

Abstract
Meizothrombin is the physiologically active intermediate generated by a single cleavage of prothrombin at R320 to separate the A and B chains. Recent evidence has suggested that meizothrombin, like thrombin, is a Na+-activated enzyme. In this study we present the first X-ray crystal structure of human meizothrombin desF1 solved in the presence of the active site inhibitor PPACK at 2.1 Å resolution. The structure reveals a Na+ binding site whose architecture is practically identical to that of human thrombin. Stopped-flow measurements of Na+ binding to meizothrombin desF1 document a slow phase of fluorescence change with a kobs decreasing hyperbolically with increasing [Na+], consistent with the existence of three conformations in equilibrium, E*, E and E:Na+, as for human thrombin. Evidence that meizothrombin exists in multiple conformations provides valuable new information for studies of the mechanism of prothrombin activation.
Keywords: Thrombin, meizothrombin, allostery, linkage, Na+ binding
Introduction
Blood coagulation is initiated by exposure of tissue factor that forms a complex with factor VIIa and results in the generation of small quantities of factors IXa and Xa [1, 2]. Small quantities of Xa generate minute concentrations of thrombin that result in the activation of factor XI and the cofactors VIII and V. At this point, the VIIIa-IXa complex generates sufficient quantities of Xa to form the prothrombinase complex, composed of factors Va, Xa, Ca2+ and phospholipids, which leads to the generation of thrombin from prothrombin by cleavage at R320 and R271 [3, 4]. The presence of factor Va directs cleavage first at R320 to generate the active intermediate meizothrombin [5], followed by cleavage at R271 to produce thrombin through a kinetic mechanism that remains controversial and presumably involves a conformational transition of prothrombinase [6] and/or a conformational rearrangement of meizothrombin [7].
A remarkable property of thrombin is its allosteric nature linked to Na+ binding [8-11]. In the absence of Na+, thrombin exists in equilibrium between an inactive form E* and a low activity form E that is converted to a high activity conformation E:Na+ upon binding of the cation [9-11]. Na+ binding is required for optimal cleavage of fibrinogen, but is dispensable for cleavage of protein C [12, 13]. Na+ promotes cleavage of PAR1, PAR3 and PAR4 [14, 15], and promotes activation of factors V [16], VIII [17] and XI [18]. Na+ binds 16-20 Å away from residues of the catalytic triad and within 5 Å from D189 in the S1 site, nestled between the 220- and 186-loops [19-21]. The long-range effect of Na+ on the catalytic S195 producing the transition from E to E:Na+ is mediated by a network of water molecules [20], but the transition from E* to E is more substantial from a structural standpoint and involves rearrangement of the β-strands shaping access to the active site [22, 23]. The activating effect of Na+ is not limited to thrombin [8, 10, 11, 24, 25] and specific sequence markers have been identified to predict its occurrence in the entire family of trypsin-like proteases to which thrombin belongs [24-26]. Several groups have shown that Na+ has a significant influence on the activity of other vitamin K-dependent clotting factors such as factors VIIa [24, 27], IXa [28], Xa [29-35] and activated protein C [36-40]. A Na+ binding site analogous to that of thrombin [19, 20] has been identified structurally in factor Xa [41, 42], factor VIIa [43] and activated protein C [44]. Surprisingly, however, the effect of Na+ on the activation of prothrombin to thrombin along the meizothrombin pathway has remained unexplored, although Na+ is likely to play an important role in the activity of the prothrombinase complex, its assembly and its ability to direct cleavage of prothrombin to generate physiological intermediates. Indeed, a recent study has documented that meizothrombin and meizothrombin desF1 are Na+-activated enzymes and express functional linkage between exosite I, the active site and the Na+ site much like the mature enzyme thrombin [45]. Interestingly, the study also estimated a Na+ affinity for meizothrombin desF1 comparable to that of thrombin, but significantly higher than that of meizothrombin [45]. These findings raise a number of important questions about the architecture of the Na+ binding site of meizothrombin and the kinetic signatures of Na+ binding that would enable a quantitative comparison with the wealth of information available for thrombin [9-11, 20, 23]. In this study we address these timely and important issues directly with X-ray crystallography and stopped-flow kinetics.
Materials and Methods
Meizothrombin is the physiological intermediate generated by a single cleavage of prothrombin at R320 to separate the A and B chains yielding an active enzyme [5]. The enzyme is unstable due to autoproteolytic cleavage at R155, R271 and R284 [46, 47] that generates first meizothrombin desF1, devoid of prothrombin fragment 1 (residues 1-155) and then thrombin with and without the additional residues 272-284 in the A chain. Because meizothrombin and meizothrombin desF1 are functionally equivalent [5], we used our expression system with BHK cells for prethrombin-1 (residues 156-579 of prothrombin) to produce a recombinant form of meizothrombin desF1 resistant to autoproteolysis by virtue of the double substitution R271A/R284A. Activation of the construct with ecarin produced an active form of meizothrombin desF1 R271A/R284A (meizoIIRRΔF1) in high yields. MeizoIIRRΔF1 is stable for both functional and X-ray crystallization studies and has kinetic properties (Table 1) consistent with published data [5, 46, 47]. Active site concentrations were determined by titration with the active site inhibitor H-D-Phe-Pro-Arg-CH2Cl (PPACK) and found to be > 95 %.
Table 1.
Properties of human meizoIIRRΔF1 compared to thrombin
| Fibrinogena | PAR1a | Protein Ca,b | FPRc | FPRd | Na+e | rf | |
|---|---|---|---|---|---|---|---|
| Thrombin | 17±1 | 39±1 | 0.22±0.01 | 88±3 | 3.2±0.2 | 160±20 | 0.72±0.07 |
| MeizoIIRRΔF1 | 0.23±0.04 | 0.75±0.08 | 0.044±0.02 | 91±2 | 7.4±0.1 | 91±9 | 0.83±0.07 |
All values are kcat/Km in μM-1 s-1, except Na+ and r.
5 mM Tris, 145 mM NaCl, 0.1 % PEG, pH 7.4, 37 °C.
In the presence of 5 mM CaCl2 and 100 nM thrombomodulin.
5 mM Tris, 200 mM NaCl, 0.1 % PEG, pH 8.0, 25 °C.
5 mM Tris, 200 mM ChCl, 0.1 % PEG, pH 8.0, 25 °C.
Values refer to the intrinsic equilibrium association constant, KA, in M-1 or the ratio r = [E*]/[E] (see eq. 1) determined by stopped-flow measurements under conditions of 5 mM Tris, 0.1 % PEG, pH 8.0, 15 °C.
Values refer to the intrinsic equilibrium association constant, KA, in M-1 or the ratio r = [E*]/[E] (see eq. 1) determined by stopped-flow measurements under conditions of 5 mM Tris, 0.1 % PEG, pH 8.0, 15 °C.
