

Abstract
Objectives
Establish role and effects of glucocorticoids and the endogenous Annexin A1 (AnxA1) pathway in inflammatory arthritis.
Methods
Ankle joint mRNA and protein expression of AnxA1 and its receptors were analysed in naïve and arthritic mice, by real time PCR and immunohistochemistry. Inflammatory arthritis was induced with the K/BxN arthritogenic serum in AnxA1+/+ and AnxA1−/− mice; in some experiments, animals were treated with dexamethasone (Dex) or with human recombinant AnxA1 or a protease-resistant mutant (termed SuperAnxA1). Readouts were arthritic score, disease incidence, paw oedema and histopathology, together with proinflammatory gene expression.
Results
All elements of the AnxA1 pathway could be detected in naïve joints, with augmentation during on-going disease, due to the infiltration of immune cells. No difference in arthritis intensity of profile could be observed between AnxA1+/+ and AnxA1−/− mice. Treatment of mice with Dex (10 μg i.p. daily from day 2) afforded potent anti-arthritic effects highly attenuated in the knockouts: macroscopic changes were mirrored by histopathological findings and proinflammatory gene (e.g. Nos2) expression. Presence of proteinase 3 mRNA in the arthritic joints led us to test AnxA1 and the mutant SuperAnxA1 (1 μg i.p. daily in both cases; from day 2), with the latter one being able to accelerate the resolving phase of the disease.
Conclusion
AnxA1 is an endogenous determinant for the therapeutic efficacy of Dex in inflammatory arthritis. Such an effect can be partially mimicked by application of SuperAnxA1 which may represent the starting point for novel anti-arthritic therapeutic strategies.
Keywords: K/BxN arthritis, Glucocorticoids, Dexamethasone, Neutrophils, Resolution
INTRODUCTION
There is genuine interest in detailing cellular and molecular events regulating the phase of the resolution of inflammation, with the characterisation of specific players and targets.[1] This interest stems from the appreciation that, in ideal settings, a strong pro-inflammatory response, orchestrated by cytokines, adhesion molecules and chemoattractants[2] must be followed by an anti-inflammatory and pro-resolving phase, which assures spatial and timely control of the response the host organises upon encounter with an insult,[3] leading to resolution and regain of tissue functions.
Annexin A1 is an effector of resolution.[4] Highly expressed in immune cells (e.g. polymorphonuclear cells [PMN] and macrophages), this protein is externalised to exert paracrine and juxtacrine effects, the vast majority of which are mediated by the formyl-peptide receptor type 2, (FPR2/ALX or Fpr2, in rodents).[5] Intriguingly, FPR2/ALX is also the lipoxin A4 receptor[6] indicating the existence of important – yet not fully appreciated - networks in resolution.[7] Another receptor also advocated to mediate the effects of AnxA1, the formyl-peptide receptor type 1 or FPR1 (Fpr1 in rodents),[8] though it is unclear if and how this receptor binds the full-length protein[9] or whether - more selectively – it would mediate effects of the AnxA1 peptidomimetic peptide Ac2-26.[10]
The majority of experimental approaches used to define properties of proresolving mediators have been reliant on models of acute inflammation, characterised by rapid PMN influx followed by inflammatory monocytes which differentiate into tissue macrophages[3, 11] to phagocytose debris and apoptotic cells and favour tissue repair, a conclusive act in resolution that may require re-epitheliation[12] or a shift in fibroblast phenotype[13]. In this context, the proresolving nature of AnxA1 has been demonstrated in a variety of experimental settings, noting induction of apoptosis, promotion of efferocytosis and tissue repair, spanning from models of acute inflammation (e.g. see[14, 15]) to a model of colitis where delayed resolution was observed in AnxA1−/− mice.[16] An important aspect of AnxA1 biology is its second messenger role in the actions of glucocorticoids, a function mainly addressed in cellular settings [e.g.[17]] or in models of acute inflammation.[18, 19] However, Morand’s group applied antigen-induced arthritis to observe reduction afforded by Dex on synovitis, soft tissue inflammation and cartilage erosion was attenuated in AnxA1−/− mice.[20]
The K/BxN serum induced model of inflammatory arthritis is ideal to investigate the impact of specific proresolving pathways. Using this model of active phase of rheumatoid arthritis, Kronke et al reported a higher degree of joint inflammation in 12/15-lipoxygenase−/− mice, associated to their inability to synthesise lipoxin A4.[21] Similarly, animals nullified for proresolving melanocortin receptor type 3 displayed higher degree of arthritis and delayed resolution, accompanied by augmented cytokine expression and osteoclast responsiveness.[22] An equally interesting study investigated the effect of absence of glucocorticoid signal in osteoblasts, by means of restricted transgenic expression of 11β-hydroxysteroid dehydrogenase type 2, in this model of inflammatory arthritis. The outcome was that of a more attenuated arthritis in the transgenic mice when assessed macroscopically and histologically, selectively in the delayed phase, after peak disease[23].
The present study made use of the serum-induced model of inflammatory arthritis to: i) monitor potential modulation of endogenous AnxA1, Fpr1 and Fpr2; ii) determine the effect of Dex in wild type and AnxA1−/− mice and; iii) establish the pharmacological potential of exogenously administered AnxA1.
MATERIALS AND METHODS
Animals
BALB/c male 8-week old wild type AnxA1+/+ and AnxA1−/− mice[24] were bred at Charles River (Kent, UK). All experiments were approved and performed under the guidelines of the Ethical Committee for the Use of Animals, Barts and the London School of Medicine and Home Office regulations (Scientific Procedures Act, 1986).
Model of Inflammatory Arthritis
Arthritis induction
Mice received either 50+50 μl (day 0 and 2) or 200 μl (day 0) of arthritogenic K/BxN serum.[22] Disease was monitored by assessing the clinical score where a maximum of 12 points could be given per animal: 0, no evidence of inflammation; 1 point, subtle inflammation on metatarsal phalanges joints, individual phalanx; 2 points, swelling on dorsal or ventral surface of paw; 3 points, major swelling on all aspects of paw.[25] Paw volume was assessed by water plethysmometry (Ugo Basile, Milan, Italy).
