

Abstract
Metallo-β-Lactamases (MBLs) protect bacteria from almost all β-lactam antibiotics. VIM enzymes are among the most clinically important MBLs, with VIM-1 increasing in carbapenem-resistant Enterobacteriaceae (Escherichia coli, Klebsiella pneumoniae) that are amongst the hardest bacterial pathogens to treat. VIM enzymes display sequence variation at residues (224 and 228) that in related MBLs are conserved and participate in substrate binding. How they accommodate this variability, while retaining catalytic efficiency against a broad substrate range, has remained unclear. Here we present crystal structures of VIM-1 and its complexes with a substrate-mimicking thioenolate inhibitor, ML302F, that restores meropenem activity against a range of VIM-1 producing clinical strains, and the hydrolysed product of the carbapenem meropenem. Comparison of these two structures identifies a water-mediated hydrogen bond, between the carboxylate group of substrate/inhibitor and the backbone carbonyl of the active site zinc ligand Cys221, that is common to both complexes. Structural comparisons show that the responsible Cys221-bound water is observed in all known VIM structures, participates in carboxylate binding with other inhibitor classes, and thus effectively replicates the role of the conserved Lys224 in analogous complexes with other MBLs. These results provide a mechanism for substrate binding that permits the variation at positions 224 and 228 that is a hallmark of VIM MBLs.
Keywords: antibiotic resistance, X-ray crystallography, hydrolase, metalloenzyme, zinc, metallo-β-lactamase, VIM-1, carbapenem
Graphical Abstract
VIM metallo-β-lactamases protect bacteria from most β-lactam antibiotics while escaping clinical inhibitors, but vary at residues (224, 228) interacting with substrate in related enzymes. Structures of VIM-1 complexes with hydrolysed meropenem (carbapenem antibiotic) and a thioenolate inhibitor identify hydrogen bonding, via a conserved water molecule (Wat3), that enables retention of activity despite sequence variation at apparently essential residues.
Introduction
Antibiotic resistance is of immediate and growing concern to global public health [1]. Resistance in opportunistic Gram-negative bacterial pathogens is of pressing importance as these organisms cause an increasing frequency of healthcare-associated infections (HCAIs) in immunocompromised individuals and treatment options are limited by both intrinsic and acquired resistance mechanisms [2]. β-Lactams, which continue to form over half of the global antibacterial market [3], remain key agents for treatment of such infections, with the carbapenems in particular rapidly supplanting third generation cephalosporins as first choice drugs. In pathogens such as the Enterobacteriaceae (e.g. Escherichia coli; Klebsiella pneumoniae) or non-fermenting organisms (e.g. Pseudomonas aeruginosa; Acinetobacter baumannii) production of β-lactamases that inactivate β-lactams via hydrolysis of the β-lactam ring [4], is the major form of β-lactam resistance [5]. Over 1300 such enzymes have now been identified in isolates of diverse clinical origin [6]. While the spectrum of activity against different β-lactam classes varies between enzymes, carbapenem-hydrolyzing β-lactamases are attracting increasing attention due to the efficiency with which they hydrolyze most β-lactam classes and the growing frequency with which they are isolated from patients [7].
β-Lactamases comprise two distinct groups - serine (SBL) and metallo- (MBL) β-lactamases - which differ in their structure and catalytic mechanisms [8]. Clinically available inhibitors (e.g. clavulanic acid, avibactam) are active against many, though not all, SBLs but all are ineffective against the MBLs [9]. In addition, while carbapenems effectively inhibit most SBLs [10], all known MBLs effectively hydrolyze these antibiotics [11]. In consequence, MBL dissemination poses a particular challenge to the continued effectiveness of β-lactams, especially carbapenems, against Gram-negative bacteria.
MBLs possess a conserved protein fold that forms an αββα sandwich with the active site zinc center found in a central groove on one edge of the two β-sheets [12]. Based upon differences in sequence and active site structures, the MBLs are divided into three sub-groups (B1, B2 and B3 [13]) with the most clinically relevant IMP, VIM, and NDM enzymes [14] contained within the subclass B1. The B1 active site binds two zinc ions, located respectively in the (usually tetrahedral) Zn1 site formed by the conserved histidines 116, 118 and 196; and the Zn2 site comprising Asp120, Cys221 and His263 [15]. (The standard MBL numbering scheme [16] is used throughout.) It is generally accepted that B1 enzymes require both zinc sites for maximal catalytic efficiency [17, 18].
The VIM (Verona Integron-encoded MBL) enzymes are one of the most widely distributed MBL families and are a leading cause of carbapenem failure, particularly in P. aeruginosa [19]. blaVIM-1 was identified in 1999 as a chromosomally encoded gene on a cassette within a class I integron in a P. aeuruginosa clinical isolate from Italy [20]. Subsequently, VIM enzymes have been identified on ‘multiresistance’ plasmids from multiple Gram-negative species; over 50 different VIM variants have now been identified [21]. Of these VIM-1 is noteworthy as it is most frequently encountered in the Enterobacteriaceae (K. pneumoniae and E. coli [22]); most other VIM types are primarily associated with the less common non-fermenting pathogens, in particular P. aeruginosa. VIM (and other B1) MBLs have a broad spectrum of activity encompassing penicillins, cephalosporins and carbapenems ([23] and references therein).
Sequence identity between VIM variants ranges from ~75% to >99%, with VIM-1 and VIM-2 progenitors of two dominant phylogenetic clusters and VIM-7 as a uniquely divergent variant [23]. Sequence substitutions at residues 224 and 228 are a hallmark of VIM variants and of particular interest as these positions interact with β-lactam substrates in other MBLs [24, 25]. Although several crystal structures of VIM enzymes are now available [24] [26] [27] [28], including of the VIM-1 point variants VIM-4 [29],and VIM-26 [25], structures of the progenitor enzyme VIM-1 have not previously been reported. Notably, from both inhibitor design and mechanistic perspectives, there is as yet no structural information describing how any VIM enzyme binds β-lactams. Here we focus on structural investigations of VIM-1 and its interactions with a non-β-lactam inhibitor that mimics β-lactam binding, ML302F[30], as well as a clinically relevant carbapenem substrate, meropenem. Our results reveal how substrate/inhibitor binding by VIM enzymes is tolerant of substitutions at sequence positions that in other MBLs make interactions, notably those involving the β-lactam carboxylate, that are crucial for activity.
Results
Crystal Structure of VIM-1 -
Crystals of VIM-1 formed after several days’ incubation at 20 °C and diffracted to near atomic resolution (1.29 Å; Table 1). Initial analysis showed these to be of space group P 21 with a solvent content of 41.4 %, indicating one molecule in the asymmetric unit. Structure solution by molecular replacement, and subsequent model building, were straightforward, with the complete chain traced without interruption between residues Gly25 and His293 giving a final model containing 233 residues. The structure displays the overall αβ/βα fold of the MBL superfamily, with the binuclear zinc center that forms the active site situated in a shallow groove formed by the interface of the two αβ domains (Figure 1A). Two extended loops, termed L3 (residues 60 – 66) and L10 (residues 221 – 241) border the active site. Superposition of the VIM-1 structure with those of other VIM family members using PDBeFOLD [31] yields RMSD values for Cα-atoms of between 0.28 Å (VIM-26, pdb 4UWP [25]) and 0.83 Å (VIM-31 (oxidized) pdb 4FSB [28]) Å, demonstrating that only small differences are evident between the overall fold of VIM-1 and other structurally characterized VIM variants (Figure 1B). These differences are largely localized to the L3, and to a lesser extent the L10, loops.