Values of s = kcat/Km for the hydrolysis of the chromogenic substrate H-D-Phe-Pro-Arg-p-nitroanilide (FPR) were determined as reported [20] under experimental conditions of 5 mM Tris, 0.1 % PEG8000, pH 8.0 at 25 °C in the presence of 200 mM NaCl or choline chloride (ChCl) to study the properties of the E:Na+ and E forms, respectively [10]. The interaction with fibrinogen leading to release of fibrinopeptides A (FpA) and B (FpB), cleavage of the protease activated receptor 1 (PAR1) and activation of protein C in the presence of saturating amounts of thrombomodulin were studied as reported [13, 15, 48] under experimental conditions of 5 mM Tris, 0.1 % PEG8000, 145 mM NaCl, pH 7.4 at 37 °C. Activity toward the chromogenic substrate FPR is slightly higher than that of thrombin and is influenced by the presence of Na+. Cleavage of fibrinogen and PAR1 is compromised about 50-fold, but activity toward protein C is reduced only seven-fold in the presence of saturating amounts of thrombomodulin (Table 1). Stopped-flow fluorescence measurements were carried out with an Applied Photophysics SX20 spectrometer, with excitation at 280 nm and a cutoff filter at 305 nm [9]. Samples of meizoIIRRΔF1 at a final concentration of 50 nM in 5 mM Tris, 0.1 % PEG8000, pH 8.0 at 15 °C were mixed 1:1 with 60 μl solutions of the same buffer containing variable amounts of NaCl (up to 400 mM) kept at constant ionic strength of 400 mM with ChCl. The baseline was measured with 400 mM ChCl in the mixing syringe. Each trace was determined in quadruplicate. Na+ binding to human thrombin causes a significant increase in intrinsic fluorescence of the enzyme with an initial rapid phase too fast to measure within the dead time (0.5 ms) of the instrument, followed by a slow phase with a kobs in the ms time scale that decreases hyperbolically as [Na+] increases [9, 10]. The fast phase resolved by continuous flow measurements reveals a linear dependence of kobs on [Na+] in the ms time scale [49]. These are signatures of a two-step mechanism for Na+ binding as shown in scheme 1 below Thrombin exists in equilibrium between two forms, E* and E, that interconvert with kinetic rate constants k1 and k-1 in the ms time scale. E* is inactive and cannot bind Na+ or substrate. E interacts with Na+ with a rate constant kA to populate E:Na+, that may dissociate into the parent components with a rate constant k-A. The fast phase of fluorescence increase is due to the E-E:Na+ interconversion involving Na+ binding/dissociation, and the slow phase is due to the E-E* interconversion that precedes Na+ binding. The kobs for the fast phase increases linearly with [Na+] [49], and the kobs for the slow phase decreases hyperbolically with increasing [Na+] according to the expression [9]
| (1) |
where the rates for the E*→E transition (k1) and backward (k-1) define r = k-1/k1 as the ratio [E*]/[E], and KA is the intrinsic equilibrium association constant for Na+ binding. Scheme 1 explains the mechanism of Na+ activation of thrombin. E and E:Na+ are the two active forms that account for the dependence of kcat on [Na+] [8, 10, 11, 50, 51] and are responsible for the anticoagulant and procoagulant functions of the enzyme, respectively [10-13]. E* is an inactive form of thrombin in equilibrium with the active E form, which in turn interconverts with the more active E:Na+ form. E* is a third thrombin form and the E*-E equilibrium exists in solution (1:1.4 ratio at 15 °C) regardless of Na+ or any other allosteric effector. Rapid kinetic measurements of the binding of Na+ [9, 23, 52], or of binding of ligands to the active site and exosite I [53] detect the presence of E* and its conversion to the active form E directly. Stopped-flow measurements of Na+ binding to meizoIIRRΔF1 gave rise to an increase in fluorescence of 6 %, which is about half the size as that of thrombin. Importantly, the amplitude of the fast phase of fluorescence increase was too small to detect at low [Na+], which prevented further analysis with continuous flow methods [49], but the slow phase was well resolved and evolved with a kobs decreasing hyperbolically with increasing [Na+] according to eq 1. These signatures corroborated the use of Scheme 1 as capturing the kinetic mechanism of Na+ binding to meizoIIRRΔF1 as in the case of thrombin.
Scheme 1.
Crystals of human meizoIIRRΔF1 bound to PPACK were obtained using the hanging drop vapor-diffusion method. MeizoIIRRΔF1 and PPACK were mixed in 1:11 molar ratio at 4 °C for 2 h before crystallization. A 2 μl solution of 5 mg/ml meizoIIRRΔF1 in 50mM ChCl, 20 mM MES, pH 6.0 was mixed with a 2 μl reservoir solution containing 23 % PEG3350, 200 mM Na2SO4 and left to equilibrate at 23° C. Single crystals grew to an approximate size of 0.6 × 0.3 × 0.2 mm3 in one-two weeks. Crystals were tetragonal, space group P41212, with unit cell parameters a = b = 121.3 Å, c = 100.2 Å, and contained one molecule in the asymmetric unit. Crystals were cryo-protected in a solution containing 15 % glycerol, 200 mM Na2SO4, 27 % PEG3350 for 3 min and frozen in liquid nitrogen to 100 K. X-ray data were collected to 2.1 Å resolution on an ADSC Quantum-315 CCD detector at the Biocars Beamline 14-BM-C of the Advanced Photon Source, Argonne National Laboratories (Argonne, IL). Data processing, indexing, integration and scaling were performed using the HKL2000 package [54]. The crystal structure of meizoIIRRΔF1 bound to PPACK was solved by molecular replacement with MOLREP from the CCP4 package [55] using the coordinates of the PPACK-inhibited form of human thrombin (Protein Data Bank ID code 1SHH) [20] with all inhibitors, sugars and solvent molecules omitted as the starting model. Fragment 2 was solved using bovine meizothrombin (Protein Data Bank ID code 1A0H). Refinement and electron density map generation were performed with the CCP4 package package [55] and 5 % of the reflections were randomly selected as a test set for cross validation. Model building and analysis of the structure were carried with Coot [56]. Ramachandran plots were calculated using PROCHECK [57]. Results of data collection, processing and refinement are listed in Table 2. Coordinates of the structure of the meizoIIRRΔF1-PPACK complex have been deposited to the Protein Data Bank (ID code 3E6P).
Table 2.
