

Abstract
Atrial fibrillation (AF) causes substantial morbidity and mortality. It may be triggered and sustained by either re-entrant or non-re-entrant electrical activity. Human atrial cellular refractory period is shortened in chronic AF, likely aiding re-entry. The ionic and molecular mechanisms are not fully understood, and may include increased inward rectifier K+ current and altered Ca2+-handling. Heart failure, a major cause of AF, may involve arrhythmogenic atrial electrical remodelling, but the pattern is unclear in humans. Beta-blocker therapy prolongs atrial cell refractory period; a potentially anti-arrhythmic influence, but the ionic and molecular mechanisms are unclear. The search for drugs to suppress AF without causing ventricular arrhythmias has been aided by basic studies of cellular mechanisms of AF. It remains to be seen whether such drugs will improve patient treatment.
Keywords: Atrial fibrillation, Arrhythmias (mechanisms), Refractory period, Transmembrane action potential, Ion current, Heart failure, Beta-blocker, Electrical remodelling
Electrophysiological mechanisms of human AF, and their study in single atrial cells
Atrial fibrillation (AF) is the most common sustained cardiac arrhythmia. It causes substantial morbidity and mortality. The majority of atrial premature beats that initiate AF originate from focal ectopic electrical activity in the pulmonary veins (PV). AF is sustained by single or multiple circuit intra-atrial re-entry and/or focal ectopy, and the latter may be re-entrant or non-re-entrant.1 Non-re-entrant mechanisms include abnormal automaticity (AA) and triggered activity. Abnormal automaticity is the premature firing of action potentials (AP) due to abnormal diastolic membrane depolarisation (Fig 1A), and is favoured by, e.g., β-adrenergic stimulation or decreased vagal activity. Triggered activity is premature firing due to afterdepolarisations. These may be early (EAD), occurring during repolarisation and favoured by AP-prolongation, or delayed (DAD), occurring after an AP and favoured by intracellular Ca2+-overload (Fig 1A). Re-entry is rapid circuitous activation caused by unidirectional conduction block, and favoured by premature impulses, heterogeneity and shortening of effective refractory period (ERP), and slowing of conduction velocity, θ (Fig 1B). Several electrophysiological parameters which may affect AF genesis and maintenance have been measured in human atrial isolated cells. The cellular ERP2 and AP maximum upstroke velocity, Vmax (Fig 1C) contribute to myocardial ERP and θ, respectively, so their reduction could promote re-entry by shortening its wavelength, λ (Fig 1B). “Cellular arrhythmic depolarisations”, CADs3 (Fig 1D) may represent AA, EADs or DADs, with potential involvement in non-re-entrant mechanisms.
Figure 1.
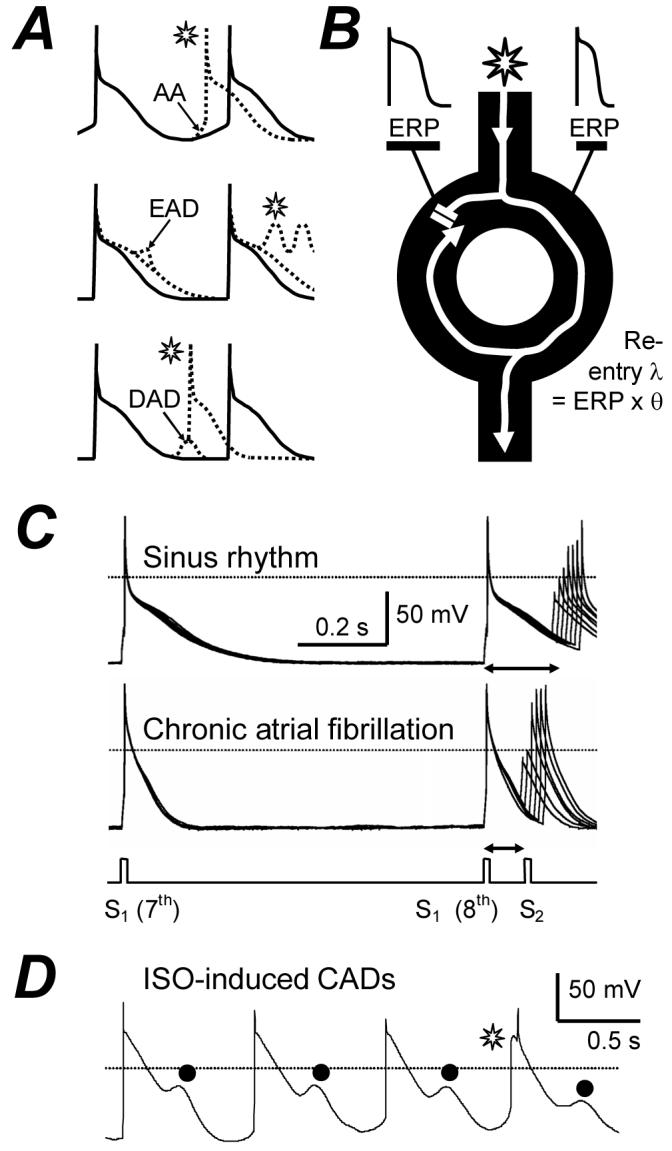
Electrophysiological mechanisms of arrhythmias and their study in human atrial cells. A, Representation of premature action potentials ( ) from abnormal automaticity (AA), early (EAD) or delayed (DAD) afterdepolarisations. B, Premature impulse divides at functional or anatomical obstacle, blocks at tissue with normal (left side) but conducts with short (right) ERP, and re-enters previously inexcitable zone. λ=wavelength. θ=conduction velocity. C, Original APs stimulated in an atrial cell from a patient in SR, and in AF, by conditioning pulses (S1) and premature test pulses (S2). ERP (↔) =longest S1-S2 failing to produce S2 response of amplitude >80% of S1. D, Original APs stimulated by a pulse-train in the presence of 0.05 μM isoproterenol (ISO), producing “cellular arrhythmic depolarisations”, CADs (•).
