

Abstract
Membrane depolarization triggers Ca2+ release from the sarcoplasmic reticulum (SR) in skeletal muscles via direct interaction between the voltage-gated L-type Ca2+ channels (the dihydropyridine receptors; VGCCs) and ryanodine receptors (RyRs), while in cardiac muscles Ca2+ entry through VGCCs triggers RyR-mediated Ca2+ release via a Ca2+-induced Ca2+-release (CICR) mechanism. Here we demonstrate that in phasic smooth muscle of the guinea-pig small intestine, excitation evoked by muscarinic receptor activation triggers an abrupt Ca2+ release from sub-plasmalemmal (sub-PM) SR elements enriched with inositol 1,4,5-trisphosphate receptors (IP3Rs) and poor in RyRs. This was followed by a lesser rise, or oscillations in [Ca2+]i. The initial abrupt sub-PM [Ca2+]i upstroke was all but abolished by block of VGCCs (by 5 μM nicardipine), depletion of intracellular Ca2+ stores (with 10 μM cyclopiazonic acid) or inhibition of IP3Rs (by 2 μM xestospongin C or 30 μM 2-APB), but was not affected by block of RyRs (by 50 – 100 μM tetracaine or 100 μM ryanodine). Inhibition of either IP3Rs or RyRs attenuated phasic muscarinic contraction by 73 %. Thus, in contrast to cardiac muscles, excitation-contraction coupling in this phasic visceral smooth muscle occurs by Ca2+ entry through VGCCs which evokes an initial IP3R-mediated Ca2+ release activated via a CICR mechanism.
1. Introduction
Smooth muscle cells (SMCs) are stimulated to contract either by depolarization of the cell membrane (electromechanical coupling), often in the form of an action potential (AP), or by activation of a variety of receptors (pharmacomechanical coupling) usually coupled to G-proteins, or by a combination of these mechanisms [1]. Most smooth muscles exhibit voltage-gated Ca2+ channels (VGCCs) which are often manifest in the generation of APs, although in many vascular smooth muscles Ca2+ is believed to ‘leak’ into the cell through these VGCCs giving rise to ‘sparklets’ [2, 3]. The entry of Ca2+ into the SMC through VGCCs and by reverse Na+/Ca2+ exchange [4] is believed to load the Ca2+ stores largely in the sarcoplasmic reticulum (SR). From here Ca2+ can be released either by a process of Ca2+-induced Ca2+ release (CICR) through ryanodine receptors (RyRs) upon Ca2+ entry during an AP [5] or by the action of inositol 1,4,5 trisphosphate (IP3) generated by a Gq/11-link from an activated stimulant receptor to phospholipase C. The entry of Ca2+ through VGCCs is believed to load SR calcium stores which, upon reaching a critical level of loading [6] discharge packets of Ca2+ which in the majority of smooth muscles generate transient high local concentrations of sub-PM Ca2+ that open Ca2+-dependent potassium (BK) channels, present in abundance in the cell membrane of smooth muscles. Bursts of openings of these BK channels are seen as spontaneous transient outward currents (STOCs) in voltage-clamped SMCs [7-9]. In vascular smooth muscles loaded with a fluorescent Ca2+ indicator, the transient localised sub-plasmalemmal (sub-PM) increase in Ca2+concentration ([Ca2+]i) is seen as a flash of emitted light (“spark”, [10, 11]). The transient BK channel currents evoked have been observed to hyperpolarize the membrane so reducing Ca2+ entry through VGCCs, reducing the rate of Ca2+ loading of the SR stores, and so reducing tension [11, 12]. It seems that Ca2+ sparks are a negative feed-back mechanism, triggered by store overload, which regulate vascular myocyte tension as they barely increase global [Ca2+]i, while the high local sub-PM [Ca2+]i they create trigger a membrane-potential dependent relaxation of tension through the opening of BK channels [13].
The respective role of SR RyRs and IP3 receptors (IP3Rs) in the generation of smooth muscle myocyte tension is an area of active investigation. As smooth muscles do not have a sarcomeric organisation of their contractile proteins and calcium stores, the sites of Ca2+ release and re-storage are necessarily very different from those of striated muscles. In vas deferens and bladder SMCs an AP generates Ca2+ release from sub-PM ‘hot spots’ which then generate a cell-wide Ca2+ wave presumably by CICR. This is followed by SMC contraction [5]. Activation of stimulant receptors, such as muscarinic receptors in intestinal SMCs, triggers additional spark activity at spontaneously discharging ‘frequent discharge sites’ and recruits additional spark discharge sites [14]. The role of IP3Rs seems to be to facilitate CICR within and between RyRs domains [15-18]. In colonic SMCs IP3R-evoked Ca2+ release did not activate RyRs but RyR blockers inhibited IP3R-mediated Ca2+ signals [19]. In vascular SMCs of small arteries Lamont and Wier [20] concluded that if RyRs were blocked, cell-wide Ca2+ waves could still be evoked by strong activation of adrenoceptors leading to myocyte and vessel contraction; thus CICR release apparently can occur among IP3Rs alone although normally, at low levels of adrenoceptor activation RyRs were necessary to trigger IP3R dependent Ca2+ waves [12]. Thus at physiologically important levels of tension the interplay of RyR and IP3R Ca2+ release is important as in visceral smooth muscles. The present work examines the relative roles of RyRs, IP3Rs and VGCCs in intracellular Ca2+ mobilisation in longitudinal intestinal smooth muscle myocytes in response to strong activation of muscarinic receptors. A preliminary account of some of this work has previously been published in an abstract form [21].
2. Materials and methods
Experiments were performed on preparations of the longitudinal muscle layer of the guinea-pig ileum: (1) freshly isolated SMCs or (2) freshly dissected strips of smooth muscles (see below). Adult male guinea-pigs (300 – 500 g) were killed by decapitation after cervical dislocation as approved under Schedule 1 of the UK Animals (Scientific Procedures) Act 1986.
2.1. Cell preparation
The longitudinal muscle layer of the ileum was dissected and cut into small pieces, which were placed in Ca2+- free physiological saline solution (PSS, see below). The pieces of the tissue were transferred into the same solution supplemented with (mg/ml): protease (Type X) 0.5, collagenase (Type 1A) 1.5, soybean trypsin inhibitor 1 and bovine serum albumin 1, and incubated for 20 min at 37 °C. The pieces of the tissue were then rinsed for 10 min in an enzyme-free Ca2+- free solution and triturated with a wide bore glass pipette. Small aliquots of the cell suspension were transferred to the experimental chambers and diluted with PSS composed of (mM): NaCl 120, KCl 6, CaCl2 2.5, MgCl2 1.2, glucose 12, HEPES 10; pH adjusted to 7.4 with NaOH. Experiments on isolated cells were conducted at 22 – 24 °C within 8 h of cell isolation.
2.2. Isometric tension recording
Smooth muscle strips (∼10 mm in length) were dissected from longitudinal layer of guinea-pig ileum, transferred to a home-made organ bath (3 ml volume) and attached to a isometric force transducer at a resting tension load of 5 mN and bathed in the PSS at 37°C. The output of the force transducer was connected to a custom-made amplifier. The signals were digitized using a Digidata 1320 AD/DA converter (Molecular Devices Co., CA, USA) hosted by a PC running Axoscope 8.0 software (Molecular Devices Co., CA, USA).
2.3. Visualisation of [Ca2+]i changes
Changes in intracellular concentration of ionised calcium ([Ca2+]i) in isolated SMCs were imaged using the high-affinity (kd(Ca) = 345 nM) fluorescent Ca2+ indicator fluo-4, which was loaded by 20-min incubation of the SMCs with 5 μM fluo-4 acetoxymethyl ester (fluo-4 AM) followed by a 40-min wash in PSS to allow time for de-esterification. To minimise SMC contraction, 40 μM of wortmannin was added to the bathing solution 10 min before imaging commenced.
The myocytes were stimulated with 10 μM carbachol (CCh) which was either superfused through the experimental bath or applied as a brief (≤ 600 ms) pulse through a glass micropipette (located within 100 – 200 μm of the cell) attached to the outlet of pressure ejector Picospritzer III (Intracel Ltd., UK). Similar application of the control solution (without agonist) had no effect on [Ca2+]i. In the experiments where the same SMC was stimulated with CCh and caffeine, CCh was superfused through the experimental bath, while 5 mM caffeine was applied through a glass micropipette. Small volumes of antagonists were added to the bath solution in amounts calculated to achieve the indicated concentrations.
In the figures the intensity of fluo-4 fluorescence was normalised to the average fluorescence intensity in the images captured before CCh application and colour coded as indicated by the bars (F/F0). The temporal profiles of CCh –induced [Ca2+]i mobilisation are illustrated by the plots showing (1) the time course of the normalised fluo-4 fluorescence intensity (F/F0) averaged within multiple sub-PM regions (outlined in the corresponding images) where F/F0 changes were initiated and rose above 2.5 or (2) the time course of F/F0 averaged within entire confocal optical slice.
2.4. Visualisation of intracellular calcium stores
Distribution of intracellular calcium stores within isolated SMCs was visualised using the low-affinity (kd(Ca) = 42 μM) fluorescent Ca2+ indicator fluo-3FF, which was loaded by 90-min incubation of the SMCs with 5 μM fluo-3FF AM followed by 60-min wash in PSS.
2.5. Immunostaining of RyRs and IP3Rs
Isolated SMCs were fixed by 5-10-min incubation with 4 % (w/v) paraformaldehyde. Nonspecific binding was blocked by incubating the SMCs with 3% (w/v) bovine serum albumin (BSA) and 0.3 % (w/v) Triton X-100 (a cell permeabilising agent) for 1 h at room temperature. Primary and secondary antibodies were diluted in PSS supplemented with 3% (w/v) BSA, 0.3 % (w/v) Triton X-100, 20 U/ml penicillin and 20 μg/ml streptomycin. To visualise the distribution of IP3Rs, we used an IP3R type 1 - specific antibody, since type 1 IP3R was shown to be ubiquitous in various tissues [22]. This antibody was developed (Sigma-Aldrich Co., RBI, Natick, MA, USA) in rabbit and has been shown to selectively recognise type 1 IP3Rs in other types of SMCs [23]. RyRs were detected with a monoclonal anti-RyR antibody derived (Sigma-Aldrich Co., RBI, Natick, MA, USA) from the 34C hybridoma (produced by the fusion of P3X 63 Ag8.653 myeloma cells and spleen cells from Balb/c mice). This antibody reacts strongly with RyR type 1, type 2 and type 3. The SMCs were incubated in the presence of the primary anti-IP3R type 1 and anti-RyR antibodies (at 1:300 and 1:480 dilution, respectively) for 16 h at 4°C. Following a 10-min rinse (4 times) in PSS supplemented with 3% (w/v) BSA, primary antibody-specific binding was visualised by incubating SMCs for 2 h at room temperature with Alexa Fluor 488 conjugated to chicken anti-rabbit IgG (1:300 dilution, Invitrogen Ltd., UK) and Alexa Fluor 633 conjugated to goat anti-mouse IgG (1:300 dilution, Invitrogen Ltd., UK). In controls, primary antibodies were omitted from the experimental media during the first (16 h) incubation.
2.6. Confocal microscopy
Experimental chambers were placed on the stage of an Axiovert 100M inverted microscope attached to a LSM 510 laser-scanning unit (Zeiss, Oberkochen, Germany). The x-y confocal images of fluo-4 fluorescence were acquired at 19 - 42 Hz using a Zeiss plan-Apochromat 40× 1.3 N.A. oil-immersion objective. Fluo-4 and fluo-3FF fluorescence was excited by the 488 nm line of a 200 mW argon ion laser (Laser-Fertigung, Hamburg, Germany) and was captured at wavelengths above 505 nm. The pinhole was set to provide a confocal optical slice below 1.2 μm.
To avoid any bleed-through in immunofluorescence experiments, SMCs were double labelled using fluorophores with extremely well separated emission spectra: Alexa Fluor 488 (Em = 519 nm) and Alexa Fluor 633 (Em = 647 nm). Dual excitation, using the multitrack mode of an LSM 510, was performed by the 488 nm line of a 200 mW argon ion laser and the 633 nm line of a 15 mW helium/neon ion laser, respectively. The emitted fluorescence signal was filtered using 505 nm - 550 nm bandpass filter (for the green IP3R label) and 650 nm longpass filter (for the red RyR label). The adequacy of the imaging protocol applied to the double-labelled SMCs was confirmed by control experiments on the single-labelled cells.
