

Abstract
The measurement of changes in NAADP (nicotinic acid adenine dinucleotide phosphate) levels in cells has been, and remains, key to the investigation of the functions of NAADP as a Ca2+-releasing second messenger. Here we provide details of how to isolate NAADP from cells by extracting with perchloric acid, and then measure the NAADP using a radioreceptor assay. . Information is provided in sufficient detail to make the measurement of NAADP accessible to a wider range of laboratories as the field grows. We demonstrate that NAADP is neither generated nor broken down during our sample processing conditions, and recover ∼65% of the NAADP. The radioreceptor assay is shown to be highly selective for the detection of NAADP in cell extract conditions. Furthermore a number of improvements, such as solid-state detection of the radioactivity, are incorporated to enhance the safety of the procedure, as well as the detection of very small concentrations of NAADP, and the swift processing of samples. Finally, we have developed a method to prevent the endogenous metabolism of NAADP, by chelating Ca2+ with BAPTA, thus reducing the difficulty of catching a small transient rise in NAADP levels. In summary, we have refined and validated a method for measuring NAADP levels and presented it in an accessible manner that will enable even inexperienced researchers to measure NAADP levels. It is hoped that this will speed research, and broaden the NAADP field.
Keywords: NAADP, mammalian, second messenger, radioreceptor
Introduction
The cytosolic concentration of Ca2+ is very closely regulated in cells: the resting concentration is usually maintained at ∼10−7 M. Changes in Ca2+ concentration play an important role in a wide range of processes, from fertilisation, throughout life, and in cell death [1]. This resting concentration may be increased by means of influx of Ca2+ from the extracellular matrix through a channel in the plasma membrane, or release of Ca2+ from intracellular stores. Release of Ca2+ from intracellular stores is usually as a result of the binding of a primary messenger, such as a hormone or neurotransmitter, to a receptor in the plasma membrane. This then results in the generation of a second messenger inside the cell that is able to release Ca2+ from the intracellular store. The best understood Ca2+-releasing second messenger is inositol 1,4,5-trisphosphate (IP3). It is synthesised following the activation of a receptor that is coupled to Gq, which in turn activates phospholipase C to cleave IP3 from phosphatidylinositol bisphosphate. IP3 binds to the IP3 receptor and releases Ca2+ from the endoplasmic reticulum [2].
NAADP (nicotinic acid adenine dinucleotide phosphate) was first reported as a Ca2+-releasing agent in 1995 [3]. However, in contrast to IP3, very little is known about the physiological roles of NAADP. It is believed that NAADP releases from an acidic lysosome-like store [4] [5], rather than the endoplasmic reticulum, though there are some reports of it affecting ryanodine receptors [6]. NAADP has now been categorised as a second messenger, according to the conditions laid down by Sutherland [7], in both the sea urchin [8], and model mammalian systems [9]. The ability to measure changes in NAADP levels in cells has been key to this categorisation, providing evidence that NAADP is synthesised in response to the binding of an agonist to its cell-surface receptor. This remains very important in characterising NAADP in a wider range of cell types, enabling a clearer picture of the functions of NAADP as a second messenger to be built up. Basal NAADP levels have been measured in a variety of sea urchin [10], plant [11] and mammalian [12, 13] cell types. Only recently, however, have agonist-induced changes in NAADP levels have been reported in sea urchin sperm [8], smooth muscle cells [14], pancreatic β cells [9], and pancreatic acinar cells [15]. The majority of these measurements have been made using a radioreceptor binding assay [12], though a sea urchin bioassay [16] and a cycling assay [13, 17] have also been described.
It is possible to use a bioassay, similar to that used for cADPR [18], to detect NAADP. However, there are no reports of NAADP measurements using this technique. Two variations on a cycling assay have been reported [13, 17]. These assays offer more sensitive detection of NAADP than the bioassay, with 50 fmol quantities being detectable with one method [13], and have the benefit that they do not require the preparation of a radioligand. However, the processing of samples is technically demanding and time-consuming. Cell extracts require purification by anion exchange chromatography, and subsequent enzyme treatment prior to determination of the NAADP concentration, which is actually determined indirectly from the resulting concentration of NAD or NADP. The radioreceptor binding assay that we report here is capable of directly detecting 100 amol quantities of NAADP, without the risk of interference from other compounds. Furthermore, it is possible to quickly process a large number of samples prepared using a straightforward acid extraction technique, without the need for further purification.
Here we report in detail a method for measuring changes in NAADP levels in a wide variety of cell types. Previously-expressed concerns, encompassing the generation or degradation of NAADP, as well as the selectivity of the assay, and the recovery of NAADP in a cell extract, are addressed in detail. Furthermore, a number of improvements to the assay are described. These improve the detection of low concentrations of NAADP in cell samples, and allow more timely analysis of the NAADP responses in cells in response to a variety of agonists. In addition, we are now able to prevent the metabolism of NAADP in cells, thus removing the difficulties associated with detecting a small transient rise in NAADP levels. Furthermore, the procedure may be conducted with a high degree of safety, and produces a minimal quantity of radioactive waste. . In summary, we have refined and validated a powerful method for measuring NAADP levels and presented it in an accessible manner that will enable even inexperienced researchers to measure NAADP levels. The importance of these measurements at this stage in our understanding of the functions of NAADP cannot be overlooked. It is hoped that this will speed research, and broaden the NAADP field.
Materials and Methods
Materials
All chemicals were purchased from Sigma (UK) except where otherwise indicated below.
High performance liquid chromatography
All HPLC was carried out on an anion-exchange resin (AGMP1, Biorad, USA), packed in 150 × 2.5 mm omnifit columns, using a concave upwards gradient of trifluoroacetic acid (TFA), delivered by a Waters 600e pump, as described previously [19]. Detection was by a UV detector (Waters 2487, UK) at 254 nm.
Stability of nucleotides under different pH conditions
NADP, NAADP or NADPH (1 mM) was incubated in the following buffers: 50 mM Hepes (pH 7.2); 0.75 M HClO4 (pH 0.88); 1 M KHCO3 (pH 9.1); 0.75 M HClO4/1 M KHCO3 (pH 8.6); or 1 M K2CO3 (pH 11) for 6 h. Samples were then neutralised with their base equivalence of either Tris base, for acidic solutions, or Hepes acid, for basic solutions, as applicable. Samples were analysed by HPLC as described above, with elution times being determined from standard runs with known compounds.
Acid extraction of NAADP
Cells are prepared At the appropriate time points, the reaction in cells was stopped by the addition of 0.75 M ice-cold HClO4 added as an equal volume of 1.5 M solution. Sonication (Jencons vibracell amplitude 60) was carried out to disrupt the cells, which were then placed on ice for 15 min. The denatured protein was pelleted by centrifugation at 9,000 xg for 10 min and stored at −80°C for later analysis. Supernatant was neutralised with 1 M KHCO3, added as an equal volume of 2 M solution, and vortexed. The resulting KClO4 precipitate was removed by centifugation at 9,000 xg for 10 min. Samples were stored at −80°C for later analysis.
Determination of the percentage recovery of NAADP by acid extraction
Lytechinus pictus sea urchins were given intracoloemic injections of 1 ml 0.5 M KCl. Eggs were harvested into ASW and sperm collected ‘dry’ on a Petri dish. Samples consisted of 20 μl sperm and 80 μl eggs in ASW. The samples were spiked with [32P]NAADP, and then processed as described under “acid extraction” below. The resulting solution was then counted, and the total percentage recovery determined.
