pyroptosis is recognized as a distinct form of cell death in large part due to its fast nature (compared to apoptosis) and its accompanying “fiery” inflammatory response [59]. Pyroptosis is commonly detected by monitoring downstream membrane damage that leads to release of intracellular proteins like lactate dehydrogenase (LDH). Of note, LDH assays have been the standard assay used by many investigators to monitor T. vaginalis lysis of vaginal epithelial cells and prostate epithelial cells [12]. To test how T. vaginalis- induced inflammasome activation may influence macrophage pyroptosis, we examined the effect of inflammasome inhibitors on macrophage LDH release following infection with the parasite. Co-incubation of cells in the presence the caspase-1 inhibitor, Ac-YVAD-CMK, led to a significant reduction in host cell lysis at each MOI (Fig. 4A). Similar reductions in THP-1 macrophage lysis by T. vaginalis were also obtained with the NLRP3/AIM2 inflammasome inhibitor CRID3 (Fig. 4B). A complete reduction in cell death is not unexpected, as inhibition of pyroptosis can activate apoptosis [60], and caspase-1 peptide inhibitors may yield incomplete inhibition of LDH release after inflammasome activation [61, 62]. Therfore, to further investigate the induction of pyroptosis by T. vaginalis at a molecular level, we generated two independent knockouts of gasdermin D in THP-1 macrophages using CRISPR-Cas9 as confirmed by western blot (Fig. 4C) and PCR evidence of genome editing. When the gasdermin D knockout macrophages were exposed to T. vaginalis, we observed a 40%-60% reduction in LDH release (cell death) compared to WT THP-1 cells (Fig. 4D). These results reveal that T. vaginalis can kill host macrophages through pyroptosis.
Fig. 4: Inflammasome activation by T. vaginalis contributes to pyroptosis.
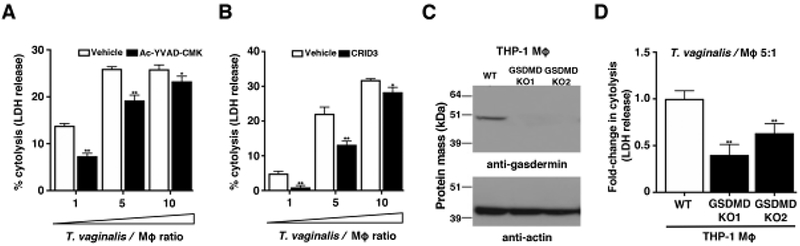
Cytolysis of host cells was assessed by measuring lactate dehydrogenase release from dying macrophages. (A-B) THP-1 macrophages were incubated with T. vaginalis using different multiplicities of infection (T. vaginalis: host cell) for 4 h in the presence of vehicle or (A) the caspase-1 peptide inhibitor Ac-YVAD-CMK or (B) the NLRP3/AIM2 inhibitor CRID3. Data are representative from 3 experiments. Graphs show the mean with standard deviations, *p-value<0.05, **p-value<0.01 compared to vehicle treatment at that multiplicity of infection. (C-D) Gasdermin D (GSDMD) was knocked out in THP-1 cells using CRISPR-Cas9. (C) Confirmation of gasdermin D knockout in two clones by western blot analysis using an anti-gasdermin D antibody. Actin loading control and molecular weight markers in kDa are also shown. (D) GSDMD KO cells or wildtype THP-1 cells were exposed to T. vaginalis at an MOI of 5 for 4 h. The average fold change in percent cytolysis compared to THP-1 wildtype cells from three independent experiments with standard errors of the mean are shown, **p-value<0.01 compared to THP-1 wildtype cells.