) from abnormal automaticity (AA), early (EAD) or delayed (DAD) afterdepolarisations. B, Premature impulse divides at functional or anatomical obstacle, blocks at tissue with normal (left side) but conducts with short (right) ERP, and re-enters previously inexcitable zone. λ=wavelength. θ=conduction velocity. C, Original APs stimulated in an atrial cell from a patient in SR, and in AF, by conditioning pulses (S1) and premature test pulses (S2). ERP (↔) =longest S1-S2 failing to produce S2 response of amplitude >80% of S1. D, Original APs stimulated by a pulse-train in the presence of 0.05 μM isoproterenol (ISO), producing “cellular arrhythmic depolarisations”, CADs (•). 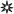 may represent AA. C&D based on data in2&3 with permission from Elsevier.
may represent AA. C&D based on data in2&3 with permission from Elsevier.
Atrial cellular electrical remodelling in AF
Atrial myocardial electrical and mechanical activity and structure adapt, or remodel, in response to a variety of diseases and other stimuli. For example, congestive heart failure (CHF) may involve electrical remodelling, atrial dilation and interstitial fibrosis, each potentially predisposing to AF. Once AF occurs, the rapid atrial rate causes atrial electrical remodelling which promotes AF, so AF is auto-perpetuating. In goats, induced AF progressively shortened atrial ERP and AF-interval over 24 hr, which reduced the re-entry λ and increased AF vulnerability.4 Maximal ERP-shortening may precede maximal AF duration, but the ERP-shortening contributes to the AF substrate. In our laboratory, a similar ERP-shortening was found in atrial cells isolated from patients with chronic AF (Fig 1C). This was associated with impaired ERP rate-adaptation, shortening and triangulation of the AP, and no change in Vmax.2 The shortened AP permitted full repolarisation at the fast rates typically encountered in AF, and thus prevented the depolarisation of the maximum diastolic potential (MDP) which was observed in sinus rhythm (SR).2 This effect on MDP might limit Ca2+-overload in the remodelled atrium, but the ERP changes favour re-entry. The ERP is largely determined by the AP duration (APD), which depends on a delicate balance of inward and outward ion currents flowing through a variety of membrane protein channels, pumps and exchangers. Therefore, an understanding of the mechanisms of human AF-induced atrial electrical remodelling requires knowledge about precise changes in each of these currents, and their contributions to the AP, in AF.
Potential ionic mechanisms of electrical remodelling in AF
Many human atrial ion currents have so far been studied in AF; collated in the Table. The inward rectifier K+ current (IK1) is the main determinant of the resting potential (Vm). Other currents contribute, including acetylcholine-activated K+ current (IKACh), Na+, K+ pump current (Ip), and possibly ATP-sensitive K+ current (IKATP). IK1 and IKACh also contribute to terminal repolarisation. There is consensus that chronic AF is associated with increased density of IK1 (Table). Furthermore, despite decreased parasympathetic-regulated IKACh, a constitutively active (CA) IKACh, not requiring its endogenous agonist, is induced in AF (Fig 2). A single study on Ip, from our laboratory, showed no change in AF, and changes in IKATP are variable. The reported increases in IK1 and CA IKACh were most prominent (with enhanced inward current) at voltages more negative than the AP voltage range. However, enhanced outward IK1 has also been reported, within the AP voltage range,5,6,8 which may contribute to the APD- and ERP-shortening in AF. Increased IK1 should also hyperpolarise Vm; whilst difficult to ascertain in human atrial isolated cells since the “chunk” isolation method may depolarise them, this has been reported in atrial trabeculae.6 The AP fires when depolarisation sufficient to drive Vm to threshold activates inward Na+ current (INa), causing the regenerative and rapid AP upstroke; the larger INa, the faster Vmax. A single study reported no change in INa density in AF, consistent with Vmax (Table), though its inactivation voltage-dependency was altered. Partial, or early, repolarisation follows the AP upstroke, via activation of a transient outward K+ current (ITO) and the ultra-rapid delayed rectifier K+ current (IKur). AF consistently and markedly reduced ITO, but data for IKur are equivocal (Table). The ITO reduction may contribute to AP triangulation in AF, as shown by blocking ITO with 4-aminopyridine (4-AP).2 However, its contribution to the APD90 and ERP is unclear since 4-AP also blocks IKur, though mathematical modelling suggested a negligible role.34 The AP plateau is maintained by inward, L-type Ca2+, current (ICaL), which is consistently and markedly reduced in chronic AF (Table), despite increased single channel open probability.35 Such ICaL reduction depresses the AP plateau, consistent with acute effects of nifedipine2 or simulated ICaL reduction,34 though its contribution, alone, to the APD902,34 - or ERP2- shortening may be small. Mid/late repolarisation results from activation of IKur, as well as the rapid (IKr) and slow (IKS) delayed rectifiers, balanced by inward Na+-Ca2+ exchange current (INa/Ca) following the [Ca2+]i transient. INa/Ca also underlies the transient inward current responsible for DADs. However, any role for these currents in human AF-remodelling is presently unclear, since data are either equivocal or unavailable (Table). Abnormal automaticity results from decreased outward and/or increased inward diastolic currents, including INa/Ca and the “funny” current (If). However, data on If are also lacking.