Isolation and use of pancreatic acinar cells
Pancreatic acinar cells were prepared as described previously [15]. Briefly, pancreata were excised from male CD1 mice of 8-10 weeks old, and finely chopped before incubation with 200 units/ml collagenase for 15 min at 37 °C. The digested tissue was then washed and resuspended to 10 ml with buffer of the following composition (mM): NaCl 140, KCl 4.7, MgCl2 1, CaCl2 1, Hepes 10, glucose 10. Protease inhibitor cocktail tablets (EDTA-free, Roche Diagnostics, UK), one tablet per 50 ml, were added to this solution before use. The suspension was triturated and supernatant that contained dispersed cells was collected into a new tube. After centrifugation at 800 xg for 3 min at 4 °C, the supernatant was discarded, and the cell pellet resuspended for further trituration and centrifugation. The resulting supernatant was discarded, and the cells were kept in buffer supplemented with 1 mM CaCl2 at room temperature on a rocking board for 30 min before use. Prior to use, the cells were centrifuged at 800 xg for 1 min at 4 °C. The cells were suspended in the above buffer, without the addition of protease inhibitors. Pancreatic acinar cells, were aliquoted into tubes. These aliquots were incubated with 20 μM BAPTA-AM, a cell permeant Ca2+-chelator, for 30 min at room temperature. Cholecystokinin (CCK), or buffer control was then added to each aliquot by pipette. Further sample preparation was carried out as described under “acid extraction” above.
[32P]NAADP synthesis
[32P]NAADP was synthesised in a two-step reaction as described previously [18, 20]. Firstly, [32P]NADP was synthesised by incubating 25 μl/9.25 MBq [32P]NAD (GE Healthcare, UK) with 0.5 U/ml human NAD kinase (formerly kindly provided by Matthias Ziegler, now obtained commercially from Alexis, UK), 5 mM MgATP, and 100 mM Hepes for 1 h. Nicotinic acid 100 mM and ADP-ribosyl cyclase 1 μg/ml (kindly provided by H.C. Lee, University of Hong Kong, Hong Kong) were added to commence the second step, which was allowed to proceed for 1 h. The resulting mixture was pumped onto a HPLC column with a peristaltic pump. Separation was carried out on an anion-exchange resin (AGMP1, Biorad, USA) using a concave upwards gradient of trifluoroacetic acid (TFA) as described previously [20-23] with detection using an in-line Geiger counter (Bioscan Flow Count B-FC-1000, Bioscan, USA) integrated to the system using a Waters bus SAT/IN unit. The NAADP fraction was then collected and stored in 10 mM Tris at 4°C for use in the assay. The 14-day half-life of 32P limits use of the [32P]NAADP to approximately six weeks from synthesis.
Sea urchin homogenate preparation
Sea urchin egg homogenate was prepared as described previously [18]. Briefly, eggs were harvested from Lytechinus pictus by intracoelemic injection of approximately 1 ml 0.5 M KCl. Artificial sea water (ASW) was prepared to have the following composition (mM): NaCl 435, MgSO4 15, CaCl2 11, KCl 10, NaHCO3 2.5, Tris-HCl 20, pH was adjusted to 8.0 using HCl. Homogenate was then prepared as described by Dargie et al. [24]. Briefly, the eggs were washed several times by centrifugation at 800 xg in the following solutions: Ca2+ -free ASW with EGTA (twice), Ca2+ -free ASW (twice), and ‘intracellular medium’. The intracellular medium had the following composition (mM): N-methyl-D-glucamine, 250; potassium gluconate, 250; Hepes, 20; MgCl2, 1; pH adjusted to 7.2 with acetic acid. Homogenisation was performed in intracellular medium, to which the following were added: 2 mM ATP, 20 mM phosphocreatine, 20 U/ml creatine phosphokinase, 50 μg/ml leupeptin (EDTA-free cocktail, Roche, UK), 20 μg/ml aprotinin and 100 μg/ml soya bean trypsin inhibitor, with a dounce glass tissue homogeniser with a size A pestle. Cortical granules were removed by centrifugation at 13,000 xg for 10 s at 4°C. Homogenate was stored in 0.5-1 ml aliquots at −80°C, for up to 5 years, and used immediately after thawing.
NAADP binding
NAADP binding protein from sea urchin (L. pictus) egg homogenate was used, which is highly selective for NAADP [20, 23, 25]. Firstly, 25 μl of test sample was added to each tube. Secondly, 125 μl of 1% (v/v) sea urchin egg homogenate in intracellular medium was added and allowed to incubate for 10 min at 25 °C. Finally, 100 μl [32P]NAADP diluted into intracellular medium was then added so as to generate around 50,000 scintillation counts per tube. The tubes were then incubated at room temperature for a further 10 min at 25 °C. Note that the order of addition is important to maximise the sensitivity of the assay [12]. Bound NAADP was then trapped onto Whatmann GF/B filter papers using a Brandel Cell Harvester. Washing was carried out 3 times using 1 ml of a buffer containing 20 mM Hepes, 500 mM potassium acetate, pH 7.4.
Scintillation counting
Radioactivity was determined by placing the filter circles in a scintillation vial containing 10 ml H20, followed by Cerenkov scintillation counting.
Storage phosphor detection
Filter papers were wrapped in cling film and then placed in a cassette to expose a storage phosphor screen (GE Healthcare, UK) for 1-4 h. The screen was then scanned using a Typhoon 9400 scanner (GE Healthcare, UK) at a resolution of 100 microns. The resulting image was then analysed using ImageQuant (GE Healthcare, UK).
Results and Discussion
Measurement of NAADP levels, both at rest and in response to agonist stimulation, has been central to the initial demonstration of NAADP as a Ca2+-releasing second messenger. It is now becoming an ever more important process, as the roles of NAADP are investigated in more systems, and the agonists that are coupled to increases in NAADP levels are fully defined. The measurement of NAADP in cells may be divided into two steps: firstly the NAADP is isolated from the cells by means of acid extraction (Fig. 1), and secondly the NAADP in these samples is measured using the radioreceptor binding assay (Fig. 2). Cells are isolated from the relevant animal: here we have used pancreatic acinar cells from CD1 mice, and sea urchin (L. pictus) eggs and sperm. They are then exposed to agonist, and the reaction stopped at the appropriate time point by the addition of HClO4. Cultured cells may be used as an alternative to a freshly prepared cell suspension, in which case they should be scraped off the tissue culture plate at this stage. Sonication is used to disrupt the cells, and the protein is then pelleted. The resulting supernatant is neutralised with KHCO3 prior to analysis by the radioreceptor binding assay. It is therefore first necessary to establish that this acid extraction is indeed a valid method for isolation of NAADP from cells, and to determine how much NAADP is recovered using this method. The radioreceptor binding assay itself will then be described and validated in detail. Finally a number of improvements to the process will be described.
Figure 1.

Schematic diagram of the acid extraction process. Sperm or eggs are harvested from L. pictus or cells are prepared from mouse pancreas. Agonist is incubated with the cell preparation, then HClO4 is added to stop the reaction at the required time point. The sample is then sonicated to disrupt the cells and centrifuged to pellet the protein for subsequent assay. The supernatant is neutralised with an equal volume of 2 M KHCO3, or other bases where indicated, in preparation for analysis using the radioreceptor binding assay.
Figure 2.
