

Abstract
Immune homeostasis in tissues is achieved through a delicate balance between pathogenic T cell responses directed at tissue-specific antigens and the ability of the tissue to inhibit these responses. The mechanisms by which tissues and the immune system communicate to establish and maintain immune homeostasis are currently unknown. Clinical evidence suggests that chronic or repeated exposure to self antigen within tissues leads to an attenuation of pathologic autoimmune responses, possibly as a means to mitigate inflammatory damage and preserve function. Many human organ-specific autoimmune diseases are characterized by the initial presentation of the disease being the most severe, with subsequent flares being of lesser severity and duration1. In fact, these diseases often spontaneously resolve, despite persistent tissue autoantigen expression2. In the practice of antigen-specific immunotherapy (antigen-SIT), allergens or self antigens are repeatedly injected in the skin, with a diminution of the inflammatory response occurring after each successive exposure3. Although these findings suggest that tissues acquire the ability to attenuate autoimmune reactions upon repeated responses to antigens, the mechanism by which this occurs is unknown. Here we show that upon expression of self antigen in a peripheral tissue, thymus-derived regulatory T cells (Treg cells) become activated, proliferate and differentiate into more potent suppressors, which mediate resolution of organ-specific autoimmunity. After resolution of the inflammatory response, activated Treg cells are maintained in the target tissue and are primed to attenuate subsequent autoimmune reactions when antigen is re-expressed. Thus, Treg cells function to confer ‘regulatory memory’ to the target tissue. These findings provide a framework for understanding how Treg cells respond when exposed to self antigen in peripheral tissues and offer mechanistic insight into how tissues regulate autoimmunity.
We hypothesized that exposure of immune cells to self antigen in peripheral tissues induces stable regulatory mechanisms that limit autoimmune injury. To test this and to define the nature of these control mechanisms, we created a novel mouse model of inducible tissue-specific self antigen expression. We crossed transgenic mice expressing a membrane-bound form of Ovalbumin (Ova) under the control of a tetracycline response element to transgenic mice expressing the tetracycline transactivator protein under the control of the keratin 5 promoter (Fig. 1a)4. In the resultant K5/TGO double transgenic mice, Ova expression in the skin is tightly controlled, as adoptively transferred Ova-specific (DO11.10) CD4+ T cells become activated and proliferate in skin-draining lymph nodes (SDLNs) of recipient mice only after treatment with doxycycline (Fig. 1b). In addition, Ova mRNA is detected in epidermal cell suspensions only after induction with doxycycline (Supplementary Fig. 1).
Figure 1. Characterization of K5/TGO/DO11 mice.
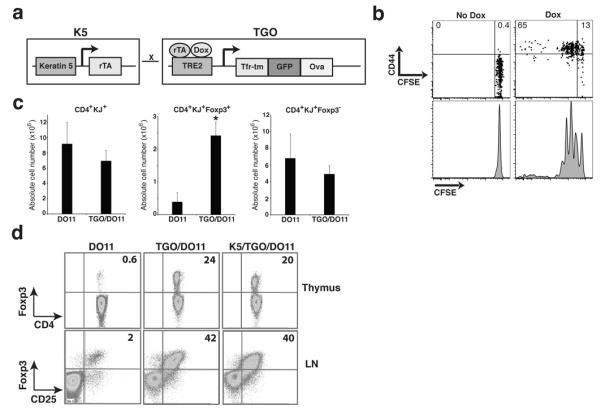
(a) Construct for double transgenic mice expressing ovalbumin (Ova) driven by the cytokeratin-5 (K5) promoter in a tetracycline-inducible fashion. (b) Lymph node cells from DO11 TCR-transgenic mice were labeled with CFSE and injected into K5/TGO mice. Recipient mice were fed doxycycline chow and DO11 cell proliferation (CFSE dilution) and CD44 expression was measured 3 days later by flow cytometry. (c) SDLN cell numbers from DO11 and TGO/DO11 mice in the absence of doxycycline treatment. (d) Thymus and SDLN cells from DO11, TGO/DO11, and K5/TGO/DO11 mice in the absence of doxycycline treatment. Thymocytes are gated on CD4+KJ+CD8− cells. Lymph node cells are gated on CD4+KJ+ cells. *P < 0.05 (t-test). Error bars represent s.d. Results are representative of 3 replicate experiments with n=3-4 mice/group.