As a pilot study to determine if activation of IL-1β release is observed in vivo, we modified a previously established mouse model of T. vaginalis infection [63]. Unfortunately, T. vaginalis mouse models have required estrogen treatment together with the synthetic glucocorticoid and immunosuppressant dexamethasone [64] prior to and after T. vaginalis infection in order to facilitate colonization by the parasite [63]. To investigate inflammasome activation by T. vaginalis under minimal immune suppression conditions, we inoculated mice with one of the most virulent clinical strains available, called MSA1132 [12], using an optimized protocol illustrated schematically in Fig. 5A. T. vaginalis strain MSA1132 also yielded bioactive IL-1β production in our in vitro studies (data not shown). Three days after parasite or mock (PBS) infection, we collected vaginal lavages and examined IL-1β protein levels by ELISA. T. vaginalis infection led to a statistically significant increase in IL-1β secretion in vivo (Fig. 5B), consistent with our in vitro results.
Fig. 5: T. vaginalis infection leads to IL-1β production in a mouse model of infection.
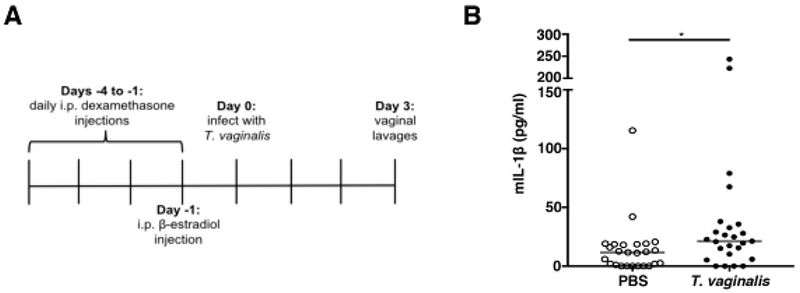
(A) Timeline shows the pre-treatment strategy of female C57BI/6 mice prior to intravaginal T. vaginalis inoculation or PBS mock infection. (B) Concentrations of IL-1β protein levels in vaginal lavages collected 3 days post-infection quantified by ELISA. Data shown is from 3 independent experiments combined (n=25 mice per treatment group). Median and range are shown. *p<0.05 compared to PBS uninfected control.
Discussion
In this work we show that upon contact with THP-1 human macrophages, T. vaginalis can activate NLRP3 inflammasomes, lead to release of bioactive IL-1β, and induce pyroptotic cell death. We thus infer that direct contact of T. vaginalis with macrophages is necessary to deliver both signals required for NLRP3 inflammasome activation. Inflammasome activation by T. vaginalis is mediated in part by sensing extracellular ATP via P2X7 receptors which lead to K+ efflux. Mildej and Benchimol [65] have shown that T. vaginalis inflicts membrane damage upon host cell contact, a likely requisite for the release of ATP and other DAMPs that can serve as Signal 2 for inflammasome activation. 100 μM oxidized ATP can inhibit the ability of 5 mM ATP to stimulate macrophage IL-1β production [66]. Similarly, we observed inhibition of IL-1β signaling with 100 μM oxATP, but higher doses of oxATP yielded still greater inhibition, so it is likely that significant ATP quantities are generated locally by T. vaginalis-induced cell damage allowing macrophage P2X7 receptor activation. We hypothesize that the general mechanism of host cell injury by T. vaginalis and subsequent release of DAMPs may underpin our preliminary observation of T. vaginalis-induced IL-1β secretion in our murine short-term challenge model.
Several groups have reported T. vaginalis activation of NF-κB in vaginal epithelial cells, human monocyte-derived macrophages, murine macrophages, and THP-1 macrophages [16, 17, 67, 68], establishing a precedence for the parasite’s ability to provide a Signal 1 required for NLRP3 inflammasome priming. To date, the only T. vaginalis cell surface component shown to contribute to NF-κB activation is the parasite’s glycocalyx [69], now known as Tv lipoglycan [70]. Toll-like receptor (TLR) 2 contributes to sensing of T. vaginalis by murine macrophages leading to NF-κB signalling [68]. It remains to be mechanistically investigated what other TLRs contribute to sensing T. vaginalis. TLR4 invovement has been implicated by indirect evidence, as cervico-vaginal lavages from T. vaginalis-infected women stimulated TNF-α production from TLR4 responsive murine splenocytes at higher levels than splenocytes nonresponsive to TLR4 ligands [71]. In HeLa cells, TLR2, TLR4, and TLR9 expression is increased upon infection with T. vaginalis [72] and TLR4 upregulation has also been reported in a prostate stromal cell line exposed to the parasite [73]. Further dissection of how T. vaginalis contact with human macrophages leads to generation and delivery of both Signals 1 and 2 is a subject of ongoing investigation in our laboratory.
