

Abstract
Tumourigenesis caused by the Bcr/Abl oncoprotein is a multi-step process proceeding from initial to tumour-maintaining events and finally results in a complex tumour-supporting network. A key to successful cancer therapy is the identification of critical functional nodes in an oncogenic network required for disease maintenance. So far, the transcription factors Stat3 and Stat5a/b have been implicated in bcr/abl-induced initial transformation. However, to qualify as a potential drug target, a signalling pathway must be required for the maintenance of the leukaemic state. Data on the roles of Stat3 or Stat5a/b in leukaemia maintenance are elusive. Here, we show that both, Stat3 and Stat5 are necessary for initial transformation. However, Stat5-but not Stat3-deletion induces G0/G1 cell cycle arrest and apoptosis of imatinib-sensitive and imatinib-resistant stable leukaemic cells in vitro. Accordingly, Stat5-abrogation led to effective elimination of myeloid and lymphoid leukaemia maintenance in vivo. Hence, we identified Stat5 as a vulnerable point in the oncogenic network downstream of Bcr/Abl representing a case of non-oncogene addiction (NOA).
Keywords: Bcr/Abl, leukaemia, leukaemic stem cells, Stat5
INTRODUCTION
Tumourigenesis is a multi-step process that requires the expression of oncogenes or the downregulation of tumour suppressor genes (Hanahan & Weinberg, 2000). The tumour initiation process is driven by continuous oncogene expression which frequently develops into oncogene addiction during tumour maintenance (e.g. in the case of Bcr/Abl, EGFR, Flt3-ITD, Jak2V617F, c-Kit) (Weinstein, 2002). The process of tumour maintenance is associated with rewiring of signalling pathways and by acquiring additional genetic mutations (e.g. mutations or deletions of Trp53 or Bcl-2 over-expression) (Letai et al, 2004; Sherr, 2004; Ventura et al, 2007). Such additional mutations may complicate the success of therapies directed against the transforming oncogene as has been shown for bcr/abl-driven disease (Moon et al, 2009; Wendel et al, 2006).
Bcr/abl-induced leukaemia is characterized by a t(9;22) (q34;q11) translocation leading to the expression of a chimeric fusion gene product (Bcr/Abl) representing a constitutively active tyrosine kinase. This translocation is mainly linked to two distinct haematopoietic disorders: acute lymphoid leukaemia (ALL) and chronic myelogenous leukaemia (CML) (Deininger et al, 2000). Targeting the Bcr/Abl oncoprotein by the kinase inhibitor imatinib-mesylate and related substances has been a major breakthrough. This success is only compromised by treatment-insensitive mutations within the transforming oncogene itself (Chu et al, 2005; Griswold et al, 2006; Shah et al, 2002). Thus, despite the success story of Bcr/Abl kinase inhibitors, additional therapeutic strategies are required. Among the signalling pathways that may allow therapeutic interference is the Jak-Stat signalling pathway that has been implicated in tumourigenesis (Bromberg, 2002; Ho et al, 1999; Levine et al, 2007; Yu & Jove, 2004). Particularly, activated Stat5 and Stat3 were found in various types of solid cancer and haematological malignancies (Bromberg, 2002; Buettner et al, 2002; Kornfeld et al, 2008; Ling & Arlinghaus, 2005; Yu & Jove, 2004) allowing the tumour cells to overcome their dependence on cytokines and growth factors. Moreover, Stat3 has recently been shown to support malignant transformation by regulating metabolic functions in mitochondria (Gough et al, 2009). Prominent examples for the tumourigenic effects of Jak/Stat activation are myeloid and lymphoid malignancies associated with constitutively active forms of Jak2 (Schwaller et al, 1998; Tefferi & Gilliland, 2005; Tefferi et al, 2005). In these tumours the constitutive activation of Jak2 drives the phosphorylation and activation of Stat1, Stat3 and Stat5 (Ho et al, 1999; Schwaller et al, 2000). A constitutive activation of the Jak/Stat pathway was also found in leukaemic cells of patients suffering from bcr/abl-induced leukaemia (Benekli et al, 2003; Lin et al, 2000; Steelman et al, 2004). In both, bcr/abl+ CML and ALL, Stat5 is highly activated (Carlesso et al, 1996; Chai et al, 1997; Coppo et al, 2003; Ilaria & Van Etten, 1996; Shuai et al, 1996; Spiekermann et al, 2002).
So far, several studies verified a driving role for Stat3 and Stat5 in the leukaemia initiation phase in vivo: transduction of bone marrow (BM) cells with a constitutively active mutant of Stat5 induces multi-lineage leukaemia in mice (Kato et al, 2005; Moriggl et al, 2005). In contrast, transduction and reconstitution with BM with a constitutive active Stat3 induces a highly aggressive lymphoid leukaemia (Ecker et al, 2009). However, no information is available whether Stat3 or Stat5 are only required for leukaemia initiation but also for disease maintenance in vivo. Initial experiments using mice expressing N-terminally deleted Stat5 (Stat5ΔN/ΔNmice) underestimated the importance of Stat5 since these animals still succumbed to leukaemia (Sexl et al, 2000). Recently we clarified that Stat5 is indeed absolutely essential for the leukaemia initiation process mediated by v-abl and bcr/ablp185 oncogenes in vitro and in vivo using a complete Stat5 knockout model (Stat5null) (Cui et al, 2004; Hoelbl et al, 2006). These in vivo studies are complemented by in vitro studies that showed that RNAi-mediated knock-down of Stat5 reduced survival of leukaemic cell lines and diminished the capacity of primary CML cells to form colonies in a cytokine-containing soft agar assay (Scherr et al, 2006). Similar conclusions were reached using dominant negative forms of Stat5 (de Groot et al, 1999; Sillaber et al, 2000). Whether Stat5 is needed for leukaemia maintenance in vivo remained unclear.
Thus, we extended our previous studies and show here that Stat3 and Stat5 are required for disease initiation. In addition, Stat5 is unequivocally required for leukaemia maintenance in both, lymphoid and myeloid bcr/abl+ leukaemia.
RESULTS
Initial myeloid and lymphoid transformation require Stat5 and Stat3
We have previously shown that initial lymphoid transformation by bcr/ablp185 and v-abl oncogenes critically depends on Stat5 in vitro and in vivo ((Hoelbl et al, 2006) and Fig 1A). Here, we investigated whether initial myeloid transformation induced by the Bcr/Ablp210 oncogene, also depends on Stat5. Hence, bcr/ablp210-induced colony formation in growth-factor free methylcellulose was investigated using Stat5null/null foetal livers (FLs) (ED 14). Importantly, the frequency of HSC numbers in Stat5null/null FLs is relatively normal (Hoelbl et al, 2006; Li et al, 2007; Yao et al, 2006) resulting in a comparable target population for transformation. We found that myeloid transformation critically depends on Stat5 in a gene-dosage dependent manner (Fig 1B). Next, we investigated the role of Stat3 in initial myeloid and lymphoid transformation. Since Stat3-deficiency results in early embryonic lethality (ED 6.5) (Takeda et al, 1997) we relied on BM cells derived from Stat3fl/flMx1Cre mice that were treated with polyinosinic: polycytidylic acid (p(I:C)) to induce Stat3 deletion in vivo. Similarly, significant reductions of v-abl+ and bcr/ablp210+ colony numbers were observed for Stat3Δ/Δ cells upon transduction of v-abl and bcr/ablp210, respectively (Fig 1C,D). Numbers of target cells were comparable within each experimental group as determined by flow cytometric analysis of HSC-enriched Lin−c-kit+Sca-1+, Lin−c-kit+Sca-1− (including myeloid and lymphoid progenitors) and CD43+CD19+B220+ pro-B cell populations, respectively (data not shown). The lymphoid or myeloid origin of v-abl- and bcr/ablp210-derived colonies was verified by light microscopy (Supporting Information Fig 1).
Figure 1. V-abl and bcr/ablp210-induced transformation depend on Stat5 or Stat3 in vitro.
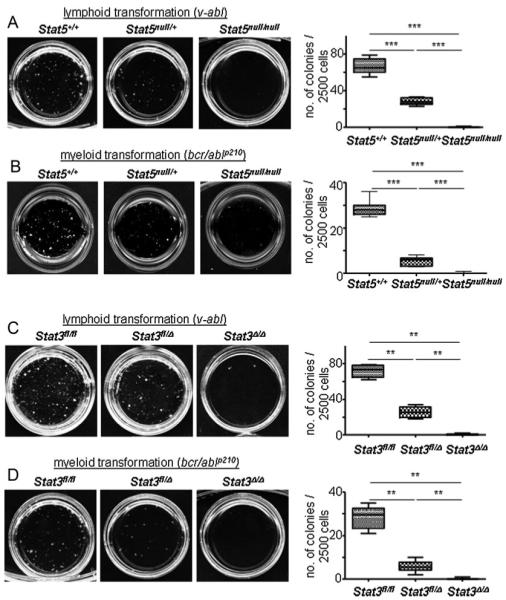
Asterisks indicate the degree of statistical significance (**p < 0.005, ***p < 0.001).