Crystallographic data for human meizoIIRRΔF1 bound to PPACK (PDB ID 3E6P)
| Data collection: | |
| Wavelength (Å) | 0.9 |
| Space Group | P41212 |
| Unit cell dimension (Å) | a = b = 121.3, c = 100.2 |
| Molecules/asymmetric unit | 1 |
| Resolution range (Å) | 40.0-2.1 |
| Observations | 479436 |
| Unique observations | 40469 |
| Completeness (%) | 92.4 (79.0) |
| Rsym (%) | 11.2 (43.8) |
| I/σ(I) | 17.7 (2.0) |
| Refinement: | |
| Resolution (Å) | 40.0-2.1 |
| |F|/σ(|F|) | > 0 |
| Rcryst, Rfree | 0.211, 0.249 |
| Reflections (working/test) | 38428/2026 |
| Protein atoms | 2928 |
| Solvent molecules | 208 |
| PPACK/Na+ | 1/1 |
| Rmsd bond lengthsa (Å) | 0.012 |
| Rmsd anglesa (°) | 1.4 |
| Rmsd ΔB (Å2) (mm/ms/ss)b | 0.86/0.47/2.06 |
| <B> protein (Å2) | 44.4 |
| <B> solvent (Å2) | 49.9 |
| <B> PPACK/Na+ (Å2) | 32.0/36.0 |
| Ramachandran plot: | |
| Most favored (%) | 99.2 |
| Generously allowed (%) | 0.4 |
| Disallowed (%) | 0.4 |
Root-mean-squared deviation (Rmsd) from ideal bond lengths and angles and Rmsd in B-factors of bonded atoms.
mm, main chain-main chain; ms, main chain-side chain; ss, side chain-side chain.
Results and Discussion
The effect of Na+ on thrombin structure and function has been studied in considerable detail [9, 10, 20, 23], but the effect of Na+ on the physiological intermediate leading to mature thrombin has remained unexplored until the recent seminal observation that meizothrombin and meizothrombin desF1 are Na+-activated enzymes like thrombin [45]. These recent findings make a direct analysis of the structural and kinetic underpinnings of Na+ binding to meizoIIRRΔF1 both timely and important.
There is currently only one structure of meizothrombin desF1 deposited in the PDB and pertains to the bovine enzyme [58]. The structure was solved at 3.2 Å resolution and shows no evidence of bound Na+.The first structure of human meizothrombin desF1 in what is likely the E:Na+ form bound to PPACK is shown in Figures 1-3. The structure, solved at 2.1 Å resolution, reveals a low B-factor, high-occupancy, strong electron density peak in the Na+ site that can be assigned unambiguously to Na+ (Fig. 1). The cation is coordinated octahedrally by two carbonyl O atoms from the protein contributed by R221a and K224, and four buried water molecules. The coordination shell is essentially identical to that of thrombin [20] and ensures communication between Na+ and the Oδ2 atom of D189 to afford higher catalytic activity and substrate binding in the E:Na+ form. The presence of PPACK obliterates much of the water network present in the free form between the primary specificity pocket and the active site [20], so no conclusion can be drawn on the longrange effect that the bound Na+ has on the orientation of the side chain of the catalytic S195. The architecture of the Na+ coordination shell explains the basic similarity of Na+ affinity and kinetic activation between meizoIIRRΔF1 and thrombin (Table 1). Although it is intrinsically difficult to predict cation binding energetics from protein structural information [59-62], a basic structural similarity among Na+ binding sites observed in vitamin K-dependent clotting factors [19-21, 41-44] accounts for a relatively small (< 3-fold) variability in their apparent binding affinities [24, 27-40, 63, 64]. The structure resolves fragment 2 in almost its entirety and reveals the expected fold for the kringle domain making extensive interactions with exosite II (especially R93 and R101) and the region above the 60-loop of the enzyme (Figs. 2 and 3). Three disulfide bonds involving the Cys pairs 170-248, 219-243 and 191-231 (prothrombin numbering) are fully resolved in fragment 2, and so are the Trp residues W194 and W230 that are stacked, with W230 engaging the guanidinium group of R93 of the B chain in a cation-π interaction (Fig. 3). Slight differences exist with the conformation reported previously for the bovine enzyme [58], with fragment 2 in our structure making more pronounced contacts with R93 and R101 of the B chain and folding as seen in the structure of human fragment 2 noncovalently bound to PPACK-inhibited human thrombin [65]. A patch of negatively charged residues of fragment 2 composed of D223, D225, E226 and E227 is analogous to the Lys-binding kringle present in plasminogen and tissue-type plasminogen activator [65]. This anionic patch and Y242 nearby engage the side chains of R93, R101 and R175 in strong ionic interactions and make polar contacts with the backbone of D178. It should be pointed out that R93 and R101 are major components of the epitope of thrombin recognizing heparin [66] and the γ -peptide of fibrinogen [67]. Other ionic interactions involve D239 and the side chain of R97 that also engages the backbone O atom of G238. The backbone N atom of G238, on the other hand, H-bonds to the backbone O atom of P92 and P237 makes a significant interaction with W96 right next to the 60-loop of the B chain. K236 makes a strong interaction with the sugar moiety attached to N60g in the 60-loop. Finally, on the other side of the surface of interaction between fragment 2 and the B chain, K204, H205 and Q206 make polar contacts with K240 and Q244 (Fig. 3).
Figure 1.
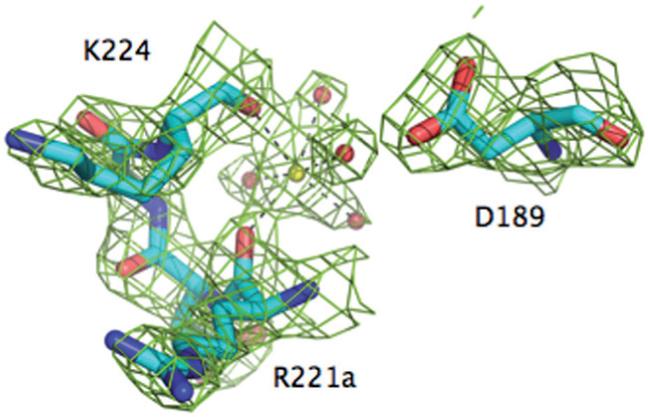
The Na+ binding site of meizoIIRRΔF1. Shown is the 221a-224 segment of the 220-loop, along with the side chain of D189. Na+ (yellow ball) is coordinated octahedrally by the backbone O atoms of R221a (2.4 Å) and K224 (2.5 Å) and four buried water molecules (red balls, 2.5-2.6 Å). One of these water molecules connects to the Oδ2 atom of D189. The Na+ coordination shell of meizoIIRRΔF1 is practically identical to that of thrombin [20], underscoring the similarity of Na+ affinity and kinetic activation between the two enzymes (Table 1). The electron density 2F0-Fc map (green mesh) is contoured at 2 σ.
Figure 3.