2.7. Electrical recordings
The cell membrane potential was monitored using perforated-patch (200 μg/ml amphotericin B) tight seal recording in the current-clamp mode of an Axopatch 200A (Molecular Devices Co., CA, USA) patch-clamp amplifier. This allowed low resistance access to the cell while minimally interfering with [Ca2+]i. The cell was bathed in PSS and dialysed with solution composed of (mM): KCl 120, KH2PO4 5, MgSO4 5, Na2ATP 5, Li2GTP, Hepes 10; pH was adjusted to 7.4 with KOH. Recording of the cell membrane potential was synchronised with confocal imaging using a TTL synchronising pulse generated by the confocal scanner at the beginning of the time series protocol.
Membrane currents were recorded using the conventional whole-cell patch clamp technique using an Axopatch 200A or a Multiclamp 700A patch-clamp amplifier (Molecular Devices Co., CA, USA). Liquid junction potential was nulled with the offset circuit of the amplifier before seal formation. Pipette and cell capacitance and series resistance were compensated electronically using corresponding amplifier controls. No electronic compensation for the leakage conductance was introduced. The electrical signals were filtered at 1 kHz (−3 dB frequency) by a four-pole low-pass Bessel filter. Voltage protocols were generated and electrical signals were digitised at 5 kHz using a DigiData 1200 or a Digidata 1322A hosted by a Pentium PC running either pCLAMP 6.0 or pClamp 8.2 software (Molecular Devices Co., CA, USA).
Muscarinic cationic current (mIcat) activated by external application of 10 μM CCh (see above) was recorded at holding potential of −50 mV. Background holding current was measured before CCh application. In these experiments, the composition of extracellular solution was (mM): CsCl 120, CaCl2 2.5, MgCl2 1.2, glucose 12, Hepes 10 (pH 7.4 adjusted with CsOH) and the composition of the pipette solution was (in mM): CsCl 80, MgATP 1, creatine 5, glucose 5, Hepes 10 (pH adjusted to7.4 with CsOH). To unmask any direct effect of the IP3R inhibitor 2-APB on mIcat, the effect of 2-APB on mIcat mediated via inhibition of IP3R-mediated Ca2+ release was eliminated by clamping [Ca2+]i at 100 nM with 4.6 mM CaCl2 / 10 mM BAPTA buffer added to the pipette solution [24].
Voltage-gated Ca2+ currents were evoked by 500-ms voltage steps to 0 mV applied from a holding potential of −80 mV every 30 s. In these experiments, the composition of the extracellular solution was (mM): NaCl 135, CsCl 6, CaCl2 2.5, MgCl2 1.2, glucose 12, Hepes 10 (pH 7.4 adjusted with NaOH) and the composition of the pipette solution was (in mM): CsCl 126, MgSO4 5, Na2ATP 5, Li2GTP 1 (pH adjusted to7.4 with CsOH and [Ca2+]i was clamped at 20 nM with 2.6 mM CaCl2 / 10 mM EGTA buffer).
2.8. Reagents
Protease (Type X), collagenase (type 1A), soybean trypsin inhibitor (Type II-S), bovine serum albumin, adenosine 5′ triphosphate (ATP, magnesium salt), guanosine 5′ triphosphate (GTP, disodium salt), creatine, N-2-hydroxyethylpiperazine-N′-2-ethanesulphonic acid (HEPES), 1,2-bis(2-aminophenoxy)-ethane-N,N,N′,N′-tetraacetic acid (BAPTA), ethylene glycol-bis(2-aminoethylether)- N,N,N′,N′-tetraacetic acid (EGTA), carbamylcholine chloride (carbachol), 1,3,7-trimethylxanthine (caffeine), dimethyl sulfoxide (DMSO), paraformaldehyde and Triton X-100 were obtained from Sigma Chemical Co., Poole, Dorset, UK. Fluo-4 acetoxymethyl ester, Alexa Fluor 488 conjugated chicken anti-rabbit IgG (H+L), Alexa Fluor 633 conjugated goat anti-mouse IgG (H+L) and pluronic F-127 were obtained from Invitrogen Ltd., Paisley, UK. Fluo-3FF acetoxymethyl ester was from TefLabs, Austin, Texas, USA. All other chemicals were from BDH Laboratory Supplies (AnalaR grade), Pool, UK.
2.9. Data analysis
Image processing was carried out using an Indy workstation (Silicon Graphic, Inc., Mountain View, CA, USA) with custom routines written in IDL (Research Systems, Inc., Boulder, CO, USA). The final figures were produced using MicroCal Origin (MicroCal Software Inc., Northampton, MA, USA) and CorelDraw 7.0 (Corel Corporation, Canada). Where appropriate, data are expressed as mean values ± SEM for the number of cells (n) analysed. Comparative analysis of the data groups was performed using Student's t-test for paired or unpaired samples, as appropriate, with the threshold for statistical significance set at the 0.05 level.
3. Results
3.1. Action potential discharge following muscarinic receptor activation is coupled to a sub-PM [Ca2+]i upstroke (SPCU)
The dynamics of the change in intracellular Ca2+ concentration ([Ca2+]i) following muscarinic receptor activation were related to the changes in the cell membrane potential (Vm). SMCs freshly isolated from the guinea-pig ileum were preloaded with the high affinity Ca2+ indicator fluo-4 and bathed in PSS (see Methods). High speed x-y confocal Ca2+ imaging (acquisition rate varied between 19 and 42 Hz) was combined with recording of Vm in perforated-patch configuration to minimally perturb endogenous [Ca2+]i (n = 4). Muscarinic receptors were stimulated by brief applications of 10 μM carbachol (CCh) through a glass micropipette positioned within 100 – 200 μm of the cell surface. This revealed that the discharge of each action potential (AP) following muscarinic receptor activation was associated with a [Ca2+]i transient which was initiated by an abrupt increase of [Ca2+]i at multiple subplasmalemmal (sub-PM) regions. The dynamics of [Ca2+]i changes in these sub-PM regions of initiation was therefore analysed. In the example shown in Fig. 1, the first 60-ms application of 10 μM CCh triggered three APs but no sustained membrane depolarization (probably because equilibrium CCh concentration near the call membrane was not achieved). Even though after each action potential the cell membrane was repolarized to a resting level (black trace, Fig. 1A and B), each AP was associated with a stepwise increase of the global [Ca2+]i (green trace Fig. 1A and B). The second 600-ms application of 10 μM CCh caused membrane depolarization, which was associated with increased frequency of APs. Within 1.5 second depolarization became so extreme that AP discharge ceased (black trace, Fig. 1A and C). The global [Ca2+]i response caused by the 600-ms CCh application (green trace Fig. 1A and C) consisted of an initial large-amplitude transient followed by a sustained phase showing small amplitude oscillations. In both cases, however, each AP was coupled to a very brief abrupt [Ca2+]i transient (red trace, Fig. 1) at multiple sub-PM regions (illustrated by (ii) and outlined in (i), Fig. 1A). The spatio-temporal profiles of intracellular Ca2+ mobilisations associated with the first AP following the first CCh application, and with the two APs following the second CCh application, are illustrated by two galleries of 18 sequential confocal images (Fig. 1 B and C, respectively). Comparing the images in the galleries revealed a remarkable constancy of the positions of the sites of initiation of Ca2+ mobilisation associated with each AP. The lack of spatial uniformity suggests that rather than Ca2+ entry through voltage-gated Ca2+ channels (VGCCs) these signals reflect Ca2+ release from non-uniformly distributed sub-PM Ca2+ stores. Because of their location and robust onset kinetics, we refer to these events hereafter as the “sub-PM [Ca2+]i upstroke (SPCU)”.
Fig. 1.
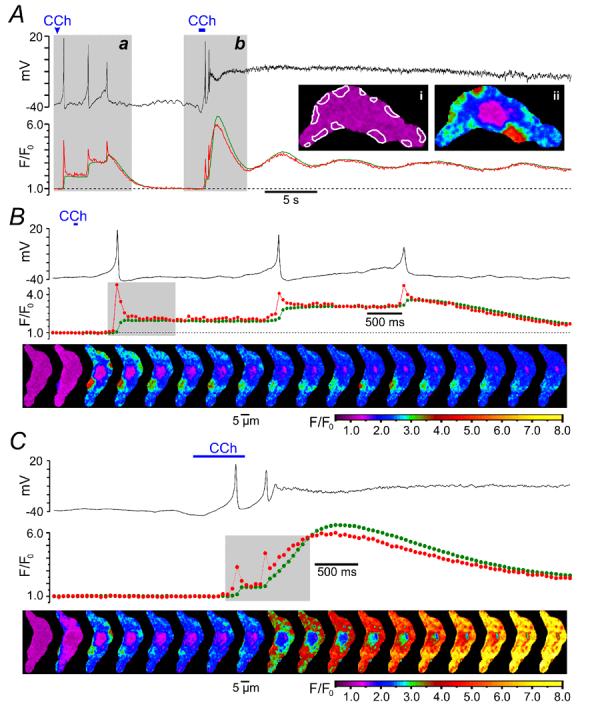
Carbachol (CCh) - induced action potentials are associated with sub-plasmalemmal (sub-PM) [Ca2+]i upstroke (SPCU). (A) The record of the cell membrane potential (black trace) is superimposed on the time course of the normalised intensity of fluo-4 fluorescence averaged (red trace) within 12 sub-PM regions outlined in (i) and (green trace) within the total confocal optical slice of SMC. The outlined regions in this and in all subsequent figures were the sites of CCh-induced Ca2+ wave initiation, as illustrated in (ii). The response of the SMC was triggered by 10 μM CCh applied twice from a glass micropipette: as 60-ms pulse (Aa and B) and as 600-ms pulse (Ab and C). The confocal images were acquired 54 ms apart. The fluorescence intensity was normalised to the average fluorescence intensity in a series of images captured before CCh application and colour coded as indicated (F/F0). Two periods of interest highlighted (grey background) in (A) are presented on an expanded time scale in (B) and (C). In (B) and (C) the gallery below the plot shows 18 sequential confocal images (after rotation by 90°) captured during the period highlighted in the corresponding plot of the normalised fluorescence signal. See also video clips in supplementary material on line.
Spatio-temporal patterns of CCh-induced [Ca2+]i mobilisation in non-patched SMCs are illustrated by Fig. 2 showing the results obtained in 5 different cells. Muscarinic receptors were activated by 10 μM CCh either superfused through the experimental bath (Fig. 2A) or applied to the cell as a 600-ms pulse through a glass micropipette (Fig. 2B-E). In all plots, the green traces show the temporal profile of the global [Ca2+]i changes, while red traces show the dynamics of [Ca2+]i changes averaged at multiple sub-PM regions (outlined in the images; insets on the plots) where CCh-induced [Ca2+]i transients were initiated. The galleries show sequential confocal images of fluo-4 fluorescence acquired during the periods marked by a grey background in the plots, respectively. In all cases CCh-induced [Ca2+]i transients were initiated by a SPCU (depicted by the arrowheads on the galleries). Muscarinic receptor activation evoked a SPCU independently of the extent of myocyte contraction (n = 147), suggesting that this phenomenon is not a result of the increase in the local density of Ca2+-release units, which could be caused by change in SMC geometry. In the vast majority of cases (97%) two phases can be clearly distinguished in the Ca2+ responses to CCh: (1) an initial high-amplitude [Ca2+]i transient and (2) a delayed increase of global [Ca2+]i characterised by a smaller amplitude and a tendency to oscillation.
Fig. 2.
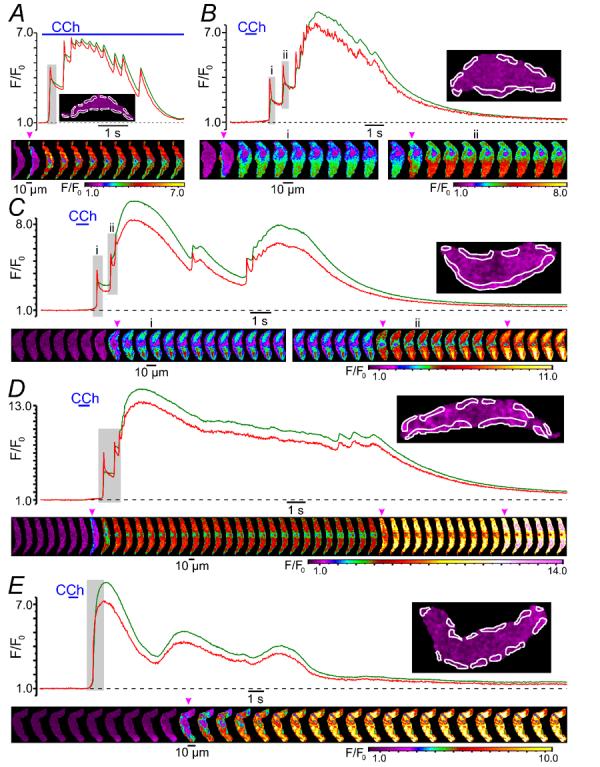
Spatio-temporal patterns of CCh-induced [Ca2+]i transients. The x-y confocal Ca2+ imaging was performed at 32 Hz (A and B), 40 Hz (C), 44 Hz (D) and 30 Hz (E). For each cell, the time course plot of the normalised fluo-4 fluorescence intensity was averaged (red trace) within sub-PM regions of interest where Ca2+ waves (induced by 10 μM CCh) were initiated (insets), and (green trace) within the total confocal optical slice of the SMC. The galleries below the plots show sequential confocal images (after rotation by 90°) captured during the periods highlighted by grey background in the plots. Magenta arrowheads in the galleries indicate sub-PM [Ca2+]i upstrokes.