A, B. V-abl (A) and bcr/ablp210 (B)-induced transformation of Stat5+/+, Stat5null/+ and Stat5null/null FL cells in growth factor-free methylcellulose. One representative set of n = 4 for each genotype is depicted. Numbers of factor-independent v-abl+ and bcr/ablp210+ Stat5null/+ and Stat5null/null colonies were reduced compared to wt (2.4- and 400-fold for v-abl, 5.4- and 172-fold for bcr/ablp210, respectively).
C, D. V-abl (C) and bcr/ablp210 (D)-induced transformation of Stat3fl/fl, Stat3fl/Δ and Stat3Δ/Δ BM cells in growth factor-free methylcellulose (n = 3 for each genotype). Numbers of factor-independent v-abl+ and bcr/ablp210+ Stat3fl/Δ and Stat3Δ/Δ colonies were reduced compared to wt (2.4- and 105-fold for v-abl, 4.8- and 85-fold for bcr/ablp210, respectively). Data are summarized in box blots (right panel) and represent means ± SD.
Taken together, these data reveal the absolute requirement of the transcription factors Stat3 and Stat5 for the initial transformation event downstream of v-Abl and Bcr/Ablp210.
Stat5, but not Stat3, is essential for cell cycle progression and survival of lymphoid leukaemic cells in vitro
Transformed cells rewire pathways for growth and survival. Only signalling pathways essential for the maintenance of the oncogenic state qualify as useful therapeutic targets. Hence, we next investigated the consequences of Stat3- or Stat5-deletion on already established leukaemic cells. Since bcr/ablp210+ myeloid cells do not give rise to stable, growth-factor free cell lines in vitro, we used v-abl+ lymphoid cell lines for the following in vitro studies. V-abl+-transformed cells readily become growth-factor independent in vitro. Thus, we retrovirally transduced Stat3fl/flMx1Cre and Stat5fl/flMx1Cre derived BM and control cells with v-abl, a murine variant of bcr/ablp185. V-abl and bcr/ablp185 transformed cells are phenotypically identical and share comparable disease kinetics in vivo (Supporting Information Fig 2). Stable cell lines of all genotypes were generated (CD19+, B220+, CD43+), analysed for proliferation rate, growth factor independent colony formation and homing to haematopoietic organs in vivo with comparable results (data not shown). We used recombinant IFN-β to activate Cre-recombinase in Stat3fl/flMx1Cre and Stat5fl/flMx1Cre cells in vitro (Fig 2A and B).
Figure 2. Stat5- but not Stat3-deletion blocks cell proliferation and induces apoptosis in vitro.
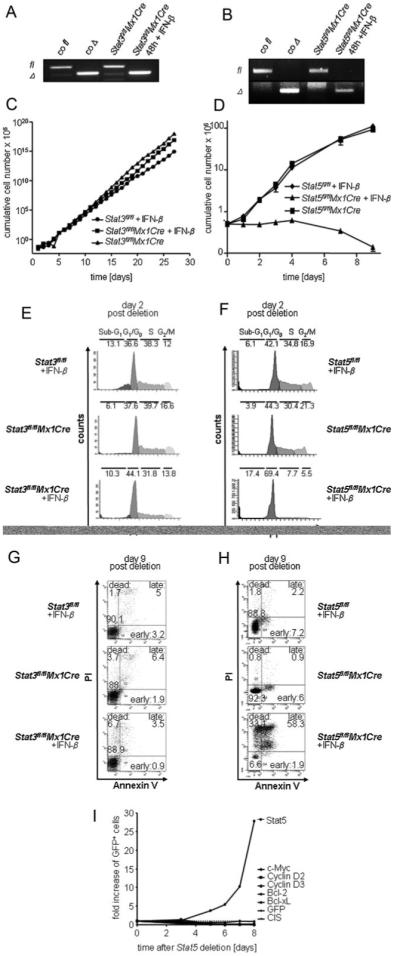
A, B. PCR analysis of gene deletion after IFN-β treatment. Deletion efficiency was determined by a PCR for floxed (fl) and deleted (Δ) Stat3 (A) and Stat5 (B) alleles 48 h after IFN-β treatment.
C, D. Effect of Stat5 or Stat3 deletion on cell proliferation in vitro. (C) Stat3fl/flMx1Cre cell lines were either treated with 1000U IFN-β or mock-treated (n = 3 each). Stat3fl/fl cell lines were used as a control (n = 3). (D) Stat5fl/flMx1Cre cell lines (n = 3 each) were treated analogously to (C).
E–H. Cell cycle and apoptosis analysis of IFN-β treated v-abl-transformed cell lines, 2 and 9 days after IFN-β treatment, respectively. Deletion of Stat5 induces a cell cycle arrest (F) and apoptosis (H), while deletion of Stat3 has no significant effect on evaluated parameters (E, G). Numbers show percentages of cells in indicated cell cycle phases (E, F) or in different stages of apoptosis (G, H). Re-expression of Stat5 target genes (D-type cyclins, c-myc, bcl-xL or bcl-2) failed to rescue Stat5-deficiency (I).
As depicted in Fig. 2, IFN-β treatment had no effect on cell proliferation in Stat3fl/flMx1Cre cells. In contrast, we observed changes in cell cultures of Stat5fl/flMx1Cre cells (Fig 2C and D). Cell cycle profiles obtained 48 h after the initiation of IFN-β treatment revealed a profound G0/G1 cell cycle arrest in Stat5fl/flMx1Cre cells (67.7 ± 1.7% compared to 45.4 ± 5.2% of untreated cells within G0/G1 phase) whereas the cell cycle profiles of Stat3fl/flMx1Cre cells remained unaltered (44.9 ± 4.8% compared to 38.5 ± 3.9% of untreated cells within G0/G1 phase) (Fig 2E and F). The cell cycle arrest in IFN-β-treated Stat5fl/flMx1Cre cells was followed by apoptosis analysed by Annexin V/PI stains 9 days post deletion. 58.3 ± 6.2% and 32 ± 1.3% of IFN-β treated Stat5fl/flMx1Cre cells were double-positive for Annexin V/PI and single-positive for PI, respectively. The time-span between IFN-β treatment and cell death results from the long half life of Stat5 in these cells (data not shown). No changes in the viability of IFN-β treated Stat3fl/flMx1Cre cells were detectable (Fig 2G and H) even after 30 days. After 10 days in the presence of IFN-β no viable cells were detected in IFN-β treated Stat5fl/flMx1Cre cell cultures. Our attempts to rescue Stat5 deficiency by re-expression of Stat5 target genes such as D-type cyclins, c-myc, bcl-xL or bcl2 failed (Fig 2I). Only re-expression of wild-type (wt) Stat5, but not of transcriptionally inactive mutants (Stat5Δ749 and Stat5Y694F), was able to protect cells from proliferation arrest and apoptosis upon deletion of endogenous Stat5 (Supporting Information Fig 3). Hence, we concluded that Stat5, but not Stat3, is required for the maintenance of the malignant state of transformed lymphoid leukaemic cells in vitro.
Stat5 is required for lymphoid leukaemia maintenance in vivo
To study the role of Stat5 in lymphoid leukaemia maintenance in vivo, we transplanted Stat5fl/flMx1Cre v-abl+ cells into Rag2−/−γc−/− mice (1 × 105 cells/mouse). Rag2−/−γc−/− mice lack lymphoid cells and are therefore particularly suited to monitor lymphoid leukaemia. Recipient mice were subsequently divided into two groups (Fig 3A) with one group receiving p(I:C) to induce type I IFN responses and to delete Stat5 within the leukaemic cells. The second group was mock-injected with PBS. Preliminary experiments had revealed that 7 days post-transplantation mice display first signs of sickness with elevated numbers of leukaemic cells in the peripheral blood (Supporting Information Fig 2D). We therefore chose this time point to initiate p(I:C) treatment which was repeated every 4 days in order to efficiently target this highly proliferating ALL-like disease (scheme in Fig 3B).
Figure 3. Lymphoid leukaemia maintenance depends on Stat5.
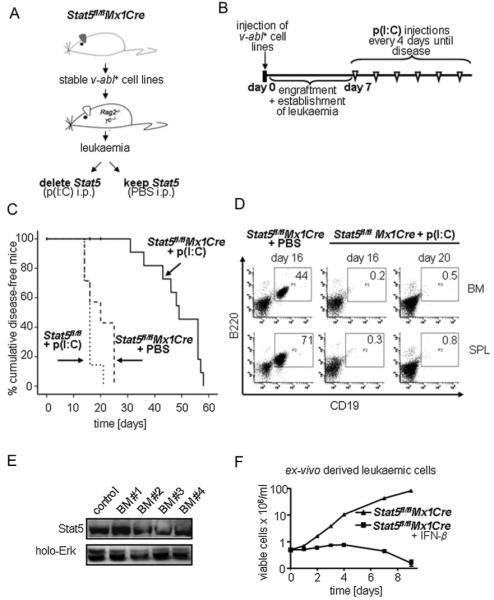
A. Scheme of the transplantation experiment. V-abl-transformed cell lines from BM of Stat5fl/flMx1Cre mice were transplanted into Rag2−/−γc−/− mice that were then treated with p(I:C) or PBS.