Table.
Human atrial cellular electrophysiological changes associated with AF, ventricular dysfunction and drug therapy. Arrows show direction of change relative to “controls”. *=atrial dilation only. NDA=no data available. See text for definitions
| Human atrial cell electrophysiological measurement | Chronic AF | Post-cardiac surgery AF | CHF, LVSD and/or atrial dilation | Chronic β1-blocker therapy |
|---|---|---|---|---|
| ERP | ↓ 2 | ↔ 22 | NDA | ↑3,22,31 |
| APD90 | ↓2,5-7 | ↔ 22 | ↑23,24 ↔25,26 | ↑22,31 |
| APD50 | ↔2,6 ↑7 | ↔ 22 | ↔23,24 ↓26 | ↔3,31 |
| AP Vmax | ↔ 2 | ↔ 22 | NDA | ↔ 31 |
| IK1 | ↑2,5,6,8-12 | ↔9,22 | ↓23 ↔*27 | ↓ 32 |
| IKACh | ↑5 ↓6,9,10,12 | ↔ 9 | ↓ 23 | NDA |
| CA IKACh | ↑10,12 | NDA | NDA | NDA |
| Ip | ↔ 13 | NDA | NDA | NDA |
| IKATP | ↓14 ↑15 | NDA | ↔ 24 | NDA |
| INa | ↔ 5 | NDA | NDA | NDA |
| ITO | ↓2,5,8,16,17 | ↔ 22 | ↑26 ↓*27,28 | ↓31,32 |
| IKur (ISUS) | ↓8,16 ↔2,5,17 | ↔ 22 | ↔25,26 ↔*28 ↓*27 | ↔31,32 |
| ICaL | ↓2,5,7,11,18-21 | ↑18 ↔22 | ↓29 ↔25,30 ↓*27 | ↔3,7,22,31,33 |
| ICaL %↑ by β-stim | ↑18-21 | ↔ 22 | ↓29,30 | ↔3,22 |
| ICaL %↑ by 5-HT | ↓ 7 | NDA | ↓ 29 | ↑7,33 |
| INa/Ca, INa/H, IKr, IKs, ICl(swell), ICaB, If | NDA | NDA | NDA | NDA |
Figure 2.
Induction of constitutively active acetylcholine-activated K+ current, CA IKACh, in human AF. Single channel IK1 and CA IKACh currents (A) and mean (±SE) open probabilities (B), recorded at -120 mV in atrial cells from patients in SR (□) and AF (■). *=P<0.05 vs SR. C, Potential signalling mechanism of increased CA IKACh in AF. In SR, IKACh may require channel phosphorylation by calmodulin-dependent protein kinase (PK) II (CaMKII), PKG and PKC.12 In AF, CA IKACh may result from upregulation of PKCε.12 A&B based on data in10 with permission from Lippincott Williams & Wilkins.
Human PV isolated cell electrophysiology has not yet been studied. Chronic atrial tachypacing (AT) in dogs produced qualitatively similar APD-shortening in PV cells to atrial cells, and also similar changes in IK1, ITO and ICaL.36 However, a current which may be analogous to human CA IKACh was increased more strongly in PV than atrial cells, perhaps favouring PV re-entry.37 The relative importance of re-entrant versus non-re-entrant activity to PV arrhythmogenesis, either before or after AF-remodelling, is unknown.1
Atrial electrical activity is intricately linked with cellular and sub-cellular Ca2+ fluxes, particularly via INa/Ca. Intracellular Ca2+-handling is altered in AF, though human data are sparse. In canine atrial cells, acute AT, analogous to a paroxysm of AF, abruptly increases diastolic [Ca2+]i, a potential trigger of the remodelling process. Chronic AT, by contrast, markedly decreased the [Ca2+]i transient amplitude,38 perhaps reflecting protection from [Ca2+]i-overload. This may result from a deficient trigger function of the markedly reduced ICaL, since sarcoplasmic reticular Ca2+ content was preserved.38 Human AF was associated with a potentially arrhythmogenic increase in the frequency of Ca2+ sparks and waves.39 This may represent sarcoplasmic reticular Ca2+ leak due to ryanodine receptor hyperphosphorylation.40
Whether the combined ionic changes so far established in human AF can account for the associated AP changes is unclear, and will require the aid of mathematical models. One such model suggested that the combined IK1, ITO and ICaL changes could explain the AP changes,34 though in dog, concurrent [Ca2+]i changes were required.38 Another suggested a major contribution from the IK1 increase to the stabilisation of re-entry.41
Potential molecular mechanisms in AF: genetic and non-genetic
Many atrial ion current changes in human AF are accompanied by, and often considered to be caused by, altered tissue expression of the ion channel pore-forming α-subunits that carry them; e.g., increased Kir2.1 (carries IK1) and decreased Kv4.3 (ITO).11 However, there are some intriguing and controversial exceptions. Protein levels of ICaL α-subunits were decreased by 40-55% in three studies,35,42,43 in line with ICaL reduction, but unchanged in four others.11,20,21,44 Also, despite increased CA IKACh in AF (Table), Kir 3.1 protein level was decreased.43 The apparent discrepancies between changes in ion current density and protein expression suggest post-translational modification or altered channel regulation. The magnitude of ICaL is influenced by a balance between channel phosphorylation by kinases and de-phosphorylation by phosphatases. Chronic AF upregulated phosphatase type-2A-C, reducing ICaL without requiring reduced channel protein.20 Similarly, induction of CA IKACh in human AF resulted from abnormal protein kinase C function.12 (Fig 2C).