3.2. Genesis of SPCU depends on both voltage-gated Ca2+ entry and Ca2+ release from the SR
Muscarinic cationic channels in gastro-intestinal smooth muscles have very low, if any, permeability for Ca2+ [25-27], and the major physiological role of muscarinic cationic current (mIcat) is to depolarise the cell membrane and to trigger the opening of voltage-gated Ca2+ channels (VGCCs), pharmacological blockade of which virtually completely abolishes muscarinic contractile responses [28, 29]. We therefore tested the effect of VGCC block on CCh-induced [Ca2+]i mobilisation. In this and all subsequent experiments we analysed the dynamics of [Ca2+]i changes at multiple sub-PM regions of initiation of CCh-induced [Ca2+]i mobilisation. In control, the response to CCh was initiated by a SPCU (Fig. 3A, red trace) and then rapidly propagated through the entire cell volume (Fig. 3A, gallery a). The initial [Ca2+]i transient was followed by a lower-amplitude sustained phase with two [Ca2+]i oscillations (Fig. 3A, red trace). Block of voltage-gated Ca2+ channels with 5 μM nicardipine (30-s incubation) eliminated the SPCU (Fig. 3A, gallery b) and substantially attenuated, but did not abolish, both the initial and delayed phase of the CCh-induced [Ca2+]i transient (Fig. 3A, green trace). In the presence of nicardipine, the peak of CCh-induced [Ca2+]i transient (green trace) was reduced by 60%, time-to-peak was increased from 0.8 s to 1.3 s and the Ca2+ wave propagated more slowly than in control (Fig. 3A, gallery b). These results suggest that: (1) the SPCU requires voltage-gated Ca2+ entry and (2) Ca2+ entry through VGCCs is not the only source of Ca2+ upon muscarinic [Ca2+]i mobilisation.
Fig. 3.
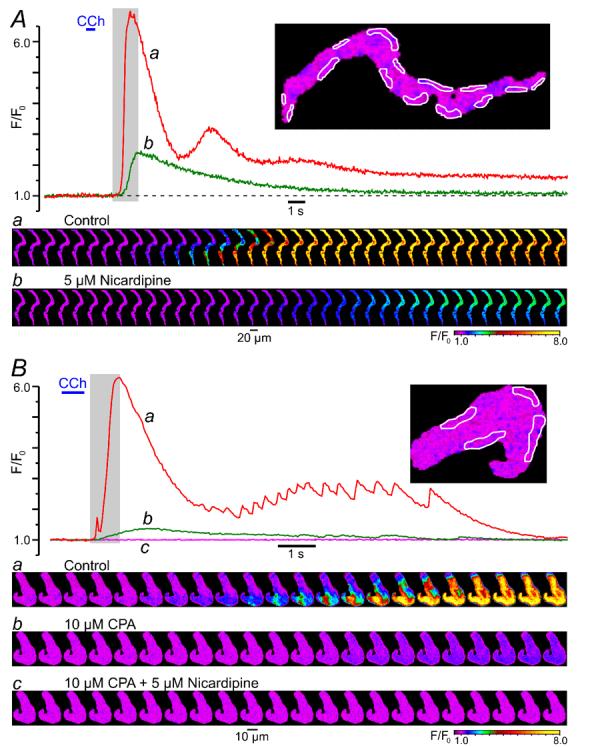
CCh-induced SPCU depends on both voltage-gated Ca2+ entry and Ca2+ release from intracellular stores: effect of block of voltage-gated Ca2+ channels with 5 μM nicardipine (A) and depletion of intracellular Ca2+ stores with 10 μM CPA (B). The imaging was performed at 26 Hz (A) and 23 Hz (B). The plot shows the time course of the normalised fluo-4 fluorescence intensity averaged within sub-PM regions (outlined) in control (a), after incubation with 5 μM nicardipine or 10 μM CPA (b) and after incubation with 5 μM nicardipine in the presence of 10 μM CPA (c). A 10-min period was allowed between subsequent 600-ms pulses of 10 μM CCh. The galleries below the plots show sequential confocal images (after rotation by 90°) captured during the highlighted periods.
To evaluate the contribution of Ca2+ release from intracellular stores to CCh-induced [Ca2+]i mobilisation, the effect of Ca2+ store depletion was tested. In control, CCh induced a rapidly propagating [Ca2+]i wave (Fig. 3B, gallery a) which was initiated by a SPCU (regions of initiation are outlined in the image, Fig. 3B). The initial [Ca2+]i transient was followed by a lower-amplitude sustained phase showing multiple small-amplitude but high-frequency oscillations (Fig. 3B, red trace). A 10-min incubation of the ileal SMCs with 10 μM cyclopiazonic acid (CPA), a reversible inhibitor of the sarco/endoplasmic reticulum Ca2+-ATPase (SERCA), resulted in complete depletion of the intracellular Ca2+ stores in this cell type, as we have previously demonstrated using flash release of “caged” IP3 preloaded into the cells through the patch pipette [24]. Depletion of Ca2+ stores with CPA treatment eliminated the SPCU, reduced the peak amplitude of the initial CCh-induced [Ca2+]i transient by 80 %, increased time-to-peak from 0.7 s to 1.4 s and virtually completely abolished the delayed phase of the response (Fig. 3B, green trace). With Ca2+ stores depleted, the residual [Ca2+]i response was spatially uniform (Fig. 3B, gallery b), consistent with Ca2+ entry through VGCCs. This was further confirmed when subsequent block of VGCCs with 5 μM nicardipine (while keeping Ca2+ stores depleted) completely abolished [Ca2+]i mobilisation in response to CCh (Fig. 3B, magenta trace and gallery c). This also directly demonstrates that muscarinic cationic channels in SMCs of the guinea-pig ileum are virtually impermeable to Ca2+.
Summing up, the above results show that both Ca2+ entry through VGCCs and Ca2+ release from intracellular Ca2+ stores contribute to the SPCU and full scale [Ca2+]i mobilisation in response to muscarinic stimulation.
3.3. SPCU results from IP3R - mediated Ca2+ release facilitated by Ca2+ entry through VGCCs
To examine what type of the SR Ca2+ release channels is involved in SPCU and what the mechanisms underlie the subsequent transient and delayed phases of CCh-induced [Ca2+]i mobilisation, we studied the effects of successive cumulative inhibitions of RyRs, IP3Rs and VGCCs on Ca2+ responses to CCh. The SMCs were incubated for 10 min with 50 – 100 μM tetracaine [30-32] to block RyRs and with 2 μM xestospongin C [16, 33, 34] to block IP3Rs. In the example shown in Fig. 4, control response to CCh consisted of a [Ca2+]i transient initiated by a SPCU and followed by numerous [Ca2+]i oscillations of gradually decreasing amplitude (Fig.4, red trace and gallery a). Nevertheless, block of RyRs by tetracaine did not abolish the SPCU and even slightly (5 %) augmented the initial [Ca2+]i transient, but abolished the delayed phase of the response to CCh and the [Ca2+]i oscillations (Fig.4, green trace and gallery b). The slight increase of the initial [Ca2+]i transient may result from the increase in the SR Ca2+ load following tetracaine treatment [31]. The overall effect of tetracaine on CCh-induced [Ca2+]i mobilisation implies that RyRs play little role, if any, in the SPCU, but are important for the sustained response to CCh and in [Ca2+]i oscillations.
Fig. 4.
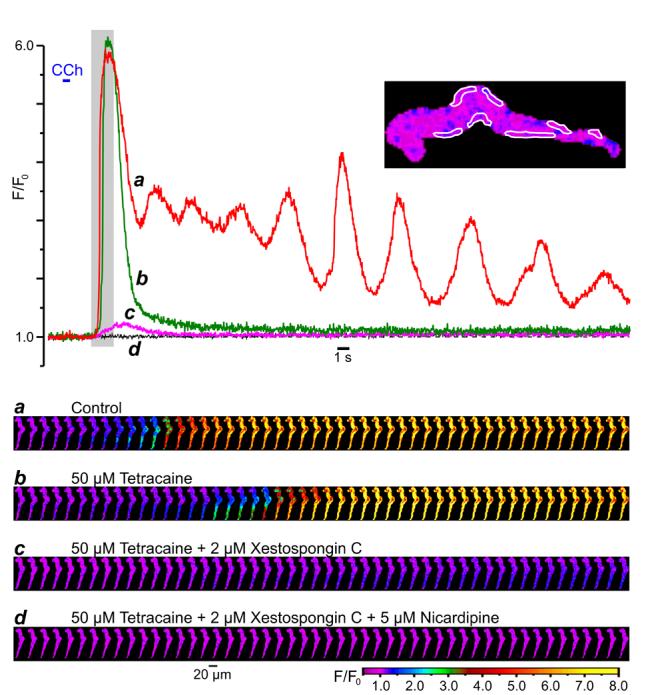
Components of the CCh-induced [Ca2+]i transient: successive cumulative inhibitions of RyRs, IP3Rs and VGCCs. The fluo-4 loaded SMC was stimulated with 10 μM CCh (600-ms pulse) and imaged at 31 Hz. A 10-min period was allowed between CCh pulses. The time course of the normalised fluo-4 fluorescence averaged within seven sub-PM regions (outlined) was plotted in control (a), and following successive cumulative inhibitions by 50 μM tetracaine (b), 2 μM xestospongin C (c) and 5 μM nicardipine (d). The galleries below the plot show sequential images (after rotation by 90°) taken during the highlighted period.
A subsequent incubation of the myocyte with 2 μM xestospongin C (in the presence of tetracaine) abolished the SPCU, inhibited the initial [Ca2+]i transient by 95 % and increased the time-to-peak from 1 s to 2.5 s (Fig.4, magenta trace c). The remaining CCh-induced [Ca2+]i transient was characterised by a slow and spatially uniform rising phase, consistent with Ca2+ entry through VGCCs (Fig.4, gallery c). This observation indicates that the SCPU strongly depends on the ability of IP3Rs to release Ca2+.
Subsequent block of VGCCs with 5 μM nicardipine (1-min incubation) completely abolished the residual response to CCh (Fig. 4, black trace and gallery d), thus confirming the idea that the uniform transient increase of [Ca2+]i triggered by CCh in the presence of tetracaine and xestospongin C was caused by Ca2+ entry through VGCCs.
It is known, however, that commercially available inhibitors of IP3R-mediated Ca2+-release, namely xestospongin C and 2-aminoethoxy-diphenylborate (2-APB), may affect some other mechanisms of intracellular Ca2+ homeostasis. Since the key event in muscarinic [Ca2+]i mobilisation seems to be the Ca2+ entry through VGCCs, we tested the effect of 2 μM xestospongin C and 30 μM 2-APB on the voltage-gated Ca2+ current (ICa) evoked by a 500-ms voltage steps from −80 to +0 mV under whole-cell voltage clamp conditions (Fig. 5). The currents were recorded in Cs+/Na+ -containing solutions (see Methods) while the pipette solution was supplemented with 5 mM ATP and 1 mM GTP to minimise rundown of ICa. The peak amplitude of ICa (Fig. 5A) evoked by repetitive (applied at 30-s intervals) voltage steps was normalised to the peak amplitude of ICa evoked by the first voltage step (Itest/Ifirst) and plotted over time (Fig. 5B) in control (n = 7) and following application of 2 μM xestospongin C (n = 4) or 30 μM 2-APB (n = 5). This revealed that after allowing for ICa rundown, the reduction of the current amplitude was by a further 74% during 10 min (from Itest/Ifirst = 0.779 ± 0.058 in control to Itest/Ifirst = 0.184 ± 0.013 in xestospongin C; significant difference: p = 0.00018) can account for the inhibitory effect of xestospongin C on VGCCs. In contrast, 2-APB during the same period augmented ICa (taking into account the ICa rundown) on average by 35% (from Itest/Ifirst = 0.779 ± 0.058 in control to Itest/Ifirst = 1.056 ± 0.159 in 2-APB; not significantly different: p = 0.073). Thus, the inhibitory effect of 2 μM xestospongin C on CCh-induced [Ca2+]i mobilisation may partially (since CCh-induced [Ca2+]i transient was not abolished by xestospongin C but was eliminated by subsequent block of VGCCs with nicardipine; Figs. 4) result from inhibition of VGCCs by this compound.