Pharmacological intervention
Mice were injected intraperitoneally with Dex (disodium salt; Sigma, Dorset, UK) prepared in sterile PBS (Sigma) and given at a dose-range of 3-30 μg once daily after disease onset (from day 2). AnxA1 and SuperAnxA1 were generated as recently described[26] and administered intraperitoneally, from day 2, at 1 μg per mouse.[26]
Histology and Immunohistochemistry
Histology
Joints were decalcified and paraffin embedded. Sections (4 micrometers) were stained with haematoxylin & eosin (H&E) or SafraninO with fast green counterstain. Standard light microscopy was used to determine the degree of synovitis, pannus formation, cartilage and bone erosion which were graded from 0 (no disease) to 5 (severe) by two blinded examiners, as reported.[22]
Immunohistochemistry
An antigen retrieval step using citrate buffer (pH6.0) was performed and then sections were incubated with rabbit polyclonal anti-AnxA1 (1:1000) (Zymed Laboratories, Cambridge, UK) or anti-FPR2 (1:4000).[27] After washing and the secondary biotinylated antibody (Dako, Cambridge, UK), staining was detected using a peroxidase conjugated streptavidin complex (Dako) and colour developed using DAB substrate (Sigma).
Molecular Analyses
Ankle joints were snap frozen in liquid nitrogen and homogenised in Qiagen RLT plus buffer (Qiagen, Crawley, UK) using Precellys24 ceramic bead homogenisation technology (Bertin Technologies, Montigny-le-Bretonneux, France). RNA was extracted using RNeasy® Plus mini kit (Qiagen) and genomic DNA contamination eliminated with Turbo DNA-free kit (Applied Biosystems, Foster City, USA). Complementary DNA was synthesised using SuperScriptIII reverse transcriptase and OligoDt primers (Invitrogen). Quantitative real-time PCR was performed using QuantiTect primers (Qiagen) and ABI Prism 7900 sequence detector system (Applied Biosystems). RQ values were calculated using the 2−( ΔΔCt) method and normalised to an individual naïve AnxA1+/+ or AnxA1−/− mouse (calibrator sample).
All Ct values were normalised to endogenous Gapdh gene product. Since factors such as hypoxia can influence Gapdh gene levels [28] a second house-keeping gene; Rpl32 was used to normalise and confirm data (see Table S2).
Statistics
Data are expressed as means ± SE. Student’s t test was used to compare two groups. Comparison of clinical scores and paw volumes were made using 2-way ANOVA. A value of P<0.05 was considered significant.
RESULTS
Characterisation of joint inflammation
We began this study to optimise the model by comparing two different protocols: 200 μl of serum at day 0 (as in[22]) or 50+50 μl at day 0 and day 2. A remarkably similar arthritic response could be measured with respect to disease profile and intensity (Figure 1A). High levels of disease were achieved as early as day 2 post-injection with maximal response by day 6, after which disease resolved to almost normal by day 20. Incidence of disease was similar between the two volumes of K/BxN serum (Table S1). The arthritic response was mirrored by changes in paw volume with similar profiles with no difference between the two protocols (Figure 1B).
Figure 1. Characterisation of the arthritic response to low volume K/BxN serum.

Mice (n=8) were given an i.p. injection of 50 μl at day 0 and day 2 (50+50 μl) or a single injection of 200 μl at day 0 of K/BxN serum. (A) Arthritic scores and (B) paw edema were monitored over the time course. No significant difference (two-way ANOVA) was observed between the two K/BxN serum groups. (C) Ankles of naïve, 50+50 μl and 200 μl K/BxN serum group. Decalcified paraffin embedded joints were staining by H&E and SafraninO. Representative images are shown; scale bars, 50 μm. (D) Histomorphometric analyses of joint sections; S, synovitis; CE, cartilage erosion; PF, pannus formation. All results are expressed as mean ± SE.
Microscopic analyses of non-arthritic ankle joints showed an acellular articular cavity with intact unicellular synovial lining and uniform cartilage structure (Figure 1C, top panel). In contrast, arthritic joints from both 50+50 μl and 200 μl K/BxN arthritis groups displayed marked cell infiltration within a thickened synovial lining. Pannus invasion leading to cartilage erosion was observed and proteoglycan loss was confirmed with SafraninO (Figure 1C, middle and bottom panels). Quantitative histological scoring showed no significant difference between the two groups of arthritic mice (Figure 1D).
The ankle is the predominant joint to be affected in this model of inflammatory arthritis, yet we monitored structural changes in the knee, as perhaps more relevant to human disease; we observed that knee joints displayed mild synovitis accompanied with thickening of synovial lining, pannus formation and marked cartilage erosion (see Figure S1). Based on these results, in agreement with a recent published study,[29] the 50+50 μl serum protocol was selected for subsequent experimentation.
Expression of AnxA1 and Fpr2 in the joint
At the mRNA level, a low degree of expression for both Fpr1 and Fpr2 could be measured, with high Anxa1 message content (see Table S2). These results were partially confirmed by protein analyses, since AnxA1 was mildly expressed and localised predominantly in the synovial lining, whilst Fpr2 was localised to the periosteal lining with very little detection in synovium (Figure 2A). In arthritic joints, AnxA1 and Fpr2 expression was highly detectable within the synovial lining (Fpr2) and tissue (AnxA1 and Fpr2) (Figure 2B). Antibody controls and AnxA1−/− joints did not show any staining (Figure 2C and 2D).
Figure 2. Protein expression of AnxA1 and Fpr2 in ankle joints of naïve mice.