B. Time course of p(I:C) injections. Seven days after transplantation mice were injected i.p. with 400 μg p(I:C) every 4 days until the mice appeared moribund.
C. Transplantation of Stat5fl/flMx1Cre and Stat5fl/fl cell lines (n = 3 each) into Rag2−/−γc−/− mice. Kaplan–Meier analysis reveals a statistically significant difference in survival after Stat5-deletion (p < 0.001 for Stat5fl/flMx1Cre + p(I:C) (n = 14) versus Stat5fl/flMx1Cre + PBS (n = 7) and p > 0.05 for Stat5fl/flMx1Cre PBS versus Stat5fl/fl + p(I:C) (n = 7)). Vertical bars indicate censored events.
D. Presence of transplanted leukaemic precursor B-cells (B220+CD19+) in BM and spleens of p(I:C)- and PBS-treated mice at indicated time points. Numbers in boxes indicate percentages of leukaemic cells.
E. Immunoblot analysis of ex vivo derived BM cells from p(I:C)-treated Stat5fl/flMx1Cre diseased mice for the presence of Stat5. Four representative samples are shown.
F. Ex vivo derived cell lines are sensitive to a secondary deletion of Stat5 by IFN-β. Cell numbers were determined by Trypan-blue exclusion.
We observed a prolonged survival of mice that had received Stat5fl/flMx1Cre leukaemic cells and p(I:C) treatment compared to mice that were mock-injected with PBS. To control for effects of p(I:C) per se, mice that had received Stat5fl/fl cells were also p(I:C) treated (Fig 3C). Whereas mice from the ‘Stat5fl/flMx1Cre + PBS’ and ‘Stat5fl/fl + p(I:C)’ groups displayed obvious severe signs of sickness from day 16 on, animalsharbouring Stat5Δ/Δ leukaemic cells appeared healthy with normal mobility, fur and weight. Mice where Stat5 had been deleted in the leukaemic cells survived significantly longer (mean survival of 49 days compared to 20 and 16 days in the ‘Stat5fl/fl + p(I:C)’ and ‘Stat5fl/flMx1Cre + PBS’ groups, respectively). We compared diseased animals sacrificed on days 16 and 20 (‘Stat5fl/flMx1Cre + PBS’ group) to healthy appearing mice of the ‘Stat5fl/fl Mx1Cre + p(I:C)’ group. Whereas BMs and spleens of the ‘Stat5fl/flMx1Cre + PBS’ group were densely infiltrated with B220+CD19+ cells, we hardly detected leukaemic cells in mice of the ‘Stat5fl/flMx1Cre + p(I:C)’ group (Fig 3D). Similar results were obtained when immunocompetent mice were used as recipient animals (Supporting Information Fig 4).
However, finally all mice succumbed to leukaemia. Examination of the leukaemic cells revealed lack of genomically Stat5 deleted cells and Stat5 protein expression (Fig 3E and data not shown). We reasoned that p(I:C)-induced deletion was incomplete in vivo and that some cells escaped deletion. This scenario is supported by the fact that we still could induce cell cycle arrest and apoptosis in the ex vivo derived leukaemic cells by IFN-β treatment (Fig 3F). This rules out that the cells have acquired secondary mutations overcoming the Stat5-requirement.
Stat5 is required for bcr/ablp210-induced myeloid leukaemia maintenance in vivo
Transplantation of bcr/ablp210+ BM cells is a reliable method to develop a fatal rapidly progressing myeloproliferative illness in mice which is commonly determined ‘CML-like’ disease (Pear et al, 1998; Van Etten, 2001). We retrovirally transduced BM cells from 5-fluorouracil (FU) pretreated Stat5fl/flMx1Cre and Stat5fl/fl mice with bcr/ablp210 IRES GFP and injected them i.v. into lethally irradiated wt mice (1 × 106 cells). In order to determine the optimal time point to initiate p(I:C) injection preliminary experiments were performed (n = 6). Under our experimental conditions 6 weeks after the initial transplantation the animals displayed first signs of disease indicated by 12.3 ± 3.7% bcr/abl+/GFP+ cells in the BM accompanied by the doubling of peripheral white blood cell counts (WBCs) and a decrease in eosinophil cell numbers (Fig 4A and Supporting Information Fig 5). A single p(I:C) injection at that time eradicated bcr/abl+/GFP+ cells in the BM when analysed 10 days thereafter, whereas increasing numbers were detected in p(I:C)-treated controlanimals (Fig 4A). This indicates that p(I:C) treatment and thus Stat5 deletion was capable to eradicate the disease at that given time point. Our attempts to wait with the p(I:C) application till the animals displayed more severe signs of leukaemia such as weight loss or reduced mobility failed. After displaying severe signs of leukaemia the animals die rapidly within less than 5 days. This time frame is insufficient to deplete the leukaemic cells of Stat5 protein despite the successful genomic deletion. The long half life of the Stat5 protein is also comprehensible from the fact that it takes 9 days to eradicate v-abl transformed cells in vitro after IFN-β treatment (Fig 2 and data not shown). Accordingly, the following experimental protocol was set up (Fig 4B). Peripheral WBCs were monitored weekly as internal control. P(I:C) treatment and Stat5 reduction resulted in a significantly prolonged survival of the Stat5fl/flMx1Cre mice compared to all other groups (p < 0.001 compared to every other group, Fig 4C). Untreated mice succumbed to disease from week 7 on. Any time when a group of control mice severely diseased and had to be sacrificed, one animal of the ‘Stat5fl/flMx1Cre + p(I:C)’ group was analysed in parallel to allow a direct comparison. Sixteen weeks post-transplantation, the number of leukaemic cells in the peripheral blood was clearly reduced in the ‘Stat5fl/flMx1Cre + p(I:C)’ compared to the ‘Stat5fl/flMx1Cre + PBS’ group. Reduction of Stat5 in the Stat5fl/flMx1Cre + p(I:C) group was verified by immunoblotting of peripheral blood leukocytes 13, 14, 16 and 18 weeks post-treatment (indicated censored events in Fig 4C and D). Similarly, histological stainings revealed a reduced infiltration of BM and spleen with leukaemic cells paralleled by a diminished pStat5 staining (Fig 4E). These findings indicated a reduction of the leukaemic cell load after p(I:C) treatment and Stat5 deletion. Measuring spleen sizes underlined the effect of p(I:C) treatment (Fig 4F). Whereas the diseased mice showed significantly enlarged spleens, the spleens of the p(I:C) treated Stat5fl/flMx1Cre mice were of regular size and comparable to age-matched control mice. The BMs of all animals were cultured in a medium supplemented with SCF, Flt3-ligand (Flt3-L), IgF-1, IL-3, IL-6, GM-SCF and dexamethasone as described previously (Kieslinger et al, 2000). As listed in Supporting Information Table 1, no bcr/abl+/GFP+ cells grew out from BMs of p(I:C) treated-, Stat5fl/flMx1Cre-transplanted mice. In contrast, after 4 weeks significant numbers of bcr/abl+/GFP+ cells were detected in control cultures, transplantation of these cells in lethally irradiated mice re-initiated leukaemia (Supporting Information Fig 6).
Figure 4. Myeloid leukaemia maintenance depends on Stat5.

A. FACS analysis of BM cells 10 days after a single p(I:C) treatment. Six weeks post-transplantation of Stat5fl/fl or Stat5fl/flMx1Cre bcr/abl+/GFP+ cells, mice received p(I:C) i.p.
B. Experimental setup for the deletion of Stat5 in myeloid leukaemia.
C. Kaplan–Meier plot displaying overall survival of p(I:C)-treated and control recipient mice harbouring Stat5fl/flMx1Cre and Stat5fl/fl myeloid leukaemia. One single p(I:C) treatment was performed 6 weeks after transplantation in order to delete Stat5 in leukaemic cells. At the time point of analysis (18 weeks post-injection), 100% (10/10) of Stat5fl/flMx1Cre p(I:C)-treated mice are alive, while recipients from all other groups succumb to leukaemia (n = 8 for Stat5fl/flMx1Cre + PBS, n = 3 for Stat5fl/fl + p(I:C) and n = 3 for Stat5fl/fl + PBS; p < 0.001; mean survival time of 12.5, 11.3 and 12 weeks, respectively).
D. Immunoblot analysis of Stat5 expression in peripheral blood cells 7 (#1), 8 (#2), 10 (#3) and 12 (#4) weeks after p(I:C) treatment. These censored events are indicated in (C).
E. Blood smears and histological sections of spleens and BM. Treatment with p(I:C) leads to a massive reduction of WBCs (left panel) and phospho-Stat5 expression (right panels) in Stat5fl/flMx1Cre–bcr/ablp210+ transplanted mice compared to controls (PBS).