Fig. 5.

Effect of xestospongin C and 2-APB on voltage-gated Ca2+ current (ICa). ICa was recorded under whole-cell voltage clamp in response to 500-ms steps to 0 mV, applied every 30 s from Vh = −80 mV, in Cs+/Na+ - containing solutions (see Methods). The traces (A) show ICa evoked by the first step (black) and the step applied 10 min after (grey) in control (a), in 2 μM xestospongin C (b) and in 30 μM 2-APB (c). The normalised peak ICa (Itest/Ifirst) is plotted over time (B) in control (circle, n = 7), in 2 μM xestospongin C (triangle, n = 4) and in 30 μM 2-APB (square, n = 5). The drug application moment is depicted by the arrow.
Taking into account that activation of VGCCs in response to muscarinic receptor activation is caused by depolarisation evoked by mIcat, and thus will not occur if the muscarinic cationic channels are blocked, the effects of 30 μM 2-APB on mIcat (Fig 6A) were tested. The current induced by 10 μM CCh was recorded under whole-cell voltage clamp at holding potential of −50 mV using Cs+-containing pipette and bathing solutions (see Methods). Since mIcat is Ca2+-sensitive [24, 26, 35-37], to eliminate the effect of 2-APB caused by inhibition of IP3R-mediated Ca2+ release, [Ca2+]i was clamped at 100 nM (see Methods). Each SMC was stimulated twice with a 10-min interval between subsequent CCh applications. The response to the second CCh application (Test) was related to the response to the first CCh application (Control). In control external solution, the peak amplitude of the test mIcat (Fig. 6Aa) constituted on average 78 ± 4 % (n = 6) of the control mIcat (Fig. 6Ac). Incubation with 30 μM 2-APB for 8 min prior to the second CCh application reduced the peak amplitude of the test mIcat (Fig. 6Ab) to the 30 ± 2 % (n = 6) of the control mIcat (Fig. 6Ac). Thus, 30 μM 2-APB caused inhibition of mIcat by 61.5 % (significant difference: p = 0.00019).
Fig. 6.
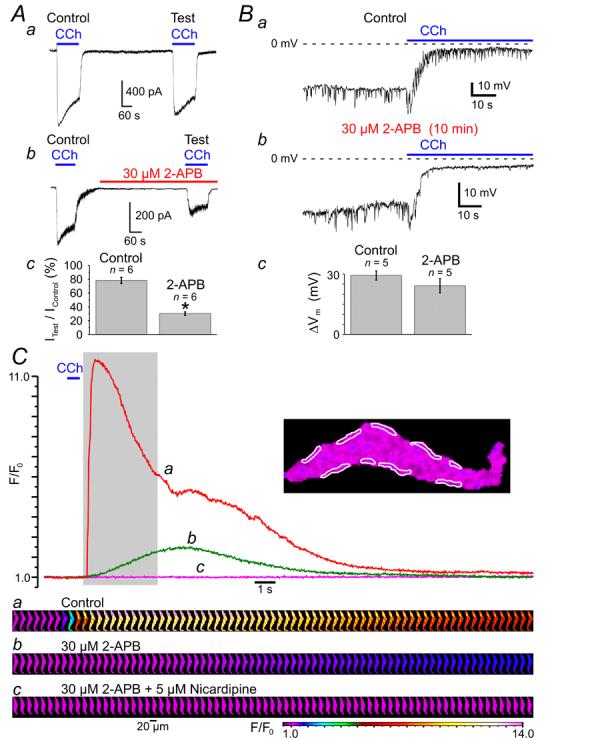
Effect of 2-APB on (A) muscarinic cationic current (mIcat), (B) CCh-induced membrane depolarization and (C) CCh-induced [Ca2+]i mobilisation. (A) mIcat was activated at −50 mV by 10 μM CCh in SMC with [Ca2+]i clamped at 100 nM with Ca2+/BAPTA buffer and was recorded in Cs+-containing solutions (see Methods). Peak mIcat (a) triggered by the second CCh application (Test) was related to that triggered by the first CCh application (Control). 30 μM 2-APB reduced the peak mIcat (b) on average (n = 6) by 61.5 % (c). *Significant difference (p < 0.0002) between control and 2-APB. Application of 10 μM CCh depolarized the cell membrane from −33 ± 2 mV to −4 ± 1 mV in control (Ba) and from −29 ± 4 mV to −5 ± 1 mV (n = 5) in 30 μM 2-APB (Bb). Summarised in (Bc). The fluo-4-loaded SMC was stimulated with 600-ms pulses of 10 μM CCh at 10-min intervals and imaged at 39 Hz (C). The time course of the normalised fluo-4 fluorescence averaged within nine sub-PM regions (outlined) was plotted in control (a), after incubation with 30 μM 2-APB (b) and after incubation with 5 μM nicardipine in the presence of 30 μM 2-APB (c). The galleries below the plot show sequential images (after rotation by 90°) taken during the period highlighted in the plot.
However, in terms of CCh-induced [Ca2+]i mobilisation, the amplitude of mIcat is not of as much importance (since this current is not conducted by Ca2+ under quasi-physiological conditions, see above) as the level of the cell membrane depolarization caused by this current. Indeed, any ionic current cannot change the cell membrane potential (Vm) beyond the level of the reversal potential of the current. In the case when the input resistance of the cell is relatively high (such as at resting condition) even a small amplitude current can produce a substantial shift in Vm. We therefore tested the effect of 30 μM 2-APB on CCh-induced membrane depolarization (Fig. 6B). The SMCs were bathed in PSS and dialysed with K+/Na+-containing solution (see Methods). Application of 10 μM CCh shifted Vm from −33 ± 2 mV to −4 ± 1 mV (n = 5) in control (Ba) and from −29 ± 4 mV to −5 ± 1 mV (n = 5) in 30 μM 2-APB (Bb), (n = 5). The values of the magnitude (ΔVm) of membrane depolarization (Bc) caused by CCh in control (29 ± 2 mV) and in 2-APB (24 ± 4 mV), as well as extent of the CCh-induced depolarization in control and in 2-APB were not significantly different (p = 0.23 and p = 0.52, respectively). This validates 2-APB as pharmacological tool for study of the role of IP3Rs in CCh-induced [Ca2+]i mobilisation.
Inhibition of IP3Rs by 10-min incubation of the SMC with 30 μM of 2-APB abolished the SPCU, inhibited the CCh-induced [Ca2+]i transient by 79% and increased time-to-peak from 0.5 s to 4.5 s (Fig.7C, green trace). Subsequent block of VGCCs with 5 μM nicardipine (30-s incubation) completely abolished the residual response to CCh (Fig. 6C, magenta trace and gallery c). This observation confirmed our conclusion that following muscarinic receptor activation Ca2+ entry through VGCCs facilitates the initial IP3R-mediated Ca2+ release at sub-PM regions leading to a SPCU, which in turn engages full scale [Ca2+]i mobilisation.
Fig. 7.

Summary of the effects of VGCC/SERCA/RyR/IP3R inhibitors on the initial phase of CCh-induced [Ca2+]i transient. Experimental protocol is illustrated in (A). The fluo-4 loaded SMCs were stimulated with 600-ms pulses of 10 μM CCh applied with a10 min interval. The response (ΔF/F0 averaged at multiple sub-PM regions) to the second CCh application (Test) was related to the response to the first CCh application (Control). The Test response was obtained either in the control (a), or following incubation with a drug (b-d) or a combination of several drugs. Two parameters were examined and summarised: (B) relative change in peak amplitude, (ΔF/F0)Test/(ΔF/F0)Control, and (C) relative change in time-to-peak, (tpeak)Test/(tpeak)Control. *Significant difference (p<0.0000002) between the parameters in control external solution and in the presence of drug (or drug combination), as indicated.
The effects of the Ca2+ store depletion, inhibition of VGCCs, RyRs and IP3Rs on the CCh-induced [Ca2+]i transient initiated by SPCU are summarised in Fig. 7. In all experiments the fluo-4 loaded SMCs were stimulated with 600-ms pulses of 10 μM CCh, which were applied to the same cell at least twice with a 10-min interval between subsequent CCh applications (Fig. 7A). The increase in the normalised intensity of fluo-4 fluorescence (ΔF/F0) averaged at multiple sub-PM regions of initiation of the CCh-induced [Ca2+]i transient was then plotted over time. The response to the succeeding CCh application (Test) was related to the response to the first CCh application (Control). The test response was obtained either in the control external solution (to evaluate reproducibility of CCh-induced [Ca2+]i transients) or following a 10-min incubation of the SMC with a drug or combination of several drugs. When the effect of VGCC block on muscarinic [Ca2+]i mobilisation was tested, nicardipine was applied 30 s – 1 min before pulse of CCh to minimise possible effect of VGCC block on the SR Ca2+ load. Two parameters were examined and are summarised in the bar diagram plots: (1) relative change in peak amplitude, (ΔF/F0)Test/(ΔF/F0)Control (Fig. 7B) and (2) relative change in time-to-peak, (tpeak)Test/(tpeak)Control (Fig. 7C). This revealed that: (i) in control external solution, the peak amplitude of the test response (Fig. 7Aa) constituted on average 92 ± 5 % (n = 29) of the control response (Fig. 7B), while time-to-peak was on average 102 ± 5 % of that in control response (Fig. 7C); (ii) block of RyRs with either 100 μM ryanodine ((ΔF/F0)Test/(ΔF/F0)Control = 84 ± 10%, (tpeak)Test/(tpeak)Control = 116 ± 15 %, n = 7) or with 50 – 100 μM tetracaine ((ΔF/F0)Test/(ΔF/F0)Control = 85 ± 6%, (tpeak)Test/(tpeak)Control = 108 ± 5 %, n = 14) had no significant effect on either amplitude (p = 0.47 and p = 0.37, respectively) or kinetics (p = 0.25 and p = 0.44, respectively) of the initial [Ca2+]i transient; (iii) depletion of intracellular Ca2+ stores with 10 μM CPA ((ΔF/F0)Test/(ΔF/F0)Control = 11.5 ± 6%, (tpeak)Test/(tpeak)Control = 284 ± 18 %, n = 6) reduced the peak amplitude by 87.5 % (p = 1.3×10−7) and increased time-to-peak by 178 % (p = 1.7×10−15); (iv) block of VGCCs with 5 μM nicardipine ((ΔF/F0)Test/(ΔF/F0)Control = 9 ± 1%, (tpeak)Test/(tpeak)Control = 334 ± 27 %, n = 18) reduced the peak amplitude by 90 % (p = 5.7×10−18) and increased time-to-peak by 227 % (p = 5.2×10−14); (v) simultaneous block of VGCCs and RyRs with 5 μM nicardipine / 50 – 100 μM tetracaine ((ΔF/F0)Test/(ΔF/F0)Control = 9 ± 3%, (tpeak)Test/(tpeak)Control = 360 ± 60 %, n = 6) reduced the peak amplitude by 90 % (p = 2.2×10−9) and increased time-to-peak by 253 % (p = 8.7×10−11); (vi) block of IP3Rs with 2 μM xestospongin C ((ΔF/F0)Test/(ΔF/F0)Control = 22 ± 6%, (tpeak)Test/(tpeak)Control = 261 ± 26 %, n = 15) reduced the peak amplitude by 76 % (p = 1.2×10−11) and increased time-to-peak by 159 % (p = 3.3×10−10); block of IP3Rs with 30 μM 2-APB ((ΔF/F0)Test/(ΔF/F0)Control = 23 ± 3%, (tpeak)Test/(tpeak)Control = 283 ± 22 %, n = 16) reduced the peak amplitude by 75 % (p = 2.0×10−13) and increased time-to-peak by 177 % (p = 3.6×10−13); (vii) simultaneous block of RyRs and IP3Rs with 50 – 100 μM tetracaine / 2 μM xestospongin C ((ΔF/F0)Test/(ΔF/F0)Control = 10 ± 4%, (tpeak)Test/(tpeak)Control = 287 ± 16 %, n = 6) reduced the peak amplitude by 89 % (p = 3.4×10−9) and increased time-to-peak by 181 % (p = 2.1×10−16); (viii) simultaneous block of RyRs and IP3Rs with 50 – 100 μM tetracaine / 30 μM 2-APB ((ΔF/F0)Test/(ΔF/F0)Control = 9 ± 2%, (tpeak)Test/(tpeak)Control = 291 ± 46 %, n = 6) reduced the peak amplitude by 90 % (p = 2.1×10−8) and increased time-to-peak by 185 % (p = 2.1×10−10); (ix) block of VGCCs (with 5 μM nicardipine) following intracellular Ca2+ store depletion (with 10 μM CPA), or simultaneous block of VGCCs and IP3Rs (with either 5 μM nicardipine / 2 μM xestospongin C or 5 μM nicardipine / 30 μM 2-APB) completely abolished [Ca2+]i mobilisation in response to CCh. Thus, significant inhibition of the CCh-induced [Ca2+]i transient was associated with significant reduction in its rate of rise, which is indicative of the elimination of the SPCU. Altogether these results strongly suggest that following muscarinic stimulation, SPCU results from Ca2+ release mediated via IP3Rs, which are activated synergistically by (1) IP3 mobilised via the M3 – Gq/11 - PLCβ pathway, and (2) Ca2+ entering the cell through VGCCs activated via the M2 - Go – mIcat – membrane depolarization pathway.