Our work has also demonstrated that T. vaginalis activation of the NLRP3 inflammasome in macrophages contributes to their physical lysis via pyroptosis, the first direct demonstration that the parasite can kill host cells through this inflammatory cell death pathway. Gu et al. found that T. vaginalis can activate NLRP3 inflammasomes in a prostate epithelial cell line [30], but measures of cell viability, pyroptosis or gasdermin D involvement were not reported. Nevertheless, our results and that of Gu et al. add T. vaginalis to the growing number of sexually-transmitted pathogens that lead to inflammasome activation [30, 74, 75]. Studying the outcomes of inflammasome activation by STIs is of clinical importance, as the pro-inflammatory microenvironment in the reproductive tract may affect susceptibility to other STIs or facilitate co-infections [74, 75], and T. vaginalis is already associated with gonorrhea, chlamydia, syphilis, and herpes simplex virus-Types 1 and 2, and increased risk and transmission of HIV [10, 11, 76]. While our work was performed with THP-1 cells, an established cell line previously used for the study of vaginal colonizing bacteria [77, 78], future experimentation with our murine model and human primary vaginal macrophages is warranted to fully evaluate how T. vaginalis activates inflammasomes in the vaginal mucosal environment.
Bioactive IL-1β can exert effects on virtually all cell types and promote a multitude of functional outcomes in innate immunity and the shaping of adaptive immunity [79, 80]. These functions include inducing fever, promoting increased leukocyte recruitment to the infection site, and enhancing effector functions and cell survival [79]. Increased neutrophil numbers are reported in vaginal lavages of T. vaginalis-infected women [14, 81], but it remains unknown whether in human infections the increased leukocyte recruitment contributes to controlling the infection or to inducing pathology. An example of the latter adverse effects occurs with the extracellular fungus C. albicans, in which NLRP3 inflammasome activation contributes to pathogenesis by promoting increased neutrophil influx and vaginitis in a murine model of infection [82, 83].
The physical lysis of host macrophages through pyroptosis, likely mitigates the magnitude of the host innate immune response to T. vaginalis. For example, although we detected increased IL-1β production in our murine infection model, we did not observe any gross visible phenotypes. Similarly, a majority of infected individuals are also asymptomatic [3]. This dichotomy highlights complexity in the biology of T. vaginalis infections, in which inflammasome activation occurs in response to the parasite; however, the lysis of IL-1β producing cells by the parasite may affect the amounts and effects of the bioactive IL-1β produced. The parasite is also highly motile with 5 flagella [84, 85]. This property likely contributes to how long the parasite remains in contact with host cells to allow for parasite sensing, how effective released host effector molecules are at targeting the parasite, and the effect of recruited leukocytes, if the parasite can physically swim away. Other parasite virulence factors also help dampen the local immune response and potentially the effects of inflammasome activation on adaptive immunity. For example, T. vaginalis proteases can degrade complement components and antibodies [86], and the parasite can also lyse B cells and T cells [15]. T. vaginalis also expresses ectoATPases which can degrade extracellular ATP [87], which we identified to serve as a DAMP for NLRP3 inflamamsome activation. Future work testing the effect of parasite mutants and uncovering such parasite virulence factors will allow us to broaden our understanding of the overall innate immune response, its impact on T. vaginalis survival in the host, and the observation that T. vaginalis frequently causes persistent and recurrent infections [88].