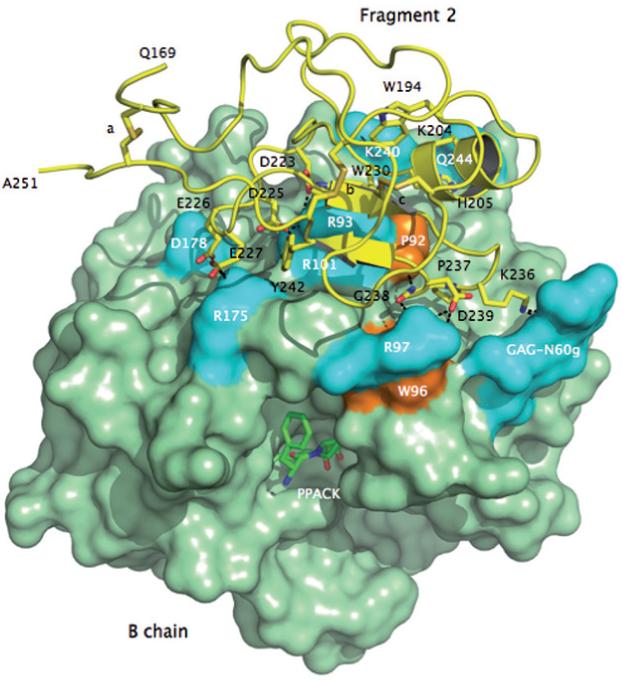
Crystal structure of meizoIIRRΔF1 at 2.1 Å resolution showing the architecture of fragment 2 (ribbon and sticks in yellow) and its docking on the surface of the B chain (green). Three disulfide bonds stabilizing the kringle domain of fragment 2 are indicated with letters a (C170-C248), b (C219-C243) and c (C191-C231). Shown are the residues of fragment 2 that make direct interactions with the B chain. The anionic cluster DGDEE (residues 223-227), corresponding to the Lys-binding center of other kringles in plasminogen and tissue-type plasminogen activator, makes extensive ionic interactions (cyan) with R93, R101 and R175. R93 and R101 are critical residues for the binding of heparin [66] and the γ’-peptide of fibrinogen [67]. Other ionic interactions involve D239 with R97 of the B chain, K236 with the GAG moiety linked to N60g of the 60-loop and Q206 with K240. The only relevant hydrophobic interactions (orange) of fragment 2 with the B chain involve the environment of P92 and W96 with P237 and G238. W194 and W230 are in stacking interaction with each other and presumably function as a single fluorophore. Perturbation of W96 and the presence of W194 and W230 may be responsible for the different fluorescence response to Na+ binding observed in meizoIIRRΔF1 compared to thrombin (see Fig. 4).
Figure 2.
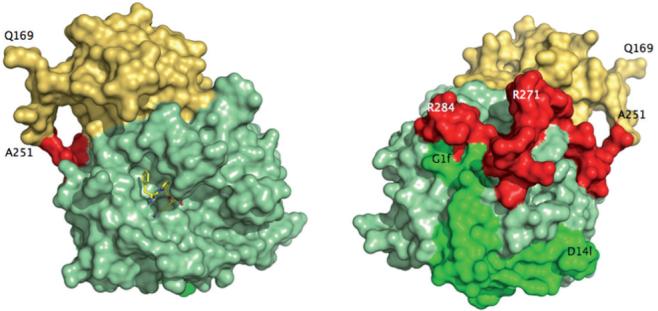
Crystal structure of meizoIIRRΔF1 at 2.1 Å resolution. Shown is the arrangement of the B chain (light green) relative to the A chain (green) and fragment 2 (gold). The linker region connecting the A chain to the kringle domain of fragment 2 (red) had poor electron density and was modeled in the final refinement assigning zero occupancy to residues 252-286. Also shown (left) is the active site inhibitor PPACK (CPK in yellow). The portion 168-251 of fragment 2 (spanning residues 156-271 of prothrombin in its entirety) folds in the expected kringle configuration (three disulfide bonds connecting 170-248, 219-243, 191-231) docked on exosite II and above the 60-loop (left, see also Fig. 3). The A chain is fully visible in the electron density map only from G1f to D14l (residues 287-318 of prothrombin). The autolysis loop is fully resolved, with the exception of the T149-G149d segment. The orientation at right shows the back of the molecule and corresponds to a 180° rotation along the y-axis relative to the standard orientation at left.
Not all residues connecting fragment 2 to the A chain, the so-called linker domain, are traceable in the electron density map. The first residue resolved in the A chain is G1f (residue 287 of prothrombin) and the last residue resolved in fragment 2 is A251 located almost 40 Å away (Figs. 2 and 3). That leaves 36 residues missing in the electron density map, including R271 and R284 mutated to Ala in meizoIIRRΔF1. Inspection of crystal packing revealed no significant contacts in the region presumably occupied by the linker in a disordered conformation. We therefore modeled the linker region and assigned an occupancy of zero to the 36 residues missing in the electron density map in the final structure (residues in red in Fig. 3). The linker assumes a twisted conformation, similar to that found in the structure of bovine meizothrombin desF1 [58]. Of interest is the position of R271 and R284, mutated to Ala in our construct. The Cα atoms of these two residues are 20.8 Å away from each other. The position of R155, whose cleavage generates meizothrombin desF1 from meizothrombin, can only be inferred from that of the first residue detected in the structure of meizoIIRRΔF1, Q169. The distance between the Cα atoms of R284 and Q169 is 45.7 Å and R271 is even further away. This implies that cleavage at R155 would require a substantial translation of prothrombinase to access R284 or R271 for further processing of substrate leading to thrombin. We hope that crystallization of human meizoIIRRΔF1 under different conditions will provide more detailed information of the architecture of the linker region. The limiting factor seems to be the intrinsic disorder of this region, rather than poor resolution of the structure. Paradoxically, the linker was resolved in its entirety for the bovine enzyme at much lower resolution [58], presumably because of the sequence differences and lower intrinsic disorder. The B chain of meizoIIRRΔF1 shows no significant differences compared to PPACK-inhibited thrombin in the E:Na+ form [20] (rmsd 0.30 Å).
In addition to crystallographic evidence of Na+ binding (Fig. 1), we have collected strong evidence that meizoIIRRΔF1 exists in the E*, E and E:Na+ forms like thrombin (Figs. 4 and 5). MeizoIIRRΔF1 responds to Na+ binding with a 6 % change in intrinsic fluorescence that is smaller than the 10 % change seen for thrombin [9]. The overall change is biphasic, but the smaller amplitude makes it difficult to resolve the fast phase, especially at low [Na+] (Fig. 4). The single-exponential slow phase is well resolved and its kobs decreases hyperbolically with increasing [Na+] (Fig. 5), thereby demonstrating an equilibrium between two forms that precedes the binding of Na+ [9]. The values of the apparent and intrinsic equilibrium association constants for Na+ binding to meizoIIRRΔF1 are only slightly smaller than those of thrombin (Table 1), as expected from the results of recent studies [45] and the structural similarity of the Na+ site between the two enzymes (Fig. 1). The rate constants k1 and k-1 predict a ratio [E*]/[E] = 0.83, similar to the value of 0.72 in thrombin. In conclusion, meizoIIRRΔF1 undergoes an equilibrium involving E*, E and E:Na+. The binding of Na+ produces a smaller fluorescence change compared to thrombin, with the fast phase being affected the most at low [Na+].
Figure 4.