To define functional Ova expression in the thymus, K5/TGO mice were crossed with the DO11.10 T cell receptor (TCR)-transgenic strain5. Deletion of CD4+ DO11 T cells or increased production of Foxp3+CD4+DO11 Treg cells in K5/TGO/DO11 mice is a sensitive indicator of thymic Ova expression. K5/TGO/DO11 triple transgenic mice as well as TGO/DO11 double transgenic mice have modest deletion of CD4+ DO11 cells (Fig. 1c) and a pronounced increase in DO11 Treg cells in both the thymus and SDLNs (Fig. 1c, d). Between 30-40% of antigen-specific CD4+ DO11 T cells in the SDLNs of K5/TGO/DO11 (and TGO/DO11 mice) co-express CD25 and Foxp3 (Fig. 1d). These results demonstrate that Ova is constitutively expressed in the thymus, independent of doxycycline treatment and dependent only on the presence of the TGO transgene. Thus, K5/TGO/DO11 mice represent a unique model in which antigen is continuously expressed in the thymus and tightly controlled in the periphery, mimicking the pattern of tissue-specific self antigen expression in mice and humans6,7
Despite the presence of a large percentage of Ova-specific Treg cells, induction of cutaneous Ova expression in K5/TGO/DO11 mice results in a pronounced inflammatory dermatitis (Fig. 2a). Disease peaks at 10-14 days after antigen induction and is characterized by marked erythema, scaling and alopecia (Fig. 2b). The skin infiltrate at the height of disease is composed primarily of antigen-specific DO11 T cells and GR-1+CD11b+ myeloid cells (Fig. 2c). Ova-specific skin-infiltrating CD4+ T cells produce IFNγ and IL-17 (Fig 2j).
Figure 2. K5/TGO/DO11 mice develop autoimmune skin disease that resolves spontaneously.

(a) Mean clinical scores of K5/TGO/DO11 mice left untreated or fed doxycycline chow. (b) Skin lesions in a representative K5/TGO/DO11 mouse at the height of clinical disease. (c) Flow cytometry of skin-infiltrating cells in K5/TGO/DO11 mice treated with doxycycline for 12 days. (d) Representative mice from the height of disease (Day 12) and after disease resolution (Day 86). (e) Skin histology of K5/TGO/DO11 mice after beginning doxycycline. (f) Flow cytometry of skin-infiltrating cells in K5/TGO/DO11 mice at 11 and 43 days after beginning doxycycline. (g) Disease scores of K5/TGO/DO11 mice treated with anti-CD25 monoclonal antibody (PC61) or control (PBS or isotype control antibody) at 10 days and 3 days prior to beginning doxycycline. (h) Flow cytometry of skin-infiltrating cells in K5/TGO/DO11 mice untreated or treated with PC61 at 23 days after beginning doxycycline. (i) Disease scores of K5/TGO/DO11 mice treated with PC61 or isotype control antibody at the height of disease. Results are pooled data from two replicate experiments with n=3-4 mice/group. (j) Intracellular cytokine stains of SDLN cells and skin-infiltrating cells from PC61- or isotype control-treated (prior to antigen induction) K5/TGO/DO11 mice at 11 days after beginning doxycycline. Gated on CD4+KJ+ cells. Error bars represent s.d. Results are representative of 3 replicate experiments with n=3-4 mice/group. Ŧ denotes euthanization of two PC61-treated mice secondary to non-resolving skin disease.
Interestingly, skin inflammation spontaneously resolves. Twenty to 30 days after antigen induction, mice begin to show signs of clinical and histological improvement, and by 40 to 60 days, there is complete resolution of cutaneous inflammation (Fig. 2d-f). Disease resolves despite continued doxycycline treatment, with ongoing Ova expression in the skin and continuous thymic output of Ova-specific T cells (Supplementary Fig. 2).
Given the large percentage of antigen-specific Treg cells present in the SDLNs of K5/TGO/DO11 mice, it is surprising that cutaneous inflammation results when antigen is induced. We were further intrigued by the spontaneous resolving nature of the autoimmune response, as it suggests that an immune regulatory mechanism(s) is initiated in an attempt to minimize tissue damage. To determine if Treg cells play a role in resolving skin inflammation, we depleted these cells prior to antigen induction and at the height of disease. Treatment of K5/TGO/DO11 mice with CD25-depleting antibody (PC61) prior to antigen induction resulted in pronounced disease exacerbation with delayed resolution of skin inflammation (Fig. 2g, h). In some experiments, depletion of Treg cells resulted in dehydration, weight loss and death, most likely from severely disrupted skin barrier function (Supplementary Fig. 3). Depletion of Treg cells at the height of disease (i.e., 10 days after antigen induction) resulted in delayed kinetics of resolution of skin inflammation, with some mice having to be euthanized secondary to non-resolving skin disease (Fig. 2i). Higher percentages of both IFNγ- and IL-17-secreting DO11 cells were observed in the skin and SDLN following Treg cell depletion (Fig. 2j). Anti-CD25 antibody was utilized in these studies because CD25 is constitutively expressed on Treg cells and antibody treatment can be accurately titrated to achieve optimal deletion of these cells (Supplementary Fig. 4). A caveat of this approach is that other CD25-expressing cells (which may be either tolerance-promoting or pathogenic) might also be deleted. The finding that anti-CD25 antibody treatment inhibits or prevents disease resolution strongly suggests that Treg cells are the major targets of the antibody and play an essential role in resolving skin disease in our model; however, depletion of other CD25-expressing regulatory populations cannot be definitively ruled out.