Ankles of naïve and K/BxN serum (50+50 μl, day 0 and day 2) were processed (see Methods) and stained for AnxA1 and Fpr2, with a haematoxylin counterstain. Images representative of three distinct analyses in 4 mice per status are shown. (A) Naïve mouse ankle joints. (B) Serum-treated mouse ankle joint. (C) Same joints but tested without the primary antibody. (D) Joint of an AnxA1−/− mouse, acting as negative control for AnxA1 staining. Scale bars, 20 μm.
Dexamethasone is inactive in AnxA1−/− mice
Despite wide application of this model of inflammatory arthritis, the bioactions of glucocorticoids have been scantly investigated. We selected the 10-μg dose of Dex from initial studies (Figure S2). Administration of Dex to AnxA1+/+ mice exerted profound inhibitory effects with ~60% inhibition of the severity of arthritis at day 6 and ~80% at day 10 compared to vehicle-treated mice (Figure 3A). AnxA1−/− mice displayed a similar profile of arthritis to AnxA1+/+ animals. However, the anti-arthritic effect of Dex was absent in animals nullified for AnxA1 (Figure 3A). Analysis of presence of severe disease (score ≥8) indicated a trend for higher incidence in AnxA1−/− mice and a marked reliance on endogenous AnxA1 for the protective effects of Dex (Table S3).
Figure 3. Dexamethasone attenuates K/BxN arthritis in AnxA1+/+ but not AnxA1−/− mice.

AnxA1+/+ and AnxA1−/− mice were given an intraperitoneal injection of K/BxN serum (50 μl at day 0 and day 2) and then received vehicle or Dex (10 μg i.p. daily). (A) Arthritic scores as monitored over a 10-day time course. *p<0.05 vs. appropriate vehicle control (two-way ANOVA). (B) Right ankles of mice from each experimental group were taken at day 10 and joints processed for staining by H&E and SafraninO. Representative images are shown. Scale bars, 50 μm. (C) Histomorphometric analyses of joint sections; S, synovitis; CE, cartilage erosion; PF, pannus formation. *p<0.05 vs. appropriate vehicle control (Student’s t test). (D) Quantitative real-time PCR was performed on cDNA from left ankle joints; proinflammatory genes were analysed with all Ct values normalised to endogenous Gapdh (4 mice per group). RQ values were calculated using 2−(ΔΔCt) and data shown here as mean % gene inhibition by Dex relative to vehicle control groups ± SE. Naïve joints were set as the calibrator samples. *p<0.05 vs. respective vehicle control (Student’s t test).
Histological analyses were in line with arthritic scores. AnxA1+/+ mice displayed marked synovitis accompanied by pannus formation and cartilage destruction (Figure 3B, left panels). Treatment with Dex prevented signs of disease (Figure 3B, middle panels), whilst AnxA1−/− mice treated with vehicle control (not shown) and Dex (Figure 3B, right panels) showed similar disease pathology to AnxA1+/+ mice. These structural changes were reflected in the histological scores with ~75% inhibition afforded by Dex only in AnxA1+/+ mice (Figure 3C).
Analyses of inflammatory gene messages, normalised to Gapdh and Rpl32 (see Figure S3) and selected from our previous study [22], revealed that in wild type mice; Dex inhibited Cxcl10, Nos2, Il-1β, and Ccl2. Tnfα was inhibited by ~25% whilst expression of the chemokine receptor Cxcr3 was not modulated (Figure 3D). These inhibitory effects of Dex were absent or significantly reduced in AnxA1−/− mice.
Next, we monitored expression of AnxA1 and its receptors, normalised to Gapdh and Rpl32 (see Figure S4). In arthritic AnxA1+/+ mice, both Fpr1 and Fpr2 mRNA expression was enhanced over naïve joints (Figure 4), a finding which is in line with Fpr2 protein expression (see Figure 2). Dex decreased both Fpr1 and Fpr2 message with little comparative effect on Anxa1 (Figure 4). In mice devoid of AnxA1, a >10-fold increase in Fpr1 expression was observed comparing arthritic to naïve joints, with Fpr2 remaining at a similar expression. In absence of AnxA1, Dex could not regulate either Fpr1 or Fpr2 gene expression (Figure 4).
Figure 4. Profile of Anxa1, Fpr1 and Fpr2 gene product expression in ankle joints of AnxA1+/+ and AnxA1−/− mice treated with Dex.
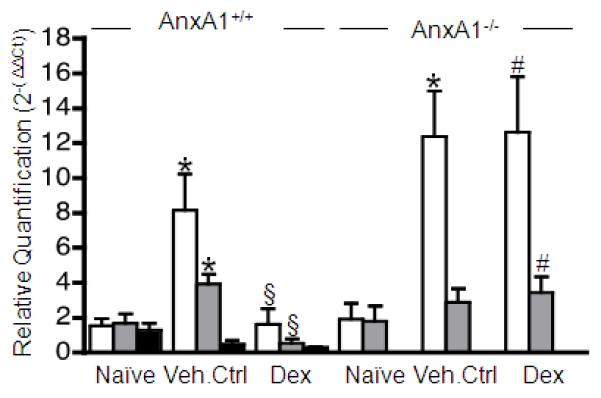
AnxA1+/+ and AnxA1−/− mice were given an intraperitoneal injection of K/BxN serum (50 μl at day 0 and day 2) and then received vehicle or Dex (10 μg i.p. daily). Quantitative real-time PCR was performed on cDNA from left ankle joints of mice from each experimental group (Day 10). The Anxa1, Fpr1 and Fpr2 genes were analysed with all Ct values normalised to endogenous Gapdh (4 mice per group). Naïve joints were set as the calibrator samples. Mean RQ values ± SE shown here were calculated using 2−(ΔΔCt) method. *p<0.05 vs. naïve; §p<0.05 vs. appropriate vehicle control; #p<0.05 vs. AnxA1+/+ group (Student’s t test).