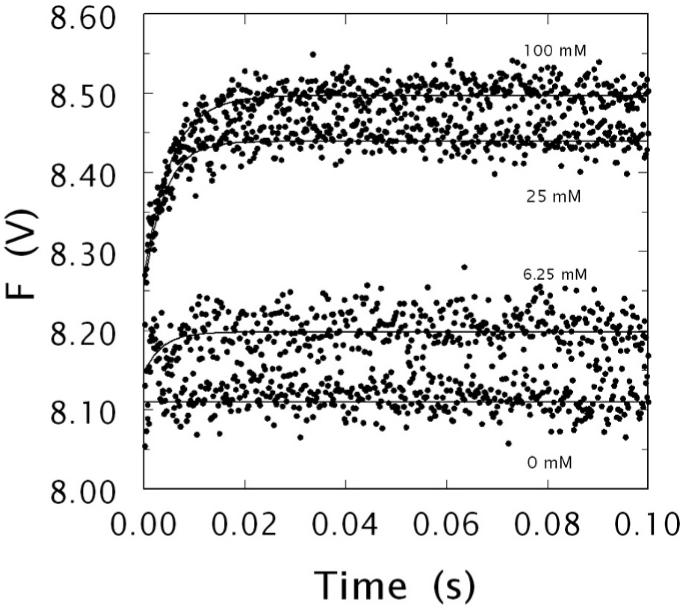
Kinetic traces of Na+ binding to human meizoIIRRΔF1 in the 0-100 ms time scale. Shown are the traces obtained at 0, 6.25, 25 and 100 mM Na+, as indicated. The binding of Na+ obeys a two-step mechanism, with a fast phase completed within the dead time (< 0.5 ms) of the spectrometer that is evident only at high [Na+ ], followed by a single-exponential slow phase. The kobs for the slow phase decreases hyperbolically with increasing [Na+ ] (see also Fig. 5). Experimental conditions are: 50 nM meizoIIRRΔF1, 5 mM Tris, 0.1 % PEG, pH 8.0 at 15 °C. The [Na+ ] was changed by keeping the ionic strength constant at 400 mM with ChCl. Continuous lines were drawn using the expression a exp (-kobs t) + b with best-fit parameter values: (0 mM) a = 0±0 V, kobs = 0±0 s-1, b = 8.11±0.01 V; (6.25 mM) a = -0.05±0.01 V, kobs = 276±9 s-1, b = 8.20±0.01 V; (25 mM) a = -0.18±0.02 V, kobs = 241±8 s-1, b = 8.44±0.01 V; (100 mM) a = -0.23±0.01 V, kobs = 191±8 s-1, b = 8.50±0.01 V.
Figure 5.
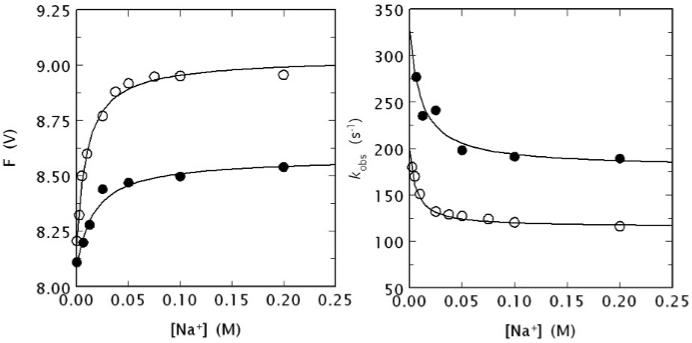
(Left) Na+ binding curve of human meizoIIRRΔF1 (black circles) obtained from the total change in intrinsic fluorescence determined by stopped flow kinetics (see Fig. 4). Similar results were obtained from direct fluorescence titration (data not shown) performed as reported elsewhere [9, 64]. The binding curve of human thrombin is also shown for comparison (open circles). Experimental conditions are: 50 nM enzyme, 5 mM Tris, 0.1 % PEG, pH 8.0 at 15 °C. The [Na+] was changed by keeping the ionic strength constant at 400 mM with ChCl. Continuous lines were drawn according to the equation F = (Fo + F1 Kapp [Na+])/(1 + Kapp [Na+]), described in detail elsewhere [9], with best-fit parameter values: (meizoIIRRΔF1) F0 = 8.09±0.01 V, F1 = 8.58±0.01 V, Kapp = 59±9 M-1; (thrombin) F0 = 8.19±0.01 V, F1 = 9.03±0.01 V, Kapp = 100±10 M-1 [9]. Note the larger fluorescence change induced by Na+ binding to thrombin compared to meizoIIRRΔF1 and also the slightly higher Na+ affinity measured as an apparent binding constant Kapp. (Right) Values of kobs for the slow phase of fluorescence increase (see Fig. 4) due to Na+ binding to human meizoIIRRΔF1 (black circles) and thrombin (open circles). Experimental conditions are: 50 nM thrombin or meizoIIRRΔF1, 5 mM Tris, 0.1 % PEG, pH 8.0 at 15 °C. Continuous lines were drawn according to eq. 1 in the text, with best-fit parameter values: (meizoIIRRΔF1) k1 = 179±9 s-1, k-1 = 148±8 s-1, KA = 91±9 M-1; (thrombin) k1 = 115±3 s-1, k-1 = 83±6 s-1, KA = 160±20 M-1 [9]. The value of r = k-1/k1 is practically identical for the two enzymes (0.83 vs. 0.72), but the intrinsic Na+ affinity is about two-fold higher for thrombin compared to meizoIIRRΔF1. The value of Kapp (see data at left) can be derived from the equation Kapp = KA/(1 + r) [9] and is in excellent agreement with that determined directly from the titration data shown at left.
Dissection of the contribution of all nine Trp residues of thrombin to the fluorescence change associated with the binding of Na+ (E to E:Na+ transition) and the E*-E interconversion has revealed that all Trp residues distributed on the thrombin surface up to 35 Å away from the bound cation contribute to the fluorescence signal [9]. Hence, the allosteric E*-E and E-E:Na+ interconversions affect the structure of thrombin globally. In addition to the nine Trp residues of the B chain, meizoIIRRΔF1 possesses two additional Trp residues, W194 and W230, both in fragment 2. Our crystal structure (Fig. 3) shows these residues in stacking interaction, perhaps acting as a single fluorophore. Interestingly, W230 is engaged in a cation-π interaction with R93 of the B chain and the entire fragment 2 docks on a surface of recognition of the B chain that involves W96 and W237. The possibility exists that the interactions involving W194 and W230 in fragment 2, W96 and W237 in the B chain may dominate the differences in fluorescence response seen between meizoIIRRΔF1 and thrombin. Future mutagenesis studies will help address this issue directly.
