

Abstract
Autoantibodies and autoreactive T lymphocytes directed against several pancreatic β cell proteins such as GAD65 have been identified in the circulation before and at the onset of clinical type 1 (insulin-dependent) diabetes. Using GAD65 synthetic peptides, we studied the proliferative response of peripheral blood mononuclear cells (PBMC) either from recently diagnosed type 1 diabetic patients, of whom the majority share the disease-associated HLA class II haplotype (DR4-DQB1*0201 or DR3-DQB1*0302), or from HLA-matched control subjects. We found that 67% (14/21) of the type 1 diabetic patients and 39% (9/23) of the control subjects exhibited a positive proliferative response. Compared with control subjects, however, PBMC from diabetic patients proliferated more frequently (P < 0.05) in the presence of peptide pools from the C-terminal region of GAD65 (amino acids 379–585). Diabetic patients with the same HLA-DQ or HLA-DR alleles showed partially identical T cell reactivity, but no clear correlation could be made between MHC class II specificity and T cell epitopes because of multiple combinations of class II alleles. In addition, by flow cytometry, we studied the direct binding of GAD65 peptides to MHC class II molecules of Epstein–Barr virus (EBV)-transformed B (EBV-B) cells obtained from a diabetic patient. We found that 11 GAD peptides were able to bind to the highly susceptible haplotype DRB1*0301/0401-DQA1*0301/0501-DQB1*0302/0201 on the surface of EBV-B cells in partial correlation with the results obtained in the proliferation assays.
Keywords: glutamic acid decarboxylase, type 1 diabetes, peptide, T cell epitope, MHC class II
INTRODUCTION
The autoimmune process of type 1 diabetes leading to the destruction of insulin-secreting β cells involves autoreactive T cells. Several autoantigens, including GAD65, have been identified as targets of patients' autoantibodies [1–3]. Previous reports have demonstrated that peripheral blood mononuclear cells (PBMC) from newly diagnosed type 1 diabetic patients [4–6] as well as from asymptomatic first-degree relatives show reactivity to these multiple islet cell antigens [5, 7]. In the murine model for type 1 diabetes (non-obese diabetic (NOD) mice), disease protection by administration of GAD has further highlighted the relevance of GAD65 as a major autoantigen [8, 9]. A better knowledge of this autoantigen and of its immunogenic epitopes is thus necessary to understand its potential role in the disease process. MHC–antigen–T cell receptor interactions involve T lymphocytes that recognize peptide fragments of antigen bound to MHC molecules expressed on antigen-presenting cells (APC). The frequent occurrence of certain histocompatibility antigens in type 1 diabetic patients [10, 11] suggests a defect of immunoregulation which might be expressed at the level of these interactions. Mapping of T cell epitopes within protein antigens can be achieved by using protein fragments or synthetic peptides [12].
In the present study, we focused on the GAD65 T cell reactivity from patients with recent-onset diagnosed type 1 diabetes. PBMC reactivity to overlapping 12mer synthetic GAD65 peptides was studied to understand further GAD65 recognition in type 1 diabetes. In contrast, with the experimental approaches used by others [13, 14], we studied the responses of PBMC from several individuals, rather than specific T cell lines, to map T cell responses to GAD65 associated with type 1 diabetes and identify immunodominant regions. Then, we examined the results in the context of the patients' HLA class II haplotypes. To dissect further the GAD65 T cell response at the level of HLA class II, we tried to determine which peptides could bind specifically the highly type 1 diabetes-susceptible haplotype DRB1*0301/0401-DQB1*0201/0302 [10] on human Epstein–Barr virus (EBV)-transformed B cells (EBV-B).