SuperAnxA1 accelerates resolution of the arthritic joint
In the final set of experiments we assessed the pharmacological potential of these findings. Proteinase 3 (PR3) may limit AnxA1 functions in neutrophilic inflammation, cleaving it at the N-terminal region. Thus we monitored the profile of Pr3 gene, in the arthritic joint and found it significantly rose from baseline (day 0) to peak of disease (day 6) (Figure 5 and Figure 5S). This observation, coupled to abundant expression of Fpr1 and Fpr2 in the joints, justified the testing of SuperAnxA1. Dosed at 1 μg per mouse, AnxA1 was not particularly effective in reducing disease (Figure 5B). On the other hand, cleavage-resistant SuperAnxA1 did not affect disease progress (e.g. from day 2 to day 6) but significantly accelerated resolution (Figure 5B), with 50% less disease incidence compared to controls (Table 1).
Figure 5. SuperAnxA1 accelerates resolution of K/BxN arthritis.

AnxA1+/+ mice (n=6) were given an i.p. injection of K/BxN serum (50 μl at day 0 and day 2) and then received vehicle, human recombinant AnxA1 or SuperAnxA1 (1 μg i.p. daily from day 2). (A) Pr3 gene product expression was analysed with Ct values normalised to endogenous Gapdh. RQ values were calculated using 2−( ΔΔCt) and data shown here as mean ± SE. Naïve mice (D0) were set as the calibrator samples. *p<0.05 vs. naïve (Student’s t test). (B) Arthritic score. *p<0.05 vs. vehicle control (two-way ANOVA). (C) Right ankles of mice from each experimental group were taken at day 10 and joints processed for staining by H&E and SafraninO. Representative images are shown. Scale bars, 50 μm. (D) Histomorphometric analyses of joint sections; S, synovitis; CE, cartilage erosion; PF, pannus formation. *p<0.05 vs. vehicle control (Student’s t test).
Table 1.
SuperAnxA1 reduces the severity of inflammatory arthritis.
| Time (days) |
Vehicle control | SuperAnxA1 | AnxA1 |
|---|---|---|---|
| 0 | 0% (0/6) | 0% (0/6) | 0% (0/6) |
| 6 | 100% (6/6) | 83.3% (5/6) | 100% (6/6) |
| 10 | 100% (6/6) | 50% (3/6) | 100% (6/6) |
Data report the number of mice with a severe arthritic score, taking a threshold of ≥ 8. SuperAnxA1 or AnxA1 were given from day 2 at daily doses of 1 μg i.p.; the K/BxN arthritogenic serum was given on day 0 and day 2 at volumes of 50 μl each time.
The macroscopic joint protection afforded by SuperAnxA1 was confirmed microscopically, with preservation of cartilage structure with little loss of proteoglycans (Figure 5C, middle panels). Synovitis was apparent within these joints, but substantially lower than controls (Figure 5C, top panels). The joints of AnxA1 treated mice were disrupted to a similar extent as controls (Figure 5C, bottom panels). Quantitative analysis of damage within the ankle joints confirmed the observational studies (Figure 5D). Finally, ankle joints from vehicle- and SuperAnxA1-treated mice demonstrated reduced expression of the Cxcl10 and Cxcr3 pair, as well as Nos2, Il-1β, Ccl2, Il-6 but not Il-10 and Tnfα (Figure S6).
Discussion
AnxA1 is the prototype of a new class of mediators able to potently modulate both the innate and adaptive immune response; whilst effecting inhibitory properties on inflammatory cells, setting in motion proresolving responses, this protein acts as a positive relay in T cells activation.[4] As an example, exogenous and endogenous AnxA1 potentiate T cell receptor-downstream signalling [30] and assure proper dendritic cell-mediated T cell clonal expansion.[31] In agreement with these dual functions, AnxA1−/− mice experience augmented acute inflammatory responses,[14, 15] yet display reduced degree of tissue damage in an experimental model of multiple sclerosis.[32] Less clear is the role of the protein in models of arthritis.
In the collagen-induced arthritis model, AnxA1 injection during the immune phase (within week 1 post-collagen immunization) led to higher disease as emerged three weeks later.[30] In a model of antigen-induced arthritis, assessed at a single time-point (day 7), endogenous AnxA1 modulated some of the anti-inflammatory effects of Dex, including inhibition of pro-inflammatory genes including IL-1β and Fα.[20] No studies have yet investigated the role of endogenous or exogenous AnxA1 in the serum-induced inflammatory arthritis model.
K/BxN serum-induced arthritis,[33, 34] is a versatile model that can be applied to transgenic mice with little dependence on strain.[35] This model addresses the contribution of neutrophils and macrophages to inflammatory arthritis, as evident from depletion studies.[36, 37] Conversely, this experimental arthritis does not rely on T and B cells as demonstrated with the RAG1−/− mice[28] which, in contrast, display delayed disease onset and severity in collagen-induced arthritis.[38] Here we describe that low dosage arthritogenic serum provokes a high degree of arthritis and disease incidence, data in agreement with a recent study by Lee’s group[29]. We noted a remarkable joint disruption and report a novel parallelism between the ankle and knee joint. Marked synovitis associated with disruption of cartilage occurs along a well-defined time-profile with peak disease at day 6 followed by a resolution phase.[22, 35] Use of transgenic mice has allowed some definition of the disease pathogenesis, revealing a fundamental role for myeloid cells[36, 37] and IL-1.[39, 40] More recently this model of arthritis has been applied to proresolving pathways, noting that 12/15-lipoxygenase or melanocortin type-3 receptor deficient mice experience a higher degree of disease.[21, 22]
AnxA1 is highly abundant in myeloid and stromal cells and its expression can be modulated by glucocorticoids and other anti-inflammatory drugs.[41] In addition, cell activation such as adhesion or extravasation also engages the AnxA1 system as a way to activate an inhibitory circuit to modulate the inflammatory response.[27] After several pharmacological investigations with AnxA1 or mimetics, AnxA1 null mice were produced and tested in models of acute inflammation[15, 24] revealing important modulatory functions.[4] As discussed above, AnxA1 role in chronic inflammatory settings in less defined.