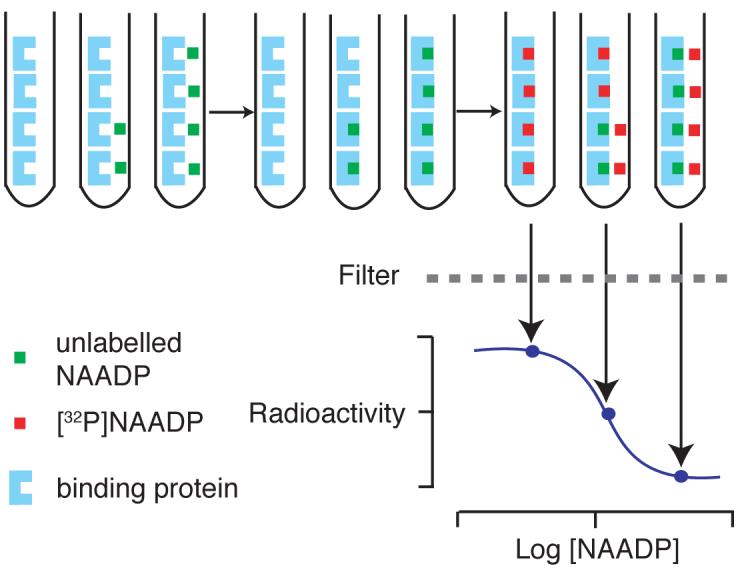
Schematic diagram of the NAADP radioreceptor binding assay. Firstly, known concentrations of NAADP (green boxes), or cell extracts, are added, followed by sea urchin egg homogenate (blue shapes) in intracellular medium, and a 10 min incubation period. NAADP binds irreversibly to the receptors in the homogenate. [32P]NAADP (red boxes) is then added; this binds to the remaining available receptor sites. Bound NAADP is separated from the mixture by filtration, and the radioactivity determined. Sample NAADP concentrations may be determined from the standard curve.
Stability of NAADP during the acid extraction process
During the acid extraction process, HClO4 is added to the cells, producing an acidic pH (0.88), and this solution is later neutralised, producing an alkaline pH (8.6). It must therefore be confirmed that NAADP is stable under the conditions used, and that no other nucleotides may be converted to NAADP in these conditions. It is commonly stated that in their oxidized form NAD and NADP are stable in acidic solutions but unstable in alkaline ones [26]. In contrast, their reduced forms, NADH and NADPH, are stable in alkaline solutions but unstable in acidic ones [26]. Additionally, alkaline treatment of NADP has been used to convert it to NAADP [27]. Moreover, Churamani et al. [12] reported that acid extraction using trichloroacetic acid caused conversion of NADP to NAADP. Therefore, we investigated the effect of our extraction conditions, on the stability of NADP, NADPH and NAADP.
NADP, NAADP and NADPH were incubated for 6 h in solutions with a range of pH. Where necessary, these solutions were neutralised with their base equivalence of Hepes acid or Tris base, prior to analysis by anion exchange HPLC (Fig. 3A). NADP, NAADP and NADPH remained stable at pH 7.2 (Fig. 3B). However, at pH 0.88, 8.6 and 9.1 NADPH became unstable, though NADP and NAADP remained stable (Fig. 3C-E); NADPH broke down to NADP (15 min) and uncharged products (0-2 min): these would not interfere with the detection of NAADP and hence were not considered a problem. At pH 11, NADPH proved similarly unstable, some conversion of NAADP to NADP occurred, and, more worryingly, NADP converted to a variety of products, including NAADP (25 min) (Fig 3F). It was therefore decided to thoroughly investigate the range of pH that might be encountered in the extraction conditions of the assay, with particular reference to the possibility of overshooting the intended pH when neutralising with base.
Figure 3.
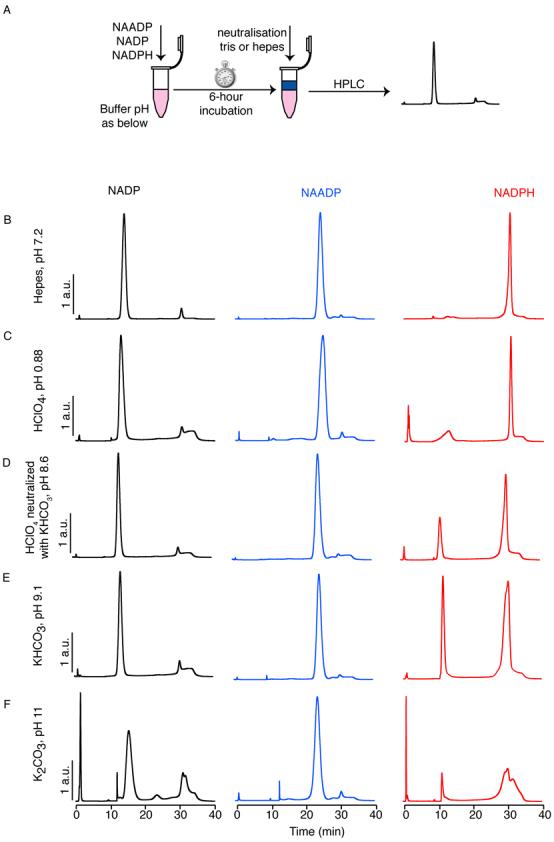
Stability of nucleotides during the acid extraction procedure. (A) Schematic showing the procedure: NAADP, NADP or NADPH (1 mM) was incubated for 6 h in the solutions indicated. Samples were then neutralised by addition of Hepes acid for basic samples, or Tris base for acidic samples and analysed by HPLC. HPLC traces show the stability of NAADP, NADP and NADPH after 6 h in (B) Hepes pH 7.2, (C) HClO4 pH 0.88, (D) HClO4 neutralised with KHCO3 pH 8.6, (E) KHCO3 pH 9.1 and (F) K2CO3 pH 11. HPLC traces are scaled to a common peak height for comparison (maximum 20% scaling). Note that NADP is stable except at pH 12, when a number of products result: in particular NAADP is generated, which would interfere with correct determination of NAADP levels from a cell extract. NADPH is stable only at pH 7.2, but NADP is the principle breakdown product; NAADP is not produced under the conditions tested.
Titration of HClO4 with different bases
Perchloric acid is a strong monoprotic acid and a 0.75 M solution has a pH of about 0.7. There are several choices of potassium bases that can be used for neutralization, producing a precipitate of KHClO4. Given the sensitivity of both NADP and NAADP to alkaline pH, we investigated a range of these possibilities. Titration of 1.5 M HClO4 with 1 M KOH, which has been reported as a neutralising base (*Guse?), gave a sharp equivalence point and resulted in a maximum pH of between 13 and 14 (Fig. 4). The sharp equivalence point makes it difficult to add precisely the correct amount to neutralise the solution, without generating an alkaline pH. This base was therefore considered unsuitable for use in this technique. Other groups have employed K2CO3 for neutralizations ?*. When 1.5 M HClO4 was titrated with 2 M K2CO3 the equivalence point was broader but a small addition of base beyond this still results in overshooting to a maximum above pH 10 (Fig. 4), which is outside the confirmed range for stability of the nucleotides. Neutralisation with KHCO3 also produced a broader equivalence point (Fig. 4). More importantly, even a vast excess of KHCO3 resulted in a pH of less than 9. Hence the nucleotides remain stable even when an excess concentration of base is added. This allows for straightforward neutralisation: it is possible to be certain of complete neutralisation, without the concern of high pH resulting in instability. The condition we use, where the cell extract containing 0.75 M HClO4 are neutralised with an equivalent volume of 2 M KHCO3 are therefore justified: at no point are conditions produced that induce breakdown of NAADP, or conversion of NADP to NAADP.
Figure 4.