F. Macroscopic anatomy of spleens from Stat5fl/flMx1Cre mice treated with p(I:C) and control mice. A spleen of one age-matched healthy wt mouse is depicted as control.
Stat5 is required for engraftment and repopulation of bcr/ablp210+ leukaemia in secondary recipients
These data suggested that Stat5 is required for the maintenance of leukaemia initiating cells driving bcr/ablp210-induced myeloid disease. CML-like disease in mice is driven by a bcr/abl+c-kit+Lin− leukaemia initiating or leukaemic stem cell (LSC) defined by the ability to carry on disease to a secondary recipient and to replenish the leukaemic cell pool (Krause et al, 2006; Wang & Dick, 2005). A definite cure of CML can only be achieved when LSCs are successfully eliminated. Therefore, we tested the effects of Stat5-deletion in LSCs by investigating their ability to engraft and repopulate leukaemia in a secondary recipient. We used bcr/ablp210 infected Stat5fl/flMx1Cre BM cells to transplant lethally irradiated wt recipient mice. When first signs of disease evolved, indicated by elevated WBCs, the BM of the affected animals was prepared (data not shown). At that time, 13.4 ± 3.3% of the BM cells were bcr/abl+/GFP+ (Fig 5, upper panel) which mainly consisted of Mac1+Gr1+ (28.5 ± 8.9%), CD19+ (19.7% ± 5.6) and Lin− (25.1 ± 15.3%) cells being mainly stem/progenitor cells (Lin−c-kit+Sca-1−; 86.7 ± 20.5%). Deletion of Stat5 was induced by treating the BM cells ex vivo with recombinant IFN-β (1000 U/ml). We had to omit our initial plan to transplant a pure Stat5Δ/Δ population into secondary recipients since we never completely deleted Stat5 despite intense efforts using various concentrations of IFN-β (Fig 5, middle panel). We reasoned that Stat5Δ/Δ cells might have a severe disadvantage in vitro and decided to transplant an IFN-β treated ‘mixed’ population of Stat5fl/flMx1Cre and Stat5Δ/ΔMx1Cre cells. This attempt has the advantage that the co-transplanted non-deleted LSCs served as internal control for successful transplantation when investigating whether the Stat5Δ/Δ LSCs contribute to CML in vivo.
Figure 5. Stat5 is required for engraftment and repopulation of bcr/ablp210+ leukaemia in secondary recipients.

Stat5-deleted LSCs do not contribute to secondary leukaemia formation. Stat5fl/flMx1Cre–bcr/ablp210+ leukaemic cells isolated from BM of primary transplants (1st tp, left panel, n = 3) and subjected to Stat5-deletion by IFN-β in vitro, do not participate in leukaemia formation. Leukaemia arising in secondary recipients (lower right panel) lacks Stat5-deleted (Δ) tumour cells (lower middle panel).
Two weeks after the secondary transplant, recipient mice displayed clear signs of disease including decreased mobility and weight loss. All mice had developed leukaemia with enlarged spleens and livers and significant numbers of bcr/abl+/GFP+ cells in the BM (Fig 5, lower panel). 45.1±31.5% of BM cells were bcr/abl+/GFP+ comprising of Lin− cells (20.5 ± 7.5%) being mainly stem/progenitor cells (Lin−c-kit+Sca-1−, 91 ± 5.5%). Only few Mac1+Gr1+ and CD19+ cells were detected (1.1 ± 1.7% and 2.1 ± 1.0, respectively) whereas 7.2±4.1% of cells were Mac1+Gr1−. These findings indicated an accelerated stage of the disease. In genotyping PCR analysis of the BM-derived leukaemic cells we only detected a PCR product corresponding to the floxed Stat5 allele. The Stat5Δ allele was not found in any leukaemic sample. Thus, the co-transplanted Stat5Δ/Δ LSCs did not contribute to leukaemia repopulation (Fig 5, lower panel).
As illustrated in Fig 1B, already Stat5-heterozygosity profoundly affected bcr/ablp210-induced myeloid colony formation. To test the effect of Stat5 heterozygosity on bcr/ablp210-induced disease in vivo, we repeated the experiments shown in Fig 5 using Stat5fl/+Mx1Cre mice as donors of the primary transplant. In this setting, a complete deletion of the floxed Stat5 allele of primary transplant-derived cells in vitro was achieved (Supporting Information Fig 7, middle panel). Transplantation of the resulting heterozygous Stat5Δ/+ population—comprised of leukaemic and non-leukaemic cells—failed to induce a bcr/ablp210+ leukaemia. No bcr/abl+/GFP+ cells were detectable in BMs and all other organs investigated including lymph nodes, spleen and liver of secondary recipients (Supporting Information Fig 7 and data not shown). However, the presence of the Stat5Δ allele was confirmed in the BM of the recipient animals proving successful transplantation and reconstitution by non-leukaemic Stat5Δ/+ cells (Supporting Information Fig 7, lower panel). To substantiate this finding we also cultivated ex vivo derived BM cells of the Stat5Δ/+ transplanted animals under stem-cell-supporting conditions (Zhang & Lodish, 2005). Whereas LSCs from the Stat5fl/flMx1Cre control BMs readily grew out we failed to detect any outgrowth of leukaemic cells from BM of the Stat5Δ/+ group even after 6 weeks (data not shown). Thus, we concluded that Stat5Δ/+ cells contribute to haematopoietic reconstitution of lethally irradiated mice but do not allow the outgrowth of bcr/ablp210+ leukaemic cells.
Normal haematopoiesis is not significantly affected by Stat5 loss
Therapeutic agents must display a sufficiently large therapeutic window that allows killing tumour cells while sparing normal cells. Since Stat5 has been implicated in particularly in foetal haematopoietic development (Burchill et al, 2003; Grebien et al, 2008; Hoelbl et al, 2006; Yao et al, 2006), we were interested in the impact of Stat5 deletion on normal adult haematopoiesis.
Stat5-deletion was induced in adult (4 weeks old) Stat5fl/flMx1Cre, Stat5fl/+Mx1Cre and wt Mx1Cre mice (n = 7 each) by p(I:C) treatment (Fig 6A). Seven days after the initial p(I:C) treatment the deletion of Stat5 in sorted HSC-enriched Lin−c-kit+Sca-1+ and Lin−c-kit+Sca-1− (including myeloid and lymphoid progenitors) cells was verified by PCR (Fig 6B). In line with published data, despite the successful deletion of Stat5 the total numbers of these cellular fractions was unaltered (Fig 6C) (Wang et al, 2009). Similarly, the frequency of common myeloid progenitors (CMPs), granulocyte-macrophage progenitors (GMPs) and megakaryocyte-erythroid progenitors (MEPs) were unaffected (data not shown). We then collected blood once a week for a total observation period of 8 weeks. The analysis of body weight, WBCs, red blood cell counts (RBCs), haemoglobin (HGB) and haematocrit (HCT) did not reveal any significant effects (Fig 6D and E and data not shown). Minor but not significant alterations were detected in the absolute number of platelets (PLTs) and a slight decrease of monocytes (MO) and granulocytes (GRA) (Fig 6E and F). In addition, analysis of several clinical parameters (triglycerides, glucose, cholesterol, bilirubin, creatine, α-amylase, γ-GT, ALT, AST) did not show any noticeable changes after Stat5-deletion (data not shown). Whereas percentages of CD3+ cells remained unaltered, ‘Stat5fl/flMx1Cre + p(I:C)’ mice displayed significantly lower levels of CD19+ cells (Fig 6F). Moreover, 8 weeks post-Stat5 deletion we did not observe changes in frequencies of LT-HSCs (Lin−c-kit+Sca-1+Flt-3−Thy1+), ST-HSCs (Lin−c-kit+Sca-1+Flt-3+Thy1+) or MPPs (Lin−c-kit+Sca-1+Flt-3+Thy1−) (Passegue et al, 2004, 2005). However, we observed slight decreases in numbers of cells undergoing erythroid differentiation (Fig 6H). In summary, the deletion of Stat5 for 8 weeks was generally well taken by the adult animals.
Figure 6. Lack of detrimental long-term effects of Stat5-deletion on haematopoiesis.

A. Time course of p(I:C) injections and analysis of mice during 8 weeks (n = 7/group).
B. Lin−c-kit+Sca-1+ (HSC-enriched population) and Lin−c-kit+Sca-1− (include myeloid and lymphoid progenitors) cells were purified by FACS-sorting and the deletion efficiency was determined by PCR. Numbers indicate samples from individual mice.
C. Bar graphs summarize the quantifications of Lin−c-kit+Sca-1+ (left panel) and Lin−c-kit+Sca-1− cells (right panel) as a percentage of total BM cells. Data are means ± SD.
D. Variation of body mass during 8 weeks after p(I:C) treatment. Data are means ± SD.
E. Blood count analysis during 8 weeks after p(I:C) treatment (WBC, white blood cells; RBC, red blood cells; PLT, platelets; HGB, haemoglobin). Data are means ± SD.