It is well recognized that the immune response to T. vaginalis is complex and that the outcomes of infection vary [3, 89]. The potency of inflammasome activation and differential macrophage lysis through pyroptosis by different strains may contribute to the observed differences in host responses. Our work has identified ATP sensing and K+ efflux as activators of NLRP3 inflammasomes in macrophages responding to T. vaginalis infection. These general processes are likely to be elicited in other cell types such as the vaginal and cervical cells first encountered by the parasite and immune cells recruited to the infection site. Our work has set the foundation to further investigate how T. vaginalis activates inflammasomes and how this molecular pathway contributes to IL-1β-mediated immune responses and proinflammatory host cell death.
Acknowledgements
We would like to thank Joshua Olson and the UCSD vivarium staff for help with establishment of our Animal Protocol and animal experiments. We would also like to thank Dr. Patricia J. Johnson for providing T. vaginalis strains and for helpful discussion of the manuscript.
Funding Sources AMR was supported by the UC President’s Postdoctoral Fellowship and the San Diego I RAC DA Fellowship (NIH/NIGMS K12 GM06852). KAP was supported by the UC Chancellor’s Postdoctoral Fellowship. Research was supported by NIH grants FID090259 and GM114139 (VN).
Footnotes
Statement of Ethics
Animal work was approved by and conducted in accordance with the UC San Diego Institutional Animal Care and Use Committee (IACUC), protocol number S00227M. UCSD is acrredited by the American Association for Accreditation of Laboratory Animal Care (AAALAC).
Disclosure Statement
The authors have no conficts of interest to declare.
References
- 1.Secor WE, et al. , Neglected parasitic infections in the United States: trichomoniasis. Am J Trop Med Hyg, 2014. 90(5): p. 800–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.WHO Global incidence and prevalence of selected curable sexually transmitted infections-2008. 2012. 1–20. [Google Scholar]
- 3.Kissinger P, Trichomonas vaginalis: a review of epidemiologic, clinical and treatment issues. BMC Infect Dis, 2015. 15: p. 307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Petrin D, et al. , Clinical and microbiological aspects of Trichomonas vaginalis. Clin Microbiol Rev, 1998. 11(2): p. 300–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cotch MF, et al. , Trichomonas vaginalis associated with low birth weight and preterm delivery. The Vaginal Infections and Prematurity Study Group. Sex Transm Dis, 1997. 24(6): p. 353–60. [DOI] [PubMed] [Google Scholar]
- 6.Gram IT, et al. , Trichomonas Vaginalis (TV) and Human Papillomavirus (HPV) Infection and the Incidence of Cervical Intraepithelial Neoplasia (CIN) Grade III. Cancer Causes & Control, 1992. 3(3): p. 231–236. [DOI] [PubMed] [Google Scholar]
- 7.Zhang ZF, et al. , Trichomonas vaginalis and cervical cancer. A prospective study in China. Ann Epidemiol, 1995. 5(4): p. 325–32. [DOI] [PubMed] [Google Scholar]
- 8.Sutcliffe S, et al. , Plasma antibodies against Trichomonas vaginalis and subsequent risk of prostate cancer. Cancer Epidemiol Biomarkers Prev, 2006. 15(5): p. 939–45. [DOI] [PubMed] [Google Scholar]
- 9.Stark JR, et al. , Prospective study of Trichomonas vaginalis infection and prostate cancer incidence and mortality: Physicians’ Health Study. J Natl Cancer Inst, 2009. 101(20): p. 1406–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mavedzenge SN, et al. , Epidemiological synergy of Trichomonas vaginalis and HIV in Zimbabwean and South African women. Sex Transm Dis, 2010. 37(7): p. 460–6. [DOI] [PubMed] [Google Scholar]