AF may be a heritable disorder: positive family history was identified in 5% of patients with AF.45 Several genetic mutations have been associated with familial AF, mainly for K+ channels. Most are “gain of function”, increasing IKs, IKr or IK1 and expected to shorten ERP and promote re-entry, though an IKur loss of function mutation might prolong ERP.45 However, such mutations occur in other diseases, e.g., dilated cardiomyopathy, long-QT, short-QT and Brugada syndromes, some of which are co-morbidities for AF. Nevertheless, it seems that genetic variants are involved in the pathogenesis of AF in a proportion of cases.
Neurohumoral involvement in AF
AF can result from a sympathetic/parasympathetic imbalance. Furthermore, neurohumoral activation in CHF, an important cause of AF, increases circulating levels of catecholamines, angiotensin and endothelin (ET-1). Beta-adrenergic-stimulation from catecholamines may promote DADs, by increasing ICaL and Ca2+-induced Ca2+ release. AF-remodelling potentiated the relative increase in ICaL produced by β-stimulation (Table). We demonstrated that ET-1 had no direct effect on ICaL, APD or ERP in human atrial cells. However, it abolished isoproterenol-induced increases in ICaL, APD50 and CADs (Fig 1D), with no effect on ERP.3 Thus, ET-1 might exert an anti-adrenergic anti-arrhythmic influence in the atria of patients with CHF. Serotonin (5-HT) is released from platelets aggregating in static blood in fibrillating atria. We demonstrated that 5-HT may be arrhythmogenic in human atrium, by increasing ICaL and producing CADs, without affecting ERP.33 Atrial remodelling by AF may protect from these effects, however, since they were attenuated in cells from patients with chronic AF7 (Table).
Post-operative AF: is there a predisposing atrial cellular electrophysiological substrate?
AF is common in patients following cardiac surgery (CS). Post-CS AF is independently predicted by old age, pre-CS AF and pre-CS P-wave changes. Therefore, pre-CS atrial cellular electrophysiology could influence the propensity for new onset AF post-CS; an issue presently under debate. An early study showed an association between post-CS AF and an enhanced pre-CS ICaL;18 potentially arrhythmogenic post-CS, when catecholamines are elevated. However, we recently demonstrated, by contrast, that neither pre-CS ICaL, AP parameters or ERP were predictive of post-CS AF.22 Furthermore, no other ion current measured, nor the ICaL response to β-stimulation, were different between patients with and without post-CS AF (Table). Some currents remain to be studied, but it appears that the electrically remodelled state caused by chronic AF (Table) is not present pre-CS in the atrial cells of patients who develop new onset post-CS AF.
Heart failure-induced atrial remodelling
AF is common in patients with CHF, and left ventricular systolic dysfunction (LVSD) substantially increases the risk of AF. It is unclear whether atrial cellular electrical remodelling, in patients in SR, contributes to this predisposition to AF. The available human data are scarce and inconsistent (Table), and compounded by inevitable variability in patients’ disease states and drug treatments. Atrial cellular electrical remodelling has been demonstrated in canine models of chronic ventricular tachypacing (VTP)-induced CHF. AF was invariably promoted, but the remodelling pattern differed from AF: atrial ERP was unchanged or increased, IK1 was not increased, both ITO and IKS were decreased, ICaL was only moderately decreased, and INa/Ca was increased.46,47 The increased INa/Ca might favour a triggered origin of AF in this model. CHF also caused atrial fibrosis, and whilst the ionic remodelling reversed after ceasing VTP, the fibrosis and AF persistence did not.46 Thus, atrial electrical remodelling may contribute to AF genesis, but was not necessary for its maintenance in this model. Human CHF or LVSD were associated with variable changes in APD, and cellular ERP has not been studied (Table). Human atrial ionic changes in CHF or LVSD may be expected to differ from those in chronic AF, with decreased IK1 and increased ITO, decreased or unchanged ICaL, and a decreased ICaL response to β-stimulation, so far reported (Table). The pattern may depend on the degree of atrial dilation, which itself may cause ionic remodelling (Table). Moreover, CHF- and AF-induced atrial remodelling interact. In dogs, this interaction was complex, not summative: chronic AT, imposed on a CHF-remodelled atrium, caused moderate ERP-shortening, IK1-increase and ICaL-decrease, but did not further remodel ITO, IKS or INa/Ca47. No comparative human atrial data could be found.
Atrial remodelling by chronic drug therapy
Atrial electrophysiology remodels in response not only to diseases and ageing, but also to long-term drug treatments; so called “pharmacological remodelling”.31 This was originally demonstrated in rabbits: treatment with the β1-blocker metoprolol caused an adaptational prolongation of the atrial APD, maximally after 6 days.48 Beta-blockers are increasingly used to treat AF and HF. We demonstrated that in patients in SR, β1-blocker treatment for ≥7 days was independently associated with prolonged atrial cell APD90 and ERP22,31 (Fig 3A&B), and not with ICaL.22 The ITO was reduced (Fig 3C), and ISUS was unchanged (Table). Preliminary data from our group suggest that the ITO reduction does not involve altered voltage-dependency or kinetics,32 nor altered ion channel expression,49 and that IK1 is also reduced.32 Recent sub-group analysis revealed a significant correlation between ERP and atenolol dose (Fig 3D), suggesting that the ERP prolongation is at least partly caused, directly or indirectly, by the atenolol treatment. Such ERP-prolongation might contribute to the anti-arrhythmic effects of beta-blockers, though a potentiation by chronic β-blockade of effects of 5-HT on ICaL (Table) and CADs33 could also oppose them.