Table 1.
Data Collection and Refinement Statistics
| Data Collection | Native VIM-1 | VIM-1:ML302F | VIM-1:Meropenem |
|---|---|---|---|
| X-ray Source | DLS (I04) | DLS (I02) | DLS (I24) |
| Wavelength (Å) | 0.9795 | 0.9795 | 0.9686 |
| Space Group | P21 | P21 | P21 |
| Cell Dimensions | |||
| a, b, c (Å) | 39.76, 67.94, 40.36 | 39.68, 67.65, 40.22 | 38.94, 68.08, 39.82 |
| α, β, γ | 90, 94.01, 90 | 90, 91.36, 90 | 90, 90, 90 |
| Molecules/ asymmetric unit | 1 | 1 | 1 |
| Resolution (Å) | 40.26 – 1.29 (1.31 – 1.29) | 28.58 – 1.30 (1.32 – 1.30) | 27.84 – 2.20 (2.28 – 2.20) |
| No. of unique reflections | 61346 | 52058 | 10547 |
| Redundancy | 3.6 (3.4) | 6.4 (6.1) | 5.8 (5.7) |
| Rpim | 0.039 (0.326) | 0.033 (0.201) | 0.081 (0.240) |
| CC1/2 | 0.997 (0.763) | 0.997 (0.881) | 0.988 (0.870) |
| I/σ | 12.5 (2.9) | 21.5 (7.9) | 8.5 (4.2) |
| Completeness (%) | 99.3 (98.7) | 99.8 (99.4) | 99.2 (98.6) |
| Refinement | |||
| Resolution (Å) | 34.25 – 1.29 | 28.58 – 1.30 | 27.84 – 2.20 |
| No. of reflections | 53159 | 52032 | 10543 |
| Rwork/Rfree | 0.1445/0.1563 | 0.1545/0.1678 | 0.1526/0.2250 |
| Atoms | |||
| Protein | 3432 | 3496 | 1755 |
| Ligand | N/A | 19 | 27 |
| Zinc | 3 | 2 | 3 |
| Solvent | 328 | 237 | 85 |
| B factor (Å2) | |||
| Protein | 15.19 | 15.32 | 16.99 |
| Ligand | N/A | 15.96 | 39.13 |
| Zinc | 16.15 | 8.34 | 25.96 |
| Solvent | 29.62 | 28.42 | 22.35 |
| RMSD | |||
| Bonds (Å) | 0.008 | 0.020 | 0.006 |
| Angles (°) | 1.262 | 1.672 | 1.076 |
| PDB accession | 5N5G | 5N5H | 5N5I |
5% of reflections were set aside for Rfree calculation. iMosflm, XDS, Aimless and Phaser MR were used for structure solution and Phenix for refinement.
Figure 1. Crystal Structure of VIM-1.

A. View of the crystal structure of di-zinc VIM-1 showing the overall fold and location of active site. Protein main chain is colour-ramped from blue (N-) to red (C-terminus). Active site residues are rendered as sticks; zinc ions as grey spheres. Secondary structure elements are labeled. B. Superposition of di-zinc VIM-1 with structures of other VIM enzymes (VIM-2, pdb 4BZ3; VIM-4, pdb 2WHG; VIM-5, pdb 5A87; VIM-7, pdb 4D1T; VIM-26, pdb 4UWO; VIM-31, pdb 4FR7; coloured as shown). C. Stereoview of di-zinc VIM-1 active site. Electron density shown is 2|Fo| - |Fc| Φcalc contoured at 1.5 sigma. This Figure was created using PyMol (www.pymol.org).
Metal ions observed in both metal sites were refined as zinc ions, with occupancies of 0.94 (Zn1) and 0.73 (Zn2), respectively. Although reduced occupancy was observed for the Zn2 site, the Cys221 ligand was refined as the fully reduced form. An additional zinc ion was present (occupancy 0.54) bound to a bicine buffer molecule at the protein surface. The two active site zinc ions are separated by a distance of 3.62 Å with Zn1 in a tetrahedral geometry and Zn2 showing octahedral coordination with one vacant position ([32]Figure 1C). Two well defined water molecules are present in the active site: a bridging water/hydroxide (Wat1, B-factor 14.65 Å2) positioned asymmetrically between the two zinc ions, lying closer (1.89 Å) to Zn1 than to Zn2 (2.18 Å); and a second water molecule (Wat2, B-factor 21.67 Å2) apparently tightly (2.06 Å) bound to Zn2.
Functionally significant differences between VIM variants have been associated primarily with substitutions at positions 224 and 228 on loop L10 [23, 26] where VIM-1 possesses His and Ser residues, respectively (Figure 2A). Notably, in our VIM-1 structure His224 is oriented by a strong hydrogen bond (2.92 Å) between Oγ of Ser228 and Nδ1 of His224, such that the side chain imidazole ring forms one wall of the active site cleft (Figure 2A). In contrast, in structures of enzymes such as VIM-4, VIM-7 and VIM-31, His224 Nδ1 instead is H-bonded to Nω1 of Arg228, apparently causing rotation of the His224 side chain to lie perpendicular to its position in VIM-1 [27–29]. In VIM-1, the orientation of the His224 side chain more closely resembles that of the phenolic ring of Tyr224 in VIM-2 (Figure 2B). Moreover, in the VIM-2, −4, −7 and −31 structures, the side chain of Arg228 protrudes into the active site groove towards the side chain of Tyr67, which is located at the base of the L3 loop on the opposite side of the cleft. Thus, substitution of Arg228 for Ser in VIM-1 (and in VIM-26 [25] where His224 is substituted by Leu) expands the active site cleft compared to other VIM enzymes of known structure.
Figure 2. Comparison of VIM Active Sites.

A. Active site of VIM-1, showing positions of His224 and Ser228 and location of Cys221-bound water Wat3. Hydrogen bonding interactions are shown as dashed lines. B. Active site superpositions of VIM-2 (pdb 4BZ3, blue) and VIM-4 (pdb 2WHG, red) showing variations at positions 224 and 228.
Inhibition of VIM-1 by the Thioenolate ML302F -
Despite their sequence differences, and the growing clinical importance of VIM-1, many studies of MBL inhibitors have used VIM-2 as a representative VIM enzyme. Recently, we reported that the thioenolate hydrolysis product, ML302F, of the rhodanine ML302, is a potent (sub-micromolar) inhibitor of both VIM-1 and VIM-2 in vitro [30], and proposed that ML302F binding to MBLs mimics that of β-lactam substrates/intermediates. Accordingly, we sought to extend these findings by studying the interactions of ML302F with VIM-1 in producer bacteria and by X-ray crystallography.
To investigate interactions of ML302F with VIM-1 in bacteria, we determined minimal inhibitory concentrations (MICs) for the clinically important carbapenem antibiotic meropenem by broth microdilution for 27 VIM-1 expressing K. pneumoniae and E. coli clinical strains in the presence and absence of 10 μg/ml ML302F (Table 2). All 20 K. pneumoniae strains could be considered as resistant to meropenem according to either CLSI or EUCAST clinical breakpoints; however only 2/7 E. coli could be classed as resistant, with the rest showing meropenem susceptibility that, while substantially reduced compared to control, was still within the susceptible range. For 8/20 K. pneumoniae strains the meropenem MIC in the presence of ML302F changes from resistant to susceptible according to EUCAST breakpoints, with a further 6 strains changing from resistant to intermediate susceptibility. Of the remaining 6 strains tested, 5 showed MIC reductions of one (2-fold) to two (4-fold) dilutions, with one being unaffected. ML302F reduced the meropenem MICs of the 2 resistant E. coli strains to susceptible in one case and to intermediate in the other. In the 5 susceptible strains, reductions of at least 2 dilutions (4-fold or more) were observed in 4 cases, with one strain apparently unaffected. These results show that ML302F can potentiate the activity of meropenem against clinical isolates of VIM-1 expressing Enterobacteriaceae, and thus that this compound can penetrate clinically relevant bacteria to act as an effective inhibitor of VIM-1 in the bacterial host.