The emergence of disease despite the presence of Treg cells and the Treg cell-dependent suppression of disease after antigen induction suggested that expression of tissue antigen activates Treg cells and enhances their suppressive function. To test this, we induced Ova expression in K5/TGO/DO11 mice and characterized Treg cells in both the skin and SDLNs. Foxp3+ DO11 cells robustly expanded in SDLNs upon antigen induction (Fig. 3a). Expansion peaked at 12-14 days, when these cells were increased ~15-fold over baseline numbers. In contrast, the peak expansion of Foxp3− DO11 effector cells occurred earlier and these cells only expanded ~4-fold over baseline (Fig. 3a). At the peak of expansion, more than 50% of Foxp3+ DO11 cells had markedly reduced CD25 on the cell surface (Fig. 3b). Loss of CD25 was transient, associated only with the proliferative burst, as Foxp3+ DO11 cells in SDLNs during the resolution phase of disease (>20 days) express CD25 (Fig. 3b). Loss of CD25 expression on in vivo proliferating Treg cells (with preservation of suppressive capacity) has been reported by others8. It is possible that proliferating Tregs cells reduce expression of CD25 in a negative feedback loop to limit proliferation and regulate Treg cell numbers. SDLN Treg cells in the resolution phase of disease have increased expression of Foxp3 and CD25 when compared to Treg cells from non-induced mice (Fig. 3b and Supplementary Fig. 5). These results suggest that Treg cells are activated upon exposure to peripheral antigen. To further test this, we analyzed expression levels of various markers shown to be involved in Treg cell function early after antigen induction. Six days after inducing Ova expression in the skin, >80% of Treg cells have entered the cell cycle, as evidenced by Ki67 expression, and proliferating CD25+ and CD25− Treg cells express higher levels of CTLA-4 compared to Treg cells from non-induced mice (Fig. 3c).
Figure 3. Treg cells are activated upon induction of peripheral antigen.
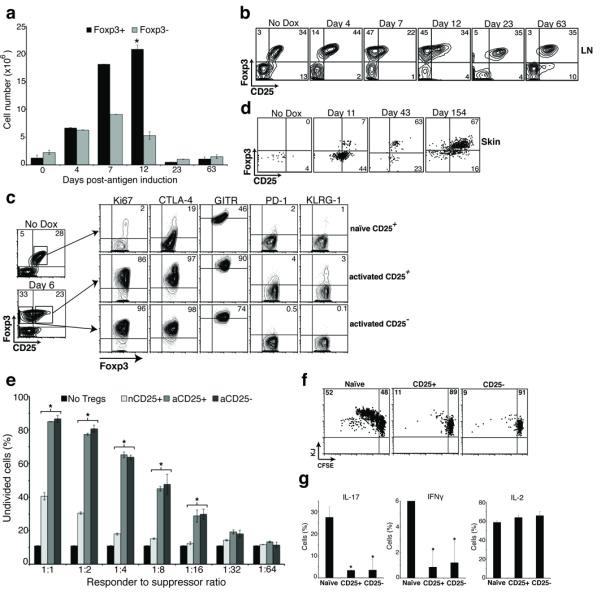
(a) Quantification of CD4+KJ+Foxp3+ and CD4+KJ+Foxp3− cell numbers from SDLNs of K5/TGO/DO11 mice after beginning doxycycline. (b) Flow cytometry of SDLN cells from K5/TGO/DO11 mice after beginning doxycycline. Gated on CD4+KJ+ cells. (c) Flow cytometry of SDLN cells from K5/TGO/DO11 mice left untreated (naïve CD25+) or treated for 6 days with doxycycline (activated CD25+ or CD25− populations). (d) Flow cytometry of skin-infiltrating cells from K5/TGO/DO11 mice after beginning doxycycline. Gated on live CD4+KJ+ cells. (e) In vitro suppression assay using sorted CD4+KJ+Foxp3+CD25+ cells from non-induced naïve mice (nCD25+), CD4+KJ+Foxp3+CD25+ cells from doxycycline-treated mice (activated CD25+, aCD25+), or CD4+KJ+Foxp3+CD25− cells from doxycycline-treated mice (aCD25−) as suppressors and CFSE-labeled DO11 LN cells as responders. (f) In vivo suppression assay using suppressors (as in part e) mixed 1:1 with naïve CFSE-labeled Thy1.1+ DO11 LN cells and adoptively transferred into K5/TGO mice. Recipient mice were treated with doxycycline and DO11 cell proliferation and intracellular cytokine expression (g) were assayed 3 days later. Gated on Thy1.1+CD4+KJ+ cells. *P < 0.05 (t-test). Error bars represent s.d. Results are representative of 3 replicate experiments with n=3-4 mice/group except for in vitro and in vivo suppression assays, which are representative of 2 replicate experiments with n=2-3 mice/group.