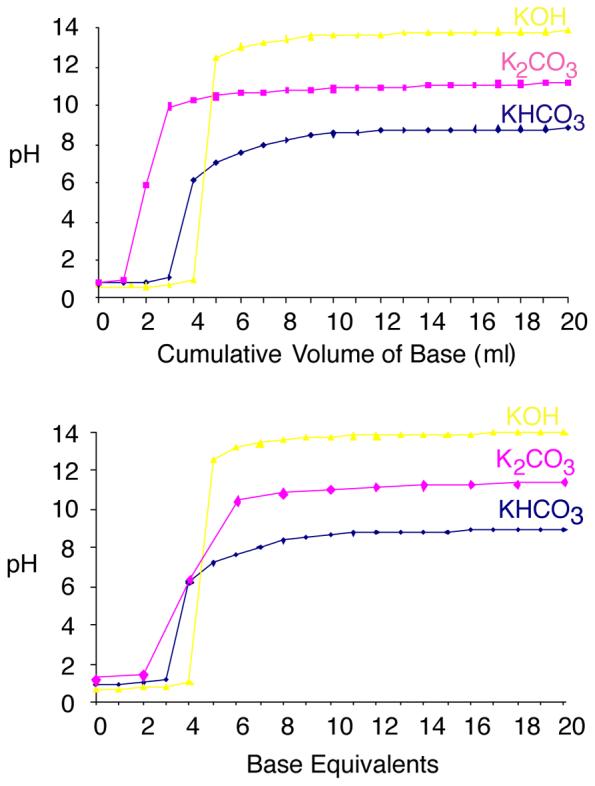
Titration of HClO4 with a variety of bases. With KHCO3, the maximum pH is 8.3, even in vast excess, ensuring that NAADP remains stable even in excess KHCO3. The addition of other bases, however, results in a higher pH; under these conditions NAADP becomes unstable.
Recovery of NAADP during acid extraction
To determine NAADP recovery in our system, we spiked the samples with known amounts of [32P]NAADP and then scintillation counted the samples after extraction. The sample was spiked with 251,100 ± 31,424 cpm and the final neutralized extract contained 162,140 ± 27,191 cpm, yielding a recovery of 65 ± 3.7%. Experiments with cold NAADP show that it is stable during acid extraction and neutralization (see above); therefore, counts in final neutralized sample are likely to be NAADP. Other groups have reported cADPR recovery of 48% using 3 M perchloric acid [28], 64% using ATP and perchloric acid [29], and 95% with perchloric acid as judged by immunoreactivity [30].
Synthesis of [32P]NAADP
Prior to commencement of the radioreceptor binding assay itself, the appropriate radio-ligand must be prepared. We routinely synthesize high-specific activity [32P]NAADP using a variation of the procedure first reported by Aarhus et al [19]. We have modified the reactions and now commonly get total yields of ≥95% based on total radioactivity.
In the first step of the reaction, the substrates are NAD and ATP. The ATP contains a small amount of contaminating ADP, but is not further purified as the ADP does not interfere with the reaction. After a 1-h incubation with human NAD kinase, all the NAD is converted to NADP and an equimolar amount of ATP is converted to ADP (Fig. 5A). Without purifiying any intermediates, the reaction is acidified by addition of nicotinic acid pH 4.0 to a final concentration of 100 mM and Aplysia ADP-ribosyl cyclase is added to a final concentration of 0.1 μg/mL to exchange the nicotinamide for nicotinic acid. After a 1-h incubation, there is complete conversion of the NADP to NAADP (Fig. 5A). Note that the presence of a high amount of nicotinic acid during the HPLC separation alters the elution times of the compounds slightly. This sequence of reactions should be completed each time, in order to confirm enzyme activity.
Figure 5.
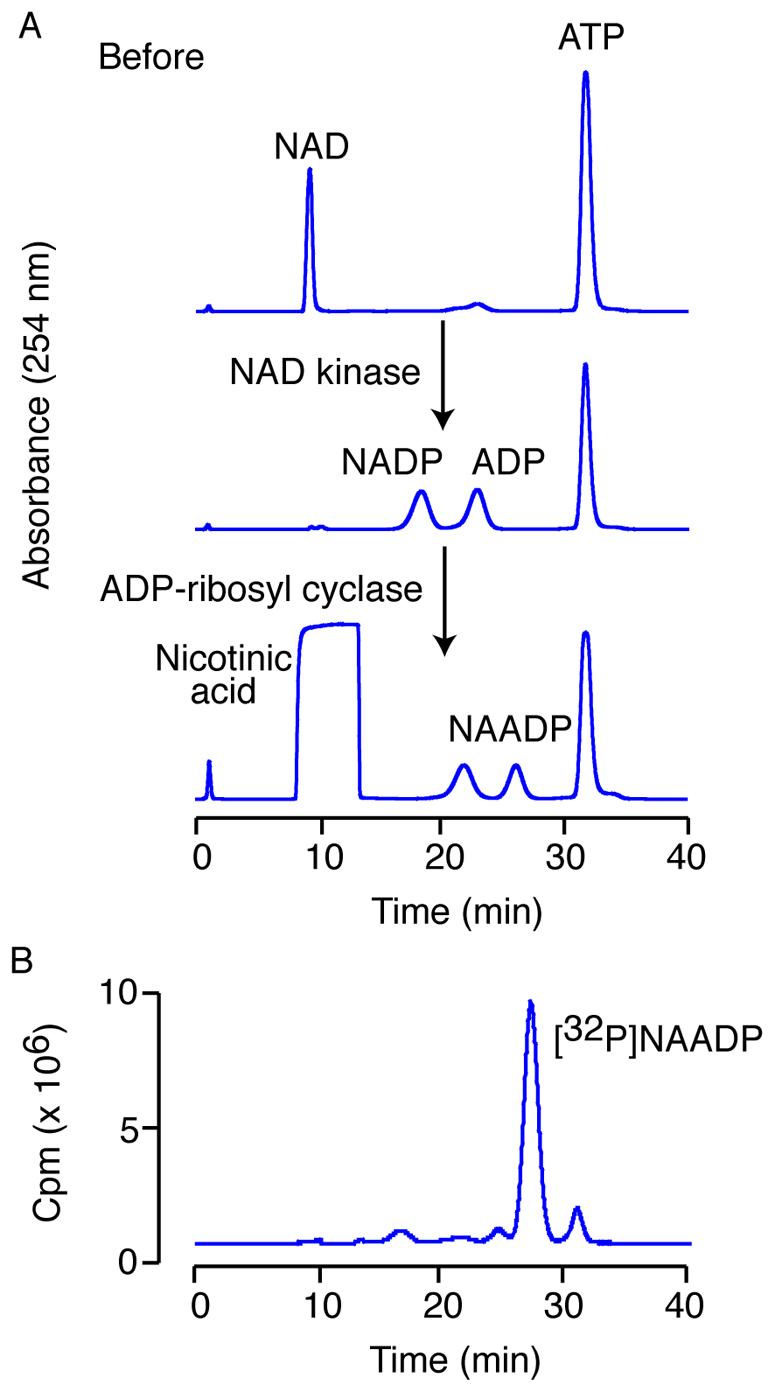
Synthesis of [32P]NAADP. (A) HPLC traces of the test reactions using unlabelled compounds. Firstly NAD is converted to NADP using NAD kinase, and secondly NADP is converted to NAADP using ADP-ribosyl cyclase. (B) The separation of the [32P]NAADP from the reaction mixture. Detection of unlabelled compounds is by UV detection at 254 nm. 32P-labelled compounds are detected using an in-line Geiger counter.
For the synthesis of radiolabelled NAADP, the entire sequence of reactions is repeated but with smaller volumes. An important consideration was that the human NAD kinase has a high Km for NAD (0.8 mM [26]). Therefore the volume of the [32P]NAD-containing reaction must be minimized to keep the NAD concentration as high as possible. Sea urchin NAD kinase has been used [20], but it must be freshly isolated. Sea urchin egg homogenates, without purification, have been used in the past *?, but both NAD and ATP are metabolised, greatly limiting the efficiency of conversion and the final yield. Human NAD kinase [26] is currently our choice due to the reproducibility and availability. Judicious choice of substrate concentrations, however, must be made, for example, too much ATP will compete with NAD at its binding site and prevent the reaction. We optimised this conversion by keeping volumes low to maintain a high [32P]NAD concentration and chose an intermediate concentration of ATP (5 mM).