Figure 3.
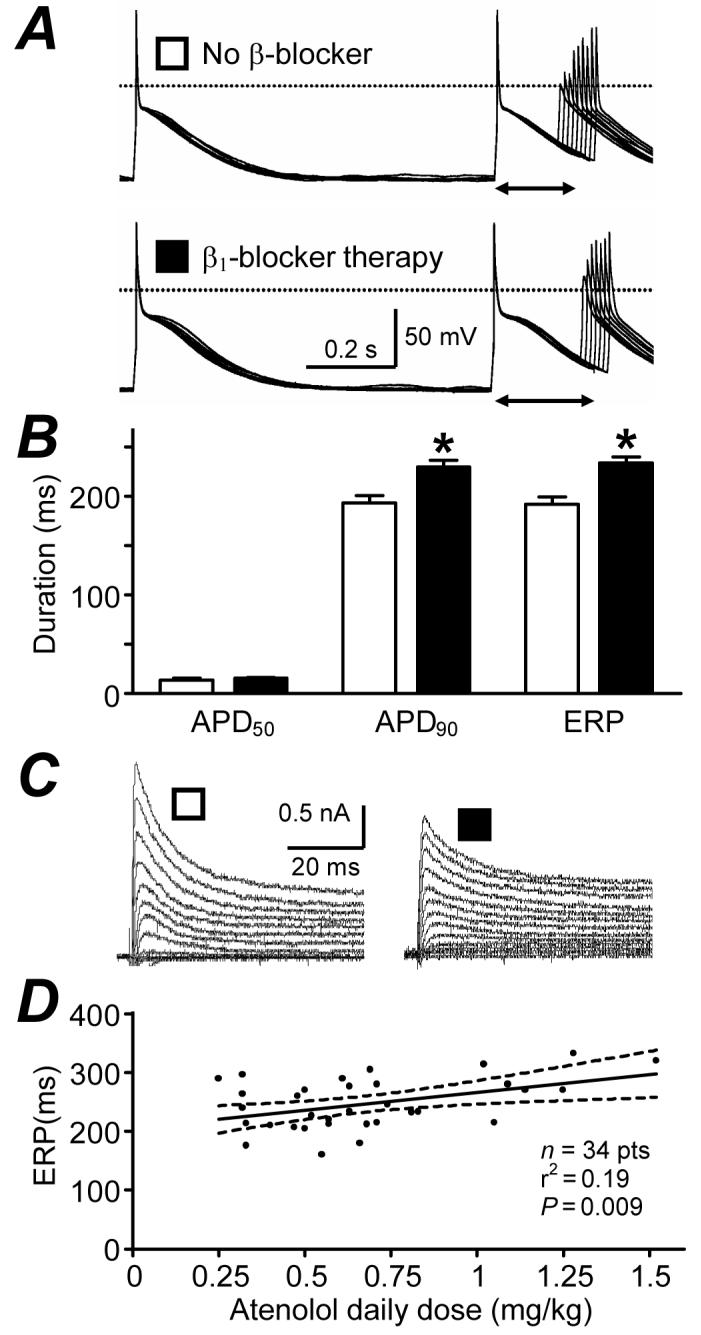
“Pharmacological remodelling” of human atrial cell electrophysiology by β1-blocker therapy. A, Action potentials and ERP (↔); B, mean (±SE) AP duration at 50 and 90% repolarisation (APD50 & APD90) and ERP; C, transient outward K+ currents, recorded in single atrial myocytes from patients in SR, treated with a β1-blocker ≥7 days (■) vs those in SR, not treated with a β-blocker (□). *=P<0.05 vs□. D, Correlation between atrial cell ERP and patient’s atenolol dose/body weight. Heart rate ≤75 beats/min. Dashed lines: 95% confidence interval. A&C based on data in31 with permission from Elsevier.
How research on cellular bases for human AF is driving new therapeutic strategies
Traditional ERP-prolonging drugs, to inhibit re-entry, do so by blocking IKr. This is problematic because IKr exists in ventricle as well as atrium, risking ventricular EADs and fibrillation. IKur and IKACh are considered to be atrium-specific, so their block might prolong ERP in atrium only, depending on secondary ionic effects. However, targeting ion channel regulation may be preferable to ion channel block. Altering the PKC pathway which induces CA IKACh in chronic AF12 might avoid undesirable effects of inhibiting parasympathetic-regulated IKACh on sinoatrial node and bladder. Blocking the phosphatase-induced ICaL decrease caused by AF20 is another possibility. Moreover, “de-remodelling”, in theory might be better than such “anti-remodelling”, since blocking potentially protective adaptations may be risky. Pharmacological targeting of non-re-entrant mechanisms of AF also may be considered.
AF is a highly complex, multifactorial and dynamic disorder with differing characteristics and aetiologies among individuals. As such, it presents an enormous challenge for the development of drugs for its effective and safe treatment. Current basic research is driving the search for new drugs. Several, including IKur and IKACh blockers, are entering clinical trials. It remains to be seen whether they will improve patient treatment.
Acknowledgments
Source/s of financial support.
AJ Workman received British Heart Foundation Basic Science Lectureship award BS/06/003.