Expansion of Treg cells in SDLNs upon antigen induction is associated with an accumulation of these cells in the skin (Fig. 3d). Prior to antigen induction, CD4+ DO11 cells are barely detectable in the skin. At the height of disease, the majority of skin-infiltrating DO11 cells are Foxp3−. However, resolution of inflammation is associated with a preferential accumulation of Foxp3+ cells in the skin, with >60% of DO11 cells in the skin expressing Foxp3 in mice maintained on doxycycline that have resolved disease (i.e., clinically and histologically normal appearing skin) (Fig. 3d).
We next tested whether Treg cells acquire a more suppressive phenotype after exposure to tissue antigen. To do so, we crossed K5/TGO/DO11 mice with Foxp3GFP reporter mice9. K5/TGO/DO11/Foxp3GFP mice were treated with doxycycline to induce antigen expression in the skin, and 6 days later, CD4+Foxp3+CD25+ and CD4+Foxp3+CD25− DO11 cells were isolated from SDLNs and their suppressive capacity tested in vitro and in vivo. Both CD25+ and CD25− Treg cells from antigen-induced mice were more potent suppressors of effector T cell proliferation in vitro when compared to Treg cells isolated from non-induced mice (i.e., peripheral antigen naïve) at all effector to suppressor ratios examined (Fig. 3e). In addition, Treg cells from antigen-induced mice were more potent suppressors of cutaneous autoimmune responses in vivo. Upon co-transfer with naïve DO11 responder cells into K5/TGO double transgenic mice (and treatment of recipient mice with doxycycline to induce Ova expression in the skin), both CD25+ and CD25− Foxp3+ Treg cells isolated from antigen-induced K5/TGO/DO11/Foxp3GFP mice were more potent at inhibiting T cell proliferation when compared to Foxp3+ cells isolated from non-induced mice (Fig. 3f). In addition, these cells were also more potent inhibitors of IL-17 and IFNγ production from effector T cells (Fig. 3g).
The mechanism of enhanced Treg cell suppression after peripheral antigen induction may involve CTLA-4, which is expressed at markedly higher levels after activation of Treg cells (Fig. 3c) and has been shown to be a critical mediator of Treg function in suppressing tissue inflammation10. In addition, CD25+ and CD25− Treg cells had similarly high levels of CTLA-4 expression and are equally suppressive in vitro and in vivo. We were unable to detect cytokine production (including IL-10, IL-17 and IFNγ) from Treg cells before or after antigen induction, suggesting that a cytokine-mediated mechanism of suppression is unlikely (Fig. 2j and data not shown).
Taken together, our results indicate that early after exposure to tissue autoantigen, Treg cells are activated, proliferate, differentiate into more potent suppressors, and progressively accumulate in the target tissue. These results are consistent with recent findings that two major populations of Foxp3+ Treg cells exist in human peripheral blood, which have been called resting Treg cells (rTreg cells) and activated Treg cells (aTreg cells)11. Resting Treg cells are recently derived from the thymus, express low levels of CTLA-4, are not actively cycling, and are inferior suppressors when compared to aTreg cells. In contrast, aTreg cells are constitutively cycling, express higher levels of Foxp3 and CTLA-4 and are more potent suppressors. Longitudinal studies showed ongoing conversion of rTreg cells to aTreg cells in a healthy human volunteer, with T-cell receptor clonotypes initially detected in the rTreg cell subset found 18 months later in the aTreg cell subset. These results suggest that in normal healthy individuals, rTreg cells are continuously activated and proliferate into more potent aTreg cells. Taken together with results presented here, we speculate that exposure to tissue self antigen may drive this process as a mechanism to inhibit and/or suppress autoimmunity.