Historically, we monitored the elution times of the radioactive compounds by collecting 1-ml fractions and then scintillation counting 1 μl from each fraction in 10 ml water. An improvement has been implemented at this stage: we now have an in-line radioactivity detector. Therefore, we can now collect only the NAADP peak (Fig. 5B). This minimizes radiation exposure during the collection, as the fraction collection tray doesn't have to be moved for each 1-ml sample, but only for the NAADP elution. Typically, the NAADP is collected in two 1.5-ml microcentrifuge tubes. The peak before the [32P]NAADP is [32P]NAAD (Fig. 5B), synthesized by base exchange of nicotinic acid with the nicotinamide on NAD that was not phosphorylated. This is the major product in some syntheses if the phosphorylation does not proceed to completion. The small peak after the [32P]NAADP is [32P]ADPR-P (Fig. 5B) resulting from the base exchange reaction when water is the attacking nucleophile. A very detailed account of this procedure can be found in [18]. A less satisfactory alternative, though one which requires less equipment, is to use the [32P]NAADP without further purification.
Determination of sample concentrations using a binding curve
To begin the radioreceptor binding assay, a range of known concentrations of unlabelled NAADP is added to racked test tubes. Sea urchin egg homogenate, containing the binding protein for NAADP, is then added. Samples are incubated for 10 min to allow NAADP to bind irreversibly to its receptor. [32P]NAADP is then added, and samples incubated for a further 10 min to allow [32P]NAADP to bind to the remaining free binding protein. Samples are then filtered using a cell harvester. Protein is retained on the filter paper, and hence the total radioactivity bound may be determined (Fig 5). Sample concentrations may then be determined from the standard curve.
To demonstrate that changes in NAADP in cell samples are detected using this method, a cell extract was performed from mouse pancreatic acinar cells. This was then serially diluted to produce a range of more dilute samples. Changes in NAADP were correctly detected from the standard curve (Fig. 6). That is, a given fold dilution resulted in the expected fold change in the NAADP detected.
Figure 6.
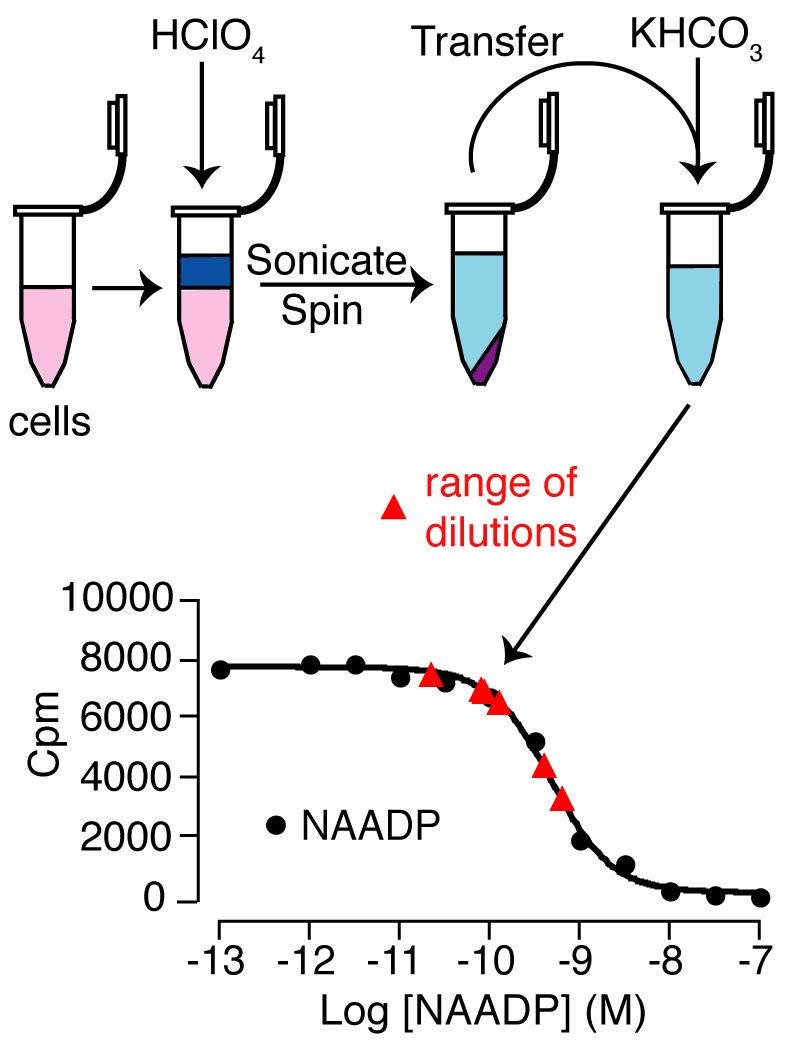
Measurement of NAADP levels in pancreatic acinar cell samples. (A) Schematic to show the preparation of pancreatic acinar cell samples. These samples were then diluted for use in the assay. (B) A competitive displacement curve was generated using standard concentrations of NAADP (black filled circles). Dilutions of the pancreatic acinar cell sample were determined from this curve and are shown on the trace (red triangles).
Inhibition of binding using related nucleotides and other Ca2+ releasing messengers
As with all binding assays a potential problem is selectivity, i.e. displacement of [32P]NAADP by compounds other than NAADP in the test samples. To get a feel for the extent of this possible problem, we screened several structurally related compounds to establish their relative ability to displace NAADP binding. The full concentration-displacement curves are shown in Fig. 6. As expected several compounds compete when present at high enough concentrations. The results can be summarized by the IC50s from the assay as follows: NAADP (540 pM), NADP (21 μM), NADPH (6.3 μM), cADPR (2.5 μM) and NAAD (51 μM). Other compounds failed to displace NAADP at the maximum concentrations tested in the assay: ATP (100 μM), ADP (100 μM), AMP (100 μM), NAD (2 mM), NADH (2 mM), ADPR (100 μM), IP3 (100 μM) and nicotinic acid (100 μM). These results are based on commercially obtained compounds (except cADPR, which was synthesized in-house, and NADP which underwent purification by HPLC). We interpreted these results by noting that all samples result from dilute cell suspensions, and are further diluted at least 100-fold in the final assay, and then calculated the maximum NAADP displacement based on known cellular concentrations of the metabolites. We concluded that none of these compounds presented any worries with regard to interference in the binding assay. For example, ATP has an intracellular concentration of between 1 and 5 mM (*Goldberg) and would be present in the assay at a maximum of 200 nM and therefore would not displace NAADP.
Validity of assay in cell extract conditions
Although none of the potential inhibitors of NAADP binding tested are able to displace NAADP at the concentrations present in a cell extract, it remains a possibility that the combination of these various compounds present in the cell would be able to displace NAADP and hence interfere with the assay. To test this possibility, a sample of chopped (McIlwain tissue chopper, 650 μm) rat brain (from 250-300 g female wistar rat) was processed by acid extraction in the manner described above. This was then spiked with two known 10-fold different concentrations of NAADP. The NAADP in these samples was then analysed using the radioreceptor binding assay (Fig. 8). The NAADP concentrations determined were 10-fold different as expected, demonstrating that changes in NAADP are accurately detected in cell extract conditions. Endogenous NAADP precludes precise detection of the original concentrations in this system.
Figure 8.
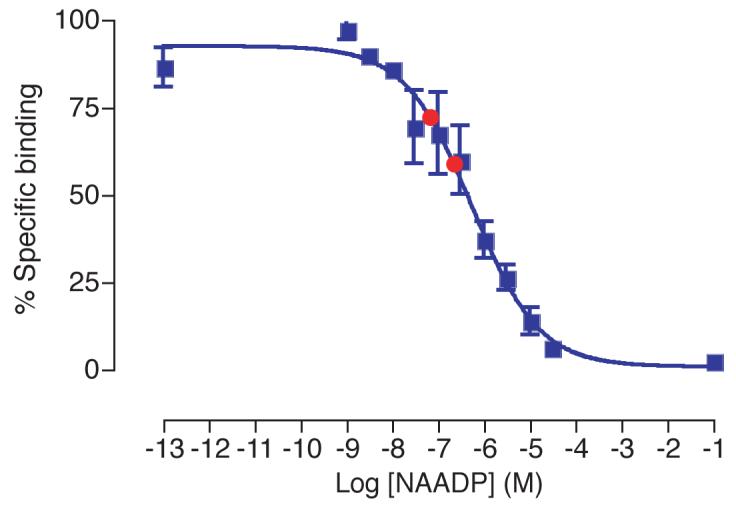
Displacement curve generated from known NAADP concentrations (blue squares). The difference between known NAADP concentrations spiked into rat brain extract (prepared as described) are correctly detected (red circles). Error bars represent standard error of the mean, n=3.