F. FACS-analysis of blood cell populations (differential) from indicated mouse genotypes (MO, monocytes; GRA, granulocytes; CD19+, CD19 positive B-cells; CD3+, CD3 positive T-cells). Data are means ± SD.
G. FACS analysis of HSC-subpopulations (LT-HSCs, ST-HSCs and MPPs) in the BM of p(I:C)-treated mice. Percentages are depicted in gates (n = 4).
H. FACS analysis of erythroid development in the BM of mice with indicated Stat5-status after p(I:C)-treatment. Numbers are relative percentages of cells in gates (n = 4).
High Bcl-2 levels or deletion of Trp53 do not relieve Stat5-dependence
Tumour cells frequently acquire additional mutations after long-term maintenance in culture. We therefore analysed our Stat5fl/fl Mx1Cre v-abl+ cell lines after 14 months of continuous culture whether any spontaneously acquired mutation could release the necessity for Stat5. When we analysed the cell lines for expression of Trp53, BclXL and Bcl-2, we found that two cell lines (#1 and #3) had completely lost the Trp53 protein (Fig 7A). Loss of Trp53 was described to result in decreased sensitivity towards imatinib (Wendel et al, 2006). Accordingly, these cells displayed a 5.7-fold reduced sensitivity towards imatinib (IC50 of 0.98 μM compared to IC50 = 0.17 μM, data not shown). Cell line #1 additionally displayed a significant up-regulation of the Bcl-2 protein and a significant decrease in Stat5 protein expression. An overexpression of the anti-apoptotic protein Bcl-2 is found in many cancers contributing to tumourigenesis and resistance to therapies (Letai et al, 2004; Moon et al, 2009; Oltersdorf et al, 2005). However, when the residual Stat5 protein was removed by activation of Cre-recombinase, all cell lines still underwent a G0/G1 cell cycle arrest followed by cell death (Fig 7B and C). Hence, even in the presence of elevated Bcl-2 protein levels, Stat5 was indispensable for proliferation and survival of leukaemic cells.
Figure 7. Leukaemic cells harbouring second hits are still sensitive to Stat5-loss.

A. Immunoblot analysis of long-term cultured (14 months) Stat5fl/flMx1Cre v-abl+ cell lines for Stat5, Trp53, Bcl-2 and BclXL protein expressions. Acquisition of secondary mutations leading to defective Trp53- and/or Bcl-2-expression in two cell lines is depicted.
B. Cell cycle analysis of two cell lines from (A) which were subjected to Stat5-deletion via IFN-β-treatment (#1, #3).
C. Stat5-deletion by IFN-β-administration suffices to induce apoptosis in secondarily mutated cell lines (#1 and #3). Percentages of apoptotic cells are determined by PI staining in a non-hypotonic buffer.
D. Stat5-deletion via IFN-β induced cell-death of imatinib-resistant bcr/ablp210T315I+ BM-derived cells. Five days after treatment, IFN-β (lower right panel)—but not imatinib (100 nM, lower middle panel)—induced apoptosis of Stat5fl/flMx1Cre bcr/ablp210T315I+ leukaemic cells. As a control, 100 nM imatinib is sufficient to eradicate wt Mx1Cre bcr/ablp210+ BM-derived cells (upper middle panel). Numbers show percentages of cells in indicated cell cycle phases.
Expression of an imatinib-resistant bcr/abl mutant (bcr/ablp210T315I) does not relieve Stat5 dependence
Treatment of bcr/abl-induced CML has been significantly improved by the availability of imatinib (Druker et al, 2001a, b). However, some patients acquire mutations in the Bcr/Abl oncoprotein which render them insensitive to imatinib (Chu et al, 2005; Griswold et al, 2006). The bcr/ablp210T315I mutation is among the biggest therapeutic challenges in CML therapy, since it mediates complete resistance to imatinib and all of the next generation Abl kinase inhibitors (Quintas-Cardama et al, 2007; Shah et al, 2002). We therefore decided to test whether cells expressing bcr/ablp210T315I require Stat5. Stat5fl/flMx1Cre and Stat5+/+Mx1Cre BM cells were infected with retrovirus encoding bcr/ablp210T315I and treated either with imatinib or IFN-β-mediated Stat5-deletion. To ensure survival and proliferation of immature progenitors, cells were main-tained in a medium supplemented with SCF, Flt3-ligand (Flt3-L), IgF-1, IL-3, IL-6, GM-SCF and dexamethasone as described previously (Kieslinger et al, 2000). Under this condition the IC50 for imatinib was 83.4 nM when tested in wt Mx1Cre cells transduced with non-mutated bcr/ablp210 (Supporting Information Fig 8). As expected wt Mx1Cre and Stat5fl/flMx1Cre cells expressing bcr/ablp210T315I did not undergo apoptosis upon imatinib treatment (100 nM) Fig 7D, middle panels). In contrast, bcr/ablp210T315I-expressing Stat5fl/flMx1Cre cells showed substantial cell death upon loss of Stat5 (Fig 7D, right panels).
DISCUSSION
A key to successful new therapeutic strategies is to identify critical functional nodes in the signalling network downstream of an oncogene. Cancer cells undergo extensive adaptations in their signalling and metabolic pathways. Thereby, they may become dependent on certain genes that are not per se canonical oncogenes. In fact, the activity of these genes may become rate limiting for a cancer cell. The term ‘non-oncogene addiction’ (NOA) has been coined recently to describe this phenomenon. The inhibition of these critical players within the signalling network is predicted to induce system failure and thus the cessation of the malignant state (Luo et al, 2009).
We show here that bcr/abl-transformed leukaemic cells are addicted to Stat5 for maintaining the leukaemic state. Thus, Stat5 fulfils the criteria of an indispensable functional node within the signalling networks downstream of Bcr/Abl. Accordingly, Stat5 represents a potential drug target. The deletion of Stat5 in leukaemic cells resulted in G0/G1 cell cycle arrest followed by apoptosis. Several signalling pathways are activated downstream of Bcr/Abl and contribute to leukaemia development; the long list includes PI3K-isoforms and Ras-dependent pathways. More recent insights highlight the significance of Hedgehog signalling (Dierks et al, 2008; Zhao et al, 2009). In spite of the complexity of the Bcr/Abl-controlled signalling network, Stat5 appears to have a privileged position that is conserved even in the absence of intact Trp53 signalling, as well as in imatinib-resistant cells. This is underscored by the finding that the mere expression of a single Stat5 target genes such as c-myc, bcl-xL, bcl-2, cyclin D2, cyclin D3 or CIS could not replace Stat5 expression. Importantly, the addiction to Stat5 extends to the LSC compartment. LSCs have been characterized in myeloid bcr/ablp210-induced leukaemia by their ability to allow for serial transplantation of the disease (Krause et al, 2006; Wang & Dick, 2005). One of the big current therapeutic challenges is to find strategies how to target and eradicate such LSCs. The most frequently used drug in CML therapy—imatinib—induces apoptosis in bcr/abl+ cells but fails to eradicate LSCs in vivo (Krause & Van Etten, 2007; Neering et al, 2007). In this context, it is worth pointing out that even one of the most dreaded imatinib-resistant mutants of Bcr/Abl—Bcr/Ablp210T315I—remains strictly dependent on Stat5. Hence neither a mutated Bcr/Abl nor the genetic instability associated with the abrogation of Trp53 allowed for the emergence of Stat5-independent leukaemic clones—even in combination with an upregulation of the anti-apoptotic protein Bcl-2.
These observations support the concept that targeting Stat5 provides new therapeutic opportunities. However, a potential therapeutic target is only useful, if upon blockage normal cells are spared at the expense of tumour cells. This issue has recently been addressed: deletion of Stat5 was reasonably well-tolerated. After 4 months WBCs and HCTs were reduced. This was paralleled by increased numbers of actively cycling HSCs (Wang et al, 2009). Our own observation covered a period over 8 weeks and confirmed the overall tolerability of Stat5 deletion, normal haematopoiesis was not compromised to an appreciable extent. We note that there is an apparent discrepancy between these observations and the strong effects associated with the non-conditional ablation of Stat5 (Grebien et al, 2008; Hoelbl et al, 2006; Yao et al, 2006). It is, however, obvious that substantial differences can exist between the phenotypic manifestations of a gene defect acting during embryonic and foetal development and the consequences of eliminating a gene in an adult animal. Foetal and adult HSCs, in particular, differ in important features (Kim et al, 2007; Mikkola & Orkin, 2006). Another observation favours the use of Stat5 as potential drug target. We observed that the mere lowering of Stat5 levels in bcr/ablp210+ Stat5fl/+Mx1Cre cells by IFN-β treatment was sufficient to prevent leukaemia engraftment in secondary recipient animals. This observation again provides evidence for a role of Stat5 in LSCs. It also further supports the concept of Stat5 as a potential drug target. The data suggest that a partial blockage of Stat5—which is well tolerated in normal tissue—may already be deleterious for the bcr/abl+ cell population. While it is difficult to extrapolate these experiments in mice to patients, at the very least these observations justify the assumption that potential side effects of Stat5 blockage will not a priori preclude their use in clinics. This conjecture is further supported by the development of an inhibitor targeting Jak2. Jak2 is essential for erythropoiesis—but nevertheless, Jak2 inhibitors have successfully entered clinical trials (Hexner et al, 2008; Wernig et al, 2008). For the past decade, the development of signal interceptor-based therapies has concentrated on the ‘druggable’ genome which represents proteins with enzymatic functions. There has been a paradigm shift more recently. Many proteins previously considered difficult or impossible to target are thought to be accessible to small molecules, because these can be designed to bind ‘hot-spots’ on contact surfaces and to disrupt protein–protein interaction. The feasibility of this approach is exemplified by binders of the Bcl-2 family such as ABT-737 (Wells & McClendon, 2007). Thus, it is conceivable that Stat proteins may also be targeted by low molecular weight compounds that target the dimeric interphase or interaction sites with specific protein partners. Alternatively, an RNAi-based strategy may also be envisaged in which levels of Stat5 are reduced to eliminate Bcr/Abl+ cells assuming that this approach is established for cancer therapy in the foreseeable future.