We hypothesized that once Treg cells have been activated and accumulate in the skin, these cells are relatively stable and persist in the tissue. They would then be capable of suppressing subsequent autoimmune reactions when antigen is re-expressed. To test this, we induced antigen expression in K5/TGO/DO11 mice and upon resolution of skin disease, removed doxycycline for >30 days to effectively ‘turn antigen off’ in the skin. We then re-induced antigen expression and followed mice clinically. To confirm the absence of antigen expression upon cessation of doxycycline, we assessed activation and proliferation of adoptively transferred DO11 cells and activation of endogenous DO11 cells in K5/TGO/DO11 mice (as a surrogate marker of antigen expression) at various times after discontinuing doxycycline. In addition, we measured Ova mRNA levels in the skin. At 20 days after discontinuing doxycycline there was no proliferation of adoptively transferred DO11 cells and by 29 days, CD44 and CD69 expression had returned to baseline levels on adoptively transferred and endogenous DO11 cells, respectively (Fig. 4a and Supplementary Fig. 6a). Expression of Ova mRNA was undetectable in all mice at 32 days after stopping doxycycline (Supplementary Fig. 6b). Taken together, these data confirm that antigen expression is effectively ‘turned off’ in all mice by 30 days after cessation of doxycycline. Analysis of DO11 T cells in the skin of K5/TGO/DO11 mice that had been off doxycycline for >30 days revealed a persistent Foxp3+ population comprising >50% of DO11 cells in the skin (Fig. 4b). The proportion of DO11 cells in the skin that were Foxp3+ was enriched relative to SDLNs, suggesting a preferential accumulation of Treg cells in the target tissue upon cessation of antigen expression (Fig. 4b). Furthermore, Treg cells in the skin continued to express high levels of CTLA-4 (Fig. 4c). When compared to Treg cells in the SDLN, cells that persisted in the skin (in the absence of antigen expression) expressed lower levels of CD25 and higher levels of CTLA-4, CD127, and KLRG-1 (Fig. 4d). The persistence of activated Treg cells in the skin suggested that there may be an attenuation of cutaneous inflammation upon antigen re-expression, as Treg cells must be present in the skin to suppress autoimmunity12. To test this, we re-started doxycycline and followed mice clinically. Upon antigen re-expression, K5/TGO/DO11 mice develop skin disease phenotypically similar to the disease that developed upon initial antigen expression; however, the severity of skin disease was significantly reduced and it resolved with accelerated kinetics (Fig. 4e). Attenuated disease upon antigen re-exposure was not secondary to reduced numbers of DO11 cells, as these cells returned to baseline levels relatively early after cessation of doxycycline (Supplementary Fig. 7). In addition, antigen-experienced DO11 T cells from K5/TGO/DO11 mice that had resolved disease and were either maintained on doxycycline or were taken off doxycycline after resolution readily induced skin disease when adoptively transferred into new antigen-expressing hosts (Supplementary Fig. 8). This suggests that cell-intrinsic anergy in responding T cells does not seem to play a major role in attenuating skin inflammation. To test whether reduced disease severity upon second antigen exposure was mediated by Treg cells, we depleted these cells prior to re-inducing antigen expression. Depletion of Treg cells resulted in a complete abrogation of the attenuated response observed upon re-expression of Ova in the skin (Fig. 4f).
Figure 4. Memory Treg cells attenuate skin disease upon re-expression of tissue antigen.
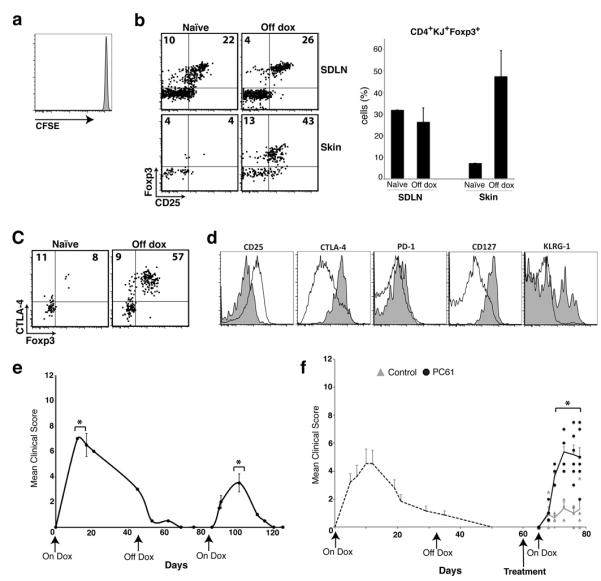
(a) Flow cytometry of CFSE-labeled Thy1.1+ DO11+ SDLN cells 3 days after adoptive transfer into K5/TGO/DO11 mice that had been on doxycycline for >30 days and off for 20 days. (b) Flow cytometry and Treg cell percentages of SDLN and skin-infiltrating CD4+DO11 cells isolated from K5/TGO/DO11 mice that were on doxycycline for >30 days and off for >30 days. Gated on CD4+KJ+ cells. (c) Flow cytometry of CD4+DO11 cells from the skin of K5/TGO/DO11 mice that have not been treated with doxycycline (naïve) or on for >30 days and off doxycycline for >30 days. (d) Phenotype of CD4+Foxp3+ DO11 cells from skin (shaded) and SDLN (unshaded) from K5/TGO/DO11 mice that were on doxycycline for >30 days and off for >40 days. (e) Clinical skin disease upon re-starting doxycycline in K5/TGO/DO11 mice that had been on doxycycline for >30 days and off for >30 days. (f) Clinical skin disease of K5/TGO/DO11 mice treated with either PC61 or isotype control antibody prior to re-starting doxycycline. In all mice, antigen was induced, skin disease developed, and disease had resolved before antibody treatment. Error bars represent standard error of samples within each group. Mean clinical scores from individual mice are shown. Results are representative of 3 replicate experiments with 3-4 mice/group except for (f) which is combined data from 2 replicate experiments with 2-4 mice/group. *P < 0.05 (t-test).