Use of the storage Phosphor screen in place of scintillation counting
We now describe a number of modifications to the procedure. These aim to reduce exposure to radioactivity, enhance detection of NAADP, particularly in response to agonist stimulation, and enable samples to be analysed as expediently as possible. Traditionally, radioactivity has been detected using Cerenkov scintillation counting. This method may be used for counting the radioactivity trapped on the filter papers in this assay, and produces accurate results. However, there are a number of disadvantages to using this technique. Each circle of filter paper must be transferred to an individual vial containing scintillation fluid, a laborious and time consuming process. Furthermore, disposal of these vials and the scintillation fluid generates a large volume of waste, and further exposure to radiation. We have therefore adopted a different technique for determining radioactivity in our samples. Whole filter papers (containing 48 samples with our harvester), are wrapped in cling film and then exposed to a storage phosphor screen for 1–4 h (dependent on the activity of the [32P]NAADP). One screen is able to expose to 4 such filter papers at a time; the times given are suggested minima as the radioactivity decays, but accurate readings may be obtained even after much longer (e.g. overnight) exposures. The storage phosphor screen contains fine crystals of BaFBr:Eu2+ in an organic binder. Exposure to radioactivity allows electrons to move within the crystals: Eu2+ is oxidised to Eu3+, and BaFBr is reduced to BaFBr−. The crystals remain in this state even when the radioactive sample is removed, so the screen stores a record of the sample. The screen is then scanned using a Typhoon scanner at a wavelength of 633 nm. The BaFBr− ions absorb light at this wavelength, and are oxidised to BaFBr; Eu3+ ions are reduced to Eu2+, but with an excited electron, which subsequently returns to its normal state. As this occurs, blue light (390 nm) is emitted and detected by the scanner. The light intensity is directly proportional to the radioactivity in the sample; in the images, darker pixels relate to a higher light intensity (Fig. 9B).
Figure 9.
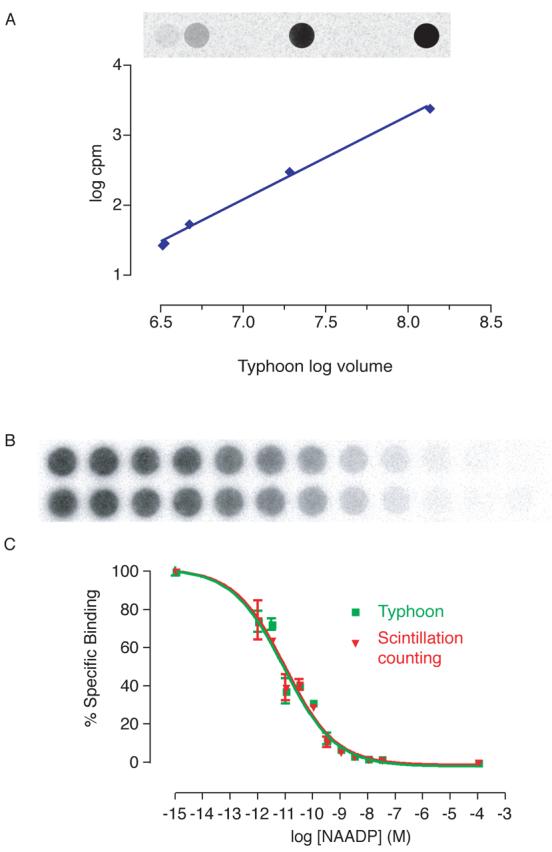
Detection of radioactivity using storage phosphor screens and a Typhoon scanner. (A) Comparison of radioactivity determined by the Typhoon scanner image versus cpm detected using Cerenkov scintillation counting. (B) Image produced for a standard NAADP displacement curve using the Typhoon scanner. (C) Comparison of standard curves with radioactivity detected using scintillation counting and the Typhoon scanner. Error bars represent standard error of the mean, n=3.
To confirm that the storage phosphor screen is able to detect the radioactivity in samples over a suitable dynamic range, and to confirm that the number of counts could be determined when required, a range of dilutions of [32P]NAADP were spotted onto filter papers. These were analysed first by typhoon scanning, and secondly by scintillation counting (Fig. 9A). This confirmed that a storage phosphor screen and typhoon scanning may be accurately used to determine radioactivity in a sample, and that it can be calibrated to determine the counts per minute should this information be required. Furthermore, the radioactivity on a filter paper using in an NAADP radioreceptor binding assay was determined using both techniques (Fig. 9C). The standard curves produced are identical, further validating the use of this method.
K+ to improve sample detection
Initially standard NAADP concentrations were made up in ‘intracellular medium’, as this provides suitable conditions for the sea urchin egg homogenate, and produces reliable binding curves. However, it was often observed that more radioactivity bound in tubes containing cell extract samples, than in the 0 NAADP (i.e. ‘intracellular medium’ only) standard. We therefore decided to make up NAADP in a solution closer to the conditions prevalent in the acid extracted cell samples. ‘Intracellular medium’ was mixed with an equal volume of 1.5 M HClO4, and this solution then mixed with and equal volume 2 M KHCO3. The resulting solution was centrifuged to remove the precipitate and then used to dilute NAADP standard for use in the assay (Fig. 10). The resulting displacement curve has an identical IC50 and Hillslope to that produced with standards in intracellular medium, but the maximum radioactivity bound was increased by approximately 3-fold. Based on previous reports [23], we hypothesised that the elevated K+ in the solution might be responsible for this effect. Intracellular medium was therefore supplemented with an approximately equivalent concentration of K+, in the form of and additional 1 M potassium acetate (pH maintained at 7.4). The binding assay was then repeated with NAADP standards made up in this medium (Fig. 10). The curve produced again had an identical IC50 and Hillslope, but with a 3-fold increase in maximum binding. Thus, it is important to use NAADP standards in a buffer with the same K+ concentration as the samples, in order to maximise detection, particularly of very small NAADP concentrations.
Figure 10.
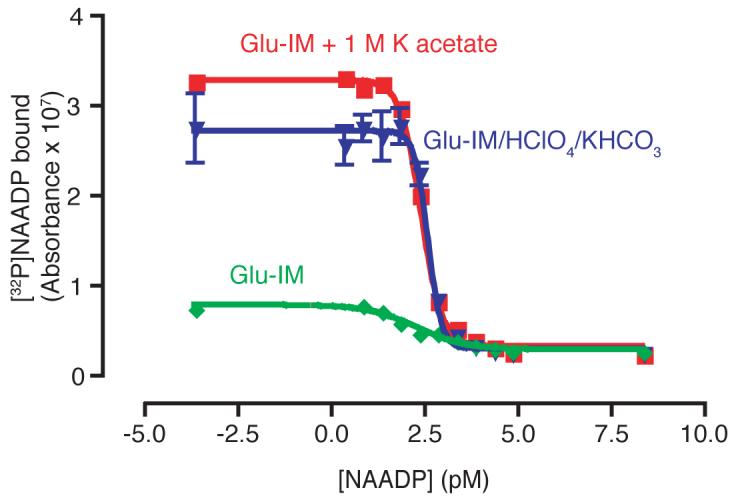
Improvement of NAADP detection in cell extract samples. Green trace shows standards made in intracellular medium (Glu-IM), the blue trace standards in ‘acid extracted’ intracellular medium, and the red trace standards in intracellular medium supplemented with 1 M potassium acetate. Error bars represent standard error of the mean, n=3.