3.4. Sub-plasmalemmal SR elements are enriched with IP3Rs
The above hypothesis suggests: (1) a sub-PM location of the SR elements in ileal SMCs and (2) the expression of IP3Rs in these SR elements.
Intracellular Ca2+ stores visualised with the low-affinity (kd(Ca) = 42 μM) Ca2+ indicator fluo-3FF (Fig. 8A) consisted of a sub-PM SR network and some central elements (n = 47). This spatial organisation of intracellular Ca2+ stores is generally similar to that we have previously demonstrated in the rabbit portal vein myocytes using DiOC6 and BODIPY TR-X ryanodine [38] and SMCs of the guinea-pig mesenteric artery using brefeldin A BODIPY 558/568 (unpublished observation).
Fig. 8.
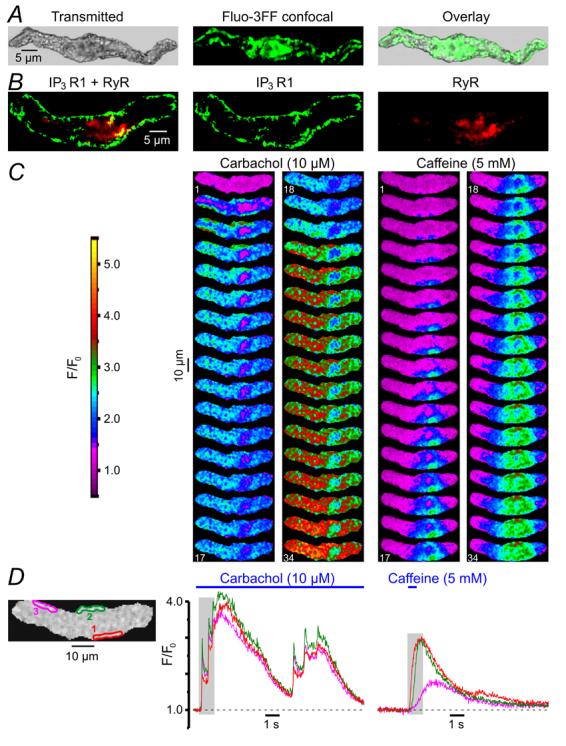
Predominant expression of IP3R type 1 in sub-PM SR encourages SPCU. (A) Ca2+ stores visualised with fluo-3FF consisted of a sub-PM SR network and some central formation. (B) Immunolocalisation of IP3R type 1 and RyRs using a double-staining protocol (see Methods). Confocal image of Alexa Fluor 488 fluorescence (green), showing type 1 IP3R distribution (middle), and confocal image of Alexa Fluor 633 fluorescence (red), showing RyR distribution (right), are overlaid (left). (C) The fluo-4-loaded SMC was stimulated with 10 μM CCh and 5 mM caffeine (with 10-min interval). Two galleries of images (acquired at 42 Hz) highlight the difference in the initial phase of the responses. (D) The temporal profiles of the fluorescence at three regions outlined in red, green and magenta (left) are shown in corresponding colour.
Immunodetection of IP3Rs (with antibody specific for type 1 IP3R) and RyRs (with antibody targeting type 1, type 2 and type 3 RyRs) by double labelling (n = 18) of the same SMC (see Methods) revealed that type 1 IP3Rs are predominantly expressed in sub-PM SR elements over the entire periphery of the cell, while RyRs are absent from the ends of the SMC, but are seen in some central sub-PM and deep SR elements (Fig. 8B). Based on the results of the cell fractionation and binding studies of the longitudinal muscle layer of the guinea-pig ileum, showing that the overall stoichiometric ratio of RyRs to IP3Rs in SMCs from this tissue is 1:9-10, the existence of a Ca2+-storage compartment devoid of RyRs but equipped with IP3Rs has been suggested previously [39]. This is in agreement with our finding showing that only a few elements of the SR in the central region of the SMC represent a Ca2+ store where the type 1 IP3Rs and RyRs are co-expressed (yellowish spots; Fig. 8B).
Close proximity of the IP3R-enriched SR elements to the cell plasma membrane facilitates an abrupt IP3R-mediated Ca2+-release when [Ca2+]i and [IP3]i rapidly rise in the restricted microvolume between the SR and plasmalemma, as observed in the case of activation of muscarinic receptors with CCh (Fig. 8C). In contrast, when the same cell was stimulated with 5 mM caffeine, which activates RyRs to release Ca2+, the Ca2+ wave developed initially in the cell centre (where RyRs are predominant) and only then spreads to the cell periphery (Fig. 8C). The difference in the dynamics of [Ca2+]i mobilisation at three regions within the SMC following stimulation with CCh and caffeine is emphasised by the plots showing the time course of the fluo-4 fluorescence averaged within each of the three regions (Fig. 8D). In response to CCh [Ca2+]i rapidly increased at all three regions: IP3R-mediated Ca2+ release leading to a SPCU. In contrast, in response to caffeine, RyRs released Ca2+ initially within the region 1, then Ca2+ wave reached the region 2 and only then, with a more substantial delay, it arrived at region 3.
3.5. IP3R-mediated Ca2+ release is essential for force generation
Several lines of evidence presented above strongly suggest that in SMCs freshly isolated from the guinea-pig ileum, [Ca2+]i mobilisation in response to muscarinic receptor activation is initiated by a SCPU resulting from an abrupt IP3R-mediated Ca2+ release at multiple sub-PM regions where it is facilitated by Ca2+ entry through VGCCs. Under conditions when the SPCU is abolished (by inhibition of IP3Rs or VGCCs), the global [Ca2+]i mobilisation within the SMC is substantially attenuated (Fig. 7). It therefore seems likely that this IP3R-mediated Ca2+ release is a key element in the chain of events resulting in the muscarinic contractile response. To test this we examined the effect of inhibition of IP3Rs and RyRs on isometric force generated in response to muscarinic stimulation of a smooth muscle strip freshly dissected from the longitudinal layer of the guinea-pig ileum (Fig. 9). The strips were stimulated with 2 μM CCh, which was transiently applied to the same strip at least twice. The response to the second CCh application was referred to as the test response, while the response to the first CCh application was referred to as the control response. The test response was obtained either in the control external solution to evaluate reproducibility of the responses to CCh (Fig. 9A), or following a 7-min incubation of the strip with either 30 μM 2-APB (Fig. 9B) or 100 μM tetracaine (Fig. 9C). The maximal isometric force (Fo) detected during the test response was then normalised to that during the control response and compared in control and following incubation with the drugs (Fig. 9D). This revealed that: (i) in control external solution, the peak amplitude of the test response constituted on average 98 ± 7 % (n = 8) of the control response; (ii) inhibition of IP3Rs with 30 μM 2-APB ((Fo)Test/(Fo)Control = 26.5 ± 5 %, n = 4) reduced the peak isometric force by 73 % (p = 0.0006); (iii) inhibition of RyRs with 100 μM tetracaine ((Fo)Test/(Fo)Control = 29 ± 10 %, n = 3) reduced the peak a isometric force by 70 % (p = 0.001). This demonstrates that both IP3R- and RyR-mediated Ca2+ release are required for full scale contractile response to 2 μM CCh. However, in contrast to tetracaine, 2-APB also abolished spontaneous oscillations of isometric tension observed in control (compare Fig. 9B and 9C).
Fig. 9.

Effect of inhibition of IP3Rs and RyRs on isometric force of muscarinic contraction. Smooth muscle strips from longitudinal layer of the guinea-pig ileum were attached to isometric force transducer at a resting tension load of 5 mN, bathed in the PSS at 37°C and stimulated with 2 μM CCh with a 10-min interval. The response to the second CCh application (Test) was related to the response to the first CCh application (Control). The Test response was obtained in the control (A), in 30 μM 2-APB (B) and in 100 μM tetracaine (C). Insets: the Test responses presented on an expanded time scale. A summary of the relative change in the maximal isometric force ((Fo)Test/(Fo)Control) is presented as bar diagram plot (D). *Significant difference (p<0.004) between the normalised maximal isometric force in control external solution and in the presence of the drug, as indicated.
On the other hand, a SPCU should activate large conductance Ca2+ -activated K+ channels (BK channels) in the cell membrane, which may lead to membrane hyperpolarization and, as a result, to a decrease of Ca2+ entry through VGCCs [11], thus slowing down [Ca2+]i mobilization and perhaps the contractile response. This, however, was not observed. Indeed, even though the peak of the CCh-induced [Ca2+]i transient was associated with some minor (about 8 mV) transient repolarization of the cell membrane (Fig. 1C), inhibition of IP3Rs with 2-APB did not accelerate the contractile response to CCh, and its kinetics remained the same in control and following 2-APB or tetracaine pre-treatment (see insets in Fig. 9A-C). Nevertheless, we tested the effect of a selective inhibitor of BK channels, paxilline [40] on CCh-induced isometric force using a similar experimental protocol (Fig. 10). Exposure of the muscle strips to 0.1 μM paxilline did not change the tonic tension, but augmented the amplitude of spontaneous oscillations of isometric tension (Fig. 10B) and increased CCh-induced isometric force ((Fo)Test/(Fo)Control = 167 ± 11 %, n = 8) by 70% (p = 0.0009; Fig. 10C). It, however, had no significant effect on the kinetics (Fig. 10D) of the muscarinic contraction: time-to-peak ((tpeak)Test/(tpeak)Control) in control (97 ± 9 %, n = 8) and following paxilline treatment (104 ± 7 %, n = 8) were not significantly different (p = 0.92).
Fig. 10.

Effect of inhibition of BK channels on isometric force of muscarinic contraction. The protocol was similar to that in Fig.9. The Test response to 2 μM CCh was obtained in control (A) and in 0.1 μM paxilline (B). Insets: the overlays of Test (black trace) and Control (grey trace) responses normalised to their maximum are presented on an expanded time scale. The bar diagram plots summarise: (C) relative change in the maximal isometric force ((Fo)Test/(Fo)Control) and (D) relative change in time-to-peak ((tpeak)Test/(tpeak)Control). *Significant (p<0.0008) increase of the maximal cholinergic force following BK channel inhibition was not associated with any significant (p = 0.92) change in the kinetics of the contraction.
4. Discussion
As an increase of [Ca2+]i is a primary signal for contraction in all types of muscles, the nature of the mechanisms linking excitation to [Ca2+]i mobilisation, as well as the spatial organisation and molecular composition of intracellular Ca2+ release units are important determinants of the contractile response. However, the mechanisms coupling excitation to [Ca2+]i mobilisation in skeletal, cardiac and smooth muscles are different. While in skeletal muscles membrane depolarization triggers Ca2+ release from the SR via direct interaction between the voltage sensors in the T-tubules (voltage-gated L-type Ca2+ channels/the dihydropyridine receptors; VGCCs) and ryanodine receptors (RyRs) expressed in the terminal sacs of the SR (reviewed in [41-43]), in ventricular cardiac muscles Ca2+ entry through VGCCs triggers RyR-mediated Ca2+ release via a Ca2+-induced Ca2+-release (CICR) mechanism (reviewed in [43-47]). In both cases the structural basis for excitation-contraction (E-C) coupling is localisation of the SR RyRs at the ends of sarcomers in close juxtaposition to T-tubular VGCCs.