Our results suggest that exposure to tissue autoantigens leads to the activation of self-reactive Treg cells that were generated by self antigen expression in the thymus. Activated Treg cells persist in the target tissue and suppress autoimmune responses upon repeated or chronic encounters with tissue autoantigen. Thus, tissues that have undergone autoimmune inflammatory reactions develop a property we have termed regulatory memory, which serves to limit the severity of future such reactions. Memory Tregs have several characteristics of effector memory cells13,14, including survival without antigen, residence in non-lymphoid tissues, phenotypic markers suggestive of prior activation, and enhanced functional activity. The life history of Tregs is fundamentally similar to that of conventional T cells, passing through defined phases that include generation in the thymus, activation in the periphery leading to proliferation and differentiation into functionally more active cells, and survival as memory populations. Although our data support an obligatory role for Treg cells in suppressing primary and memory responses in the skin, it is possible that these cells work in concert with other, Treg cell-independent, mechanisms to suppress tissue-specific autoimmunity. Identifying and harnessing the network of regulatory pathways that serve to limit tissue inflammation will undoubtedly offer new strategies for controlling autoimmune reactions and preserving end-organ function.
METHODS SUMMARY
Mouse model
TRE-TGO mice were crossed with K5-rtTA mice to generate mice with inducible epidermal expression of Ova. The TGO construct encodes a fusion protein linking the transferrin receptor transmembrane domain, GFP, and aa 230–359 of chicken ovalbumin. To create K5/TGO/DO11 mice, K5-rTA/TRE-TGO mice were crossed onto the DO11.10 TCR-transgenic background. All mice were bred and maintained in a specific pathogen-free facility in accordance with the guidelines of the Laboratory Animal Resource Center of the University of California San Francisco.
Inflammatory skin disease model
To induce expression of the TGO transgene, K5/TGO/DO11 mice were maintained on 1gm/kg doxycycline chow. A 12-point clinical scoring scale was utilized to quantify skin disease. Scaling, alopecia, erythema, and level of activity were each given a score from 0 to 3. Individual scores were summed and mean scores per group are displayed.
Treg cell depletion
PC61 (anti–CD25 monoclonal antibody) or isotype control were injected intraperitoneally at 10- and 3-days prior to doxycycline treatment. For depletion at the height of disease, PC61 mAb was injected on days 10 and 11 after starting K5/TGO/DO11 mice on doxycycline. For experiments in which CD25+ cells were deleted prior to antigen re-expression, K5/TGO/DO11 mice that had been off doxycycline for >30 days were treated with PC61 at 7- and 3-days prior to restarting doxycycline.
In vitro and in vivo suppression assays
K5/TGO/DO11 mice were crossed to Foxp3-GFP transgenic mice. K5/TGO/DO11/Foxp3-GFP mice were started on doxycycline and six days later, Treg cells were isolated. For in vitro suppression assays, Treg cells were cultured with CFSE-labeled SDLN cells isolated from DO11.10+/Rag2−/−/Thy1.1+ mice and Ova peptide-pulsed BMDCs. For in vivo suppression assays, purified Treg cells were mixed 1:1 with CFSE-labeled DO11 effector cells from DO11.10+/Rag2−/−/Thy1.1+ mice. Cell mixtures were injected intravenously into K5/TGO mice and recipient mice were started on doxycycline.