The paper explained.
PROBLEM
Acute lymphoid leukaemia (ALL) and chronic myelogenous leukaemia (CML) can be induced by the chimeric fusion gene product Bcr/Abl, a constitutively active tyrosine kinase. A complex signalling network downstream of Bcr/Abl supports proliferation and survival of the leukaemic cells. Bcr/Abl kinase inhibitors (e.g. Imatinib) can hamper these signals and induce cell death but several mutations were described that confer resistance to these inhibitors. Here we tested whether the transcription factors Stat3 and Stat5, acting downstream of Bcr/Abl are critical for leukaemia maintenance and are alternative pharmaceutical targets.
RESULTS
We developed a tumour-specific gene-deletion approach to dissect the roles of Stat5 and Stat3 in Bcr/Abl-induced leukaemia maintenance. We found that both are required for the initial transformation by Bcr/Abl. Once established, only Stat5 is crucial for viability and proliferation of leukaemic myeloid and lymphoid cells. The absolute necessity for Stat5 is conserved in Imatinib-resistant cells and is also maintained when Trp53 signalling is disrupted or Bcl-2 over-expressed.
IMPACT
In many leukaemia patients, effective treatment with Imatinib is hampered by the occurrence of mutations in Bcr/Abl. Our study identified Stat5 as an Achilles’ heel in the signalling network downstream of Bcr/Abl. Thus, inhibition of Stat5—alone or in combination with Bcr/Abl—may provide a novel therapeutic approach for treatment of leukaemia.
Thus, our observations argue for a privileged position of Stat5 in the signalling network controlled by Bcr/Abl and justify selecting Stat5 as a candidate drug target.
MATERIALS AND METHODS
Mice and genotyping
Stat5fl/flMx1Cre (mixed C57BL/6J × Sv129), Stat3fl/flMx1Cre (mixed C57BL/6J × Sv129), C57BL/6J and Rag2−/−γc−/− (C57BL/6J) mice were maintained at the Biomedical Research Institute (Medical University of Vienna) and at the NIH (Bethesda, Maryland), C57BL/6J × Sv129 F1 (here referred to as B6129F1) at the Institute of Molecular Pathology (IMP, Vienna) under specifically pathogen-free sterile conditions. Genotyping of mice and cells was performed as described previously (Cui et al, 2004). The sensitivities of the Stat5 fl and Δ PCRs were determined by limited dilution and we are able to detect 100 Stat5fl/fl or Stat5Δ/Δ cells within the starting material (data not shown). All animal experiments were carried out in accordance with protocols approved by Austrian law.
BM transplants of bcr/ablp210-infected cells and deletion of Stat5
For BM transplantation studies two different approaches were used. (i) Donor mice (6 weeks of age) were injected i.p. with 5-FU (150 mg/kg body weight). BM cells were co-cultivated on bcr/ablp210 retroviral producer cells for 48 h in the presence of IL-3 (25 ng/ml), IL-6 (50 ng/ml), SCF (50 ng/ml) and 7 μg/ml polybrene and injected via tail vein into lethally irradiated (10 Gy) wt recipients (1 × 106 cells/mouse). For in vivo deletion of Stat5, mice received 400 μg p(I:C) (Sigma) at a single dose 6 weeks post-transplantation. (ii) BM cells from 6 weeks old donor mice were co-cultivated on bcr/ablp210 retroviral producer cells as described above. BM cells (1 × 106) were injected into lethally irradiated B6129F1 recipient mice. BM cells of three diseased animals were pooled and treated with recombinant IFN-β (1000 U/ml; Serotech) for 48 h to delete Stat5. Thereafter cells were transplanted into lethally irradiated secondary recipients (B6129F1).
Deletion of Stat5 and Stat3 in lymphoid leukaemic cell lines
For in vivo deletion of Stat5 or Stat3 in lymphoid leukaemia studies, 1 × 105 or 1 × 106 v-abl+ cells were injected via tail vein into Rag2−/−γc−/− or C57BL/6J mice, respectively. From day 7 on, mice received 400 μg p(I:C) i.p. every 4 days to induce Stat5-deletion in the transplanted leukaemic cells. Mice injected with PBS served as controls. Upon signs of sickness (decreased mobility, weight loss and scrubby fur), mice were sacrificed and lymphatic organs were analysed for leukaemic cell (CD19+, B220+) infiltrations by flow cytometry.
For in vitro deletion of Stat5, Stat5fl/flMx1Cre v-abl+ cells were seeded at a density of 3 × 105 cells/ml and incubated for 48 h in 1000 U/ml recombinant IFN-β (Serotech) in complete RPMI. Stat5fl/fl v-abl+ cells treated with IFN-β and untreated Stat5fl/flMx1Cre v-abl+ served as controls. Cells were analysed by flow cytometry for cell cycle progression and apoptosis every day.
Statistical analysis
Statistics were carried out using Student’s t-test or Mann–Whitney U-test as appropriate. Transplant experiments were analysed for statistical significance using log-rank test. Data are presented as averages ± SD and were analysed by GraphPad® and SPSS® software. Additional information regarding analysis tissue culture conditions, immunoblotting, transformation and imatinib sensibility assays is available within the Supporting Information.
Acknowledgements
We are deeply indebted to M. Freissmuth, M. Busslinger, P. Valent, O. Hantschel, G. Superti-Furga and T. Decker for continuous discussion and scientific input. We also thank M. Mayerhofer for providing the Bcr/Ablp210T315I mutant. This work was made possible by financial support from the Austrian Research fund (FWF-SFB-28), the Vienna Science and Technology Fund (WWTF-LS07-037) and the GEN-AU-program DRAGON.
Footnotes
Supporting information is available at EMBO Molecular Medicine online.
The authors declare that they have no conflict of interest.