Table 2.
Effect of ML302F on Meropenem Minimal Inhibitory Concentrations for VIM-1-Expressing Enterobacteriaceae
| Strain | Meropenem MIC (μg ml−1) | Meropenem Resistant/Intermediate/Susceptible (clinical strains; EUCAST*) | Meropenem MIC (μg ml−1) + 10 μg ml−1 ML302F | Meropenem Resistant/Intermediate/Susceptible (clinical strains; EUCAST*) | Fold difference in MIC |
|---|---|---|---|---|---|
| E. coli TOP1O (control) | ≤ 0.25 | ≤ 0.25 | 0 | ||
| K. pneumoniae NCTC 5055 (control) | ≤ 0.25 | ≤ 0.25 | 0 | ||
| K. pneumoniae Kpn20 | 32 | R | 8 | R | 4 |
| K. pneumoniae 08Y70 | 32 | R | 2 | S | 16 |
| K. pneumoniae 08Z37 | 16 | R | 4 | I | 4 |
| K. pneumoniae 09A69 | 32 | R | 4 | I | 8 |
| K. pneumoniae 09B51 | 8 | R | 1 | S | 8 |
| K. pneumoniae 09B53 | 8 | R | 1 | S | 8 |
| K. pneumoniae 09B61 | 32 | R | 4 | I | 8 |
| K. pneumoniae 09B76 | 64 | R | 32 | R | 2 |
| K. pneumoniae 09C12 | 64 | R | 16 | R | 4 |
| K. pneumoniae 09C74 | 256 | R | 128 | R | 2 |
| K. pneumoniae 09C77 | 32 | R | 4 | I | 8 |
| K. pneumoniae 09D21 | 16 | R | 1 | S | 16 |
| K. pneumoniae 10D60 | 16 | R | 2 | S | 8 |
| K. pneumoniae 10F53 | 16 | R | 2 | S | 8 |
| K. pneumoniae 10F74 | 32 | R | 8 | R | 4 |
| K. pneumoniae 1–57 | 16 | R | 2 | S | 8 |
| K. pneumoniae 1–60 | 16 | R | 4 | I | 4 |
| K. pneumoniae 1–61 | 128 | R | 128 | R | 0 |
| K. pneumoniae 1–70 | 32 | R | 4 | I | 8 |
| K. pneumoniae 10I28 | 16 | R | 0.5 | S | 32 |
| E. coli08Y79 | 8 | R | 2 | S | 4 |
| E. coli 09B20 | 2 | S | 0.5 | S | 4 |
| E. coli 09D25 | 2 | S | ≤ 0.25 | S | ≥ 8 |
| E. coli 1–37 | 1 | S | ≤ 0.25 | S | ≥ 4 |
| E. coli 1–47 | 16 | R | 4 | I | 4 |
| E. coli 10F75 | 2 | S | 2 | S | 0 |
| E. coli 2–4 | 2 | S | 0.5 | S | 4 |
Crystal Structure of VIM-1:ML302F Complex -
We next sought to obtain structural information on the binding of ML302F to VIM-1. An ML302F:VIM-1 complex was obtained from a co-crystal, of the same symmetry as native VIM-1, that yielded a diffraction dataset complete to 1.30 Å resolution. 234 amino acids were built into this structure with four amino acids missing from the N-, and two from the C-, terminus. Five residues were built with alternative conformations. The Zn1 site was modeled with occupancy of 1.0, and the Zn2 site with occupancy 0.94. Initial electron density maps featured positive difference peaks in the active site that indicated the likely presence of bound ligand and into which ML302F could be readily fitted (Figure 3A). ML302F was refined with occupancy 0.91 with a B-factor (15.96 Å2) comparable to that of the protein (15.32 Å2).
Figure 3. Interactions of Thioenolate Inhibitor ML302F with VIM-1.

A. Structure of ML302F:VIM-1 Complex. VIM-1 main chain Cα atoms are colour-ramped from blue (N-) to red (C-terminus). Active site residues are rendered as sticks; zinc ions as gray spheres, water molecules as red spheres. Inhibitor carbon atoms are in cyan, side chain carbons in green. Other atom colours are as standard. Electron density shown is |Fo| - |Fc| Φcalc contoured at 3 sigma around inhibitor and calculated with the ligand omitted. Hydrogen bonding interactions are shown as dashed lines. Inset shows structure of ML302F. B. Map of VIM-1:ML302F interactions. Zinc ions are rendered in green; water molecules in cyan; other atom colours as standard. The Figure was generated using LIGPLOT [59].
Binding of ML302F (Figure 3) manifests displacement of the di-zinc ion bridging water molecule Wat1 by the inhibitor thiol, which intercalates between the two zinc ions (distances 2.32 Å and 2.38 Å from Zn1 and Zn2, respectively). The inhibitor carboxylate displaces Wat2 to make an electrostatic interaction with Zn2 (distance 2.37 Å), with the consequence that Zn2 is five-fold coordinated in a trigonal bipyramidal geometry. Water-mediated interactions reminiscent of those observed in carbapenem complexes (see below) connect one oxygen atom of the inhibitor C1 carboxylate to the Ala231 carbonyl and His224 Nδ1, and, via an additional water molecule, to the Asn233 backbone amide. A further water molecule, Wat3, connects the Zn2-bound oxygen to the Cys221 carbonyl. The trichlorine substituted phenyl ring of ML302F makes a π-stacking interaction with Phe61 at the base of loop L3, with one of the halogen atoms making a chlorine-π interaction with Trp87.
Interactions of VIM-1 with Hydrolysed Meropenem -
Although recent progress has been made in structural characterization of the interactions of B1 MBLs with hydrolysed β-lactam substrates [33–36], to date there is no reported information on how VIM enzymes bind their β-lactam substrates. Given the clinical importance of the VIM enzymes, and their lack of Lys224, a residue likely crucial in the interaction of most other B1 MBLs with the C2/C3 carboxylate group of β-lactams, information regarding the mode of β-lactam binding to VIM-1 is important both to understanding the mechanism of β-lactam hydrolysis and design of inhibitors. Hence, to investigate the interactions of VIM-1 with β-lactams we soaked native VIM-1 crystals with meropenem with the aim of obtaining structural information on enzyme-bound species.
Inspection of Fo - Fc difference density maps calculated from a diffraction data set collected from a native VIM-1 crystal after overnight exposure to meropenem powder revealed positive density peaks into which hydrolysed meropenem could be refined (Figure 4A). This yielded a structure for a complex to a resolution of 2.20 Å. The structure contains 232 amino acids with five residues missing from the N-, and three from the C-, terminus. Four residues were built with alternative side chain conformations. Zinc ions were refined in the Zn1 (occupancy 1.0) and Zn2 (occupancy 0.87) sites, with a third zinc ion (occupancy 0.55) involved in crystal contacts at the interface of VIM-1 molecules in two adjacent asymmetric units. Notably, Zn2 presented a higher than average B-factor (33.67 Å2 compared to 13.18 Å2 for Zn1).