METHODS
Mouse model
K5/rTA mice were generated as described4. The TGO construct encodes for a fusion protein linking the transferrin receptor transmembrane domain (Tfr-tm), GFP, and aa 230–359 of chicken ovalbumin (Ova)15. TGO was cloned upstream of the tetracycline response element (TRE2) and transgenic mice with stable incorporation of the TRE-TGO construct were generated. GFP is expressed at levels too low to detect in these mice. TRE-TGO mice were crossed with K5-rtTA mice to generate mice with inducible epidermal expression of Ova. To create K5/TGO/DO11 mice, K5-rTA/TRE-TGO mice were crossed onto the DO11.10 TCR transgenic background, which express Ova-specific CD4+ T cells.5 KJ1-26 monoclonal antibody (KJ) specifically recognizes the DO11 TCR. All mice were bred and maintained in a specific pathogen-free facility in accordance with the guidelines of the Laboratory Animal Resource Center of the University of California San Francisco.
Treg cell depletion
Anti–CD25 monoclonal antibody (PC61) preferentially depletes Treg cells16, especially in the Balb/c strain17. PC61 or isotype control (UCSF Monoclonal Antibody Core, CA) were injected (0.5 mg/mouse) intraperitoneally at 10- and 3-days prior to doxycycline treatment. PC61 treatment of TGO/DO11 mice results in a >90% reduction of CD4+CD25+Foxp3+ DO11 cells at 7 days after injection (Supplementary Fig. 4). For depletion of CD25+ cells at the height of disease, PC61 mAb (0.5 mg/mouse) was injected on days 10 and 11 after starting K5/TGO/DO11 mice on doxycycline chow. For experiments in which CD25+ cells were deleted prior to antigen re-expression, K5/TGO/DO11 mice that had been off doxycycline for >30 days were treated with PC61 at 7- and 3-days prior to restarting doxycycline.
Inflammatory skin disease model
To induce expression of the TGO transgene in the skin, K5/TGO/DO11 mice were maintained on 1gm/kg doxycycline chow (Bio-Serv, Frenchtown, NJ). A 12-point clinical scoring scale was utilized to quantify skin disease. The clinical parameters of scaling, alopecia, erythema, and level of activity were each given a score from 0 to 3. Scores for individual parameters were summed, and each mouse was assigned a clinical severity score out of 12. Individual mouse scores were averaged to get mean clinical scores per group.
In vitro and in vivo suppression assays
K5/TGO/DO11 mice were crossed to Foxp3-GFP transgenic mice9, kindly provided by A. Rudensky. K5/TGO/DO11/Foxp3-GFP mice were started on doxycycline chow or left untreated and six days later, SDLN cells were isolated and purified by FACS. For in vitro suppression assays, cells were cultured in varying ratios with freshly isolated CFSE-labeled SDLN cells isolated from DO11.10+/Rag2−/−/Thy1.1+ mice and Ova peptide-pulsed BMDCs. For in vivo suppression assays, FACS purified Treg cells were mixed 1:1 with CFSE-labeled DO11 effector cells from DO11.10+/Rag2−/−/Thy1.1+ mice. Cell mixtures were injected intravenously into K5/TGO mice and recipient mice were started on doxycycline chow. Three days later, SDLN cells were isolated and CFSE-dilution and intracellular cytokine expression by DO11.10+/Thy1.1+ cells was quantified by flow cytometry.
Supplementary Material
Acknowledgements
We thank Carlos Benetiz for assistance with animal husbandry, Sara Isakson for genotyping, Shu-wei Jiang and Mike Lee for cell sorting, and Dr. Katya Ravid and Greg Martin for derivation of TRE-TGO transgenic mice. We thank Dr. Steve Ziegler, Benaroya Research Institute, for transgenic mice. M.D.R. is supported by a Dermatology Foundation Career Development Award and the UCSF Department of Dermatology. This work was partially funded through NIH grants P01 AI35297, R01 AI73656, and U19 AI56388 (to A.K.A.); NIH grant AR055634 to (A.M.R.); and the Scleroderma Research Foundation (A.M.R.). I.K.G is supported by an Erwin Schroedinger Fellowship from the Austrian Science Fund (FWF), J2997-B13.
Footnotes
Supplementary Information available online
Author Contributions M.D.R. and I.K.G. contributed equally to this work and designed the studies, performed the experiments, and analyzed the data. M.D.R. and A.K.A wrote the manuscript. J.P. collected and analyzed data as well as helped with mouse husbandry. K.L. engineered and derived the TRE-TGO mice in the laboratory of A. M-R. A.K.A. oversaw all study design and data analysis. A.M-R. was involved in study design and data analysis. All authors discussed results and commented on the manuscript.