For more information
Jak-Stat Signalling—from Basics to Disease: www.jak-stat.at
References
- Benekli M, Baer MR, Baumann H, Wetzler M. Signal transducer and activator of transcription proteins in leukemias. Blood. 2003;101:2940–2954. doi: 10.1182/blood-2002-04-1204. [DOI] [PubMed] [Google Scholar]
- Bromberg J. Stat proteins and oncogenesis. J Clin Invest. 2002;109:1139–1142. doi: 10.1172/JCI15617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buettner R, Mora LB, Jove R. Activated STAT signaling in human tumors provides novel molecular targets for therapeutic intervention. Clin Cancer Res. 2002;8:945–954. [PubMed] [Google Scholar]
- Burchill MA, Goetz CA, Prlic M, O’Neil JJ, Harmon IR, Bensinger SJ, Turka LA, Brennan P, Jameson SC, Farrar MA. Distinct effects of STAT5 activation on CD4+ and CD8+ T cell homeostasis: development of CD4+CD25+ regulatory T cells versus CD8+ memory T cells. J Immunol. 2003;171:5853–5864. doi: 10.4049/jimmunol.171.11.5853. [DOI] [PubMed] [Google Scholar]
- Carlesso N, Frank DA, Griffin JD. Tyrosyl phosphorylation and DNA binding activity of signal transducers and activators of transcription (STAT) proteins in hematopoietic cell lines transformed by Bcr/Abl. J Exp Med. 1996;183:811–820. doi: 10.1084/jem.183.3.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai SK, Nichols GL, Rothman P. Constitutive activation of JAKs and STATs in BCR-Abl-expressing cell lines and peripheral blood cells derived from leukemic patients. J Immunol. 1997;159:4720–4728. [PubMed] [Google Scholar]
- Chu S, Xu H, Shah NP, Snyder DS, Forman SJ, Sawyers CL, Bhatia R. Detection of BCR-ABL kinase mutations in CD34+ cells from chronic myelogenous leukemia patients in complete cytogenetic remission on imatinib mesylate treatment. Blood. 2005;105:2093–2098. doi: 10.1182/blood-2004-03-1114. [DOI] [PubMed] [Google Scholar]
- Coppo P, Dusanter-Fourt I, Millot G, Nogueira MM, Dugray A, Bonnet ML, Mitjavila-Garcia MT, Le Pesteur D, Guilhot F, Vainchenker W, et al. Constitutive and specific activation of STAT3 by BCR-ABL in embryonic stem cells. Oncogene. 2003;22:4102–4110. doi: 10.1038/sj.onc.1206607. [DOI] [PubMed] [Google Scholar]
- Cui Y, Riedlinger G, Miyoshi K, Tang W, Li C, Deng CX, Robinson GW, Hennighausen L. Inactivation of Stat5 in mouse mammary epithelium during pregnancy reveals distinct functions in cell proliferation, survival, and differentiation. Mol Cell Biol. 2004;24:8037–8047. doi: 10.1128/MCB.24.18.8037-8047.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Groot RP, Raaijmakers JA, Lammers JW, Jove R, Koenderman L. STAT5 activation by BCR-Abl contributes to transformation of K562 leukemia cells. Blood. 1999;94:1108–1112. [PubMed] [Google Scholar]
- Deininger MW, Goldman JM, Melo JV. The molecular biology of chronic myeloid leukemia. Blood. 2000;96:3343–3356. [PubMed] [Google Scholar]
- Dierks C, Beigi R, Guo GR, Zirlik K, Stegert MR, Manley P, Trussell C, Schmitt-Graeff A, Landwerlin K, Veelken H, et al. Expansion of Bcr-Abl-positive leukemic stem cells is dependent on Hedgehog pathway activation. Cancer Cell. 2008;14:238–249. doi: 10.1016/j.ccr.2008.08.003. [DOI] [PubMed] [Google Scholar]
- Druker BJ, Sawyers CL, Kantarjian H, Resta DJ, Reese SF, Ford JM, Capdeville R, Talpaz M. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N Engl J Med. 2001a;344:1038–1042. doi: 10.1056/NEJM200104053441402. [DOI] [PubMed] [Google Scholar]
- Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, Lydon NB, Kantarjian H, Capdeville R, Ohno-Jones S, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001b;344:1031–1037. doi: 10.1056/NEJM200104053441401. [DOI] [PubMed] [Google Scholar]
- Ecker A, Simma O, Hoelbl A, Kenner L, Beug H, Moriggl R, Sexl V. The dark and the bright side of Stat3: proto-oncogene and tumor-suppressor. Front Biosci. 2009;14:2944–2958. doi: 10.2741/3425. [DOI] [PubMed] [Google Scholar]
- Gough DJ, Corlett A, Schlessinger K, Wegrzyn J, Larner AC, Levy DE. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science. 2009;324:1713–1716. doi: 10.1126/science.1171721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grebien F, Kerenyi MA, Kovacic B, Kolbe T, Becker V, Dolznig H, Pfeffer K, Klingmuller U, Muller M, Beug H, et al. Stat5 activation enables erythropoiesis in the absence of EpoR and Jak2. Blood. 2008;111:4511–4522. doi: 10.1182/blood-2007-07-102848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griswold IJ, MacPartlin M, Bumm T, Goss VL, O’Hare T, Lee KA, Corbin AS, Stoffregen EP, Smith C, Johnson K, et al. Kinase domain mutants of Bcr-Abl exhibit altered transformation potency, kinase activity, and substrate utilization, irrespective of sensitivity to imatinib. Mol Cell Biol. 2006;26:6082–6093. doi: 10.1128/MCB.02202-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Hexner EO, Serdikoff C, Jan M, Swider CR, Robinson C, Yang S, Angeles T, Emerson SG, Carroll M, Ruggeri B, et al. Lestaurtinib (CEP701) is a JAK2 inhibitor that suppresses JAK2/STAT5 signaling and the proliferation of primary erythroid cells from patients with myeloproliferative disorders. Blood. 2008;111:5663–5671. doi: 10.1182/blood-2007-04-083402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho JM, Beattie BK, Squire JA, Frank DA, Barber DL. Fusion of the ets transcription factor TEL to Jak2 results in constitutive Jak-Stat signaling. Blood. 1999;93:4354–4364. [PubMed] [Google Scholar]
- Hoelbl A, Kovacic B, Kerenyi MA, Simma O, Warsch W, Cui Y, Beug H, Hennighausen L, Moriggl R, Sexl V. Clarifying the role of Stat5 in lymphoid development and Abelson-induced transformation. Blood. 2006;107:4898–4906. doi: 10.1182/blood-2005-09-3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilaria RL, Jr, Van Etten RA. P210 and P190(BCR/ABL) induce the tyrosine phosphorylation and DNA binding activity of multiple specific STAT family members. J Biol Chem. 1996;271:31704–31710. doi: 10.1074/jbc.271.49.31704. [DOI] [PubMed] [Google Scholar]
- Kato Y, Iwama A, Tadokoro Y, Shimoda K, Minoguchi M, Akira S, Tanaka M, Miyajima A, Kitamura T, Nakauchi H. Selective activation of STAT5 unveils its role in stem cell self-renewal in normal and leukemic hematopoiesis. J Exp Med. 2005;202:169–179. doi: 10.1084/jem.20042541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieslinger M, Woldman I, Moriggl R, Hofmann J, Marine JC, Ihle JN, Beug H, Decker T. Antiapoptotic activity of Stat5 required during terminal stages of myeloid differentiation. Genes Dev. 2000;14:232–244. [PMC free article] [PubMed] [Google Scholar]
- Kim I, Saunders TL, Morrison SJ. Sox17 dependence distinguishes the transcriptional regulation of fetal from adult hematopoietic stem cells. Cell. 2007;130:470–483. doi: 10.1016/j.cell.2007.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornfeld JW, Grebien F, Kerenyi MA, Friedbichler K, Kovacic B, Zankl B, Hoelbl A, Nivarti H, Beug H, Sexl V, et al. The different functions of Stat5 and chromatin alteration through Stat5 proteins. Front Biosci. 2008;13:6237–6254. doi: 10.2741/3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause DS, Van Etten RA. Right on target: eradicating leukemic stem cells. Trends Mol Med. 2007;13:470–481. doi: 10.1016/j.molmed.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause DS, Lazarides K, von Andrian UH, Van Etten RA. Requirement for CD44 in homing and engraftment of BCR-ABL-expressing leukemic stem cells. Nat Med. 2006;12:1175–1180. doi: 10.1038/nm1489. [DOI] [PubMed] [Google Scholar]
- Letai A, Sorcinelli MD, Beard C, Korsmeyer SJ. Antiapoptotic BCL-2 is required for maintenance of a model leukemia. Cancer Cell. 2004;6:241–249. doi: 10.1016/j.ccr.2004.07.011. [DOI] [PubMed] [Google Scholar]
- Levine RL, Pardanani A, Tefferi A, Gilliland DG. Role of JAK2 in the pathogenesis and therapy of myeloproliferative disorders. Nat Rev Cancer. 2007;7:673–683. doi: 10.1038/nrc2210. [DOI] [PubMed] [Google Scholar]
- Li G, Wang Z, Zhang Y, Kang Z, Haviernikova E, Cui Y, Hennighausen L, Moriggl R, Wang D, Tse W, et al. STAT5 requires the N-domain to maintain hematopoietic stem cell repopulating function and appropriate lymphoid-myeloid lineage output. Exp Hematol. 2007;35:1684–1694. doi: 10.1016/j.exphem.2007.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin TS, Mahajan S, Frank DA. STAT signaling in the pathogenesis and treatment of leukemias. Oncogene. 2000;19:2496–2504. doi: 10.1038/sj.onc.1203486. [DOI] [PubMed] [Google Scholar]