List of abbreviations
- 4-AP
(4-aminopyridine)
- 5-HT
(5-hydroxytryptamine/serotonin)
- AA
(abnormal automaticity)
- AF
(atrial fibrillation)
- AP
(action potential)
- APD
(action potential duration)
- APD50
(action potential duration at 50% repolarisation)
- AT
(atrial tachypacing)
- CA
(constitutively active)
- CAD
(cellular arrhythmic depolarisation)
- CHF
(congestive heart failure)
- CS
(cardiac surgery)
- DAD
(delayed afterdepolarisation)
- EAD
(early afterdepolarisation)
- ERP
(effective refractory period)
- ET-1
(endothelin)
- ICaL
(L-type Ca2+ current)
- If
(funny current)
- IK1
(inward rectifier K+ current)
- IKACh
(acetylcholine-activated K+ current)
- IKATP
(adenosine triphosphate-sensitive K+ current)
- IKr
(rapid delayed rectifier K+ current)
- IKS
(slow delayed rectifier K+ current)
- IKur
(ultra-rapid delayed rectifier K+ current)
- INa
(Na+ current)
- INa/Ca
(Na+-Ca2+ exchange current)
- Ip
(Na+, K+ pump current)
- ISO
(isoproterenol)
- ITO
(transient outward K+ current)
- LVSD
(left ventricular systolic dysfunction)
- MDP
(maximum diastolic potential)
- NDA
(no data available)
- PK
(protein kinase)
- PV
(pulmonary vein)
- SR
(sinus rhythm)
- Vm
(resting potential)
- Vmax
(maximum upstroke velocity)
- VTP
(ventricular tachypacing)
- λ
(wavelength)
- θ
(conduction velocity).
Footnotes
Conflict/s of interest.
None.
References
- 1.Wit AL, Boyden PA. Triggered activity and atrial fibrillation. Heart Rhythm. 2007;4:S17–S23. doi: 10.1016/j.hrthm.2006.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Workman AJ, Kane KA, Rankin AC. The contribution of ionic currents to changes in refractoriness of human atrial myocytes associated with chronic atrial fibrillation. Cardiovasc Res. 2001;52:226–235. doi: 10.1016/s0008-6363(01)00380-7. [DOI] [PubMed] [Google Scholar]
- 3.Redpath CJ, Rankin AC, Kane KA, et al. Anti-adrenergic effects of endothelin on human atrial action potentials are potentially anti-arrhythmic. J Mol Cell Cardiol. 2006;40:717–724. doi: 10.1016/j.yjmcc.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 4.Wijffels MCEF, Kirchhof CJHJ, Dorland R, et al. Atrial fibrillation begets atrial fibrillation. A study in awake chronically instrumented goats. Circulation. 1995;92:1954–1968. doi: 10.1161/01.cir.92.7.1954. [DOI] [PubMed] [Google Scholar]
- 5.Bosch RF, Zeng X, Grammer JB, et al. Ionic mechanisms of electrical remodeling in human atrial fibrillation. Cardiovasc Res. 1999;44:121–131. doi: 10.1016/s0008-6363(99)00178-9. [DOI] [PubMed] [Google Scholar]
- 6.Dobrev D, Graf E, Wettwer E, et al. Molecular basis of downregulation of G-protein-coupled inward rectifying K+ current (IK,ACh) in chronic human atrial fibrillation: decrease in GIRK4 mRNA correlates with reduced IK,ACh and muscarinic receptor-mediated shortening of action potentials. Circulation. 2001;104:2551–2557. doi: 10.1161/hc4601.099466. [DOI] [PubMed] [Google Scholar]
- 7.Pau D, Workman AJ, Kane KA, et al. Electrophysiological and arrhythmogenic effects of 5-hydroxytryptamine on human atrial cells are reduced in atrial fibrillation. J Mol Cell Cardiol. 2007;42:54–62. doi: 10.1016/j.yjmcc.2006.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Van Wagoner DR, Pond AL, McCarthy PM, et al. Outward K+ current densities and Kv1.5 expression are reduced in chronic human atrial fibrillation. Circ Res. 1997;80:772–781. doi: 10.1161/01.res.80.6.772. [DOI] [PubMed] [Google Scholar]
- 9.Dobrev D, Wettwer E, Kortner A, et al. Human inward rectifier potassium channels in chronic and postoperative atrial fibrillation. Cardiovasc Res. 2002;54:397–404. doi: 10.1016/s0008-6363(01)00555-7. [DOI] [PubMed] [Google Scholar]
- 10.Dobrev D, Friedrich A, Voigt N, et al. The G protein-gated potassium current IK,ACh is constitutively active in patients with chronic atrial fibrillation. Circulation. 2005;112:3697–3706. doi: 10.1161/CIRCULATIONAHA.105.575332. [DOI] [PubMed] [Google Scholar]
- 11.Gaborit N, Steenman M, Lamirault G, et al. Human atrial ion channel and transporter subunit gene-expression remodeling associated with valvular heart disease and atrial fibrillation. Circulation. 2005;112:471–481. doi: 10.1161/CIRCULATIONAHA.104.506857. [DOI] [PubMed] [Google Scholar]
- 12.Voigt N, Friedrich A, Bock M, et al. Differential phosphorylation-dependent regulation of constitutively active and muscarinic receptor-activated IK,ACh channels in patients with chronic atrial fibrillation. Cardiovasc Res. 2007;74:426–437. doi: 10.1016/j.cardiores.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 13.Workman AJ, Kane KA, Rankin AC. Characterisation of the Na, K pump current in atrial cells from patients with and without chronic atrial fibrillation. Cardiovasc Res. 2003;59:593–602. doi: 10.1016/s0008-6363(03)00466-8. [DOI] [PubMed] [Google Scholar]