PATIENTS AND METHODS
Patients
Twenty-one patients (eight men and 13 women) with recent-onset type 1 diabetes were included in this study after giving their or their parents' informed consent (Table 1). Criteria for inclusion were age ≤ 35 years (mean age 15.4 ± 6.8 years; range 2–34 years), time interval between admission to the hospital and study (< 7 days), and clear manifestations of insulinopenia ≤ 6 weeks before diagnosis (fasting hyperglycaemia > 200 mg/dl, ketosis, polyuria, polydipsia, and weight loss). A control group of 23 healthy non-diabetic HLA-matched individuals (10 men and 13 women; mean age 27 ± 9 years) was tested in parallel (Table 2). The 23 control subjects were also matched for sex and ethnic origin with diabetic patients and they presented neither familial history of diabetes nor autoimmune diseases. Age inclusion of the control group was extended because 11/23 subjects were adult donors selected for their DR3 and/or DR4 phenotypes from the Saint-Etienne bone marrow registry in France.
Table 1.
Characteristics of recent-onset insulin-dependent diabetes mellitus (IDDM) patients and their T cell reactivity
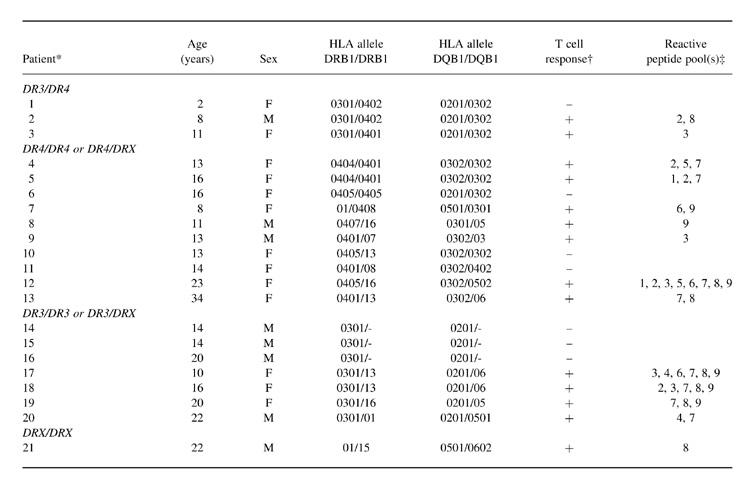
* DRX represents a DR allele different from DR3 or DR4.
† Positive proliferative response to at least one peptide pool is indicated by ‘+’ (SI > 3), a negative response by ‘−’.
‡ Peptide pools 1–9 spanned GAD65 regions [1–66], [61–132], [127–192], [187–264], [259–324], [319–384], [379–450], [445–522] and [517–585], respectively.
Table 2.
Characteristics of control subjects and their T cell reactivity

* DRX represents a DR allele different from DR3 or DR4.
† Positive proliferative response to at least one peptide pool is indicated by ‘+’ (SI > 3), a negative response by ‘−’.
‡ Peptide pools 1–9 spanned GAD65 regions [1–66], [61–132], [127–192], [187–264], [259–324], [319–384], [379–450], [445–522] and [517–585], respectively.
GAD overlapping peptides
A set of 93 12mer overlapping peptides spanning the entire human GAD65 sequence was synthesized by fluorenylmethoxycarbonyl (Fmoc) chemistry as previously described [15]. Peptides overlapped the adjoining sequences by six amino acids. Each peptide was biotinylated at its N-terminal extremity and the COOH-terminus was carboxamidated by using MBHA resin from Novabiochem (Meudon, France). Nine irrelevant 12mer peptides derived from the sequences of the p53 protein, lysozyme and a murine IgG were used as controls in the proliferation assays. Peptides were lyophilized, redissolved in DMSO and diluted 1:40 in PBMC culture medium for the stimulation assay or PBS–1% bovine serum albumin (BSA) for the binding assay described below.