This study began by detecting protein and mRNA of elements of the AnxA1 pathway during on-going inflammatory arthritis, reporting a high degree of expression for Anxa1, Fpr1 and Fpr2 both is resting and arthritic joints. At the protein level, Anxa1 localisation in the mouse synovium seems to parallel that observed in the human RA synovium[42]. However, at least in the conditions here applied, lack of AnxA1 does not seem to impact on the profile of disease. Yet it remains to be seen whether such a lack of function would also be seen in knockouts for the AnxA1 receptors.[5, 43] As an example, in preliminary observations, mice nullified for Fpr2 displayed higher arthritic score upon injection of the arthritogenic serum.[44]
Glucocorticoids have been scarcely tested in this model. Buttgereit and colleagues have tested the effect of osteoblast-restricted absence of glucocorticoid signalling on disease outcome, noting a worsening with marked joint disruption[23]. In these mice, bone cells could not respond to glucocorticoids from birth, thus it is possible that developmental aspects might be relevant and subtle long-term alterations occur. For instance it is know that glucocorticoids exert a permissive effect on importance cytokine function in the liver, by assuring proper expression of the IL-6 and IL-1 receptors[45, 46]. IL-6 is also a master-cytokine for osteoblasts (and fibroblasts) so a malfunctioning or inadequate glucocorticoid signalling, prolonged from birth, may generate responses that are not necessarily replicated by acute glucocorticoid treatment.
In keeping with this, our pharmacological analyses with doses of Dex as low as 10 μg exerted potent protective effects on the joint, evident both as inhibition of pannus formation and chondro-protection. It was striking to observe that the effects of the glucocorticoid were lost in AnxA1−/− animals. Thus, whilst AnxA1−/− mice did not experience a difference in the extent and profile of arthritis, at variance from the antigen-induced arthritis model,[20] endogenous AnxA1 still proved important in the inhibitory effects of Dex on a discrete set of proinflammatory genes, of which Il-1β, Cxcl10 and Ccl2 were of particular interest. Selective modulation of gene expression downstream AnxA1 is in keeping with the observations made in the antigen-induced arthritis model.[20] The fundamental role that IL-1β exerts in this model of inflammatory arthritis has been addressed through a variety of genetic approaches targeting directly the cytokine, its receptor[39, 47] or endogenous antagonist.[40]
Modulation of gene expression downstream of endogenous AnxA1 can also be discussed in relation to the modulation of genes of the AnxA1 pathway. In fact, Dex did not appear to induce Anxa1 or Fpr gene expression at the level of the joint, yet was highly reliant on endogenous AnxA1 both with respect to cell recruitment and inflammatory gene expression. We reason that these data indicate multiple functions of endogenous AnxA1, which become of relevance only after administration of the glucocorticoid.
The lack of correlation between AnxA1 gene product and protein is intriguing though It should be said that glucocorticoid-regulation of the AnxA1 pathway is multifaceted; thus, non-genomic modulation[48] with release of the protein, for instance, could account for apparent lower tissue levels of the AnxA1 protein. AnxA1 analysis, at a single time point, could also be confounded by genomic regulation exerted by glucocorticoids[15, 49] and other factors present in an inflamed joint such as IL-6[50] and TNF[51]. Clearly, the current lack of characterisation of the AnxA1 promoter contributes to this poor understanding of the mechanisms operative in these settings. Moreover, the fact that glucocorticoids like dexamethasone induce AnxA1 gene in innate immune cells [14, 15] whilst downregulating it in adaptive immune cells[52] add another layer of complexity to this issue which inevitably requires further experimentation.
The versatility of this model of inflammatory arthritis justifies its application to a variety of targets; in line with the major infiltration of neutrophils and mast cells,[53] we could observe dynamic expression of Pr3 gene product in the arthritic joint. Proteinase 3 is emerging as an important effector of neutrophilic inflammation[54] and can cleave, among a variety of substrates, also AnxA1.[55] Therefore, we concluded this study by testing the potential of AnxA1 mimetics, comparing the effect of native protein with that of SuperAnxA1, a mutant resistant to serine protease-induced cleavage.[26] Administered at the dose of 1 μg (corresponding to 27 pmol), SuperAnxA1, but not AnxA1, attenuated joint disease. It was interesting to note that SuperAnxA1 did not impact on the induction of arthritis yet it accelerated the resolution process so that by day 10 a preserved joint structure could be observed. We wish to propose that higher resistance to proteolysis might underlie its pharmacological efficacy, though we cannot exclude the confounding element of potential differences in pharmacokinetics. In any case, these results provide strong proof-of-concept that an AnxA1 derivative, resistant to proteolysis, could be beneficial in modulating joint inflammation. SuperAnxA1 could be the backbone for future strategies aiming at capitalising AnxA1 biology for novel anti-arthritic approaches.
Glucocorticoids are of wide clinical use because of their ability to impact several effectors of arthritis, with a recent appreciation that, besides inhibition of proinflammatory gene expression, gene induction is also of therapeutic relevance.[56, 57] In the context of chronic inflammatory diseases, induction of dual-specificity phosphatase 1[58] and glucocorticoid-induced leucine zipper[59] genes could be of mechanistic value. In addition, we show here that endogenous AnxA1 is also relevant to the anti-arthritic effect of low-dose Dex, yet we cannot demonstrate induction of this gene at the level of the arthritic joint leading to our proposal that the protein is either modulated in a non-genomic fashion by this glucocorticoid[41, 60] or the regulation occurs outside the site of the arthritic joint, an obvious site being the vasculature and the control of the egress of blood-borne neutrophils[4]. Analysis of AnxA1 expression in distant tissues (e.g. blood) during on-going arthritis and glucocorticoid treatment can shed light on this point, which has also been touched upon above in relation to lack of correlation between joint AnxA1 mRNA and protein expression.