- 14.Balana B, Dobrev D, Wettwer E, et al. Decreased ATP-sensitive K+ current density during chronic human atrial fibrillation. J Mol Cell Cardiol. 2003;35:1399–1405. doi: 10.1016/s0022-2828(03)00246-3. [DOI] [PubMed] [Google Scholar]
- 15.Wu G, Huang CX, Tang YH, et al. Changes of IK,ATP current density and allosteric modulation during chronic atrial fibrillation. Chin Med J. 2005;118:1161–1166. [PubMed] [Google Scholar]
- 16.Brandt MC, Priebe L, Bohle T, et al. The ultrarapid and the transient outward K+ current in human atrial fibrillation. Their possible role in postoperative atrial fibrillation. J Mol Cell Cardiol. 2000;32:1885–1896. doi: 10.1006/jmcc.2000.1221. [DOI] [PubMed] [Google Scholar]
- 17.Grammer JB, Bosch RF, Kuhlkamp V, et al. Molecular remodeling of Kv4.3 potassium channels in human atrial fibrillation. J Cardiovasc Electrophysiol. 2000;11:626–633. doi: 10.1111/j.1540-8167.2000.tb00024.x. [DOI] [PubMed] [Google Scholar]
- 18.Van Wagoner DR, Pond AL, Lamorgese M, et al. Atrial L-type Ca2+ currents and human atrial fibrillation. Circ Res. 1999;85:428–436. doi: 10.1161/01.res.85.5.428. [DOI] [PubMed] [Google Scholar]
- 19.Skasa M, Jungling E, Picht E, et al. L-type calcium currents in atrial myocytes from patients with persistent and non-persistent atrial fibrillation. Basic Res Cardiol. 2001;96:151–159. doi: 10.1007/s003950170065. [DOI] [PubMed] [Google Scholar]
- 20.Christ T, Boknik P, Wohrl S, et al. L-type Ca2+ current downregulation in chronic human atrial fibrillation is associated with increased activity of protein phosphatases. Circulation. 2004;110:2651–2657. doi: 10.1161/01.CIR.0000145659.80212.6A. [DOI] [PubMed] [Google Scholar]
- 21.Greiser M, Halaszovich CR, Frechen D, et al. Pharmacological evidence for altered src kinase regulation of ICa,L in patients with chronic atrial fibrillation. Naunyn-Schmiedeberg’s Arch Pharmacol. 2007;375:383–392. doi: 10.1007/s00210-007-0174-6. [DOI] [PubMed] [Google Scholar]
- 22.Workman AJ, Pau D, Redpath CJ, et al. Post-operative atrial fibrillation is influenced by beta-blocker therapy but not by pre-operative atrial cellular electrophysiology. J Cardiovasc Electrophysiol. 2006;17:1230–1238. doi: 10.1111/j.1540-8167.2006.00592.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koumi S, Arentzen CE, Backer CL, et al. Alterations in muscarinic K+ channel response to acetylcholine and to G protein-mediated activation in atrial myocytes isolated from failing human hearts. Circulation. 1994;90:2213–2224. doi: 10.1161/01.cir.90.5.2213. [DOI] [PubMed] [Google Scholar]
- 24.Koumi S, Martin RL, Sato R. Alterations in ATP-sensitive potassium channel sensitivity to ATP in failing human hearts. Am J Physiol. 1997;272:H1656–H1665. doi: 10.1152/ajpheart.1997.272.4.H1656. [DOI] [PubMed] [Google Scholar]
- 25.Schreieck J, Wang YG, Kalra B, et al. Differential rate dependence of action potentials, calcium inward and transient outward current in atrial myocytes of patients with and without heart failure. Circulation. 1998;98:611. Abstract. [Google Scholar]
- 26.Schreieck J, Wang Y, Overbeck M, et al. Altered transient outward current in human atrial myocytes of patients with reduced left ventricular function. J Cardiovasc Electrophysiol. 2000;11:180–192. doi: 10.1111/j.1540-8167.2000.tb00318.x. [DOI] [PubMed] [Google Scholar]
- 27.Le Grand B, Hatem S, Deroubaix E, et al. Depressed transient outward and calcium currents in dilated human atria. Cardiovasc Res. 1994;28:548–556. doi: 10.1093/cvr/28.4.548. [DOI] [PubMed] [Google Scholar]
- 28.Mansourati J, Le Grand B. Transient outward current in young and adult diseased human atria. Am J Physiol. 1993;265:H1466–H1470. doi: 10.1152/ajpheart.1993.265.4.H1466. [DOI] [PubMed] [Google Scholar]
- 29.Ouadid H, Albat B, Nargeot J. Calcium currents in diseased human cardiac cells. J Cardiovasc Pharmacol. 1995;25:282–291. doi: 10.1097/00005344-199502000-00014. [DOI] [PubMed] [Google Scholar]
- 30.Cheng TH, Lee FY, Wei J, et al. Comparison of calcium-current in isolated atrial myocytes from failing and nonfailing human hearts. Mol Cell Biochem. 1996;157:157–162. doi: 10.1007/BF00227894. [DOI] [PubMed] [Google Scholar]
- 31.Workman AJ, Kane KA, Russell JA, et al. Chronic beta-adrenoceptor blockade and human atrial cell electrophysiology: evidence of pharmacological remodelling. Cardiovasc Res. 2003;58:518–525. doi: 10.1016/s0008-6363(03)00263-3. [DOI] [PubMed] [Google Scholar]
- 32.Marshall G, Rankin AC, Kane KA, et al. Pharmacological remodelling of human atrial K+ currents by chronic beta-blockade. Eur Heart J. 2006;27:30. Abstract. [Google Scholar]