- 11.Shafir SC, Sorvillo FJ, and Smith L, Current issues and considerations regarding trichomoniasis and human immunodeficiency virus in African-Americans. Clin Microbiol Rev, 2009. 22(1): p. 37–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lustig G, et al. , Trichomonas vaginalis contact-dependent cytolysis of epithelial cells. Infect Immun, 2013. 81(5): p. 1411–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rendon-Maldonado JG, et al. , Trichomonas vaginalis: in vitro phagocytosis of lactobacilli, vaginal epithelial cells, leukocytes, and erythrocytes. Exp Parasitol, 1998. 89(2): p. 241–50. [DOI] [PubMed] [Google Scholar]
- 14.Cauci S and Culhane JF, Modulation of vaginal immune response among pregnant women with bacterial vaginosis by Trichomonas vaginalis, Chlamydia trachomatis, Neisseria gonorrhoeae, and yeast. Am J Obstet Gynecol, 2007. 196(2): p. 133 e1–7. [DOI] [PubMed] [Google Scholar]
- 15.Mercer F, et al. , Leukocyte Lysis and Cytokine Induction by the Human Sexually Transmitted Parasite Trichomonas vaginalis. PLoS Negl Trop Dis, 2016. 10(8): p. e0004913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Han IH, et al. , Proinflammatory cytokine and nitric oxide production by human macrophages stimulated with Trichomonas vaginalis. Korean J Parasitol, 2009. 47(3): p. 205–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fiori PL, et al. , Association of Trichomonas vaginalis with its symbiont Mycoplasma hominis synergistically upregulates the in vitro proinflammatory response of human monocytes. Sex Transm Infect, 2013. 89(6): p. 449–54. [DOI] [PubMed] [Google Scholar]
- 18.Place DE and Kanneganti TD, Recent advances in inflammasome biology. Curr Opin Immunol, 2018. 50: p. 32–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Elliott JM, et al. , Crystal structure of procaspase-1 zymogen domain reveals insight into inflammatory caspase autoactivation. J Biol Chem, 2009. 284(10): p. 6546–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Broz P, et al. , Differential requirement for Caspase-1 autoproteolysis in pathogen-induced cell death and cytokine processing. Cell host & microbe, 2010. 8(6): p. 471–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Afonina IS, et al. , Proteolytic Processing of Interleukin-1 Family Cytokines: Variations on a Common Theme. Immunity, 2015. 42(6): p. 991–1004. [DOI] [PubMed] [Google Scholar]
- 22.Mosley B, et al. , The interleukin-1 receptor binds the human interleukin-1 alpha precursor but not the interleukin-1 beta precursor. J Biol Chem, 1987. 262(7): p. 2941–4. [PubMed] [Google Scholar]
- 23.Kostura MJ, et al. , Identification of a monocyte specific pre-interleukin 1 beta convertase activity. Proc Natl Acad Sci U S A, 1989. 86(14): p. 5227–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ghayur T, et al. , Caspase-1 processes IFN-gamma-inducing factor and regulates LPS-induced IFN-gamma production. Nature, 1997. 386(6625): p. 619–23. [DOI] [PubMed] [Google Scholar]
- 25.Gu Y, et al. , Activation of interferon-gamma inducing factor mediated by interleukin-1beta converting enzyme. Science, 1997. 275(5297): p. 206–9. [DOI] [PubMed] [Google Scholar]
- 26.Cerretti DP, et al. , Molecular cloning of the interleukin-1 beta converting enzyme. Science, 1992. 256(5053): p. 97–100. [DOI] [PubMed] [Google Scholar]
- 27.Thornberry NA, et al. , A novel heterodimeric cysteine protease is required for interleukin-1 beta processing in monocytes. Nature, 1992. 356(6372): p. 768–74. [DOI] [PubMed] [Google Scholar]
- 28.He WT, et al. , Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res, 2015. 25(12): p. 1285–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu X, et al. , Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature, 2016. 535(7610): p. 153–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gu NY, et al. , Trichomonas vaginalis induces IL-1beta production in a human prostate epithelial cell line by activating the NLRP3 inflammasome via reactive oxygen species and potassium ion efflux. Prostate, 2016. [DOI] [PubMed] [Google Scholar]