Collectively, this study reveals potent anti-arthritic properties for Dexamethasone in a model of inflammatory arthritis, using very low doses perhaps reminiscent of clinical application on low-dose glucocorticoids in rheumatoid arthritis[61] and, in terms of drug discovery programmes, justifies the testing of AnxA1 mimetics to control the active phase of the disease.
Supplementary Material
Acknowledgments
Funding This work was supported by a Wellcome Trust (UK) project grant 083551. SMO is funded Fundação de Amparo à Pesquisa do Estado de São Paulo - FAPESP (Grant 2011/00128-1) and Conselho Nacional de Desenvolvimento Científico e Tecnológico - CNPq (Grant 302768/2010-6)
Footnotes
Competing interests The authors have no conflicting financial interests.
REFERENCES
- 1.Gilroy DW, Lawrence T, Perretti M, et al. Inflammatory resolution: new opportunities for drug discovery. Nat Rev Drug Discov. 2004;3:401–16. doi: 10.1038/nrd1383. [DOI] [PubMed] [Google Scholar]
- 2.Ley K, Laudanna C, Cybulsky MI, et al. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. 2007;7:678–89. doi: 10.1038/nri2156. [DOI] [PubMed] [Google Scholar]
- 3.Serhan CN, Brain SD, Buckley CD, et al. Resolution of inflammation: state of the art, definitions and terms. FASEB J. 2007;21:325–32. doi: 10.1096/fj.06-7227rev. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Perretti M, D’Acquisto F. Annexin A1 and glucocorticoids as effectors of the resolution of inflammation. Nat Rev Immunol. 2009;9:62–70. doi: 10.1038/nri2470. [DOI] [PubMed] [Google Scholar]
- 5.Ye RD, Boulay F, Wang JM, et al. International Union of Basic and Clinical Pharmacology. LXXIII. Nomenclature for the formyl peptide receptor (FPR) family. Pharmacol Rev. 2009;61:119–61. doi: 10.1124/pr.109.001578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chiang N, Serhan CN, Dahlen SE, et al. The lipoxin receptor ALX: potent ligand-specific and stereoselective actions in vivo. Pharmacol Rev. 2006;58:463–87. doi: 10.1124/pr.58.3.4. [DOI] [PubMed] [Google Scholar]
- 7.Perretti M, Chiang N, La M, et al. Endogenous lipid- and peptide-derived anti-inflammatory pathways generated with glucocorticoid and aspirin treatment activate the lipoxin A4 receptor. Nat Med. 2002;8:1296–302. doi: 10.1038/nm786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Walther A, Riehemann K, Gerke V. A novel ligand of the formyl peptide receptor: annexin I regulates neutrophil extravasation by interacting with the FPR. Mol Cell. 2000;5:831–40. doi: 10.1016/s1097-2765(00)80323-8. [DOI] [PubMed] [Google Scholar]
- 9.Hayhoe RP, Kamal AM, Solito E, et al. Annexin 1 and its bioactive peptide inhibit neutrophil-endothelium interactions under flow: indication of distinct receptor involvement. Blood. 2006;107:2123–30. doi: 10.1182/blood-2005-08-3099. [DOI] [PubMed] [Google Scholar]
- 10.Ernst S, Lange C, Wilbers A, et al. An annexin 1 N-terminal peptide activates leukocytes by triggering different members of the formyl peptide receptor family. J Immunol. 2004;172:7669–76. doi: 10.4049/jimmunol.172.12.7669. [DOI] [PubMed] [Google Scholar]
- 11.Serhan CN. Novel lipid mediators and resolution mechanisms in acute inflammation: to resolve or not? Am J Pathol. 2010;177:1576–91. doi: 10.2353/ajpath.2010.100322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Babbin BA, Lee WY, Parkos CA, et al. Annexin I regulates SKCO-15 cell invasion by signaling through formyl peptide receptors. The Journal of biological chemistry. 2006;281:19588–99. doi: 10.1074/jbc.M513025200. [DOI] [PubMed] [Google Scholar]
- 13.Muffley LA, Zhu KQ, Engrav LH, et al. Spatial and temporal localization of the melanocortin 1 receptor and its ligand alpha-melanocyte-stimulating hormone during cutaneous wound repair. The journal of histochemistry and cytochemistry : official journal of the Histochemistry Society. 2011;59:278–88. doi: 10.1369/0022155410397999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Damazo AS, Yona S, D’Acquisto F, et al. Critical protective role for annexin 1 gene expression in the endotoxemic murine microcirculation. Am J Pathol. 2005;166:1607–17. doi: 10.1016/S0002-9440(10)62471-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Damazo AS, Yona S, Flower RJ, et al. Spatial and temporal profiles for anti-inflammatory gene expression in leukocytes during a resolving model of peritonitis. J Immunol. 2006;176:4410–8. doi: 10.4049/jimmunol.176.7.4410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Babbin BA, Laukoetter MG, Nava P, et al. Annexin A1 regulates intestinal mucosal injury, inflammation, and repair. J Immunol. 2008;181:5035–44. doi: 10.4049/jimmunol.181.7.5035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O’Sullivan TP, Vallin KS, Shah ST, et al. Aromatic lipoxin A4 and lipoxin B4 analogues display potent biological activities. Journal of medicinal chemistry. 2007;50:5894–902. doi: 10.1021/jm060270d. [DOI] [PubMed] [Google Scholar]
- 18.Getting SJ, Flower RJ, Perretti M. Inhibition of neutrophil and monocyte recruitment by endogenous and exogenous lipocortin 1. British journal of pharmacology. 1997;120:1075–82. doi: 10.1038/sj.bjp.0701029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang Y, Leech M, Hutchinson P, et al. Antiinflammatory effect of lipocortin 1 in experimental arthritis. Inflammation. 1997;21:583–96. doi: 10.1023/a:1027330021479. [DOI] [PubMed] [Google Scholar]