Peptide stimulation of PBMC
Peripheral venous blood (20–30 ml, according to the patient's state) was drawn between 08.00 and 09.00 into sterile heparinized tubes and immediately processed. PBMC were isolated under sterile conditions by Ficoll–Hypaque (Sigma, St Louis, MO) density centrifugation, washed twice, and resuspended in 10% human AB + serum (Sigma) in RPMI 1640 medium (Biowhittaker, Verviers, Belgium) supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mml-glutamine, and 10 mm HEPES. T cell stimulation assays were performed using fresh cells in 96-well round-bottomed plates by incubating 1.5 × 105 PBMC/well in a final volume of 200 μl. Individual GAD65 peptides or peptide pools consisting of 10 or 11 overlapping peptides spanning GAD65 regions of 65–77 amino acids, were added to quadruplicate culture wells to provide a 5-μg/ml final concentration for each peptide. Tetanus toxoid (Institut Pasteur, Paris, France) at 10 μg/ml and recombinant IL-2 at 50 U/ml (Hoffman La Roche, Basel, Switzerland) were used as positive controls. Eight wells to which no antigen was added were used for basal proliferation determination. Cultures were incubated for 6 days at 37°C in a humidified 5% CO2 incubator, then pulsed for 16–18 h with 1 μCi 3H-thymidine (NEN, Boston, MA). The cultures were then harvested onto glassfibre filters and 3H-thymidine incorporation was measured by liquid scintillation counting. The mean value of each quadruplicate was calculated and a coefficient of variation (s.d./mean) < 50% was required for subsequent evaluation. Results were expressed in terms of a stimulation index (SI), i.e. the ratio of the radioactivity measured in cells cultured in the presence of antigen to that obtained in cells cultured without antigen. Stimulation was considered as positive if SI > 3.
Determination of GAD autoantibodies
GAD65 autoantibodies (GADA) were measured by a competitive 35S-methionine-labelled human GAD65 immunoprecipitation assay as described by Petersen et al. [16] using recombinant GAD65 prepared from cDNA obtained from T. Dyrberg (Baegsverd, Denmark). GADA were expressed as arbitrary units in comparison with a pool of antibody-positive sera that was arbitrarily assigned the value of 1.00 unit. GADA activity > 0.11 U (mean + 3 s.d. values of 95 standardized control sera) was considered positive. The sensitivity of the present assay was 80% and the specificity 96%, as assessed at the second international GAD antibody workshop [17].
Lymphoblastoid cells
The EBV-B cell clone DPC, obtained by Madec et al. [18] from EBV-transformed B lymphocytes from a type 1 diabetic patient, was used to study peptide binding to the MHC class II. DPC cells are HLA-A2/11-B8/62-C3-DRB1*0301/0401-DRB3*0101-DRB4*0103-DQB1*0201/0302-DPB1*0401/0402. These cells were cultured in RPMI 1640 containing 10% fetal calf serum (FCS). By using the appropriate labelled MoAb, expression of CD19 (Becton Dickinson, Mountain View, CA), CD40 (Immunotech, Marseille, France), HLA-DR (Becton Dickinson), and HLA-DQ (Becton Dickinson) on the surface of the DPC cells was assessed by flow cytometry. The class II-negative mutant EBV-B cell line RJ 2.2.5 was used as control. This cell line was kindly provided by Dr D. Charron (INSERM U396, Paris, France) with the authorization of Dr R. S. Accolla (Advanced Biotechnology Centre, Genoa, Italy). RJ 2.2.5 cells were derived from RAJI cells (HLA-A3/19-B51/35-C3/4-BW4/6) [19].
HLA determination
Genetic studies were performed in new-onset type 1 diabetes patients, controls and lymphoblastoïd cells by the Blood Transfusion Centres of Lyon and Saint-Etienne, France. HLA class II alleles were determined serologically on all subjects by the standard microlymphocytotoxic technique. HLA-DR and -DQ were typed by polymerase chain reaction amplification with sequence-specific primers (PCR-SSP; Dynal SSP-sets, Compiegne, France). The latest nomenclature of the World Health Organization Nomenclature Committee for Factors of the HLA System was used [20].