The effect of Na+ has provided new context for understanding thrombin allostery at the molecular level. Na+ binding is required for the procoagulant, prothrombotic and signaling functions of the enzyme and is dispensable for its anticoagulant function [10, 11]. The physiological significance of this effect is illustrated directly by the bleeding consequences of mutations that compromise Na+ binding to thrombin [10, 11]. Studies on the Na+ effect of thrombin have produced significant advances in our understanding of how trypsin-like proteases work and are regulated [25]. Perhaps the most notable of these advances has been the discovery of the E* form from stopped-flow measurements of Na+ binding [9] followed by X-ray crystallography [23]. E* is an inactive form of thrombin that is in equilibrium with the low activity E form in the absence of Na+. E is then converted to the high activity E:Na+ form upon binding of the cation. Because the Na+ affinity is not high enough to saturate the site under physiological [Na+] [10, 11], all three conformations of the enzyme, E*, E and E:Na+ contribute to the activity of thrombin in vivo. The present study demonstrates that meizothrombin, the most important intermediate along the prothrombin activation pathway [5, 46, 47], possesses the same conformational plasticity as the mature enzyme. The Na+ site of meizothrombin desF1 is structurally identical to that of thrombin, and the Na+ affinity is comparable. This implies that when meizothrombin functions as a substrate for the prothrombinase complex under physiological conditions, its various forms E*, E and E:Na+ are significantly populated and may have distinct kinetic properties. Because the interconversion between E* and E occurs on the ms time scale (Fig. 4), which is comparable to that required for substrate binding to and dissociation from the prothrombinase complex, kinetic schemes of prothrombinase action where the substrate meizothrombin is modeled as a single conformation [6] are unlikely to be correct. Alternative kinetic schemes where meizothrombin is assumed to undergo a zymogen-protease conversion when processed by the prothrombinase complex [7] correctly account for conformational plasticity of the substrate. However, these schemes confound E* for a zymogen form and do not account for the Na+ effect converting E to E:Na+. E* is not a zymogen form, because its I16-D194 ion-pair is correctly formed [23]. E and E:Na+ are both significantly populated and likely to feature distinct functional behavior as substrates of prothrombinase, although their interconversion may be considerably faster than other kinetic steps. The possible consequences of Na+ binding to factor Xa on the function of the prothrombinase complex should also be considered in a kinetic scheme of prothrombin processing, given that the activating effect of Na+ on factor Xa has been known for over 30 years [31].
Significant structural differences exist among E*, E and E:Na+ in thrombin [10], so it is realistic to expect similar differences for meizothrombin. The structure reported in this paper documents the Na+ binding site of meizothrombin desF1 bound to PPACK and represents a solid starting point for the future analysis of the free forms of this important enzyme.
Acknowledgments
This work was supported by a Postdoctoral Fellowship from the American Heart Association (to M.E.P.), a Predoctoral Fellowship from Sigma Aldrich to honor Gerty T. Cori (to A.B.) and by NIH research grants HL49413, HL58141 and HL73813 (to E.D.C.). E.D.C. is a Scientific Advisor of HumaGene. HumaGene did not support this work.
References
- 1.Gailani D, Broze GJ., Jr. Factor XI activation in a revised model of blood coagulation. Science. 1991;253:909–912. doi: 10.1126/science.1652157. [DOI] [PubMed] [Google Scholar]
- 2.Girard TJ, MacPhail LA, Likert KM, Novotny WF, Miletich JP, Broze GJ., Jr. Inhibition of factor VIIa-tissue factor coagulation activity by a hybrid protein. Science. 1990;248:1421–1424. doi: 10.1126/science.1972598. [DOI] [PubMed] [Google Scholar]
- 3.Mann KG, Butenas S, Brummel K. The dynamics of thrombin formation. Arterioscler. Thromb. Vasc. Biol. 2003;23:17–25. doi: 10.1161/01.atv.0000046238.23903.fc. [DOI] [PubMed] [Google Scholar]
- 4.Jackson CM, Nemerson Y. Blood coagulation. Annu. Rev. Biochem. 1980;49:765–811. doi: 10.1146/annurev.bi.49.070180.004001. [DOI] [PubMed] [Google Scholar]
- 5.Doyle MF, Mann KG. Multiple active forms of thrombin. IV. Relative activities of meizothrombins. J. Biol. Chem. 1990;265:10693–10701. [PubMed] [Google Scholar]
- 6.Kim PY, Nesheim ME. Further evidence for two functional forms of prothrombinase each specific for either of the two prothrombin activation cleavages. J. Biol. Chem. 2007;282:32568–32581. doi: 10.1074/jbc.M701781200. [DOI] [PubMed] [Google Scholar]
- 7.Bianchini EP, Orcutt SJ, Panizzi P, Bock PE, Krishnaswamy S. Ratcheting of the substrate from the zymogen to proteinase conformations directs the sequential cleavage of prothrombin by prothrombinase. Proc. Natl. Acad. Sci. 2005;102:10099–10104. doi: 10.1073/pnas.0504704102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wells CM, Di Cera E. Thrombin is a Na(+)-activated enzyme. Biochemistry. 1992;31:11721–11730. doi: 10.1021/bi00162a008. [DOI] [PubMed] [Google Scholar]
- 9.Bah A, Garvey LC, Ge J, Di Cera E. Rapid kinetics of Na+ binding to thrombin. J. Biol. Chem. 2006;281:40049–40056. doi: 10.1074/jbc.M608600200. [DOI] [PubMed] [Google Scholar]
- 10.Di Cera E. Thrombin. Mol Aspects Med. 2008;29:203–254. doi: 10.1016/j.mam.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Di Cera E, Page MJ, Bah A, Bush-Pelc LA, Garvey LC. Thrombin allostery. Phys. Chem. Chem. Phys. 2007;9:1292–1306. doi: 10.1039/b616819a. [DOI] [PubMed] [Google Scholar]
- 12.Dang OD, Vindigni A, Di Cera E. An allosteric switch controls the procoagulant and anticoagulant activities of thrombin. Proc. Natl. Acad. Sci. USA. 1995;92:5977–5981. doi: 10.1073/pnas.92.13.5977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dang QD, Guinto ER, Di Cera E. Rational engineering of activity and specificity in a serine protease. Nat. Biotechnol. 1997;15:146–149. doi: 10.1038/nbt0297-146. [DOI] [PubMed] [Google Scholar]