- 20.Yang YH, Morand EF, Getting SJ, et al. Modulation of inflammation and response to dexamethasone by Annexin 1 in antigen-induced arthritis. Arthritis and rheumatism. 2004;50:976–84. doi: 10.1002/art.20201. [DOI] [PubMed] [Google Scholar]
- 21.Kronke G, Katzenbeisser J, Uderhardt S, et al. 12/15-lipoxygenase counteracts inflammation and tissue damage in arthritis. J Immunol. 2009;183:3383–9. doi: 10.4049/jimmunol.0900327. [DOI] [PubMed] [Google Scholar]
- 22.Patel HB, Bombardieri M, Sampaio AL, et al. Anti-inflammatory and antiosteoclastogenesis properties of endogenous melanocortin receptor type 3 in experimental arthritis. FASEB J. 2010;24:4835–43. doi: 10.1096/fj.10-167759. [DOI] [PubMed] [Google Scholar]
- 23.Buttgereit F, Zhou H, Kalak R, et al. Transgenic disruption of glucocorticoid signaling in mature osteoblasts and osteocytes attenuates K/BxN mouse serum-induced arthritis in vivo. Arthritis and rheumatism. 2009;60:1998–2007. doi: 10.1002/art.24619. [DOI] [PubMed] [Google Scholar]
- 24.Hannon R, Croxtall JD, Getting SJ, et al. Aberrant inflammation and resistance to glucocorticoids in annexin 1-/- mouse. FASEB J. 2003;17:253–5. doi: 10.1096/fj.02-0239fje. [DOI] [PubMed] [Google Scholar]
- 25.Lee DM, Friend DS, Gurish MF, et al. Mast cells: a cellular link between autoantibodies and inflammatory arthritis. Science. 2002;297:1689–92. doi: 10.1126/science.1073176. [DOI] [PubMed] [Google Scholar]
- 26.Pederzoli-Ribeil M, Maione F, Cooper D, et al. Design and characterization of a cleavage-resistant Annexin A1 mutant to control inflammation in the microvasculature. Blood. 2010;116:4288–96. doi: 10.1182/blood-2010-02-270520. [DOI] [PubMed] [Google Scholar]
- 27.Gastardelo TS, Damazo AS, Dalli J, et al. Functional and ultrastructural analysis of annexin A1 and its receptor in extravasating neutrophils during acute inflammation. Am J Pathol. 2009;174:177–83. doi: 10.2353/ajpath.2009.080342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhong H, Simons JW. Direct comparison of GAPDH, beta-actin, cyclophilin, and 28S rRNA as internal standards for quantifying RNA levels under hypoxia. Biochemical and biophysical research communications. 1999;259:523–6. doi: 10.1006/bbrc.1999.0815. [DOI] [PubMed] [Google Scholar]
- 29.Boilard E, Lai Y, Larabee K, et al. A novel anti-inflammatory role for secretory phospholipase A2 in immune complex-mediated arthritis. EMBO molecular medicine. 2010;2:172–87. doi: 10.1002/emmm.201000072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.D’Acquisto F, Merghani A, Lecona E, et al. Annexin-1 modulates T-cell activation and differentiation. Blood. 2007;109:1095–102. doi: 10.1182/blood-2006-05-022798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huggins A, Paschalidis N, Flower RJ, et al. Annexin-1-deficient dendritic cells acquire a mature phenotype during differentiation. FASEB J. 2009;23:985–96. doi: 10.1096/fj.08-119040. [DOI] [PubMed] [Google Scholar]
- 32.Paschalidis N, Huggins A, Rowbotham NJ, et al. Role of endogenous annexin-A1 in the regulation of thymocyte positive and negative selection. Cell Cycle. 2010;9:784–93. [PubMed] [Google Scholar]
- 33.Kouskoff V, Korganow AS, Duchatelle V, et al. Organ-specific disease provoked by systemic autoimmunity. Cell. 1996;87:811–22. doi: 10.1016/s0092-8674(00)81989-3. [DOI] [PubMed] [Google Scholar]
- 34.Matsumoto I, Staub A, Benoist C, et al. Arthritis provoked by linked T and B cell recognition of a glycolytic enzyme. Science. 1999;286:1732–5. doi: 10.1126/science.286.5445.1732. [DOI] [PubMed] [Google Scholar]
- 35.Ji H, Gauguier D, Ohmura K, et al. Genetic influences on the end-stage effector phase of arthritis. The Journal of experimental medicine. 2001;194:321–30. doi: 10.1084/jem.194.3.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wipke BT, Allen PM. Essential role of neutrophils in the initiation and progression of a murine model of rheumatoid arthritis. J Immunol. 2001;167:1601–8. doi: 10.4049/jimmunol.167.3.1601. [DOI] [PubMed] [Google Scholar]
- 37.Solomon S, Rajasekaran N, Jeisy-Walder E, et al. A crucial role for macrophages in the pathology of K/B x N serum-induced arthritis. European journal of immunology. 2005;35:3064–73. doi: 10.1002/eji.200526167. [DOI] [PubMed] [Google Scholar]
- 38.Plows D, Kontogeorgos G, Kollias G. Mice lacking mature T and B lymphocytes develop arthritic lesions after immunization with type II collagen. J Immunol. 1999;162:1018–23. [PubMed] [Google Scholar]
- 39.Ji H, Pettit A, Ohmura K, et al. Critical roles for interleukin 1 and tumor necrosis factor alpha in antibody-induced arthritis. The Journal of experimental medicine. 2002;196:77–85. doi: 10.1084/jem.20020439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lamacchia C, Rodriguez E, Palmer G, et al. Articular inflammation is controlled by myeloid cell-derived interleukin 1 receptor antagonist during the acute phase of arthritis in mice. Annals of the rheumatic diseases. 2011 doi: 10.1136/annrheumdis-2011-200429. [DOI] [PubMed] [Google Scholar]