REFERENCES
- 1.James WD. Andrews’ Diseases of the Skin: Clinical Dermatology. Saunders Elsevier; Philadelphia: 2006. [Google Scholar]
- 2.Lara-Corrales I, Pope E. Autoimmune blistering diseases in children. Semin Cutan Med Surg. 2010;29:85–91. doi: 10.1016/j.sder.2010.03.005. [DOI] [PubMed] [Google Scholar]
- 3.Sabatos-Peyton CA, Verhagen J, Wraith DC. Antigen-specific immunotherapy of autoimmune and allergic diseases. Curr. Opin. Immunol. 2010;22:609–615. doi: 10.1016/j.coi.2010.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Diamond I, Owolabi T, Marco M, Lam C, Glick A. Conditional gene expression in the epidermis of transgenic mice using the tetracycline-regulated transactivators tTA and rTA linked to the keratin 5 promoter. J. Invest. Dermatol. 2000;115:788–794. doi: 10.1046/j.1523-1747.2000.00144.x. [DOI] [PubMed] [Google Scholar]
- 5.Murphy KM, Heimberger AB, Loh DY. Induction by antigen of intrathymic apoptosis of CD4+CD8+TCRlo thymocytes in vivo. Science. 1990;250:1720–1723. doi: 10.1126/science.2125367. [DOI] [PubMed] [Google Scholar]
- 6.Wada N, et al. Aire-dependent thymic expression of desmoglein 3, the autoantigen in pemphigus vulgaris, and its role in T-cell tolerance. J. Invest. Dermatol. 2011;131:410–417. doi: 10.1038/jid.2010.330. [DOI] [PubMed] [Google Scholar]
- 7.Mouquet H, et al. Expression of pemphigus-autoantigen desmoglein 1 in human thymus. Tissue Antigens. 2008;71:464–470. doi: 10.1111/j.1399-0039.2008.01020.x. [DOI] [PubMed] [Google Scholar]
- 8.Gavin MA, Clarke SR, Negrou E, Gallegos A, Rudensky A. Homeostasis and anergy of CD4(+)CD25(+) suppressor T cells in vivo. Nat. Immunol. 2002;3:33–41. doi: 10.1038/ni743. [DOI] [PubMed] [Google Scholar]
- 9.Fontenot JD, et al. Regulatory T Cell Lineage Specification by the Forkhead Transcription Factor Foxp3. Immunity. 2005;22:329–341. doi: 10.1016/j.immuni.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 10.Wing K, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science. 2008;322:271–275. doi: 10.1126/science.1160062. [DOI] [PubMed] [Google Scholar]
- 11.Miyara M, et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity. 2009;30:899–911. doi: 10.1016/j.immuni.2009.03.019. [DOI] [PubMed] [Google Scholar]
- 12.Dudda JC, Perdue N, Bachtanian E, Campbell DJ. Foxp3+ regulatory T cells maintain immune homeostasis in the skin. J. Exp. Med. 2008;205:1559–1565. doi: 10.1084/jem.20072594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kurtulus S, Tripathi P, Opferman JT, Hildeman DA. Contracting the “mus cells”--does down-sizing suit us for diving into the memory pool? Immunol. Rev. 2010;236:54–67. doi: 10.1111/j.1600-065X.2010.00920.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Akbar AN, Vukmanovic-Stejic M, Taams LS, Macallan DC. The dynamic co-evolution of memory and regulatory CD4+ T cells in the periphery. Nat. Rev. Immunol. 2007;7:231–237. doi: 10.1038/nri2037. [DOI] [PubMed] [Google Scholar]
- 15.Saff RR, Spanjaard ES, Hohlbaum AM, Marshak-Rothstein A. Activation-induced cell death limits effector function of CD4 tumor-specific T cells. J. Immunol. 2004;172:6598–6606. doi: 10.4049/jimmunol.172.11.6598. [DOI] [PubMed] [Google Scholar]
- 16.Setiady YY, Coccia JA, Park PU. In vivo depletion of CD4+FOXP3+ Treg cells by the PC61 anti-CD25 monoclonal antibody is mediated by FcgammaRIII+ phagocytes. Eur. J. Immunol. 2010;40:780–786. doi: 10.1002/eji.200939613. [DOI] [PubMed] [Google Scholar]
- 17.Tenorio EP, Fernández J, Olguín JE, Saavedra R. Depletion with PC61 mAb before Toxoplasma gondii infection eliminates mainly Tregs in BALB/c mice, but activated cells in C57BL/6J mice. FEMS Immunol. Med. Microbiol. 2011;62:362–367. doi: 10.1111/j.1574-695X.2011.00805.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.