- Ling X, Arlinghaus RB. Knockdown of STAT3 expression by RNA interference inhibits the induction of breast tumors in immunocompetent mice. Cancer Res. 2005;65:2532–2536. doi: 10.1158/0008-5472.CAN-04-2425. [DOI] [PubMed] [Google Scholar]
- Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 2009;136:823–837. doi: 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkola HK, Orkin SH. The journey of developing hematopoietic stem cells. Development. 2006;133:3733–3744. doi: 10.1242/dev.02568. [DOI] [PubMed] [Google Scholar]
- Moon JH, Sohn SK, Lee MH, Jang JH, Kim K, Jung CW, Kim DH. BCL2 gene polymorphism could predict the treatment outcomes in acute myeloid leukemia patients. Leuk Res. 2010;34:166–172. doi: 10.1016/j.leukres.2009.05.009. [DOI] [PubMed] [Google Scholar]
- Moriggl R, Sexl V, Kenner L, Duntsch C, Stangl K, Gingras S, Hoffmeyer A, Bauer A, Piekorz R, Wang D, et al. Stat5 tetramer formation is associated with leukemogenesis. Cancer Cell. 2005;7:87–99. doi: 10.1016/j.ccr.2004.12.010. [DOI] [PubMed] [Google Scholar]
- Neering SJ, Bushnell T, Sozer S, Ashton J, Rossi RM, Wang PY, Bell DR, Heinrich D, Bottaro A, Jordan CT. Leukemia stem cells in a genetically defined murine model of blast-crisis CML. Blood. 2007;110:2578–2585. doi: 10.1182/blood-2007-02-073031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- Passegue E, Wagner EF, Weissman IL. JunB deficiency leads to a myeloproliferative disorder arising from hematopoietic stem cells. Cell. 2004;119:431–443. doi: 10.1016/j.cell.2004.10.010. [DOI] [PubMed] [Google Scholar]
- Passegue E, Wagers AJ, Giuriato S, Anderson WC, Weissman IL. Global analysis of proliferation and cell cycle gene expression in the regulation of hematopoietic stem and progenitor cell fates. J Exp Med. 2005;202:1599–1611. doi: 10.1084/jem.20050967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pear WS, Miller JP, Xu L, Pui JC, Soffer B, Quackenbush RC, Pendergast AM, Bronson R, Aster JC, Scott ML, et al. Efficient and rapid induction of a chronic myelogenous leukemia-like myeloproliferative disease in mice receiving P210bcr/abl-transduced bone marrow. Blood. 1998;92:3780–3792. [PubMed] [Google Scholar]
- Quintas-Cardama A, Kantarjian H, Cortes J. Flying under the radar: the new wave of BCR-ABL inhibitors. Nat Rev Drug Discov. 2007;6:834–848. doi: 10.1038/nrd2324. [DOI] [PubMed] [Google Scholar]
- Scherr M, Chaturvedi A, Battmer K, Dallmann I, Schultheis B, Ganser A, Eder M. Enhanced sensitivity to inhibition of SHP2, STAT5, and Gab2 expression in chronic myeloid leukemia (CML) Blood. 2006;107:3279–3287. doi: 10.1182/blood-2005-08-3087. [DOI] [PubMed] [Google Scholar]
- Schwaller J, Frantsve J, Aster J, Williams IR, Tomasson MH, Ross TS, Peeters P, Van Rompaey L, Van Etten RA, Ilaria R, Jr, et al. Transformation of hematopoietic cell lines to growth-factor independence and induction of a fatal myelo- and lymphoproliferative disease in mice by retrovirally transduced TEL/JAK2 fusion genes. EMBO J. 1998;17:5321–5333. doi: 10.1093/emboj/17.18.5321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwaller J, Parganas E, Wang D, Cain D, Aster JC, Williams IR, Lee CK, Gerthner R, Kitamura T, Frantsve J, et al. Stat5 is essential for the myelo- and lymphoproliferative disease induced by TEL/JAK2. Mol Cell. 2000;6:693–704. doi: 10.1016/s1097-2765(00)00067-8. [DOI] [PubMed] [Google Scholar]
- Sexl V, Piekorz R, Moriggl R, Rohrer J, Brown MP, Bunting KD, Rothammer K, Roussel MF, Ihle JN. Stat5a/b contribute to interleukin 7-induced B-cell precursor expansion, but abl- and bcr/abl-induced transformation are independent of stat5. Blood. 2000;96:2277–2283. [PubMed] [Google Scholar]
- Shah NP, Nicoll JM, Nagar B, Gorre ME, Paquette RL, Kuriyan J, Sawyers CL. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell. 2002;2:117–125. doi: 10.1016/s1535-6108(02)00096-x. [DOI] [PubMed] [Google Scholar]
- Sherr CJ. Principles of tumor suppression. Cell. 2004;116:235–246. doi: 10.1016/s0092-8674(03)01075-4. [DOI] [PubMed] [Google Scholar]
- Shuai K, Halpern J, ten Hoeve J, Rao X, Sawyers CL. Constitutive activation of STAT5 by the BCR-ABL oncogene in chronic myelogenous leukemia. Oncogene. 1996;13:247–254. [PubMed] [Google Scholar]
- Sillaber C, Gesbert F, Frank DA, Sattler M, Griffin JD. STAT5 activation contributes to growth and viability in Bcr/Abl-transformed cells. Blood. 2000;95:2118–2125. [PubMed] [Google Scholar]
- Spiekermann K, Pau M, Schwab R, Schmieja K, Franzrahe S, Hiddemann W. Constitutive activation of STAT3 and STAT5 is induced by leukemic fusion proteins with protein tyrosine kinase activity and is sufficient for transformation of hematopoietic precursor cells. Exp Hematol. 2002;30:262–271. doi: 10.1016/s0301-472x(01)00787-1. [DOI] [PubMed] [Google Scholar]
- Steelman LS, Pohnert SC, Shelton JG, Franklin RA, Bertrand FE, McCubrey JA. JAK/STAT, Raf/MEK/ERK, PI3K/Akt and BCR-ABL in cell cycle progression and leukemogenesis. Leukemia. 2004;18:189–218. doi: 10.1038/sj.leu.2403241. [DOI] [PubMed] [Google Scholar]
- Takeda K, Noguchi K, Shi W, Tanaka T, Matsumoto M, Yoshida N, Kishimoto T, Akira S. Targeted disruption of the mouse Stat3 gene leads to early embryonic lethality. Proc Natl Acad Sci USA. 1997;94:3801–3804. doi: 10.1073/pnas.94.8.3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tefferi A, Gilliland DG. The JAK2V617F tyrosine kinase mutation in myeloproliferative disorders: status report and immediate implications for disease classification and diagnosis. Mayo Clin Proc. 2005;80:947–958. doi: 10.4065/80.7.947. [DOI] [PubMed] [Google Scholar]
- Tefferi A, Lasho TL, Gilliland G. JAK2 mutations in myeloproliferative disorders. N Engl J Med. 2005;353:1416–1417. doi: 10.1056/NEJMc051878. author reply 1416-1417. [DOI] [PubMed] [Google Scholar]
- Van Etten RA. Models of chronic myeloid leukemia. Curr Oncol Rep. 2001;3:228–237. doi: 10.1007/s11912-001-0055-y. [DOI] [PubMed] [Google Scholar]
- Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L, Newman J, Reczek EE, Weissleder R, Jacks T. Restoration of p53 function leads to tumour regression in vivo. Nature. 2007;445:661–665. doi: 10.1038/nature05541. [DOI] [PubMed] [Google Scholar]
- Wang JC, Dick JE. Cancer stem cells: lessons from leukemia. Trends Cell Biol. 2005;15:494–501. doi: 10.1016/j.tcb.2005.07.004. [DOI] [PubMed] [Google Scholar]
- Wang Z, Li G, Tse W, Bunting KD. Conditional deletion of STAT5 in adult mouse hematopoietic stem cells causes loss of quiescence and permits efficient nonablative stem cell replacement. Blood. 2009;113:4856–4865. doi: 10.1182/blood-2008-09-181107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein IB. Cancer. Addiction to oncogenes—the Achilles heal of cancer. Science. 2002;297:63–64. doi: 10.1126/science.1073096. [DOI] [PubMed] [Google Scholar]
- Wells JA, McClendon CL. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature. 2007;450:1001–1009. doi: 10.1038/nature06526. [DOI] [PubMed] [Google Scholar]
- Wendel HG, de Stanchina E, Cepero E, Ray S, Emig M, Fridman JS, Veach DR, Bornmann WG, Clarkson B, McCombie WR, et al. Loss of p53 impedes the antileukemic response to BCR-ABL inhibition. Proc Natl Acad Sci USA. 2006;103:7444–7449. doi: 10.1073/pnas.0602402103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wernig G, Kharas MG, Okabe R, Moore SA, Leeman DS, Cullen DE, Gozo M, McDowell EP, Levine RL, Doukas J, et al. Efficacy of TG101348, a selective JAK2 inhibitor, in treatment of a murine model of JAK2V617F-induced polycythemia vera. Cancer Cell. 2008;13:311–320. doi: 10.1016/j.ccr.2008.02.009. [DOI] [PubMed] [Google Scholar]
- Yao Z, Cui Y, Watford WT, Bream JH, Yamaoka K, Hissong BD, Li D, Durum SK, Jiang Q, Bhandoola A, et al. Stat5a/b are essential for normal lymphoid development and differentiation. Proc Natl Acad Sci USA. 2006;103:1000–1005. doi: 10.1073/pnas.0507350103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Jove R. The STATs of cancer—new molecular targets come of age. Nat Rev Cancer. 2004;4:97–105. doi: 10.1038/nrc1275. [DOI] [PubMed] [Google Scholar]
- Zhang CC, Lodish HF. Murine hematopoietic stem cells change their surface phenotype during ex vivo expansion. Blood. 2005;105:4314–4320. doi: 10.1182/blood-2004-11-4418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Chen A, Jamieson CH, Fereshteh M, Abrahamsson A, Blum J, Kwon HY, Kim J, Chute JP, Rizzieri D, et al. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature. 2009;458:776–779. doi: 10.1038/nature07737. [DOI] [PMC free article] [PubMed] [Google Scholar]