- 31.Lossick JG, Muller M, and Gorrell TE, In vitro drug susceptibility and doses of metronidazole required for cure in cases of refractory vaginal trichomoniasis. J Infect Dis, 1986. 153(5): p. 948–55. [DOI] [PubMed] [Google Scholar]
- 32.Clark CG and Diamond LS, Methods for cultivation of luminal parasitic protists of clinical importance. Clin Microbiol Rev, 2002. 15(3): p. 329–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sanjana NE, Shalem O, and Zhang F, Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods, 2014. 11(8): p. 783–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Okondo MC, et al. , DPP8 and DPP9 inhibition induces pro-caspase-1-dependent monocyte and macrophage pyroptosis. Nat Chem Biol, 2017. 13(1): p. 46–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kummer JA, et al. , Inflammasome components NALP 1 and 3 show distinct but separate expression profiles in human tissues suggesting a site-specific role in the inflammatory response. J Histochem Cytochem, 2007. 55(5): p. 443–52. [DOI] [PubMed] [Google Scholar]
- 36.Anderson JP, et al. , Structural, expression, and evolutionary analysis of mouse CIAS1. Gene, 2004. 338(1): p. 25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee SK, et al. , Immune cells in the female reproductive tract. Immune Netw, 2015. 15(1): p. 16–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sutterwala FS, Haasken S, and Cassel SL, Mechanism of NLRP3 inflammasome activation. Annals of the New York Academy of Sciences, 2014. 1319(1): p. 82–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bauernfeind FG, et al. , Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol, 2009. 183(2): p. 787–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Franchi L, Eigenbrod T, and Nunez G, Cutting edge: TNF-alpha mediates sensitization to ATP and silica via the NLRP3 inflammasome in the absence of microbial stimulation. J Immunol, 2009. 183(2): p. 792–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gong T, et al. , Orchestration of NLRP3 Inflammasome Activation by Ion Fluxes. Trends Immunol, 2018. [DOI] [PubMed] [Google Scholar]
- 42.Valderrama JA, et al. , Group A streptococcal M protein activates the NLRP3 inflammasome. Nat Microbiol, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Duncan JA, et al. , Cryopyrin/NALP3 binds ATP/dATP, is an ATPase, and requires ATP binding to mediate inflammatory signaling. Proc Natl Acad Sci U S A, 2007. 104(19): p. 8041–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wilson KP, et al. , Structure and mechanism of interleukin-1 beta converting enzyme. Nature, 1994. 370(6487): p. 270–5. [DOI] [PubMed] [Google Scholar]
- 45.Laliberte RE, Eggler J, and Gabel CA, ATP treatment of human monocytes promotes caspase-1 maturation and externalization. J Biol Chem, 1999. 274(52): p. 36944–51. [DOI] [PubMed] [Google Scholar]
- 46.Guo H, Callaway JB, and Ting JPY, Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med, 2015. 21(7): p. 677–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Coll RC, et al. , The cytokine release inhibitory drug CRID3 targets ASC oligomerisation in the NLRP3 and AIM2 inflammasomes. PLoS One, 2011. 6(12): p. e29539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Munoz-Planillo R, et al. , K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity, 2013. 38(6): p. 1142–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Petrilli V, et al. , Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ, 2007. 14(9): p. 1583–9. [DOI] [PubMed] [Google Scholar]
- 50.Di Virgilio F, Liaisons dangereuses: P2X(7) and the inflammasome. Trends Pharmacol Sci, 2007. 28(9): p. 465–72. [DOI] [PubMed] [Google Scholar]