Measurement of NAADP accumulation over time in agonist-stimulated cells: a novel methodology
The changes in NAADP levels thus far reported in mammalian systems consist of a very transient early spike in NAADP levels [15], which is easy to miss, especially in an uncharacterised system, dependent upon the time points chosen. However, NAADP is believed to be metabolised by a 2'-phosphatase, which is Ca2+-dependent [31]. We therefore tested whether the breakdown if NAADP could be inhibited by prior incubation of cells with the cell permeant form of the Ca2+-chelator BAPTA, BAPTA-AM (Fig. 11). Incubation with BAPTA-AM alone produced no change in NAADP levels, but subsequent exposure to CCK produced a continuous increase in NAADP levels, which lasted for at least 20 min. Thus, it is possible to detect changes in NAADP in this system, by measuring a basal level, and then accumulation over time after exposure to agonist. This removes the potential problems of missing a transient increase in NAADP levels, as any NAADP synthesised will not be broken down and will remain in the cells for measurement. The detection of agonists which produce increases in NAADP levels is thus greatly facilitated, and the problem of ‘false negatives’ nearly eliminated.
Figure 11.
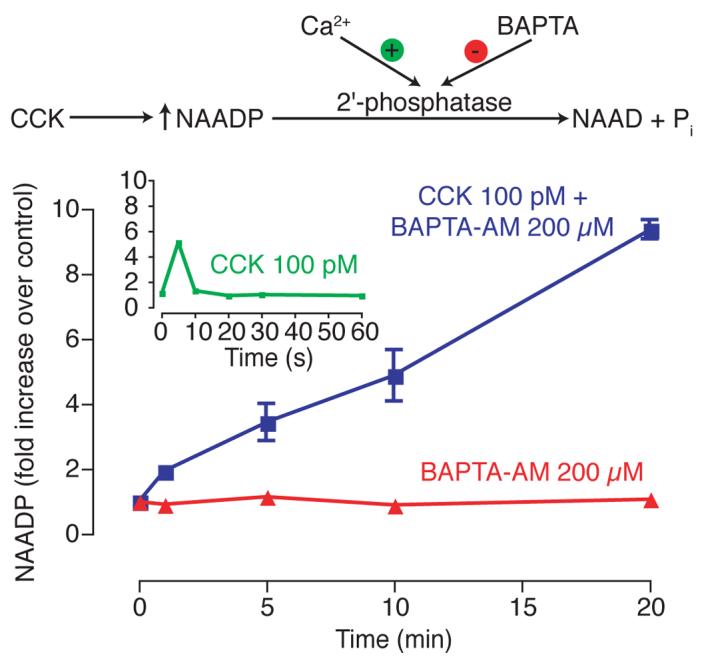
BAPTA-AM prevents NAADP metabolism and hence facilitates NAADP measurements in pancreatic acinar cells. Red trace shows cells pre-incubated with 20 μM BAPTA-AM, followed by control buffer additions and incubations for the various times. The blue trace shows cells that were pre-incubated with BAPTA-AM, followed by addition of 100 pM CCK for the times indicated. Error bars represent standard error of the mean, n=3. The inset (green trace) shows an initial peak in NAADP levels in response to CCK. This response is very transient and hence difficult to detect.
A similar technique has also proved useful in measuring changes in the levels of second messengers. For example, metabolism of cAMP may be prevented by the application of methyl xanthines [7]. Similarly, changes in IP3 may be measured using a radioreceptor binding assay [32]. However, in many systems, including pancreatic acinar cells, the changes are transient, and difficult to detect. Li+ may therefore be added to the cells to inhibit metabolism of IP3, and thus allow measurement of accumulation over time [33].
In conclusion, we report and validate a method that allows rapid and accurate measurements of NAADP concentrations. Information is provided in sufficient detail to make the measurement of NAADP accessible to a wider range of laboratories as the field grows. The acid extraction technique discussed is applicable to a wide variety of systems: it may be used in cell cultures, isolated cells, chopped tissue, and indeed whole tissues. The radioreceptor binding assay employed is highly selective for NAADP and is sufficiently sensitive to detect very small concentrations of NAADP. Finally, and most importantly, we report a number of improvements to the assay. In particular, the synthesis of [32P]NAADP has been modified to produce a very high yield with minimal exposure. Furthermore, the detection of radioactivity in samples is now faster, involves less exposure, and produces less waste. In addition, we have developed a method to prevent the metabolism of NAADP. This allows the measurement of NAADP accumulation over time, allowing more straightforward detection of even the most transient increases in NAADP. This development should prove very useful in the further investigation of the roles of NAADP, just as the use of Li+ has proved useful in the IP3 field.
Figure 7.

Selectivity of NAADP binding. Binding curves showing the effect of Ca2+-releasing messengers and related nucleotides on NAADP displacement. A curve for NAADP is provided for comparison. Error bars represent standard error of the mean, n=3.
Acknowledgements
This work was funded by project grants from the Wellcome Trust and BBSRC, and a Newton Abraham studentship to Alexander Lewis.
References
- 1.Berridge M, Lipp P, Bootman M. The versatility and universality of calcium signalling. Nature Mol Cell Biol Rev. 2000;1:11–21. doi: 10.1038/35036035. [DOI] [PubMed] [Google Scholar]
- 2.Taylor CW, Thorn P. Calcium signalling: IP3 rises again…and again. Curr Biol. 2001;11:R352–5. doi: 10.1016/s0960-9822(01)00192-0. [DOI] [PubMed] [Google Scholar]
- 3.Lee HC, Aarhus R. A derivative of NADP mobilizes calcium stores insensitive to inositol trisphosphate and cyclic ADP-ribose. J Biol Chem. 1995;270:2152–7. doi: 10.1074/jbc.270.5.2152. [DOI] [PubMed] [Google Scholar]
- 4.Churchill GC, Okada Y, Thomas JM, Armando A, Genazzani, Patel S, Galione A. NAADP mobilizes Ca2+ from reserve granules, a lysosome-related organelle, in sea urchin eggs. Cell. 2002;111:703–708. doi: 10.1016/s0092-8674(02)01082-6. [DOI] [PubMed] [Google Scholar]
- 5.Yamasaki M, Masgrau R, Morgan AJ, Churchill GC, Patel S, Ashcroft SJ, Galione A. Organelle selection determines agonist-specific Ca2+ signals in pancreatic acinar and beta cells. J. Biol. Chem. 2004;279:7234–7240. doi: 10.1074/jbc.M311088200. [DOI] [PubMed] [Google Scholar]
- 6.Cancela JM, Gerasimenko OV, Gerasimenko JV, Tepikin AV, Petersen OH. Two different but converging messenger pathways to intracellular Ca2+ release: the roles of nicotinic acid adenine dinucleotide phosphate, cyclic ADP-ribose and inositol trisphosphate. Embo J. 2000;19:2549–57. doi: 10.1093/emboj/19.11.2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Robison GA, Butcher RW, Sutherland EW, Posternak T, Hardman JG. Cyclic AMP. New York ; London: Academic Press; 1971. [Google Scholar]
- 8.Churchill GC, O'Neill JS, Masgrau R, Patel S, Thomas JM, Genazzani AA, Galione A. Sperm Deliver a New Second Messenger. NAADP. Curr Biol. 2003;13:125–8. doi: 10.1016/s0960-9822(03)00002-2. [DOI] [PubMed] [Google Scholar]
- 9.Masgrau R, Churchill GC, Morgan AJ, Ashcroft SJ, Galione A. NAADP. A New Second Messenger for Glucose-Induced Ca2+ Responses in Clonal Pancreatic beta Cells. Curr Biol. 2003;13:247–51. doi: 10.1016/s0960-9822(03)00041-1. [DOI] [PubMed] [Google Scholar]