Cytosolic Ca2+ which triggers contraction of smooth muscle cells (SMCs) is mobilised either by depolarization of the cell membrane leading to Ca2+ entry through VGCCs (electromechanical coupling, [1]), or by activation of a variety of receptors (pharmacomechanical coupling, [1]) usually coupled via Gq/11-protein to stimulation of phospholipase C (PLC), IP3 production and IP3R-mediated Ca2+ release, or by a combination of these mechanisms. Either of these events, which cause an initial rise in [Ca2+]i, may be further augmented by RyR-mediated Ca2+ release activated via CICR [5, 8, 16, 20, 34, 48-52]. The relative contribution of RyRs and IP3Rs to intracellular [Ca2+]i mobilisation and the role of these receptors in the genesis of localised Ca2+-release events (sparks or puffs), propagating Ca2+ waves and [Ca2+]i oscillations varies in different types of SMCs, and often depends on the strengths and mechanism of SMC stimulation. In some phasic SMCs, e.g. urinary bladder and vas deferens, Ca2+ entry through VGCCs is tightly coupled to RyR-mediated Ca2+ release. Electrical stimulations of these SMCs (under current- or voltage-clamp conditions) triggered an abrupt RyR-mediated Ca2+-release at multiple sub-PM regions (‘hot spots’, [5, 50, 53]). Using 3D-immunofluorescence, freeze fracture and thin section electron microscopy, it was demonstrated that in SMCs of urinary bladder VGCCs and RyRs are in close proximity to each other within the caveolar domains [54], thus forming a complex analogous to Ca2+ release units of striated muscles [55]. Destruction of caveolae in SMCs of urinary bladder with methyl-β-cyclodextrin attenuated coupling between voltage-gated Ca2+ entry and RyR-mediated Ca2+ release and reduced contractile responses elicited by electrical stimulation [52]. Nevertheless, it was demonstrated that IP3R-mediated Ca2+-release is essential for [Ca2+]i mobilisation and contraction, especially (but not exclusively) induced by stimulation of SMCs with neurotransmitters and hormones [19, 20, 30, 33, 34, 56-60]. It therefore was suggested that at least in some SMC types CICR could be initiated and facilitated by IP3R-mediated Ca2+ release [16, 34, 48, 57, 58].
In this study we have demonstrated that strong muscarinic stimulation (with 10 μM CCh) of single ileal SMCs triggers an abrupt sub-PM [Ca2+]i upstroke (SPCU) produced by IP3R-mediated Ca2+ release from sub-PM SR elements. These events were closely associated with action potentials (Fig. 1) and strongly depended on Ca2+ entry through VGCCs (Figs. 2A and 7), suggesting that in this SMC type E-C coupling involves an initial IP3R-mediated Ca2+ release facilitated by voltage-gated Ca2+ entry. A molecular basis for this is that IP3Rs in this cell type are expressed in much greater quantity than RyRs (an overall stoichiometric ratio of RyRs to IP3Rs in the guinea-pig intestinal smooth muscle was reported to be 1:9-10 [39]) and are predominantly located in sub-PM SR, while RyRs are mostly confined to the centrally located deep SR (Fig. 8B, [24]), similarly to some other types of phasic SMCs [61]. Differential distribution of RyRs and IP3Rs explains the difference between spatio-temporal patterns of Ca2+ waves induced by direct stimulation of RyRs and Ca2+ waves triggered following IP3 mobilisation in response to muscarinic stimulation under conditions when voltage-gated Ca2+ entry is either facilitated (Fig. 8C and D) or diminished (by holding the cell membrane potential at a negative level [24]).
In gastrointestinal SMCs, a mixed population of muscarinic receptors (M2 and M3) with the predominance of the M2 subtype (75-82%) are co-expressed (reviewed in [62-64]). There are several signal transduction mechanisms which link activation of these receptors to stimulation of IP3R-mediated Ca2+ release. The main universal Ca2+ signalling pathway couples activated M3 receptors via pertussis toxin (PTX)-insensitive G proteins (Gq/11) to stimulation of phospholipase C β (PLCβ) leading to formation of IP3 [65, 66]. In addition, activation of M2 receptors is coupled via PTX-sensitive G proteins (Gi) to inhibition of adenylyl cyclase activity [63, 66] leading to a decrease in cAMP level and, as a result, to suppression of the inhibitory effects of protein kinase A on the PLCβ – IP3 – IP3R signalling pathway [67, 68]. Finally, activation of muscarinic receptors produces excitation of gastro-intestinal (GI) smooth muscles causing depolarization of SMCs and an increase in the frequency of the action potentials via modulation of the activity of many different channel types (reviewed in [64, 69; 70]), of which opening of the cationic channels [35; 71] synergistically regulated via M2 – Gαo [37; 72, 73] and/or M2/M3 – Go – atypical PLC [64, 74-76] and M3 – Gq/11 – PLCβ – IP3 – Ca2+ [24, 26, 27; 35-37] pathways is one of the major mechanisms of GI smooth muscle excitation. The inward Na+ current through muscarinic cationic channels (in many smooth muscles, including guinea-pig ileum, these channels have very low, if any, permeability to Ca2+ [25-27], see also Figs. 3Bc, 4d, and 6Cc) produces membrane depolarization resulting in an increased Ca2+ influx via VGCCs. There is growing evidence that Ca2+ entering SMC is “trapped” between the SR and opposed regions of the plasmalemma [59; 77] resulting in an increase of the local [Ca2+]i up to the order of 10 μM [9, 77]. Using two-photon flash photolysis of “caged” Ca2+ it was recently demonstrated that a local increase of [Ca2+]i may trigger IP3R-mediated Ca2+ release [51].
The SPCU triggered by stimulation of SMCs of the guinea pig ileum with 10 μM CCh was virtually abolished by either block of VGCCs (Fig. 3A) or by depletion of intracellular Ca2+ stores (Fig. 3B). Thus, similarly to Ca2+ mobilization at sub-PM ‘hot spots’ evoked by electrical stimulation of SMCs from urinary bladder and vas deferens [5, 50, 51, 52], SPCU results from the SR Ca2+ release induced by Ca2+ entry (CICR) through VGCCs. After Ca2+ store depletion the residual CCh-induced [Ca2+]i transient was spatially uniform, which is characteristic of Ca2+ entry through VGCCs [8, 50, 59, 77]. Indeed, subsequent inhibition of VGCCs (while keeping Ca2+ stores depleted) completely abolished [Ca2+]i mobilisation in response to CCh (Fig. 3B), which also demonstrates that muscarinic cationic channels in SMCs of the guinea-pig ileum are virtually impermeable to Ca2+ (see also [27]).
In contrast to that reported in SMCs from urinary bladder [50] (where RyRs are predominantly expressed in sub-PM SR elements [53] in close juxtaposition to plasmalemmal VGCCs within multiple caveolar domains [54]), the SPCU in SMCs of the guinea-pig ileum (where sub-PM SR elements are enriched with IP3Rs; Fig. 8B and [24]) was not affected by block of RyRs with either 50 – 100 μM tetracaine (Fig. 4) or 100 μM ryanodine (summarised in Fig. 7). Inhibition of RyRs, however, abolished the sustained phase of the CCh-induced [Ca2+]i transient and/or [Ca2+]i oscillations (Fig. 4), thus indicating that Ca2+ release via RyRs (predominantly expressed in centrally located SR in this cell type; Fig. 8B and [24]) is also involved in CCh-induced [Ca2+]i mobilisation. However, in contrast to SMCs from urinary bladder and vas deferens [5, 50, 53], RyR-mediated Ca2+ release in ileal SMCs is ‘loosely coupled’ [49, 78] to Ca2+ entry through VGCCs. It is also evident that in ileal myocytes IP3Rs alone (Fig. 4b) may account for Ca2+ wave propagation, not unlike the situation in guinea-pig colonic myocytes [19].
In contrast with Ca2+ ‘hot spots’ elicited by membrane depolarization in urinary bladder SMCs, which were insensitive to inhibition of IP3Rs with 3 μM xestospongin C [50], CCh-induced SPCU in ileal myocytes was virtually abolished by inhibition of IP3Rs (Figs. 4 and 6B). It should be noted, however, that in both cell types an initial global [Ca2+]i transient elicited by muscarinic stimulation was substantially attenuated by IP3R inhibition, but only in ileal SMCs this also suppressed the sustained rise of [Ca2+]i (Fig. 6C). On the other hand, in ileal SMCs, block of VGCCs or cumulative block of VGCCs and RyRs reduced CCh-induced [Ca2+]i transients by 90% (Fig. 7B). Altogether these observations strongly suggest that following muscarinic receptor activation: (1) SPCU results from IP3R-mediated Ca2+ release facilitated by Ca2+ entry through VGCCs, (2) Ca2+ mobilised upon initial [Ca2+]i transient activates RyRs to release Ca2+ and (3) the sustained rise of [Ca2+]i and/or [Ca2+]i oscillations are the result of interplay between IP3Rs and RyRs.
It should be noted that pharmacological agents used in this study to assess the contribution of IP3Rs to SPCU may affect voltage-gated Ca2+ entry induced by muscarinic receptor activation, namely: (1) xestospongin C was shown to inhibit barium current through VGCCs in guinea-pig ileal myocytes but had no effect on mIcat [79], (2) 2-APB was reported to inhibit mIcat in SMCs from murine stomach [80]. We found that 2 μM xestospongin C inhibited ICa by 74% (Fig.5) and, thus, its effect on muscarinic [Ca2+]i mobilisation may partially result from inhibition of VGCCs. Nevertheless, suppression of SPCU by 30 μM 2-APB, which did not inhibit VGCCs (Fig. 5) and had no effect on CCh-induced membrane depolarization (Fig. 6B), confirms that this event results from IP3R-mediated Ca2+ release.
Under conditions when SPCU is abolished (by inhibition of IP3Rs or VGCCs), the global [Ca2+]i mobilisation within the SMC is substantially attenuated (Fig. 7). It therefore seems likely that SPCU is a key element in the chain of events resulting in the muscarinic contractile response. Indeed, isometric force generated by smooth muscle strips of the longitudinal layer of the guinea-pig ileum in response to 2 μM CCh was attenuated by 73% following inhibition of IP3Rs (Fig. 9D), similarly to that reported in the guinea-pig distal colon [30]. However, similarly to that reported in urinary bladder, where contraction evoked by electrical stimulation [50] or stimulation of muscarinic receptors [81] was shown to depend on RyR-mediated Ca2+ release activated by CICR, muscarinic contraction of the longitudinal layer of the guinea-pig ileum was attenuated by 70% following inhibition of RyRs (Fig. 9D). This suggests that the sustained phase of the CCh-induced [Ca2+]i transient, which depends on RyR-mediated Ca2+-release (Fig. 4), is crucial for force generation and that the initial IP3R-mediated Ca2+ release serves to link Ca2+ entry through VGCCs to RyR-mediated Ca2+ release. It is noteworthy that inhibition of IP3Rs also abolished spontaneous oscillations of isometric tension observed in control (Fig. 9B). This indicates that IP3Rs are also involved in spontaneous rhythmical contractile activity of gastro-intestinal smooth muscles, which is driven by interstitial cells of Cajal (reviewed in [82]). The importance of IP3R type 1 for normal activity of gastro-intestinal smooth muscles was recently illustrated by the demonstration that gastric smooth muscle from mutant mice lacking the type 1 IP3R revealed no slow wave activity and had attenuated muscarinic excitatory responses [56].
Another important target for sub-PM Ca2+ mobilisation in SMCs are Ca2+-sesitive membrane ion channels [8, 9, 11, 33, 83-85]. We have recently demonstrated that mIcat in ileal SMCs is synergistically potentiated by Ca2+ and IP3 [24]. Hence, on the one hand, SPCU serves to accelerate membrane depolarization and Ca2+ influx through VGCCs, thus providing a positive feedback mechanism whereby Ca2+ entry and Ca2+ release promote membrane depolarization and further Ca2+ influx, termed Ca2+-induced Ca2+ entry (CICE) [86]. On the other hand, activation of BK channels by SPCU should limit the rate and the extent of muscarinic excitation. Nevertheless, neither inhibition of IP3Rs with 2-APB (which eliminates SPCU) nor direct inhibition of BK channels with 0.1 μM paxilline accelerated the contractile response to CCh (Figs. 9 and 10). Paxilline (0.1 μM), however, augmented the muscarinic contractile response by 70% (Fig. 10B and C) but had no effect on tonic tension before CCh application (Fig. 10B). A likely explanation is that Ca2+ entry though VGCCs facilitated by inhibition of BK channels (via the resulting greater membrane depolarization) does not alter [Ca2+]i, but instead it increases the SR Ca2+ load, thus causing an increase of CCh-induced isometric force. Thus, it seems likely that overall physiological effect of SPCU is to engage a rapid full-scale [Ca2+]i mobilization, rather than to control contractile activity of intestinal smooth muscles via modulation of the BK channel activity.