- 14.Di Cera E, Dang QD, Ayala YM. Molecular mechanisms of thrombin function. Cell. Mol. Life Sci. 1997;53:701–730. doi: 10.1007/s000180050091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ayala YM, Cantwell AM, Rose T, Bush LA, Arosio D, Di Cera E. Molecular mapping of thrombin-receptor interactions. Proteins. 2001;45:107–116. doi: 10.1002/prot.1130. [DOI] [PubMed] [Google Scholar]
- 16.Myles T, Yun TH, Hall SW, Leung LL. An extensive interaction interface between thrombin and factor V is required for factor V activation. J. Biol. Chem. 2001;276:25143–25149. doi: 10.1074/jbc.M011324200. [DOI] [PubMed] [Google Scholar]
- 17.Nogami K, Zhou Q, Myles T, Leung LL, Wakabayashi H, Fay PJ. Exosite-interactive regions in the A1 and A2 domains of factor VIII facilitate thrombin-catalyzed cleavage of heavy chain. J. Biol. Chem. 2005;280:18476–18487. doi: 10.1074/jbc.M412778200. [DOI] [PubMed] [Google Scholar]
- 18.Yun TH, Baglia FA, Myles T, Navaneetham D, Lopez JA, Walsh PN, Leung LL. Thrombin activation of factor XI on activated platelets requires the interaction of factor XI and platelet glycoprotein Ib alpha with thrombin anion-binding exosites I and II, respectively. J. Biol. Chem. 2003;278:48112–48119. doi: 10.1074/jbc.M306925200. [DOI] [PubMed] [Google Scholar]
- 19.Di Cera E, Guinto ER, Vindigni A, Dang QD, Ayala YM, Wuyi M, Tulinsky A. The Na+ binding site of thrombin. J. Biol. Chem. 1995;270:22089–22092. doi: 10.1074/jbc.270.38.22089. [DOI] [PubMed] [Google Scholar]
- 20.Pineda AO, Carrell CJ, Bush LA, Prasad S, Caccia S, Chen ZW, Mathews FS, Di Cera E. Molecular dissection of Na+ binding to thrombin. J. Biol. Chem. 2004;279:31842–31853. doi: 10.1074/jbc.M401756200. [DOI] [PubMed] [Google Scholar]
- 21.Nayal M, Di Cera E. Valence screening of water in protein crystals reveals potential Na+ binding sites. J. Mol. Biol. 1996;256:228–234. doi: 10.1006/jmbi.1996.0081. [DOI] [PubMed] [Google Scholar]
- 22.Gandhi PS, Chen Z, Mathews FS, Di Cera E. Structural identification of the pathway of long-range communication in an allosteric enzyme. Proc. Natl. Acad. Sci. USA. 2008;105:1832–1837. doi: 10.1073/pnas.0710894105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pineda AO, Chen ZW, Bah A, Garvey LC, Mathews FS, Di Cera E. Crystal structure of thrombin in a self-inhibited conformation. J. Biol. Chem. 2006;281:32922–32928. doi: 10.1074/jbc.M605530200. [DOI] [PubMed] [Google Scholar]
- 24.Dang QD, Di Cera E. Residue 225 determines the Na(+)-induced allosteric regulation of catalytic activity in serine proteases. Proc. Natl. Acad. Sci. USA. 1996;93:10653–10656. doi: 10.1073/pnas.93.20.10653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Page MJ, Di Cera E. Serine peptidases: classification, structure and function. Cell. Mol. Life Sci. 2008;65:1220–1236. doi: 10.1007/s00018-008-7565-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krem MM, Di Cera E. Molecular markers of serine protease evolution. Embo. J. 2001;20:3036–3045. doi: 10.1093/emboj/20.12.3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Petrovan RJ, Ruf W. Role of residue Phe225 in the cofactor-mediated, allosteric regulation of the serine protease coagulation factor VIIa. Biochemistry. 2000;39:14457–14463. doi: 10.1021/bi0009486. [DOI] [PubMed] [Google Scholar]
- 28.Schmidt AE, Stewart JE, Mathur A, Krishnaswamy S, Bajaj SP. Na+ site in blood coagulation factor IXa: effect on catalysis and factor VIIIa binding. J. Mol. Biol. 2005;350:78–91. doi: 10.1016/j.jmb.2005.04.052. [DOI] [PubMed] [Google Scholar]
- 29.Rezaie AR, He X. Sodium binding site of factor Xa: role of sodium in the prothrombinase complex. Biochemistry. 2000;39:1817–1825. doi: 10.1021/bi992006a. [DOI] [PubMed] [Google Scholar]
- 30.Rezaie AR, Kittur FS. The critical role of the 185-189-loop in the factor Xa interaction with Na+ and factor Va in the prothrombinase complex. J. Biol. Chem. 2004;279:48262–48269. doi: 10.1074/jbc.M409964200. [DOI] [PubMed] [Google Scholar]
- 31.Orthner CL, Kosow DP. The effect of metal ions on the amidolytic acitivity of human factor Xa (activated Stuart-Prower factor) Arch. Biochem. Biophys. 1978;185:400–406. doi: 10.1016/0003-9861(78)90182-0. [DOI] [PubMed] [Google Scholar]
- 32.Monnaie D, Arosio D, Griffon N, Rose T, Rezaie AR, Di Cera E. Identification of a binding site for quaternary amines in factor Xa. Biochemistry. 2000;39:5349–5354. doi: 10.1021/bi9926781. [DOI] [PubMed] [Google Scholar]
- 33.Underwood MC, Zhong D, Mathur A, Heyduk T, Bajaj SP. Thermodynamic linkage between the S1 site, the Na+ site, and the Ca2+ site in the protease domain of human coagulation factor xa. Studies on catalytic efficiency and inhibitor binding. J. Biol. Chem. 2000;275:36876–36884. doi: 10.1074/jbc.M001386200. [DOI] [PubMed] [Google Scholar]
- 34.Camire RM. Prothrombinase assembly and S1 site occupation restore the catalytic activity of FXa impaired by mutation at the sodium-binding site. J. Biol. Chem. 2002;277:37863–37870. doi: 10.1074/jbc.M203692200. [DOI] [PubMed] [Google Scholar]
- 35.Levigne S, Thiec F, Cherel S, Irving JA, Fribourg C, Christophe OD. Role of the alpha-helix 163-170 in factor Xa catalytic activity. J. Biol. Chem. 2007;282:31569–31579. doi: 10.1074/jbc.M704837200. [DOI] [PubMed] [Google Scholar]
- 36.He X, Rezaie AR. Identification and characterization of the sodium-binding site of activated protein C. J. Biol. Chem. 1999;274:4970–4976. doi: 10.1074/jbc.274.8.4970. [DOI] [PubMed] [Google Scholar]
- 37.Steiner SA, Amphlett GW, Castellino FJ. Stimulation of the amidase and esterase activity of activated bovine plasma protein C by monovalent cations. Biochem. Biophys. Res. Commun. 1980;94:340–347. doi: 10.1016/s0006-291x(80)80226-9. [DOI] [PubMed] [Google Scholar]
- 38.Steiner SA, Castellino FJ. Kinetic studies of the role of monovalent cations in the amidolytic activity of activated bovine plasma protein C. Biochemistry. 1982;21:4609–4614. doi: 10.1021/bi00262a015. [DOI] [PubMed] [Google Scholar]
- 39.Steiner SA, Castellino FJ. Effect of monovalent cations on the pre-steady-state kinetic parameters of the plasma protease bovine activated protein C. Biochemistry. 1985;24:1136–1141. doi: 10.1021/bi00326a011. [DOI] [PubMed] [Google Scholar]
- 40.Steiner SA, Castellino FJ. Kinetic mechanism for stimulation by monovalent cations of the amidase activity of the plasma protease bovine activated protein C. Biochemistry. 1985;24:609–617. doi: 10.1021/bi00324a011. [DOI] [PubMed] [Google Scholar]