Binding assay by flow cytometry
Binding of the biotinylated GAD65 peptides to DPC cells was studied according to the method of Busch et al. [21] with modifications. Using 96-well round-bottomed plates, DPC cells (2 × 105/well) were incubated with a given biotinylated peptide (50 μg/ml final concentration) at 37°C for 18 h in PBS–1% BSA in a final volume of 200 μl. After two washings with 200 μl of PBS–1% BSA, the cells were incubated at 4°C for 30 min with 50 μl of streptavidin–PE diluted (Dako, Glostrup, Denmark) 1:20. As controls, DPC cells were labelled in each experiment with a MoAb specific for the HLA-DR invariant region (L243; kind gift from D. Charron, INSERM U396), then with PE-labelled goat anti-mouse IgG (Calbiochem, Meudon, France). The stained cells were washed three times in PBS–1% BSA, fixed with PBS–1% formaldehyde, and analysed immediately or after a storage period not exceeding 2 days at 4°C. Cell fluorescence was analysed by flow cytometry using a FACScan analyser (Becton Dickinson). To evaluate the relative amount of bound peptide, the mean fluorescence of 5000 stained cells was measured. The background fluorescence was determined in the absence of biotinylated peptide. Peptide binding was considered as significant if fluorescence was five-fold greater than background fluorescence, i.e. > 20% of fluorescent cells.
Statistical analysis
To assess the results of the PBMC response to GAD, we compared, for each pool of GAD peptides, the frequency of positive proliferation (SI > 3) among patients and controls with the Fisher's exact test (contingency table) using the S-PLUS program (version 3.2; StatSci 1993, Seattle). P < 0.05 was considered significant.
RESULTS
T cell proliferation to GAD65 peptides
T cell reactivity was assessed by 3H-thymidine incorporation in PBMC cultured in the presence of pooled GAD65 peptides or pooled irrelevant peptides. This study involved PBMC from 21 recent-onset (< 7 days) type 1 diabetic patients (Table 1) and 23 healthy control subjects (Table 2). Mean T cell responses to tetanus toxoid or IL-2 were in the same range in the two groups: mean SI ± s.d. for tetanus toxoid was 85.2 ± 81.5 for type 1 diabetic patients versus 73.5 ± 82.1 for control subjects, and 68.0 ± 66.8 versus 32.6 ± 44.6, respectively, for IL-2. In spite of a wide range of responses, these results confirmed the lack of abnormality of T cell reactivity to a control antigen and a mitogen in type 1 diabetic patients [22]. No inhibitory effect on PBMC proliferation was shown by peptide pools, in so far as they did not interfere with cell stimulations elicited by tetanus toxoid (Fig. 1).
Fig. 1.
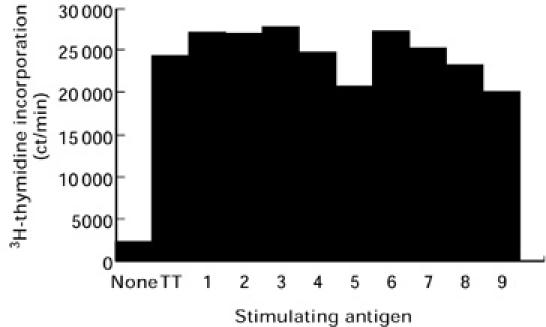
Proliferative response of peripheral blood mononuclear cells (PBMC) obtained from one diabetic patient in the presence of tetanus toxoid alone (TT) or in combination with each of the nine GAD65 peptide pools (1–9). Peptide pools numbered 1–9 correspond to GAD65 regions [1–66], [61–132], [127–192], [187–264], [259–324], [319–384], [379–450], [445–522] and [517–585], respectively. The basal 3H-thymidine incorporation was obtained without antigen stimulation. The values are the mean of quadruplicate from a single representative experiment.
PBMC of 66.6% (14/21) of type 1 diabetic patients and 39.1% (9/23) of the control subjects proliferated in the presence of at least one of the nine GAD65 peptide pools tested. The intensity of the responses was considerably variable (data not shown) and apparently not related to age or sex. In the type 1 diabetic group, the maximum SI was 44.2 versus 11.5 in the control group, but the mean SI of the positive responses in each group did not differ (11.0 ± 11.3 versus 5.7 ± 3.0). T cell proliferation occurred in response to either several or a few peptide pools, depending on the type 1 diabetic patient tested (Table 1). In general, proliferative responses of control subjects were triggered by only a few peptide pools (Table 2).