- 51.Schaefer L, Complexity of danger: the diverse nature of damage-associated molecular patterns. J Biol Chem, 2014. 289(51): p. 35237–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Steinberg TH and Silverstein SC, Extracellular ATP4-promotes cation fluxes in the J774 mouse macrophage cell line. J Biol Chem, 1987. 262(7): p. 3118–22. [PubMed] [Google Scholar]
- 53.Franchi L, et al. , Differential requirement of P2X7 receptor and intracellular K+ for caspase-1 activation induced by intracellular and extracellular bacteria. J Biol Chem, 2007. 282(26): p. 18810–8. [DOI] [PubMed] [Google Scholar]
- 54.Ferrari D, et al. , Extracellular ATP triggers IL-1 beta release by activating the purinergic P2Z receptor of human macrophages. J Immunol, 1997. 159(3): p. 1451–8. [PubMed] [Google Scholar]
- 55.Yan Z, et al. , The P2X7 receptor channel pore dilates under physiological ion conditions. J Gen Physiol, 2008. 132(5): p. 563–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Murgia M, et al. , Oxidized ATP. An irreversible inhibitor of the macrophage purinergic P2Z receptor. J Biol Chem, 1993. 268(11): p. 8199–203. [PubMed] [Google Scholar]
- 57.Lamkanfi M, et al. , Glyburide inhibits the Cryopyrin/Nalp3 inflammasome. J Cell Biol, 2009. 187(1): p. 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ding J, et al. , Pore-forming activity and structural autoinhibition of the gasdermin family. Nature, 2016. 535(7610): p. 111–116. [DOI] [PubMed] [Google Scholar]
- 59.Kovacs SB and Miao EA, Gasdermins: Effectors of Pyroptosis. Trends Cell Biol, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Taabazuing CY, Okondo MC, and Bachovchin DA, Pyroptosis and Apoptosis Pathways Engage in Bidirectional Crosstalk in Monocytes and Macrophages. Cell Chem Biol, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schneider KS, et al. , The Inflammasome Drives GSDMD-Independent Secondary Pyroptosis and IL-1 Release in the Absence of Caspase-1 Protease Activity. Cell Rep, 2017. 21(13): p. 3846–3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Broz P, et al. , Differential requirement for Caspase-1 autoproteolysis in pathogen-induced cell death and cytokine processing. Cell Host Microbe, 2010. 8(6): p. 471–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cobo ER, Eckmann L, and Corbeil LB, Murine models of vaginal trichomonad infections. Am J Trop Med Hyg, 2011. 85(4): p. 667–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Auphan N, et al. , Immunosuppression by glucocorticoids: inhibition of NF-kappa B activity through induction of I kappa B synthesis. Science, 1995. 270(5234): p. 286–90. [DOI] [PubMed] [Google Scholar]
- 65.Midlej V and Benchimol M, Trichomonas vaginalis kills and eats--evidence for phagocytic activity as a cytopathic effect. Parasitology, 2010. 137(1): p. 65–76. [DOI] [PubMed] [Google Scholar]
- 66.Grahames CB, et al. , Pharmacological characterization of ATP- and LPS-induced IL-1beta release in human monocytes. Br J Pharmacol, 1999. 127(8): p. 1915–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fichorova RN, et al. , Trichomonas vaginalis lipophosphoglycan triggers a selective upregulation of cytokines by human female reproductive tract epithelial cells. Infect Immun, 2006. 74(10): p. 5773–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li L, et al. , Trichomonas vaginalis Induces Production of Proinflammatory Cytokines in Mouse Macrophages Through Activation of MAPK and NF-kappaB Pathways Partially Mediated by TLR2. Front Microbiol, 2018. 9: p. 712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fichorova RN, et al. , Trichomonas vaginalis Lipophosphoglycan Exploits Binding to Galectin-1 and −3 to Modulate Epithelial Immunity. J Biol Chem, 2016. 291(2): p. 998–1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ryan CM, et al. , Chemical structure of Trichomonas vaginalis surface lipoglycan: a role for short galactose (beta1–4/3) N-acetylglucosamine repeats in host cell interaction. J Biol Chem, 2011. 286(47): p. 40494–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zariffard MR, et al. , Trichomonas vaginalis infection activates cells through toll-like receptor 4. Clinical Immunology, 2004. 111(1): p. 103–107. [DOI] [PubMed] [Google Scholar]