- 33.Pau D, Workman AJ, Kane KA, et al. Electrophysiological effects of 5-hydroxytryptamine on isolated human atrial myocytes, and the influence of chronic β-adrenoceptor blockade. Br J Pharmacol. 2003;140:1434–1441. doi: 10.1038/sj.bjp.0705553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang H, Garratt CJ, Zhu J, et al. Role of up-regulation of IK1 in action potential shortening associated with atrial fibrillation in humans. Cardiovasc Res. 2005;66:493–502. doi: 10.1016/j.cardiores.2005.01.020. [DOI] [PubMed] [Google Scholar]
- 35.Klein G, Schroder F, Vogler D, et al. Increased open probability of single cardiac L-type calcium channels in patients with chronic atrial fibrillation: role of phosphatase 2A. Cardiovasc Res. 2003;59:37–45. doi: 10.1016/s0008-6363(03)00357-2. [DOI] [PubMed] [Google Scholar]
- 36.Cha TJ, Ehrlich JR, Zhang L, et al. Atrial tachycardia remodeling of pulmonary vein cardiomyocytes: comparison with left atrium and potential relation to arrhythmogenesis. Circulation. 2005;111:728–735. doi: 10.1161/01.CIR.0000155240.05251.D0. [DOI] [PubMed] [Google Scholar]
- 37.Ehrlich JR, Cha TJ, Zhang L, et al. Characterization of a hyperpolarization-activated time-dependent potassium current in canine cardiomyocytes from pulmonary vein myocardial sleeves and left atrium. J Physiol. 2004;557:583–597. doi: 10.1113/jphysiol.2004.061119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kneller J, Sun H, Leblanc N, et al. Remodeling of Ca2+-handling by atrial tachycardia: evidence for a role in loss of rate-adaptation. Cardiovasc Res. 2002;54:416–426. doi: 10.1016/s0008-6363(02)00274-2. [DOI] [PubMed] [Google Scholar]
- 39.Hove-Madsen L, Llach A, Bayes-Genis A, et al. Atrial fibrillation is associated with increased spontaneous calcium release from the sarcoplasmic reticulum in human atrial myocytes. Circulation. 2004;110:1358–1363. doi: 10.1161/01.CIR.0000141296.59876.87. [DOI] [PubMed] [Google Scholar]
- 40.Vest JA, Wehrens XHT, Reiken SR, et al. Defective cardiac ryanodine receptor regulation during atrial fibrillation. Circulation. 2005;111:2025–2032. doi: 10.1161/01.CIR.0000162461.67140.4C. [DOI] [PubMed] [Google Scholar]
- 41.Pandit SV, Berenfeld O, Anumonwo JMB, et al. Ionic determinants of functional reentry in a 2-D model of human atrial cells during simulated chronic atrial fibrillation. Biophys J. 2005;88:3806–3821. doi: 10.1529/biophysj.105.060459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brundel BJJM, Van Gelder IC, Henning RH, et al. Gene expression of proteins influencing the calcium homeostasis in patients with persistent and paroxysmal atrial fibrillation. Cardiovasc Res. 1999;42:443–454. doi: 10.1016/s0008-6363(99)00045-0. [DOI] [PubMed] [Google Scholar]
- 43.Brundel BJJM, Van Gelder IC, Henning RH, et al. Ion channel remodeling is related to intraoperative atrial effective refractory periods in patients with paroxysmal and persistent atrial fibrillation. Circulation. 2001;103:684–690. doi: 10.1161/01.cir.103.5.684. [DOI] [PubMed] [Google Scholar]
- 44.Schotten U, Haase H, Frechen D, et al. The L-type Ca2+-channel subunits α1C and β2 are not downregulated in atrial myocardium of patients with chronic atrial fibrillation. J Mol Cell Cardiol. 2003;35:437–443. doi: 10.1016/s0022-2828(03)00012-9. [DOI] [PubMed] [Google Scholar]
- 45.Fatkin D, Otway R, Vandenberg JI. Genes and atrial fibrillation: a new look at an old problem. Circulation. 2007;116:782–792. doi: 10.1161/CIRCULATIONAHA.106.688889. [DOI] [PubMed] [Google Scholar]
- 46.Cha TJ, Ehrlich JR, Zhang L, et al. Dissociation between ionic remodeling and ability to sustain atrial fibrillation during recovery from experimental congestive heart failure. Circulation. 2004;109:412–418. doi: 10.1161/01.CIR.0000109501.47603.0C. [DOI] [PubMed] [Google Scholar]
- 47.Cha TJ, Ehrlich JR, Zhang L, et al. Atrial ionic remodeling induced by atrial tachycardia in the presence of congestive heart failure. Circulation. 2004;110:1520–1526. doi: 10.1161/01.CIR.0000142052.03565.87. [DOI] [PubMed] [Google Scholar]
- 48.Raine AEG, Vaughan Williams EM. Adaptation to prolonged β-blockade of rabbit atrial, Purkinje, and ventricular potentials, and of papillary muscle contraction. Time-course of development of and recovery from adaptation. Circ Res. 1981;48:804–812. doi: 10.1161/01.res.48.6.804. [DOI] [PubMed] [Google Scholar]
- 49.Marshall GE, Tellez JO, Russell JA, et al. Reduction of human atrial ITO by chronic beta blockade is not due to changes in ion channel expression. Proc Physiol Soc. 2007;8:PC20. Abstract. [Google Scholar]