In conclusion, SPCU is caused by an abrupt IP3R-mediated Ca2+ release from sub-PM SR elements facilitated by Ca2+ influx through VGCCs and serves to augment intracellular Ca2+ mobilization via CICE (acting on muscarinic cationic channels) and via CICR (acting on RyRs) mechanisms, and represents the key element in the chain of the signalling events in cholinergic contraction.
Supplementary Material
Acknowledgements
Supported by The Wellcome Trust (075112, 074724, 042293) and British Heart Foundation (FS/04/052 and FS/06/077).
References
- 1.Somlyo AV, Somlyo AP. Electromechanical and pharmacomechanical coupling in vascular smooth muscle. J. Pharm. Exp. Ther. 1968;159:129–145. [PubMed] [Google Scholar]
- 2.Navedo MF, Amberg GC, Votaw VS, Santana LF. Constitutively active L-type Ca2+ channels. Proc. Natl. Acad. Sci. 2005;102:11112–11117. doi: 10.1073/pnas.0500360102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Amberg GC, Navedo MF, Nieves-Cintron M, Molkentin JD, Santana LF. Calcium sparklets regulate local and global calcium in murine arterial smooth muscle. J. Physiol. 2007;579:187–201. doi: 10.1113/jphysiol.2006.124420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee CH, Poburko D, Kuo KH, Seow CY, van Breemen C. Ca2+ oscillations, gradients, and homeostasis in vascular smooth muscle. Am. J. Physiol. 2002;282:H1571–H1583. doi: 10.1152/ajpheart.01035.2001. [DOI] [PubMed] [Google Scholar]
- 5.Imaizumi Y, Torii Y, Ohi Y, et al. Ca2+ images and K+ current during depolarization in smooth muscle cells of the guinea-pig vas deferens and urinary bladder. J. Physiol. 1998;510:705–719. doi: 10.1111/j.1469-7793.1998.705bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bolton TB, Imaizumi Y. Spontaneous transient outward currents in smooth muscle cells. Cell Calcium. 1996;20:141–152. doi: 10.1016/s0143-4160(96)90103-7. [DOI] [PubMed] [Google Scholar]
- 7.Benham CD, Bolton TB. Spontaneous transient outward currents in single visceral and vascular smooth muscle cells of the rabbit. J. Physiol. 1986;381:385–406. doi: 10.1113/jphysiol.1986.sp016333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bolton TB, Gordienko DV. Confocal imaging of calcium release events in single smooth muscle cells. Acta Physiologica Scandinavica. 1998;164:567–575. doi: 10.1046/j.1365-201X.1998.00464.x. [DOI] [PubMed] [Google Scholar]
- 9.ZhuGe R, Fogarty KE, Tuft RA, Walsh JV. Spontaneous outward transient currents arise from microdomains where BK channels are exposed to a mean Ca2+ concentration on the order of 10 μM during a Ca2+ spark. J. Gen. Physiol. 2002;120:15–27. doi: 10.1085/jgp.20028571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheng H, Lederer WJ, Cannell MB. Calcium sparks: elementary events underlying excitation –contraction coupling in heart muscle. Science. 1993;262:740–744. doi: 10.1126/science.8235594. [DOI] [PubMed] [Google Scholar]
- 11.Nelson MT, Cheng H, Rubart M, et al. Relaxation of arterial smooth muscle by calcium sparks. Science. 1995;270:633–637. doi: 10.1126/science.270.5236.633. [DOI] [PubMed] [Google Scholar]
- 12.Wier WG, Morgan K. α1-adrenergic signalling mechanisms in contraction of resistance arteries. Rev. Physiol. Biochem. Pharmacol. 2003;150:91–139. doi: 10.1007/s10254-003-0019-8. [DOI] [PubMed] [Google Scholar]
- 13.Wellman GC, Nelson MT. Signaling between SR and plasmalemma in smooth muscle: sparks and the activation of Ca2+-sensitive ion channels. Cell Calcium. 2003;34:211–229. doi: 10.1016/s0143-4160(03)00124-6. [DOI] [PubMed] [Google Scholar]
- 14.Gordienko DV, Bolton TB, Cannell MB. Variability of spontaneous subcellular calcium release in guinea-pig ileum smooth muscle cells. J. Physiol. 1998;507:707–720. doi: 10.1111/j.1469-7793.1998.707bs.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boittin F-X, Coussin F, Macrez N, Mironneau G, Mironneau J. Inositol 1,4,5-trisphosphate and ryanodine-sensitive Ca2+ release channel-dependent Ca2+ signalling in rat portal vein. Cell Calcium. 1998;23:303–311. doi: 10.1016/s0143-4160(98)90026-4. [DOI] [PubMed] [Google Scholar]
- 16.Gordienko DV, Bolton TB. Crosstalk between ryanodine receptors and IP3 receptors as a factor shaping spontaneous Ca2+-release events in rabbit portal vein myocytes. J. Physiol. 2002;542:743–762. doi: 10.1113/jphysiol.2001.015966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bolton TB. Calcium events in smooth muscles and their interstitial cells; physiological roles of sparks. J. Physiol. 2006;570:5–11. doi: 10.1113/jphysiol.2005.095604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McCarron JG, Chalmers S, Bradley KN, MacMillan D, Muir TC. Ca2+ microdomains in smooth muscle. Cell Calcium. 2006;40:461–493. doi: 10.1016/j.ceca.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 19.MacMillan D, Chalmers S, Muir TC, McCarron JG. IP3-mediated Ca2+ increases do not involve the ryanodine receptor, but ryanodine receptor antagonists reduce IP3-mediated Ca2+ increases in guinea-pig colonic smooth muscle. J. Physiol. 2005;569:533–544. doi: 10.1113/jphysiol.2005.096529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lamont C, Wier WG. Different roles of ryanodine receptors and inositol (1,4,5)-trisphosphate receptors in adrenergically stimulated contractions of small arteries. Am. J. Physiol. 2004;287:617–625. doi: 10.1152/ajpheart.00708.2003. [DOI] [PubMed] [Google Scholar]
- 21.Gordienko DV, Zholos AV, Bolton TB. Coupling between receptors, channels and intracellular calcium signalling in smooth muscle of small intestine. J. Physiol. 2005;568P:SA2. [Google Scholar]
- 22.Mackrill JJ, Challiss RAJ, O'Connell DA, Lai FA, Nahorski SR. Differential expression and regulation of ryanodine receptor and myo-inositol 1,4,5-trisphosphate receptor Ca2+ release channels in mammalian tissues and cell lines. Biochem. J. 1997;327:251–258. doi: 10.1042/bj3270251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Taylor CW, Traynor D. Calcium and inositol trisphosphate receptors. J. Mem. Biol. 1995;145:109–118. doi: 10.1007/BF00237369. [DOI] [PubMed] [Google Scholar]
- 24.Gordienko DV, Zholos AV. Regulation of muscarinic cationic current in myocytes from guinea-pig ileum by intracellular Ca2+ release: a central role of inositol 1,4,5-trisphosphate receptors. Cell Calcium. 2004;36:367–386. doi: 10.1016/j.ceca.2004.02.021. [DOI] [PubMed] [Google Scholar]
- 25.Inoue R, Kitamura K, Kuriyama H. Acetylcholine activates single sodium channels in smooth muscle cells. Pflügers Arch. 1987;410:69–74. doi: 10.1007/BF00581898. [DOI] [PubMed] [Google Scholar]
- 26.Pacaud P, Bolton TB. Relation between muscarinic receptor cationic current and internal calcium in guinea-pig jejunal smooth muscle cells. J. Physiol. 1991;441:477–499. doi: 10.1113/jphysiol.1991.sp018763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zholos AV, Bolton TB. Effects of divalent cations on muscarinic receptor cationic current in smooth muscle from guinea-pig small intestine. J. Physiol. 1995;486:67–82. doi: 10.1113/jphysiol.1995.sp020791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brading AF, Sneddon P. Evidence for multiple sources of calcium for activation of the contractile mechanism of guinea-pig taenia coli on stimulation with carbachol. Br. J. Pharmacol. 1980;70:229–240. doi: 10.1111/j.1476-5381.1980.tb07928.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Parekh AB, Brading AF. The sources of calcium for carbachol-induced contraction in the circular smooth muscle of guinea-pig stomach. Br. J. Pharmacol. 1991;104:412–418. doi: 10.1111/j.1476-5381.1991.tb12444.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McCarron JG, Craig JW, Bradley KN, Muir TC. Agonist-induced phasic and tonic responses in smooth muscle are mediated by InsP3. J. Cell. Sci. 2002;115:2207–2218. doi: 10.1242/jcs.115.10.2207. [DOI] [PubMed] [Google Scholar]
- 31.Cheranov SY, Jaggar J. Sarcoplasmic reticulum calcium load regulates rat arterial smooth muscle calcium sparks and transient K Ca currents. J. Physiol. 2002;544:71–84. doi: 10.1113/jphysiol.2002.025197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zheng YM, Wang QS, Rathore R, et al. Type-3 ryanodine receptors mediate hypoxia-, but not neurotransmitter-induced calcium release and contraction in pulmonary artery smooth muscle cells. J. Gen. Physiol. 2005;125:427–440. doi: 10.1085/jgp.200409232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bayguinov O, Hagen B, Bonev AD, Nelson MT, Sanders KM. Intracellular calcium events activated by ATP in murine colonic myocytes. Am. J. Physiol. 2000;279:C126–C135. doi: 10.1152/ajpcell.2000.279.1.C126. [DOI] [PubMed] [Google Scholar]
- 34.Zhang WM, Yip KP, Lin MJ, Shimoda LA, Li WH, Sham JS. ET-1 activates Ca2+ sparks in PASMC: local Ca2+ signaling between inositol trisphosphate and ryanodine receptors. Am. J. Physiol. 2003;285:L680–L690. doi: 10.1152/ajplung.00067.2003. [DOI] [PubMed] [Google Scholar]
- 35.Benham CD, Bolton TB, Lang RJ. Acetylcholine activates an inward current in single mammalian smooth muscle cells. Nature. 1985;316:345–347. doi: 10.1038/316345a0. [DOI] [PubMed] [Google Scholar]
- 36.Inoue R, Isenberg G. Intracellular calcium ions modulate acetylcholine-induced inward current in guinea-pig ileum. J. Physiol. 1990;424:73–92. doi: 10.1113/jphysiol.1990.sp018056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang YX, Fleischmann BK, Kotlikoff MI. M2 receptor activation of nonselective cation channels in smooth muscles: Calcium and Gi/Go requirements. Am. J. Physiol. 1997;273:C500–C508. doi: 10.1152/ajpcell.1997.273.2.C500. [DOI] [PubMed] [Google Scholar]
- 38.Gordienko DV, Greenwood IA, Bolton TB. Direct visualization of sarcoplasmic reticulum regions discharging Ca2+ sparks in vascular myocytes. Cell Calcium. 2001;29:13–28. doi: 10.1054/ceca.2000.0180. [DOI] [PubMed] [Google Scholar]
- 39.Wibo M, Godfraind T. Comparative localization of inositol 1,4,5-trisphosphate and ryanodine receptors in intestinal smooth muscle: an analytical subfractionation study. Biochem. J. 1994;297:415–423. doi: 10.1042/bj2970415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sanchez M, McManus OB. Paxilline inhibition of the alpha-subunit of the high-conductance calcium-activated potassium channel. Neuropharmacology. 1996;35:963–968. doi: 10.1016/0028-3908(96)00137-2. [DOI] [PubMed] [Google Scholar]
- 41.Rios E, Stern MD. Calcium in close quarters: microdomain feedback in excitation-contraction coupling and other cell biological phenomena. Annu. Rev. Biophys. Biomol. Struct. 1997;26:47–82. doi: 10.1146/annurev.biophys.26.1.47. [DOI] [PubMed] [Google Scholar]
- 42.Lamb GD. Excitation-contraction coupling and fatigue mechanisms in skeletal muscle: studies with mechanically skinned fibres. J. Muscle Res. Cell. Motil. 2002;23:81–91. doi: 10.1023/a:1019932730457. [DOI] [PubMed] [Google Scholar]
- 43.Dulhunty AF. Excitation-contraction coupling from the 1950s into the new millennium. Clin. Exp. Pharmacol. Physiol. 2006;33:763–772. doi: 10.1111/j.1440-1681.2006.04441.x. [DOI] [PubMed] [Google Scholar]