Figure 4. Electron Density Maps for Hydrolysed Meropenem bound to VIM-1.

A. Stereoview of |Fo| - |Fc| Φcalc electron density (green; calculated with ligand omitted) contoured at 2.5 sigma around hydrolysed meropenem (cyan). Note the position of the S atom out of the plane of the pyrroline ring. B. Stereoview of 2|Fo| - |Fc| Φcalc electron density (blue) and |Fo| - |Fc| Φcalc electron density (negative peak red; both calculated with ligand included) contoured at 1.0 and 3.0 sigma respectively around hydrolysed meropenem (cyan). Note the presence of only very limited negative density close to the C7 carboxylate. Active site residues are rendered as sticks; zinc ions as grey spheres, water molecules as red spheres. Other atom colours are as standard.
Compared to the VIM-1:ML302F complex, binding of hydrolysed meropenem to VIM-2 is less well defined- in addition to the lower resolution, elevated B-factors were observed for the hydrolysed meropenem ligand (overall B-factor of 39.13 Å2 compared to 16.99 Å2 for the protein main chain). Nevertheless, refining the ligand at full occupancy yielded a real-space correlation coefficient (RSCC) of 0.85 and real-space R-value (RSR) of 0.25; parameters that indicate acceptable agreement of observed and calculated electron densities for bound ligand [37]. This is confirmed by visual inspection; omit (|Fo| - |Fc| Φcalc; Figure 4A) electron density maps define the positions of key elements of hydrolysed meropenem - the dihydropyrrole ring and associated N4 nitrogen and C3 carboxylate groups; and the S atom and pyrrolidine ring of the C2 substituent. Appropriate ligand placement is also evident based upon inspection of 2|Fo| - |Fc| Φcalc and associated difference (|Fo| - |Fc| Φcalc) maps (Figure 4B), with the latter providing no strong negative peaks indicating substantial errors in ligand placement. However, omit electron density is weaker for the methyl group attached to C1, the carbapenem C6 hydroxyethyl group and, in particular for the C7 carboxylate that is formed on hydrolysis of the β-lactam ring. This was positioned after trial refinements in several alternative orientations, including in the zinc-bridging position, with the final structure representing the mode of binding that minimised overly close contacts with Zn ions and coordinating residues, such as Asp120, and steric clashes of the C6 hydroxyethyl with the adjacent side chains of Phe61 and Trp87. Hence, as in the uncomplexed VIM-1 structure, Wat1 is closer to Zn1 (1.84 Å) than Zn2 (2.28 Å, Figure 5A, B), and the presence of hydrolysed meropenem has little effect on the Zn – Zn separation distance (3.52 Å compared to 3.62 Å in the uncomplexed structure).
Figure 5. Interactions of Hydrolysed Meropenem with VIM-1.

A. Interactions made by hydrolysed meropenem with VIM-1 active site. Hydrogen bonds are shown as dashed lines. B. Map of VIM-1 interactions with hydrolysed meropenem. Zinc ions are rendered in green; water molecules in cyan; other atom colours as standard. C. Structure of intact and hydrolysed meropenem in Δ2 (B) and Δ1 (C) pyrroline forms.
Carbapenems contain a 4:5 fused β-lactam ring system with a double bond between C2 and C3 in the five-membered pyrroline ring. This results in two possible tautomeric forms (Δ1 and Δ2 pyrroline) for the product resulting from hydrolysis of the β-lactam ring (Figure 5C [10, 38]). In intact meropenem the C2/C3 carbon atoms are sp2 hybridised, due to the double bond between them, and are coplanar with the C2-linked S atom. This configuration can be retained after β-lactam hydrolysis. However, in ring-opened carbapenems tautomerisation of the enamine double bond can occur, giving rise to the Δ1 (imine) form with a double bond between C3 and N4. Our experimental data, at 2.2 Å resolution, cannot permit unambiguous assignment of the tautomeric form present. However, experimental difference electron density maps (Figure 4A) indicate the S atom as lying out of the plane of the pyrroline ring, suggesting that the meropenem C2 is sp3, rather than sp2, hybridized. In consequence we have refined the ligand in the Δ1 (imine), rather than the Δ2, tautomer, and in the (S) (β), rather than (R) (α) stereochemistry (Figures 4, 5).
These limitations notwithstanding, the comparatively strong electron density observed for elements of the ligand interacting with the MBL metal centre (N4 nitrogen and C3 carboxylate groups) defines key interactions of VIM-1 with hydrolysed meropenem (Figure 5A, B) that involve the Zn2 metal ion. Zn2 displays octahedral co-ordination, contacting O1 of the substrate C3 carboxylate (in the Wat2 position of the native active site, 2.57 Å) and N4 of the hydrolysed β-lactam-derived amine. However, the Zn2:N4 interaction is apparently weak (3.02 Å) compared to the equivalent contacts in other complexes of MBLs with hydrolysed β-lactams (possibly reflecting more facile release of hydrolysis product by VIM-1). The C3 carboxylate is also positioned to make additional interactions with the enzyme, both directly (via its O2 atom and the backbone amide of Asn233; 2.68 Å) and indirectly (between O2 and the imidazole side chain of His224 via an additional water, and between O1 and the main chain carbonyl of Cys221 via a water molecule in the Wat3 position. Wat3 thus plays similar roles in carboxylate binding in both the meropenem and ML302F complex structures.
Discussion
Clinically relevant MBLs are largely contained within the B1 subclass. The VIM enzymes are among the most important and are noted for their wide bacterial species and geographical distribution and diversity (54 variants at the time of writing). Variation in VIM enzymes is notable as it occurs at two positions, 224 and 228, that are proposed to interact with β-lactam substrates in other MBLs [23], raising the possibility that different variants bind ligands (substrates or inhibitors) in different ways. Different variants feature Tyr, His or Leu at position 224 and Arg, Ser or Leu at 228. Thus, while VIM-2 Arg228 is proposed to contact the substrate carboxylate as a functional equivalent of Lys224 in other B1 MBLs [24, 39], other VIM variants can be expected to interact with this key β-lactam functional group in different ways.
Binding of the thioenolate inhibitor ML302F to VIM-1 gives insight into how different VIM variants might interact with both inhibitors and substrates. Previous studies [30, 40] identified ML302F as a low micromolar inhibitor of both VIM-1 and VIM-2 that potentiates meropenem activity against VIM-expressing laboratory and clinical Enterobacteriaceae in susceptibility testing and promotes survival of infected insect (Galleria) larvae [40]. The current work extends these observations by demonstrating activity of the ML302F:meropenem combination against a wider panel of VIM-1 producing clinical strains. Moreover, when considered with the existing VIM-2:ML302F structure [30], our structure of the VIM-1:ML302F complex enables, for the first time, direct comparison of how the same ligand is bound by two different VIM variants (Figure 6A).
Figure 6. Importance of Cys221-bound water to ligand binding by VIM-1.