- 10.Billington RA, Ho A, Genazzani AA. Nicotinic acid adenine dinucleotide phosphate (NAADP) is present at micromolar concentrations in sea urchin spermatozoa. J Physiol. 2002;544:107–12. doi: 10.1113/jphysiol.2002.030098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Navazio L, Bewell MA, Siddiqua A, Dickinson GD, Galione A, Sanders D. Calcium release from the endoplasmic reticulum of higher plants elicited by the NADP metabolite nicotinic acid adenine dinucleotide phosphate. Proc Natl Acad Sci U S A. 2000;97:8693–8. doi: 10.1073/pnas.140217897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Churamani D, Carrey EA, Dickinson GD, Patel S. Determination of cellular nicotinic acid adenine dinucleotide phosphate (NAADP) levels. Biochem J. 2004 doi: 10.1042/BJ20031754. Pt. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gasser A, Bruhn S, Guse AH. Second messenger function of nicotinic acid adenine dinucleotide phosphate revealed by an improved enzymatic cycling assay. J Biol Chem. 2006;281:16906–13. doi: 10.1074/jbc.M601347200. [DOI] [PubMed] [Google Scholar]
- 14.Kinnear NP, Boittin FX, Thomas JM, Galione A, Evans AM. Lysosome-Sarcoplasmic reticulum junctions: A trigger zone for calcium signalling by NAADP and endothelin-1. J Biol Chem. 2004 doi: 10.1074/jbc.M406132200. [DOI] [PubMed] [Google Scholar]
- 15.Yamasaki M, Thomas JM, Churchill GC, Garnham C, Lewis AM, Cancela JM, Patel S, Galione A. Role of NAADP and cADPR in the induction and maintenance of agonist-evoked Ca2+ spiking in mouse pancreatic acinar cells. Curr Biol. 2005;15:874–8. doi: 10.1016/j.cub.2005.04.033. [DOI] [PubMed] [Google Scholar]
- 16.Yusufi AN, Cheng J, Thompson MA, Chini EN, Grande JP. Nicotinic acid- adenine dinucleotide phosphate (NAADP) elicits specific microsomal Ca2+ release from mammalian cells. Biochem J. 2001;353:531–536. doi: 10.1042/0264-6021:3530531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Graeff R, Lee HC. A novel cycling assay for nicotinic acid-adenine dinucleotide phosphate with nanomolar sensitivity. Biochem J. 2002;367:163–8. doi: 10.1042/BJ20020644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morgan AJ, Churchill GC, Masgrau R, Ruas M, Davis LC, Billington RA, Patel S, Yamasaki M, Thomas JM, Genazzani AA, Galione A. Methods in cyclic ADP-ribose and NAADP research. In: JWP Jr., editor. Calcium Signaling. Boca Raton: CRC Press; 2006. pp. 265–264. [Google Scholar]
- 19.Aarhus R, Graeff RM, Dickey DM, Walseth TF, Lee HC. ADP-ribosyl cyclase and CD38 catalyze the synthesis of a calcium-mobilizing metabolite from NADP. Journal of Biological Chemistry. 1995;270:30327–33. doi: 10.1074/jbc.270.51.30327. [DOI] [PubMed] [Google Scholar]
- 20.Aarhus R, Dickey DM, Graeff RM, Gee KR, Walseth TF, Lee HC. Activation and inactivation of Ca2+ release by NAADP+ J Biol Chem. 1996;271:8513–6. doi: 10.1074/jbc.271.15.8513. [DOI] [PubMed] [Google Scholar]
- 21.Billington RA, Genazzani AA. Characterization of NAADP+ binding in sea urchin eggs. Biochem Biophys Res Commun. 2000;276:112–6. doi: 10.1006/bbrc.2000.3444. [DOI] [PubMed] [Google Scholar]
- 22.Galione A, Thomas J, Churchill G. Pharmacological studies of new calcium release mechanisms. In: Tepikin A, editor. Calcium signalling, a practical approach. Oxford: IRL Press; 2000. [Google Scholar]
- 23.Patel S, Churchill GC, Galione A. Unique kinetics of nicotinic acid-adenine dinucleotide phosphate (NAADP) binding enhance the sensitivity of NAADP receptors for their ligand [In Process Citation] Biochem J. 2000;352(Pt 3):725–9. [PMC free article] [PubMed] [Google Scholar]
- 24.Dargie PJ, Agre MC, Lee HC. Comparison of Ca2+ mobilizing activities of cyclic ADP-ribose and inositol trisphosphate. Cell Regul. 1990;1:279–90. doi: 10.1091/mbc.1.3.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Walseth TF, Aarhus R, Zeleznikar RJ, Jr., Lee HC. Determination of endogenous levels of cyclic ADP-ribose in rat tissues. Biochim Biophys Acta. 1991;1094:113–20. doi: 10.1016/0167-4889(91)90032-s. [DOI] [PubMed] [Google Scholar]
- 26.Pollak N, Dolle C, Ziegler M. The power to reduce: pyridine nucleotides--small molecules with a multitude of functions. Biochem J. 2007;402:205–18. doi: 10.1042/BJ20061638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Clapper DL, Walseth TF, Dargie PJ, Lee HC. Pyridine nucleotide metabolites stimulate calcium release from sea urchin egg microsomes desensitized to inositol trisphosphate. J Biol Chem. 1987;262:9561–9568. [PubMed] [Google Scholar]
- 28.da Silva CP, Potter BV, Mayr GW, Guse AH. Quantification of intracellular levels of cyclic ADP-ribose by high- performance liquid chromatography. J Chromatogr B Biomed Sci Appl. 1998;707:43–50. doi: 10.1016/s0378-4347(97)00622-1. [DOI] [PubMed] [Google Scholar]
- 29.Malaisse WJ, Kanda Y, Inageda K, Scruel O, Sener A, Katada T. Cyclic ADP-ribose measurements in rat pancreatic islets. Biochem Biophys Res Commun. 1997;231:546–8. doi: 10.1006/bbrc.1996.5715. [DOI] [PubMed] [Google Scholar]
- 30.Takahashi K, Kukimoto I, Tokita K, Inageda K, Inoue S, Kontani K, Hoshino S, Nishina H, Kanaho Y, Katada T. Accumulation of cyclic ADP-ribose measured by a specific radioimmunoassay in differentiated human leukemic HL-60 cells with all- trans-retinoic acid. FEBS Lett. 1995;371:204–8. doi: 10.1016/0014-5793(95)00914-u. [DOI] [PubMed] [Google Scholar]
- 31.Berridge G, Cramer R, Galione A, Patel S. Metabolism of the novel Ca2+-mobilizing messenger nicotinic acid-adenine dinucleotide phosphate via a 2′-specific Ca2+-dependent phosphatase. Biochem J. 2002;365:295–301. doi: 10.1042/BJ20020180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bredt DS, Mourey RJ, Snyder SH. A simple, sensitive, and specific radioreceptor assay for inositol 1,4,5-trisphosphate in biological tissues. Biochem Biophys Res Commun. 1989;159:976–82. doi: 10.1016/0006-291x(89)92204-3. [DOI] [PubMed] [Google Scholar]
- 33.Rowley WH, Sato S, Huang SC, Collado-Escobar DM, Beaven MA, Wang LH, Martinez J, Gardner JD, Jensen RT. Cholecystokinin-induced formation of inositol phosphates in pancreatic acini. Am J Physiol. 1990;259:G655–65. doi: 10.1152/ajpgi.1990.259.4.G655. [DOI] [PubMed] [Google Scholar]