- 44.Wier WG, Balke CW. Ca2+ release mechanisms, Ca2+ sparks, and local control of excitation-contraction coupling in normal heart muscle. Circ. Res. 1999;85:770–776. doi: 10.1161/01.res.85.9.770. [DOI] [PubMed] [Google Scholar]
- 45.Cheng H, Wang SQ. Calcium signaling between sarcolemmal calcium channels and ryanodine receptors in heart cells. Front. Biosci. 2002;7:d1867–d1878. doi: 10.2741/A885. [DOI] [PubMed] [Google Scholar]
- 46.Soeller C, Cannell MB. Analysing cardiac excitation-contraction coupling with mathematical models of local control. Prog. Biophys. Mol. Biol. 2004;85:141–162. doi: 10.1016/j.pbiomolbio.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 47.Song LS, Guatimosim S, Gomez-Viquez L, et al. Calcium biology of the transverse tubules in heart. Ann. N. Y. Acad. Sci. 2005;1047:99–111. doi: 10.1196/annals.1341.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.White C, McGeown JG. Carbachol triggers RyR-dependent Ca2+ release via activation of IP3 receptors in isolated rat gastric myocytes. J. Physiol. 2002;542:725–733. doi: 10.1113/jphysiol.2002.020131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kotlikoff MI. Calcium-induced calcium release in smooth muscle: the case for loose coupling. Prog. Biophys. Mol. Biol. 2003;83:171–191. doi: 10.1016/s0079-6107(03)00056-7. [DOI] [PubMed] [Google Scholar]
- 50.Morimura K, Ohi Y, Yamamura H, Ohya S, Muraki K, Imaizumi Y. Two-step Ca2+ intracellular release underlies excitation-contraction coupling in mouse urinary bladder myocytes. Am, J, Physiol. 2006;290:C388–C403. doi: 10.1152/ajpcell.00409.2005. [DOI] [PubMed] [Google Scholar]
- 51.Ji G, Feldman M, Doran R, Zipfel W, Kotlikoff MI. Ca2+-induced Ca2+ release through localized Ca2+ uncaging in smooth muscle. J. Gen. Physiol. 2006;127:225–235. doi: 10.1085/jgp.200509422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hotta S, Yamamura H, Ohya S, Imaizumi Y. Methyl-beta-cyclodextrin prevents Ca2+-induced Ca2+ release in smooth muscle cells of mouse urinary bladder. J. Pharmacol. Sci. 2007;103:121–126. doi: 10.1254/jphs.sc0060213. [DOI] [PubMed] [Google Scholar]
- 53.Ohi Y, Yamamura H, Nagano N, et al. Local Ca2+ transients and distribution of BK channels and ryanodine receptors in smooth muscle cells of guinea-pig vas deferens and urinary bladder. J. Physiol. 2001;534:313–326. doi: 10.1111/j.1469-7793.2001.t01-3-00313.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Moore ED, Voigt T, Kobayashi YM, et al. Organization of Ca2+ release units in excitable smooth muscle of the guinea-pig urinary bladder. Biophys. J. 2004;87:1836–1847. doi: 10.1529/biophysj.104.044123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Franzini-Armstrong C, Protasi F, Ramesh V. Shape, size, and distribution of Ca2+ release units and couplons in skeletal and cardiac muscles. Biophys J. 1999;77:1528–1539. doi: 10.1016/S0006-3495(99)77000-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Suzuki H, Takano H, Yamamoto Y, et al. Properties of gastric smooth muscles obtained from mice which lack inositol trisphosphate receptor. J. Physiol. 2000;525:105–111. doi: 10.1111/j.1469-7793.2000.00105.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Boittin FX, Macrez N, Halet G, Mironneau J. Norepinephrine-induced Ca2+ waves depend on InsP3 and ryanodine receptor activation in vascular myocytes. Am. J. Physiol. 1999;277:C139–C151. doi: 10.1152/ajpcell.1999.277.1.C139. [DOI] [PubMed] [Google Scholar]
- 58.Boittin FX, Coussin F, Morel JL, Halet G, Macrez N, Mironneau J. Ca2+ signals mediated by Ins(1,4,5)P3-gated channels in rat ureteric myocytes. Biochem. J. 2000;349:323–332. doi: 10.1042/0264-6021:3490323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McCarron JG, Bradley KN, MacMillan D, Chalmers S, Muir TC. The sarcoplasmic reticulum, Ca2+ trapping, and wave mechanisms in smooth muscle. News Physiol. Sci. 2004;19:138–147. doi: 10.1152/nips.01518.2004. [DOI] [PubMed] [Google Scholar]
- 60.McCarron JG, MacMillan D, Bradley KN, Chalmers S, Muir TC. Origin and mechanisms of Ca2+ waves in smooth muscle as revealed by localized photolysis of caged inositol 1,4,5-trisphosphate. J. Biol. Chem. 2004;279:8417–8427. doi: 10.1074/jbc.M311797200. [DOI] [PubMed] [Google Scholar]
- 61.Nixon GF, Mignery GA, Somlyo AV. Immunogold localization of inositol 1,4,5-trisphosphate receptors and characterization of ultrastructural features of the sarcoplasmic reticulum in phasic and tonic smooth muscle. J. Muscle. Res. Cell. Motil. 1994;15:682–700. doi: 10.1007/BF00121075. [DOI] [PubMed] [Google Scholar]
- 62.Eglen RM, Reddy H, Watson N, Challiss RAJ. Muscarinic acetylcholine receptor subtypes in smooth muscle. TIPS. 1994;15:114–119. doi: 10.1016/0165-6147(94)90047-7. [DOI] [PubMed] [Google Scholar]
- 63.Gerthoffer WT. Signal-transduction pathways that regulate visceral smooth muscle function. III. Coupling of muscarinic receptors to signaling kinases and effector proteins in gastrointestinal smooth muscles. Am. J. Physiol. 2005;288:G849–G853. doi: 10.1152/ajpgi.00530.2004. [DOI] [PubMed] [Google Scholar]
- 64.Zholos AV. Regulation of TRP-like muscarinic cation current in gastrointestinal smooth muscle with special reference to PLC/InsP3/Ca2+ system. Acta. Pharmacol. Sin. 2006;27:833–842. doi: 10.1111/j.1745-7254.2006.00392.x. [DOI] [PubMed] [Google Scholar]
- 65.Prestwich SA, Bolton TB. G-protein involvement in muscarinic receptor stimulation of inositol phosphates in longitudinal smooth muscle from small intestine of the guinea-pig. Br. J. Pharmacol. 1995;114:119–126. doi: 10.1111/j.1476-5381.1995.tb14915.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Okamoto H, Prestwich SA, Asai S, Unno T, Bolton TB, Komori S. Muscarinic agonist potencies at three different effector systems linked to the M2 or M3 receptor in longitudinal smooth muscle of guinea-pig small intestine. Br. J. Pharmacol. 2002;135:1765–1775. doi: 10.1038/sj.bjp.0704642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zizzo MG, Mule F, Serio R. Mechanisms underlying the inhibitory effects induced by pituitary adenylate cyclase-activating peptide in mouse ileum. Eur. J. Pharmacol. 2005;521:133–138. doi: 10.1016/j.ejphar.2005.08.027. [DOI] [PubMed] [Google Scholar]
- 68.Bai Y, Sanderson MJ. Airway smooth muscle relaxation results from a reduction in the frequency of Ca2+ oscillations induced by a cAMP-mediated inhibition of the IP3 receptor. Respir. Res. 2006;7:34. doi: 10.1186/1465-9921-7-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bolton TB, Prestwich SA, Zholos AV, Gordienko DV. Excitation-contraction coupling in gastrointestinal and other smooth muscles. Ann. Rev. Physiol. 1999;61:85–115. doi: 10.1146/annurev.physiol.61.1.85. [DOI] [PubMed] [Google Scholar]
- 70.Zholos AV. Muscarinic effects on ion channels in smooth muscle cells. Neurophysiology. 1999;31:212–228. [Google Scholar]
- 71.Zholos AV, Bolton TB. Muscarinic receptor subtypes controlling the cationic current in guinea-pig ileal smooth muscle. Br. J. Pharmacol. 1997;122:885–893. doi: 10.1038/sj.bjp.0701438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kim YC, Kim SJ, Sim JH, et al. Suppression of the carbachol-activated nonselective cationic current by antibody against α subunit of Go protein in guinea-pig gastric myocytes. Pflügers Arch. 1998;436:494–496. doi: 10.1007/s004240050663. [DOI] [PubMed] [Google Scholar]
- 73.Yan H-D, Okamoto H, Unno T, et al. Effects of G protein-specific antibodies and Gβγ subunits on the muscarinic receptor-operated cation current in guinea-pig ileal smooth muscle cells. Br. J. Pharmacol. 2003;139:605–615. doi: 10.1038/sj.bjp.0705289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zholos AV, Tsytsyura Ya.D., Gordienko DV, Tsvilovskyy VV, Bolton TB. Phospholipase C, but not InsP3 or DAG, dependent activation of the muscarinic receptor-operated cation current in guinea-pig ileal smooth muscle cells. Br. J. Pharmacol. 2004;141:23–36. doi: 10.1038/sj.bjp.0705584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Okamoto H, Unno T, Arima D, et al. Phospholipase C involvement in activation of the muscarinic receptor-operated cationic current in Guinea pig ileal smooth muscle cells. J. Pharmacol. Sci. 2004;95:203–213. doi: 10.1254/jphs.fp0030635. [DOI] [PubMed] [Google Scholar]
- 76.Unno T, Matsuyama H, Okamoto H, et al. Muscarinic cationic current in gastrointestinal smooth muscles: signal transduction and role in contraction. Auton. Autacoid. Pharmacol. 2006;26:203–217. doi: 10.1111/j.1474-8673.2006.00366.x. [DOI] [PubMed] [Google Scholar]
- 77.Bradley KN, Craig JW, Muir TC, McCarron JG. The sarcoplasmic reticulum and sarcolemma together form a passive Ca2+ trap in colonic smooth muscle. Cell Calcium. 2004:29–41. doi: 10.1016/j.ceca.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 78.Collier ML, Ji G, Wang Y, Kotlikoff MI. Calcium-induced calcium release in smooth muscle: loose coupling between the action potential and calcium release. J. Gen. Physiol. 2000;115:653–662. doi: 10.1085/jgp.115.5.653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ozaki H, Hori M, Kim YS, et al. Inhibitory mechanism of xestospongin-C on contraction and ion channels in the intestinal smooth muscle. Br. J. Pharmacol. 2002;137:1207–1212. doi: 10.1038/sj.bjp.0704988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lee YM, Kim BJ, Kim HJ, et al. TRPC5 as a candidate for the nonselective cation channel activated by muscarinic stimulation in murine stomach. Am. J. Physiol. 2003;284:G604–G616. doi: 10.1152/ajpgi.00069.2002. [DOI] [PubMed] [Google Scholar]
- 81.Rivera L, Brading AF. The role of Ca2+ influx and intracellular Ca2+ release in the muscarinic-mediated contraction of mammalian urinary bladder smooth muscle. BJU Int. 2006;98:868–875. doi: 10.1111/j.1464-410X.2006.06431.x. [DOI] [PubMed] [Google Scholar]
- 82.Sanders KM, Koh SD, Ward SM. Interstitial cells of cajal as pacemakers in the gastrointestinal tract. Annu. Rev. Physiol. 2006;68:307–343. doi: 10.1146/annurev.physiol.68.040504.094718. [DOI] [PubMed] [Google Scholar]
- 83.Gordienko DV, Zholos AV, Bolton TB. Membrane ion channels as physiological targets for local Ca2+ signalling. J. Microsc. 1999;196:305–316. doi: 10.1046/j.1365-2818.1999.00599.x. [DOI] [PubMed] [Google Scholar]
- 84.ZhuGe R, Sims SM, Tuft RA, Fogarty KE, Walsh JV., Jr. Ca2+ sparks activate K+ and Cl− channels, resulting in spontaneous transient currents in guinea-pig tracheal myocytes. J. Physiol. 1998;513:711–718. doi: 10.1111/j.1469-7793.1998.711ba.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pucovsky V, Bolton TB. Localisation, function and composition of primary Ca2+ spark discharge region in isolated smooth muscle cells from guinea-pig mesenteric arteries. Cell Calcium. 2006;39:113–129. doi: 10.1016/j.ceca.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 86.Lee HK, Bayguinov O, Sanders KM. Role of nonselective cation current in muscarinic responses of canine colonic muscle. Am. J. Physiol. 1993;265:C1463–C1471. doi: 10.1152/ajpcell.1993.265.6.C1463. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.