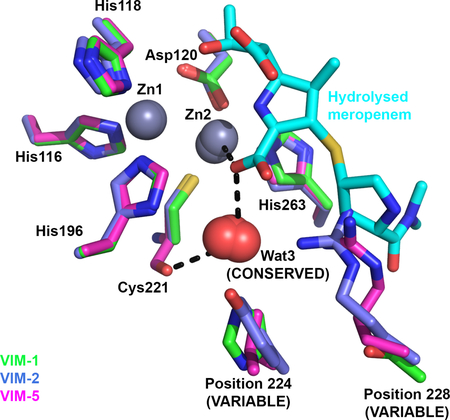
A. Mode of ML302F binding to VIM-1 (green) and VIM-2 (pdb 4PVO [30]; blue). Wat3 in VIM-2 structure is in dark red. B. Overlay of ML302F and hydrolysed meropenem complexes with VIM-1. The active site view of the ML302F complex is shown in lighter, and meropenem in darker, shades. C. Overlay of hydrolysed meropenem binding to VIM-1 (darker colours) and NDM-1 (pdb 5N0H [35] [60]; lighter colours). D. Overlay of available VIM crystal structures (details as Figure 1) showing conserved location of Wat3. Colours are as specified. Note how Wat3 mediates interactions of both substrate and inhibitor carboxylate groups with the Cys221 backbone carbonyl, and substitutes for NDM-1 Lys224 in binding the carboxylate of hydrolysed meropenem (Panel C).
ML302F is almost identically positioned in the two structures. Similar to VIM-1 binding (above) the ML302F thiol intercalates between the two zinc ions and the carboxylate interacts with the VIM-2 Zn2 ion. In addition, the ML302F carboxylate makes direct (hydrogen-bond) contact with the guanidinium side chain of VIM-2 Arg228. In VIM-1 the equivalent oxygen of the ML302F carboxylate makes a water-mediated interaction with His224. However, in both complexes the ML302F Zn2-bound oxygen atom also interacts with the backbone carbonyl of the Zn2 ligand Cys221 via a water molecule, Wat3. Wat3 adopts near-identical positions in the VIM-1 and VIM-2 complexes, implying a conserved role in substrate / inhibitor carboxylate binding. This conclusion is further supported by near-identical interactions in complexes of VIM-2 with cyclic boronate [41] and triazolylthioacetamide [42] inhibitors, that similarly involve Wat3 in binding carboxylate.
Involvement of Wat3 in ML302F binding by VIM-1 and VIM-2, along with the similarity between ML302F binding to VIM-2 and interactions of hydrolysed β-lactams with other B1 MBLs [30], suggests that VIM enzymes might make equivalent contacts with β-lactams. Indeed, comparison of our ML302F and meropenem VIM-1 complexes indicates that the oxygen atoms of the ML302F C1 and meropenem C3 carboxylates adopt near-identical positions and, importantly, make equivalent interactions with the VIM-1 active site (Figure 6B). In both cases the Zn2-bound oxygen contacts the Cys221 backbone carbonyl via Wat3, supporting involvement of Wat3 in β-lactam, as well as inhibitor, binding by VIM-1 and, by implication, other VIM enzymes. The other carboxylate oxygen hydrogen bonds, via a water molecule, to His224, thus providing a mechanism for involvement of this residue in substrate binding, although it is too distant (> 5.7 Å) from the meropenem carboxylate to make a direct interaction. Increases in KM values (i.e. reduced affinity) for carbapenem and some cephalosporin substrates for VIM-26 (VIM-1 His224Leu) compared to VIM-1 [25], also supports involvement of VIM-1 His224 in β-lactam binding. However, the fact that VIM-26 retains catalytic activity shows that a residue at position 224 able to make electrostatic interactions with substrate is not essential to activity of VIM enzymes.
Until now, structures of complexes of VIM MBLs with hydrolysed β-lactams have been elusive. However, these are available for the B1 MBL NDM-1, permitting comparison of meropenem binding to VIM-1 with β-lactam binding to NDM-1. In such structures, the C2/C3 carboxylates of hydrolysed penicillins [34–36] cephalosporins [33] and carbapenems [35, 43] contact the NDM-1 Zn2 metal ion, the backbone amide of Asn233 and the terminal amino group of Lys224. Comparing hydrolysed meropenem binding to VIM-1 and NDM-1 (Figure 6C), in each case the C2 carboxylate adopts similar orientations and makes equivalent interactions with Zn2 and the Asn233 backbone amide; but the two complexes differ in the interactions made by the Zn2-bound carboxylate oxygen, which in NDM-1 hydrogen bonds to Lys224 and in VIM-1 to the Cys221 backbone carbonyl via Wat3. Wat3 thus enables VIM-1, and by implication other VIM enzymes, to replicate interactions with substrates made by Lys224 in other B1 MBLs. The importance of these to β-lactam binding and hydrolysis is evidenced by the profound loss of activity observed in Lys224 mutants of other B1 enzymes [44–46]. Thus, in both of the structures presented here, as well as the ML302F:VIM-2 complex, the water molecule Wat3 connects the carboxylate of hydrolysed β-lactam/inhibitor to the Cys221 backbone carbonyl. These observations indicate that Wat3 enables VIM enzymes to replicate the role of Lys224 in ligand binding by other subclass B1 MBLs, while simultaneously accommodating sequence variations at positions 224 and 228. To investigate this hypothesis, we inspected crystal structures of VIM MBLs (Figure 6D). This analysis identified water molecules in the Wat3 position in all available VIM variant structures and their complexes in the active (binuclear) form. We thus propose that this active site water molecule, positioned by the backbone of the invariant Zn2 ligand Cys221, enables different VIM variants to make productive interactions with substrate even where, as in VIM-1, direct contact between the C2/C3 carboxylate and protein is impossible.
Mechanistic interpretation of the complex with bound meropenem is limited by both the resolution of our data, and the weak electron density for the C6 hydroxyethyl and C7 carboxylate groups. The situation is further complicated by the likelihood that several species are likely to be present, given that carbapenem breakdown in solution can yield a mixture of hydrolysis products, i.e. both the Δ1 (R- and S- stereoisomers) and Δ2 tautomers [47] and that trapped complexes may thus represent either species present on the late stages of the reaction pathway or rebinding of highest affinity products present in solution. However, we note that in the present structure the well-defined meropenem N4 atom lies more distant (3.02 Å) from Zn2 than in other such complexes with B1 MBLs, where this is typically a tight interaction (≤ 2.40 Å). When considered together with our inclusion of a zinc-bridging water molecule in our final model, (which is consistent with available structures for NDM-1:penicillin and cephalosporin-derived complexes [33–36]), one possible interpretation is that our complex could represent a stage in product release where water has displaced the C7 carboxylate from the Zn-bridging position. This would indicate that release of product from the MBL active site is not a single step process, and that interactions of β-lactams around the Zn2 site persist longer than those involving Zn1.
Overall, our work provides the first structural description of VIM-1, an MBL of particular relevance to the growing problem of carbapenem resistant Enterobacteriaceae. Our structures of VIM-1:meropenem and inhibitor complexes establish how VIM MBLs accommodate variation at residues 224 and 228, without loss of activity, and provide new information on carbapenem hydrolysis, specifically related to product release. Identification of interactions, involving carboxylate groups of small molecule ligands and the invariant water molecule Wat3, that are common to VIM-1, VIM-2 and, we infer, other VIM variants, will aid in structure-based rational development of effective inhibitors for the full range of these heterogeneous and clinically important enzymes.
Experimental Procedures
Materials-
General laboratory reagents were purchased from Sigma (Poole, U.K.) or VWR (Lutterworth, U.K.) and were of analytical grade. Inhibitor ML302F was synthesized as previously described [30]. Meropenem was a gift from AstraZeneca (Macclesfield, U.K).