- 72.Chang JH, Park JY, and Kim SK, Dependence on p38 MAPK signalling in the up-regulation of TLR2, TLR4 and TLR9 gene expression in Trichomonas vaginalis-treated HeLa cells. Immunology, 2006. 118(2): p. 164–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Im SJ, et al. , Inflammatory response of a prostate stromal cell line induced by Trichomonas vaginalis. Parasite Immunol, 2016. 38(4): p. 218–27. [DOI] [PubMed] [Google Scholar]
- 74.Lupfer C and Anand PK, Integrating Inflammasome Signaling in Sexually Transmitted Infections. Trends Immunol, 2016. 37(10): p. 703–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Verma V, et al. , Inflammasomes and Their Role in Innate Immunity of Sexually Transmitted Infections. Front Immunol, 2016. 7: p. 540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Allsworth JE, Ratner JA, and Peipert JF, Trichomoniasis and other sexually transmitted infections: results from the 2001–2004 National Health and Nutrition Examination Surveys. Sex Transm Dis, 2009. 36(12): p. 738–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vick EJ, et al. , Gardnerella vaginalis triggers NLRP3 inflammasome recruitment in THP-1 monocytes. J Reprod Immunol, 2014. 106: p. 67–75. [DOI] [PubMed] [Google Scholar]
- 78.Harada K, et al. , Vaginal infection with Ureaplasma urealyticum accounts for preterm delivery via induction of inflammatory responses. Microbiol Immunol, 2008. 52(6): p. 297–304. [DOI] [PubMed] [Google Scholar]
- 79.Garlanda C, Dinarello CA, and Mantovani A, The interleukin-1 family: back to the future. Immunity, 2013. 39(6): p. 1003–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Evavold CL and Kagan JC, How Inflammasomes Inform Adaptive Immunity. J Mol Biol, 2018. 430(2): p. 217–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lazenby GB, Soper DE, and Nolte FS, Correlation of leukorrhea and Trichomonas vaginalis infection. J Clin Microbiol, 2013. 51(7): p. 2323–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pietrella D, et al. , Secreted aspartic proteases of Candida albicans activate the NLRP3 inflammasome. Eur J Immunol, 2013. 43(3): p. 679–92. [DOI] [PubMed] [Google Scholar]
- 83.Peters BM, et al. , Fungal morphogenetic pathways are required for the hallmark inflammatory response during Candida albicans vaginitis. Infect Immun, 2014. 82(2): p. 532–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Benchimol M, Trichomonads under Microscopy. Microsc Microanal, 2004. 10(5): p. 528–50. [DOI] [PubMed] [Google Scholar]
- 85.Kusdian G and Gould SB, The biology of Trichomonas vaginalis in the light of urogenital tract infection. Mol Biochem Parasitol, 2014. 198(2): p. 92–9. [DOI] [PubMed] [Google Scholar]
- 86.Mercer F and Johnson PJ, Trichomonas vaginalis: Pathogenesis, Symbiont Interactions, and Host Cell Immune Responses. Trends Parasitol, 2018. 34(8): p. 683–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.De Jesus JB, et al. , Iron modulates ecto-phosphohydrolase activities in pathogenic trichomonads. Parasitol Int, 2006. 55(4): p. 285–90. [DOI] [PubMed] [Google Scholar]
- 88.Sena AC, Bachmann LH, and Hobbs MM, Persistent and recurrent Trichomonas vaginalis infections: epidemiology, treatment and management considerations. Expert Rev Anti Infect Ther, 2014. 12(6): p. 673–85. [DOI] [PubMed] [Google Scholar]
- 89.Fichorova RN, Impact of T vaginalis infection on innate immune responses and reproductive outcome. J Reprod Immunol, 2009. 83(1–2): p. 185–9. [DOI] [PMC free article] [PubMed] [Google Scholar]