- 41.Yazid S, Leoni G, Getting SJ, et al. Antiallergic cromones inhibit neutrophil recruitment onto vascular endothelium via annexin-A1 mobilization. Arteriosclerosis, thrombosis, and vascular biology. 2010;30:1718–24. doi: 10.1161/ATVBAHA.110.209536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Goulding NJ, Dixey J, Morand EF, et al. Differential distribution of annexins-I, -II, -IV, and -VI in synovium. Annals of the rheumatic diseases. 1995;54:841–5. doi: 10.1136/ard.54.10.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Perretti M. The annexin 1 receptor(s): is the plot unravelling? Trends in pharmacological sciences. 2003;24:574–9. doi: 10.1016/j.tips.2003.09.010. [DOI] [PubMed] [Google Scholar]
- 44.Dufton N, Hannon R, Brancaleone V, et al. Anti-inflammatory role of the murine formyl-peptide receptor 2: ligand-specific effects on leukocyte responses and experimental inflammation. J Immunol. 2010;184:2611–9. doi: 10.4049/jimmunol.0903526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fischer CP, Bode BP, Takahashi K, et al. Glucocorticoid-dependent induction of interleukin-6 receptor expression in human hepatocytes facilitates interleukin-6 stimulation of amino acid transport. Annals of surgery. 1996;223:610–8. doi: 10.1097/00000658-199605000-00017. discussion 18-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Granner DK. Restoration of sensitivity of cultured hepatoma cells to cyclic nucleotides shows permissive effect of dexamethasone. Nature. 1976;259:572–3. doi: 10.1038/259572a0. [DOI] [PubMed] [Google Scholar]
- 47.Nigrovic PA, Binstadt BA, Monach PA, et al. Mast cells contribute to initiation of autoantibody-mediated arthritis via IL-1. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:2325–30. doi: 10.1073/pnas.0610852103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Croxtall JD, van Hal PT, Choudhury Q, et al. Different glucocorticoids vary in their genomic and non-genomic mechanism of action in A549 cells. British journal of pharmacology. 2002;135:511–9. doi: 10.1038/sj.bjp.0704474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vishwanath BS, Frey FJ, Bradbury M, et al. Adrenalectomy decreases lipocortin-I messenger ribonucleic acid and tissue protein content in rats. Endocrinology. 1992;130:585–91. doi: 10.1210/endo.130.2.1531128. [DOI] [PubMed] [Google Scholar]
- 50.Solito E, de Coupade C, Parente L, et al. IL-6 stimulates annexin 1 expression and translocation and suggests a new biological role as class II acute phase protein. Cytokine. 1998;10:514–21. doi: 10.1006/cyto.1997.0325. [DOI] [PubMed] [Google Scholar]
- 51.Tagoe CE, Marjanovic N, Park JY, et al. Annexin-1 mediates TNF-alpha-stimulated matrix metalloproteinase secretion from rheumatoid arthritis synovial fibroblasts. J Immunol. 2008;181:2813–20. doi: 10.4049/jimmunol.181.4.2813. [DOI] [PubMed] [Google Scholar]
- 52.D’Acquisto F, Paschalidis N, Raza K, et al. Glucocorticoid treatment inhibits annexin-1 expression in rheumatoid arthritis CD4+ T cells. Rheumatology (Oxford) 2008;47:636–9. doi: 10.1093/rheumatology/ken062. [DOI] [PubMed] [Google Scholar]
- 53.Pimentel TA, Sampaio AL, D’Acquisto F, et al. An essential role for mast cells as modulators of neutrophils influx in collagen-induced arthritis in the mouse. Laboratory investigation; a journal of technical methods and pathology. 2011;91:33–42. doi: 10.1038/labinvest.2010.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Witko-Sarsat V, Reuter N, Mouthon L. Interaction of proteinase 3 with its associated partners: implications in the pathogenesis of Wegener’s granulomatosis. Current opinion in rheumatology. 2010;22:1–7. doi: 10.1097/BOR.0b013e3283331594. [DOI] [PubMed] [Google Scholar]
- 55.Vong L, D’Acquisto F, Pederzoli-Ribeil M, et al. Annexin 1 cleavage in activated neutrophils: a pivotal role for proteinase 3. The Journal of biological chemistry. 2007;282:29998–30004. doi: 10.1074/jbc.M702876200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Clark AR. Anti-inflammatory functions of glucocorticoid-induced genes. Molecular and cellular endocrinology. 2007;275:79–97. doi: 10.1016/j.mce.2007.04.013. [DOI] [PubMed] [Google Scholar]
- 57.Ayroldi E, Riccardi C. Glucocorticoid-induced leucine zipper (GILZ): a new important mediator of glucocorticoid action. FASEB J. 2009;23:3649–58. doi: 10.1096/fj.09-134684. [DOI] [PubMed] [Google Scholar]
- 58.Abraham SM, Lawrence T, Kleiman A, et al. Antiinflammatory effects of dexamethasone are partly dependent on induction of dual specificity phosphatase 1. The Journal of experimental medicine. 2006;203:1883–9. doi: 10.1084/jem.20060336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Beaulieu E, Ngo D, Santos L, et al. Glucocorticoid-induced leucine zipper is an endogenous antiinflammatory mediator in arthritis. Arthritis and rheumatism. 2010;62:2651–61. doi: 10.1002/art.27566. [DOI] [PubMed] [Google Scholar]
- 60.Solito E, Christian HC, Festa M, et al. Post-translational modification plays an essential role in the translocation of annexin A1 from the cytoplasm to the cell surface. FASEB J. 2006;20:1498–500. doi: 10.1096/fj.05-5319fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kirwan J. The origins, results and consequences of the 1995 Arthritis Research Campaign Low-Dose Glucocorticoid Study. Clinical and experimental rheumatology. 2011;29:S52–8. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.