Cloning, expression and purification-
Full length VIM-1 with a C-terminal 6His tag was expressed and purified largely according to previously published procedures [48] using the pOPINE T7 plasmid [49] containing a synthetic codon optimized gene. Modifications are detailed below. Protein was expressed in E. coli Rosetta 2 (DE3) (Merck, Watford, U.K.) with cells (500 ml in 2l conical flasks) grown in Terrific Broth autoinduction media (Formedium, Hunstanton, Norfolk, U.K.) supplemented with 100 μg ml−1 ampicillin. Cultures were grown at 37°C shaking at 160 rpm for 8 h; the temperature was subsequently lowered to 25°C for overnight growth. Cultures were harvested by centrifugation (7 200 g, 30 mins, 4°C) and pellets from 3 l cells resuspended in 200 ml of buffer A (50 mM Tris pH 7.5, 500 mM NaCl, 30 mM imidazole) supplemented with 20 μl of 5 KU benzonase (Sigma Aldrich, Poole, U.K.), 0.02% Tween 20 (Sigma) and two EDTA-free protease inhibitor cocktail tablets (Roche, Burgess Hill, U.K.). A Constant Cell Disruption System (Constant Systems, Daventry, U.K.) was used to lyse homogenized cells at 25 kpsi, the lysate was cleared by centrifugation (1 h, 38 500 g, 4 °C). 6His-tagged VIM-1 was purified on a 5 ml HisTrap HP column (GE Life Sciences, Little Chalfont, U.K.) using a 0 – 500 mM imidazole gradient, as described [48]. Protein obtained from this single step purification was estimated as being over 98 % pure as adjudged by SDS-PAGE. Fractions containing VIM-1 were pooled and concentrated by centrifugation (10 kDa molecular weight cut off Amicon Ultra-15 (Merck). Protein used to obtain the di-zinc VIM-1 structure was further purified by size exclusion (Superdex-200) as described [48]; samples used to obtain other structures were used without further purification.
Crystallization, data collection and structure solution –
Crystallization screening used commercial sparse matrix screens (Molecular Dimensions, Newmarket, U.K.; Qiagen, Manchester, U.K.) dispensed via a Hydra 96 microdispenser (Robbins Scientific, Sunnyvale. CA). A Cartesian Honeybee X8 (Digilab) robot was used to set up crystallization trials in 96 well sitting drop plates (CrystalQuick, Greiner, Stonehouse, U.K.) with drops containing 0.1 μl of protein and 0.1 μl of reservoir solution and a total reservoir volume of 95 μl. Crystals were stored and monitored at 21 °C using a Formulatrix (Bedord, MA) Rock Imager 1000 system. The di-zinc VIM-1 structure was solved using a crystal obtained from protein concentrated to 15 mg/ml (containing 7% glycerol) and crystallized under conditions (0.05 M MgCl2, 0.03 M CaCl2, 0.1 M Morpheus buffer 3 pH 8.5, 13.5% w/v PEG 3350, 9.5% w/v PEG 1000 and 12.5% w/v MPD) obtained by optimization of an initial hit from the Morpheus screen [50]. This crystal was obtained from a drop set up by hand in 24-well hanging drop format using 1 μl protein and 1 μl reservoir condition and a total reservoir volume of 500 μl.
Other VIM-1 structures were obtained from protein concentrated to ~23 mg/ml containing 10% glycerol and supplemented with 100 μM ZnCl2, 2 mM tris(2-carboxyethyl)phosphine (TCEP; Thermo Scientific). Crystals of the VIM-1:ML302F complex were obtained from 0.02 M Na-formate, 0.02 M NH4-acetate, 0.02 M Na3-citrate, 0.02 M NaK-tartrate, 0.02 M Na-oxamate, 0.1 M Morpheus Buffer 1 pH 6.5, 12.5% w/v PEG 3350, 12.5% w/v PEG 1000 and 12.5% w/v MPD. Crystallization drops were set up by hand in 96-well MRC sitting drop plates (Molecular Dimensions) using commercial screens with 0.5 μl of protein and 0.5 μl reservoir solution and a total reservoir volume of 50 μl. Experiments were incubated at 20° C. Excess inhibitor was added to the drop in the form of powder. The structure of the VIM-1:meropenem complex was obtained from an overnight soak of crystals obtained from a different Morpheus condition (0.1 M MOPS/HEPES-Na pH 7.5, 12.5% w/v PEG 1000, 12.5% w/v PEG 3350, 0.03 M CaCl2, 0.03 M MgCl2 and 12.5% v/v MPD) with excess substrate powder. Crystals were snap-frozen in liquid nitrogen prior to transportation for diffraction data collection.
Crystallographic data were collected at 100 K on beamlines of Diamond Light Source (DLS; Didcot, U.K.) using Pilatus 6M-F detectors. Diffraction data were integrated with XDS and scaled using Aimless as part of the CCP4 software suite [51–53]. Phases were solved by molecular replacement in PHASER [54] using either VIM-4 (PDB 2WGH [29]) or previously determined VIM-1 structures as search models. Rounds of refinement were carried out using Phenix [55] with manual rebuilding in Coot [56]. MolProbity [57] was used for structure validation as part of the Phenix suite. PyMOL (www.pymol.org) was used to generate figures. Data collection and refinement statistics, together with Protein Data Bank (pdb) accession codes, are presented in Table 1.
Determination of Minimal Inhibitory Concentrations-
Twenty clinical strains of K. pneumoniae and seven clinical strains of E. coli, originating from clinical specimens from Spain, were confirmed for the presence of VIM-1 by PCR assay using the primers VIM-F (5’-CCG ACA GTC ARC GAA ATT CCG-3’) VIM-R (5’-CTA CTC RRC GAC TGA GCG ATT-3’). Minimal inhibitory concentration (MIC) values were determined by broth microdilution, in triplicate, in cation adjusted Mueller Hinton broth (Sigma) according to the Clinical Laboratory Standards Institute (CLSI) guidelines [58]. Experiments were carried out in microtiter plates (Corning) containing the medium plus meropenem and inhibitor ML302F (dissolved in DMSO and added to the wells with a final concentration of 10 μg/ml (0.1 % DMSO)). Plates were incubated overnight at 37 °C for 18 – 24 h and absorbance at 600 nm read using a Polarstar Omega (BMG LabTech, Aylesbury, U.K.) plate reader.
Acknowledgements:
We acknowledge Diamond Light Source for access to beamlines I02, I04 and I24 (proposal number MX313) that contributed to the results presented here. This study was supported by the UK Medical Research Council (MR/N002679/1, RS, TRW, JB, CJS and JS; MR/K018779/1, R.J.O. and A.V.); and by the National Institute of Allergy and Infectious Diseases of the U.S. National Institutes of Health under award number R01AI100560 (PH, MK and JS). Research leading to these results has received support from the Innovative Medicines Initiative Joint Undertaking under grant agreement no 115583 (JB, MAM, JT, TRW and CJS), resources of which are composed of financial contributions from the European Union`s Seventh Framework Programme (FP7/2007–2013) and EFPIA companies` in kind contribution. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Abbreviations:
- MBL
Metallo-β-Lactamase
- VIM
Verona Integron-encoded MBL
Footnotes
Enzymes: EC 3.5.2.6
Databases: Co-ordinates and structure factors for protein structures described in this manuscript have been deposited in the Protein Data Bank (www.rcsb.org/pdb) with accession codes 5N5G (VIM-1), 5N5H (VIM-1:ML302F complex) and 5N5I (VIM-1 hydrolysed meropenem complex).