- 41.Zhang E, Tulinsky A. The molecular environment of the Na+ binding site of thrombin. Biophys. Chem. 1997;63:185–200. doi: 10.1016/s0301-4622(96)02227-2. [DOI] [PubMed] [Google Scholar]
- 42.Scharer K, Morgenthaler M, Paulini R, Obst-Sander U, Banner DW, Schlatter D, Benz J, Stihle M, Diederich F. Quantification of cation-pi interactions in protein-ligand complexes: crystal-structure analysis of Factor Xa bound to a quaternary ammonium ion ligand. Angew. Chem. Int. Ed. Engl. 2005;44:4400–4404. doi: 10.1002/anie.200500883. [DOI] [PubMed] [Google Scholar]
- 43.Bajaj SP, Schmidt AE, Agah S, Bajaj MS, Padmanabhan K. High Resolution Structures of p-Aminobenzamidine- and Benzamidine-VIIa/Soluble Tissue Factor: Unpredicted conformation of the 192-193 peptide bond and mapping of Ca2+, Mg2+, Na+, AND Zn2+ sites in Factor VIIa. J. Biol. Chem. 2006;281:24873–24888. doi: 10.1074/jbc.M509971200. [DOI] [PubMed] [Google Scholar]
- 44.Schmidt AE, Padmanabhan K, Underwood MC, Bode W, Mather T, Bajaj SP. Thermodynamic linkage between the S1 site, the Na+ site, and the Ca2+ site in the protease domain of human activated protein C (APC). Sodium ion in the APC crystal structure is coordinated to four carbonyl groups from two separate loops. J. Biol. Chem. 2002;277:28987–28995. doi: 10.1074/jbc.M201892200. [DOI] [PubMed] [Google Scholar]
- 45.Kroh HK, Tans G, Nicolaes GAF, Rosing J, Bock PE. Expression of allosteric linkage between the sodium ion binding site and exosite I of thrombin during prothrombin activation. J. Biol. Chem. 2007;282:16095–16104. doi: 10.1074/jbc.M610577200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cote HC, Stevens WK, Bajzar L, Banfield DK, Nesheim ME, MacGillivray RT. Characterization of a stable form of human meizothrombin derived from recombinant prothrombin (R155A, R271A, and R284A) J. Biol. Chem. 1994;269:11374–11380. [PubMed] [Google Scholar]
- 47.Petrovan RJ, Govers-Riemslag JW, Nowak G, Hemker HC, Tans G, Rosing J. Autocatalytic peptide bond cleavages in prothrombin and meizothrombin. Biochemistry. 1998;37:1185–1191. doi: 10.1021/bi971948h. [DOI] [PubMed] [Google Scholar]
- 48.Vindigni A, Di Cera E. Release of fibrinopeptides by the slow and fast forms of thrombin. Biochemistry. 1996;35:4417–4426. doi: 10.1021/bi952834d. [DOI] [PubMed] [Google Scholar]
- 49.Gianni S, Ivarsson Y, Bah A, Bush-Pelc LA, Di Cera E. Mechanism of Na+ binding to thrombin resolved by ultra-rapid kinetics. Biophys. Chem. 2007;131:111–114. doi: 10.1016/j.bpc.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Orthner CL, Kosow DP. Evidence that human alpha-thrombin is a monovalent cation-activated enzyme. Arch. Biochem. Biophys. 1980;202:63–75. doi: 10.1016/0003-9861(80)90406-3. [DOI] [PubMed] [Google Scholar]
- 51.Carrell CJ, Bush LA, Mathews FS, Di Cera E. High resolution crystal structures of free thrombin in the presence of K+ reveal the basis of monovalent cation selectivity and an inactive slow form. Biophys. Chem. 2006;121:177–184. doi: 10.1016/j.bpc.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 52.Papaconstantinou ME, Bah A, Di Cera E. Role of the A chain in thrombin function. Cell. Mol. Life Sci. 2008;65:1943–1947. doi: 10.1007/s00018-008-8179-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lai MT, Di Cera E, Shafer JA. Kinetic pathway for the slow to fast transition of thrombin. Evidence of linked ligand binding at structurally distinct domains. J. Biol. Chem. 1997;272:30275–30282. doi: 10.1074/jbc.272.48.30275. [DOI] [PubMed] [Google Scholar]
- 54.Otwinowski Z, Minor W. Processing of x-ray diffraction data collected by oscillation methods. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 55.Bailey S. The CCP4 suite. Programs for protein crystallography. Acta Crystallogr. D. Biol. Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 56.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D. Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 57.Morris AL, MacArthur MW, Hutchinson EG, Thornton JM. Stereochemical quality of protein structure coordinates. Proteins. 1992;12:345–364. doi: 10.1002/prot.340120407. [DOI] [PubMed] [Google Scholar]
- 58.Martin PD, Malkowski MG, Box J, Esmon CT, Edwards BF. New insights into the regulation of the blood clotting cascade derived from the X-ray crystal structure of bovine meizothrombin des F1 in complex with PPACK. Structure. 1997;5:1681–1693. doi: 10.1016/s0969-2126(97)00314-6. [DOI] [PubMed] [Google Scholar]
- 59.Di Cera E. A structural perspective on enzymes activated by monovalent cations. J. Biol. Chem. 2006;281:1305–1308. doi: 10.1074/jbc.R500023200. [DOI] [PubMed] [Google Scholar]
- 60.Nayal M, Di Cera E. Predicting Ca(2+)-binding sites in proteins. Proc. Natl. Acad. Sci. USA. 1994;91:817–821. doi: 10.1073/pnas.91.2.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Noskov SY, Berneche S, Roux B. Control of ion selectivity in potassium channels by electrostatic and dynamic properties of carbonyl ligands. Nature. 2004;431:830–834. doi: 10.1038/nature02943. [DOI] [PubMed] [Google Scholar]
- 62.Noskov SY, Roux B. Control of ion selectivity in LeuT: two Na+ binding sites with two different mechanisms. J. Mol. Biol. 2008;377:804–818. doi: 10.1016/j.jmb.2008.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Griffon N, Di Stasio E. Thermodynamics of Na+ binding to coagulation serine proteases. Biophys. Chem. 2001;90:89–96. doi: 10.1016/s0301-4622(01)00129-6. [DOI] [PubMed] [Google Scholar]
- 64.Prasad S, Wright KJ, Roy DB, Bush LA, Cantwell AM, Di Cera E. Redesigning the monovalent cation specificity of an enzyme. Proc. Natl. Acad. Sci. USA. 2003;100:13785–13790. doi: 10.1073/pnas.2333109100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Arni RK, Padmanabhan K, Padmanabhan KP, Wu TP, Tulinsky A. Structures of the noncovalent complexes of human and bovine prothrombin fragment 2 with human PPACK-thrombin. Biochemistry. 1993;32:4727–4737. doi: 10.1021/bi00069a006. [DOI] [PubMed] [Google Scholar]
- 66.Carter WJ, Cama E, Huntington JA. Crystal structure of thrombin bound to heparin. J. Biol. Chem. 2005;280:2745–2749. doi: 10.1074/jbc.M411606200. [DOI] [PubMed] [Google Scholar]
- 67.Pineda AO, Chen ZW, Marino F, Mathews FS, Mosesson MW, Di Cera E. Crystal structure of thrombin in complex with fibrinogen gamma’ peptide. Biophys. Chem. 2007;125:555–559. doi: 10.1016/j.bpc.2006.08.005. [DOI] [PubMed] [Google Scholar]