Conflicts of Interest: the authors declare no conflicts of interest
References
- 1.Davies SC (2013) Infections and the rise of antimicrobial resistance in Annual Report of the Chief Medical Officer, Volume Two, 2011, Department of Health, London. [DOI] [PubMed] [Google Scholar]
- 2.Thomson JM & Bonomo RA (2005) The threat of antibiotic resistance in Gram-negative pathogenic bacteria: beta-lactams in peril!, Curr Opin Microbiol. 8, 518–524. [DOI] [PubMed] [Google Scholar]
- 3.Elander RP (2003) Industrial production of beta-lactam antibiotics, Appl Microbiol Biot. 61, 385–392. [DOI] [PubMed] [Google Scholar]
- 4.Frere JM (1995) Beta-Lactamases and Bacterial-Resistance to Antibiotics, Molecular Microbiology. 16, 385–395. [DOI] [PubMed] [Google Scholar]
- 5.Livermore DM (1995) Beta-Lactamases in Laboratory and Clinical Resistance, Clinical Microbiology Reviews. 8, 557-&. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bush K (2013) Proliferation and significance of clinically relevant beta-lactamases, Annals of the New York Academy of Sciences. 1277, 84–90. [DOI] [PubMed] [Google Scholar]
- 7.Jean SS, Lee WS, Lam C, Hsu CW, Chen RJ & Hsueh PR (2015) Carbapenemase-producing Gram-negative bacteria: current epidemics, antimicrobial susceptibility and treatment options, Future Microbiol. 10, 407–25. [DOI] [PubMed] [Google Scholar]
- 8.Perez-Llarena FJ & Bou G (2009) beta-Lactamase Inhibitors: The Story so Far, Curr Med Chem. 16, 3740–3765. [DOI] [PubMed] [Google Scholar]
- 9.Drawz SM & Bonomo RA (2010) Three Decades of beta-Lactamase Inhibitors, Clin Microbiol Rev. 23, 160-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Papp-Wallace KM, Endimiani A, Taracila MA & Bonomo RA (2011) Carbapenems: past, present, and future, Antimicrob Agents Chemother. 55, 4943–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Queenan AM & Bush K (2007) Carbapenemases: the versatile beta-lactamases, Clin Microbiol Rev. 20, 440–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bebrone C (2007) Metallo-beta-lactamases (classification, activity, genetic organization, structure, zinc coordination) and their superfamily, Biochem Pharmacol. 74, 1686–1701. [DOI] [PubMed] [Google Scholar]
- 13.Bush K & Jacoby GA (2010) Updated Functional Classification of beta-Lactamases, Antimicrob Agents Ch. 54, 969–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walsh TR, Toleman MA, Poirel L & Nordmann P (2005) Metallo-beta-lactamases: the quiet before the storm?, Clin Microbiol Rev. 18, 306-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Crowder MW, Spencer J & Vila AJ (2006) Metallo-beta-lactamases: Novel weaponry for antibiotic resistance in bacteria, Accounts Chem Res. 39, 721–728. [DOI] [PubMed] [Google Scholar]
- 16.Galleni M, Lamotte-Brasseur J, Rossolini GM, Spencer J, Dideberg O & Frere JM (2001) Standard numbering scheme for class B beta-lactamases, Antimicrob Agents Chemother. 45, 660–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Badarau A & Page MI (2006) Enzyme deactivation due to metal-ion dissociation during turnover of the cobalt-beta-lactamase catalyzed hydrolysis of beta-lactams, Biochemistry. 45, 11012–20. [DOI] [PubMed] [Google Scholar]
- 18.Gonzalez JM, Meini MR, Tomatis PE, Medrano Martin FJ, Cricco JA & Vila AJ (2012) Metallo-beta-lactamases withstand low Zn(II) conditions by tuning metal-ligand interactions, Nat Chem Biol. 8, 698–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Edelstein MV, Skleenova EN, Shevchenko OV, D’Souza J W, Tapalski DV, Azizov IS, Sukhorukova MV, Pavlukov RA, Kozlov RS, Toleman MA & Walsh TR (2013) Spread of extensively resistant VIM-2-positive ST235 Pseudomonas aeruginosa in Belarus, Kazakhstan, and Russia: a longitudinal epidemiological and clinical study, The Lancet infectious diseases. 13, 867–76. [DOI] [PubMed] [Google Scholar]
- 20.Lauretti L, Riccio ML, Mazzariol A, Cornaglia G, Amicosante G, Fontana R & Rossolini GM (1999) Cloning and characterization of bla(VIM), a new integron-borne metallo-beta-lactamase gene from a Pseudomonas aeruginosa clinical isolate, Antimicrob Agents Ch. 43, 1584–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Naas T, Oueslati S, Bonnin RA, Dabos ML, Zavala A, Dortet L, Retailleau P & Iorga BI (2017) Beta-lactamase database (BLDB) - structure and function, J Enzyme Inhib Med Chem. 32, 917–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Papagiannitsis CC, Izdebski R, Baraniak A, Fiett J, Herda M, Hrabak J, Derde LP, Bonten MJ, Carmeli Y, Goossens H, Hryniewicz W, Brun-Buisson C, Gniadkowski M, Mosar Wp WP, groups W. P. s., Mosar Wp WP & groups W. P. s. (2015) Survey of metallo-beta-lactamase-producing Enterobacteriaceae colonizing patients in European ICUs and rehabilitation units, 2008–11, J Antimicrob Chemother. 70, 1981–8. [DOI] [PubMed] [Google Scholar]
- 23.Meini MR, Llarrull LI & Vila AJ (2014) Evolution of Metallo-beta-lactamases: Trends Revealed by Natural Diversity and Evolution, Antibiotics. 3, 285–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Garcia-Saez I, Docquier J-D, Rossolini GM & Dideberg O (2008) The three-dimensional structure of VIM-2, a Zn-beta-lactamase from Pseudomonas aeruginosa in its reduced and oxidised form., J Mol Biol. 375, 604–611. [DOI] [PubMed] [Google Scholar]
- 25.Leiros HK, Edvardsen KS, Bjerga GE & Samuelsen O (2015) Structural and biochemical characterization of VIM-26 shows that Leu224 has implications for the substrate specificity of VIM metallo-beta-lactamases, FEBS J. 282, 1031–42. [DOI] [PubMed] [Google Scholar]
- 26.Makena A, Duzgun AO, Brem J, McDonough MA, Rydzik AM, Abboud MI, Saral A, Cicek AC, Sandalli C & Schofield CJ (2015) Comparison of Verona Integron-Borne Metallo-beta-lactamase Variants Reveals Differences in Stability and Inhibition Profiles, Antimicrob Agents Chemother. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Borra PS, Leiros HKS, Ahmad R, Spencer J, Leiros I, Walsh TR, Sundsfjord A & Samuelsen O (2011) Structural and Computational Investigations of VIM-7: Insights into the Substrate Specificity of VIM Metallo-beta-Lactamases, Journal of Molecular Biology. 411, 174–189. [DOI] [PubMed] [Google Scholar]
- 28.Kupper MB, Herzog K, Bennink S, Schlomer P, Bogaerts P, Glupczynski Y, Fischer R, Bebrone C & Hoffmann KM (2015) The three-dimensional structure of VIM-31--a metallo-beta-lactamase from Enterobacter cloacae in its native and oxidized form, FEBS J. 282, 2352–60. [DOI] [PubMed] [Google Scholar]
- 29.Lassaux P, Traore DA, Loisel E, Favier A, Docquier JD, Sohier JS, Laurent C, Bebrone C, Frere JM, Ferrer JL & Galleni M (2011) Biochemical and structural characterization of the subclass B1 metallo-beta-lactamase VIM-4, Antimicrob Agents Chemother. 55, 1248–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brem J, van Berkel SS, Aik W, Rydzik AM, Avison MB, Pettinati I, Umland KD, Kawamura A, Spencer J, Claridge TD, McDonough MA & Schofield CJ (2014) Rhodanine hydrolysis leads to potent thioenolate mediated metallo-beta-lactamase inhibition, Nature chemistry. 6, 1084–90. [DOI] [PubMed] [Google Scholar]
- 31.Krissinel E & Henrick K (2004) Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions, Acta Crystallogr D Biol Crystallogr. 60, 2256–68. [DOI] [PubMed] [Google Scholar]
- 32.Zheng H, Cooper DR, Porebski PJ, Shabalin IG, Handing KB & Minor W (2017) CheckMyMetal: a macromolecular metal-binding validation tool, Acta Crystallogr D Struct Biol. 73, 223–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Feng H, Ding J, Zhu D, Liu X, Xu X, Zhang Y, Zang S, Wang DC & Liu W (2014) Structural and Mechanistic Insights into NDM-1 Catalyzed Hydrolysis of Cephalosporins, J Am Chem Soc. 136, 14694–7. [DOI] [PubMed] [Google Scholar]
- 34.Kim Y, Cunningham MA, Mire J, Tesar C, Sacchettini J & Joachimiak A (2013) NDM-1, the ultimate promiscuous enzyme: substrate recognition and catalytic mechanism, FASEB J. 27, 1917–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.King DT, Worrall LJ, Gruninger R & Strynadka NC (2012) New Delhi metallo-beta-lactamase: structural insights into beta-lactam recognition and inhibition, J Am Chem Soc. 134, 11362–5. [DOI] [PubMed] [Google Scholar]
- 36.Zhang HM & Hao Q (2011) Crystal structure of NDM-1 reveals a common beta-lactam hydrolysis mechanism, Faseb J. 25, 2574–2582. [DOI] [PubMed] [Google Scholar]
- 37.Smart OS, Horsky V, Gore S, Svobodova Varekova R, Bendova V, Kleywegt GJ & Velankar S (2018) Validation of ligands in macromolecular structures determined by X-ray crystallography, Acta Crystallogr D Struct Biol. 74, 228–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Easton CJ & Knowles JR (1982) Inhibition of the RTEM beta-lactamase from Escherichia coli. Interaction of the enzyme with derivatives of olivanic acid, Biochemistry. 21, 2857–62. [DOI] [PubMed] [Google Scholar]
- 39.Yamaguchi Y, Jin W, Matsunaga K, Ikemizu S, Yamagata Y, Wachino J, Shibata N, Arakawa Y & Kurosaki H (2007) Crystallographic investigation of the inhibition mode of a VIM-2 metallo-beta-lactamase from Pseudomonas aeruginosa by a mercaptocarboxylate inhibitor, J Med Chem. 50, 6647–53. [DOI] [PubMed] [Google Scholar]
- 40.Betts JW, Phee LM, Momin MHFA, Umland KD, Brem J, Schofield CJ & Wareham DW (2016) In vitro and in vivo activity of ML302F: a thioenolate inhibitor of VIM-subfamily metallo beta-lactamases, Medchemcomm. 7, 190–193. [Google Scholar]
- 41.Brem J, Cain R, Cahill S, McDonough MA, Clifton IJ, Jimenez-Castellanos JC, Avison MB, Spencer J, Fishwick CW & Schofield CJ (2016) Structural basis of metallo-beta-lactamase, serine-beta-lactamase and penicillin-binding protein inhibition by cyclic boronates, Nat Commun. 7, 12406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Christopeit T, Yang KW, Yang SK & Leiros HK (2016) The structure of the metallo-beta-lactamase VIM-2 in complex with a triazolylthioacetamide inhibitor, Acta Crystallogr F Struct Biol Commun. 72, 813–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Feng H, Liu X, Wang S, Fleming J, Wang DC & Liu W (2017) The mechanism of NDM-1-catalyzed carbapenem hydrolysis is distinct from that of penicillin or cephalosporin hydrolysis, Nat Commun. 8, 2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Haruta S, Yamamoto ET, Eriguchi Y & Sawai T (2001) Characterization of the active-site residues asparagine 167 and lysine 161 of the IMP-1 metallo beta-lactamase, FEMS Microbiol Lett. 197, 85–9. [DOI] [PubMed] [Google Scholar]
- 45.Liang ZJ, Li LC, Wang YY, Chen LM, Kong XQ, Hong Y, Lan LF, Zheng MY, Cai GY, Liu H, Shen X, Luo C, Li KK, Chen KX & Jiang HL (2011) Molecular Basis of NDM-1, a New Antibiotic Resistance Determinant, PloS one. 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yanchak MP, Taylor RA & Crowder MW (2000) Mutational analysis of metallo-beta-lactamase CcrA from Bacteroides fragilis, Biochemistry. 39, 11330–9. [DOI] [PubMed] [Google Scholar]
- 47.Ratcliffe RW, Wildonger KJ, Di Michele L, Douglas AW, Hadju R, Goegelman RT, Springer JP & Hirshfield J (1989) Studies on the structures of imipenem, dehydropeptidase I-hydrolyzed imipenem, and related analogs, Journal of Organic Chemistry. 54, 653–660. [Google Scholar]
- 48.Makena A, van Berkel SS, Lejeune C, Owens RJ, Verma A, Salimraj R, Spencer J, Brem J & Schofield CJ (2013) Chromophore-linked substrate (CLS405): probing metallo-beta-lactamase activity and inhibition, ChemMedChem. 8, 1923–9. [DOI] [PubMed] [Google Scholar]
- 49.Berrow NS, Alderton D, Sainsbury S, Nettleship J, Assenberg R, Rahman N, Stuart DI & Owens RJ (2007) A versatile ligation-independent cloning method suitable for high-throughput expression screening applications, Nucleic Acids Research. 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gorrec F (2009) The MORPHEUS protein crystallization screen, J Appl Crystallogr. 42, 1035–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bailey S (1994) The Ccp4 Suite - Programs for Protein Crystallography, Acta Crystallogr D. 50, 760–763. [DOI] [PubMed] [Google Scholar]
- 52.Evans P (2006) Scaling and assessment of data quality, Acta Crystallogr D. 62, 72–82. [DOI] [PubMed] [Google Scholar]
- 53.Evans PR (2011) An introduction to data reduction: space-group determination, scaling and intensity statistics, Acta Crystallogr D. 67, 282–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mccoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC & Read RJ (2007) Phaser crystallographic software, J Appl Crystallogr. 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC & Zwart PH (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution, Acta Crystallogr D Biol Crystallogr. 66, 213–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Emsley P, Lohkamp B, Scott WG & Cowtan K (2010) Features and development of Coot., Acta Crystallogr D Biol Crystallogr. 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS & Richardson DC (2010) MolProbity: all-atom structure validation for macromolecular crystallography, Acta Crystallogr D Biol Crystallogr. 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.CLSI (2015) Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Approved Standard- Tenth Edition in, Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 59.Wallace AC, Laskowski RA & Thornton JM (1995) LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions, Protein Eng. 8, 127–34. [DOI] [PubMed] [Google Scholar]
- 60.Raczynska JE, Shabalin IG, Minor W, Wlodawer A & Jaskolski M (2018) A close look onto structural models and primary ligands of metallo-β-lactamases, Drug Resist Updat. 40, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.EUCAST (2017) The European Committee on Antimicrobial Susceptibility Testing Breakpoint tables for interpretation of MICs and zone diameters. Version 7.1 in, ESCMID, Basel, Switzerland. [Google Scholar]