Examination of the proliferative responses to the peptide pools shows that each GAD65 peptide pool was reactive at least once (Fig. 2). Positive proliferative responses among type 1 diabetic patients were most frequently observed with peptide pools 7–9 (GAD65 region 379–585). The frequency of proliferative response to GAD peptide pool 7 and 9 was significantly higher in the type 1 diabetic group than in the control group (Fisher's exact test, P < 0.05). Pool 7 triggered T cell reactivity in 8/21 (38%) of the type 1 diabetic patients, but did not induce reactivity in any control subject. This reactivity represented 57% (8/14) of all the positive responses of the type 1 diabetic patients (Table 1). Although to a lesser extent, diabetic patients also exhibited reactivity to peptides from region 61–192 (pools 2–3), a region to which PBMC from control subjects also reacted. Peptides from the central region of the GAD65 sequence (residues 187–324, pools 4–6) were the least reactive (< 20%) in the two groups studied.
Fig. 2.
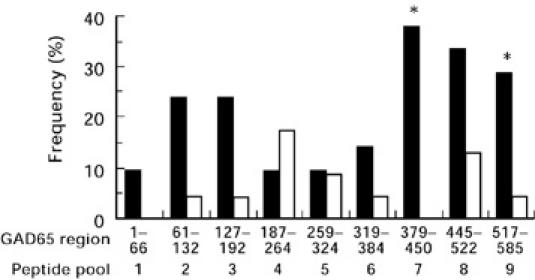
Percentage of individuals demonstrating T cell proliferation (stimulation index (SI) > 3) in the presence of GAD65 peptide pools (1–9) among the 21 newly diagnosed insulin-dependent diabetes mellitus (IDDM) patients (▪) and 23 control subjects (□). For each peptide pool (1–9), the proportion of positive responses among the patients was compared with that among the controls. A significant difference between the response of peripheral blood mononuclear cells (PBMC) from patients and controls was only observed with peptide pool 7 and 9 (*P < 0.05, Fisher's exact test).
Proliferation in the presence of the 11 individual peptides forming pool 7 (GAD residues 379–450) was measured using conserved frozen PBMC from five of the eight type 1 diabetic patients whose PBMC reacted to pool 7 and from five control subjects. No PBMC proliferation was observed in control subjects. Interestingly, all the PBMC from the diabetic patients exhibited a positive proliferative response to one or several of these GAD65 peptides (Fig. 3). Among them, peptide 379–390 initiated proliferation of PBMC from four out of the five patients tested.
Fig. 3.
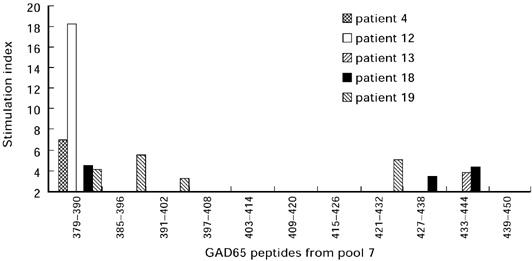
Peripheral blood mononuclear cell (PBMC) proliferation of five type 1 diabetic patients in the presence of each individual peptide forming pool 7 (GAD65 residues 379–450).
GADA and PBMC reponses to GAD65 peptides
Recent-onset type 1 diabetic patients were GADA-positive in 66.6% (14/21) of the cases, whereas all control subjects were GADA-negative (data not shown). Occurrence of both a positive T cell response and the presence of GADA was found in 47.6% (10/21) of the patients. Indeed, we found no significant positive or negative correlation between GADA and T cell stimulation by GAD peptides (r = − 0.09; P > 0.10, Fisher's exact test). In both GADA-positive and GADA-negative patients, practically the same distribution of peptide pools triggering proliferative responses was obtained, indicating that there was no association between GADA and reactivity to a given peptide pool.
HLA and PBMC reponses to GAD65 peptides
Analysis of the HLA specificities showed a very close distribution of the disease-susceptible HLA class II alleles among the type 1 diabetic patients and control subjects (Tables 1 and 2). Several groups of identical HLA class II type 1 diabetic patients could be identified (patients 1 and 2, patients 4 and 5, patients 17 and 18, and patients 14, 15 and 16). The PBMC of patients 14, 15 and 16 were not stimulated by any peptide pool. Patients 4 and 5, and patients 17 and 18 did not elicit strictly identical T cell responses, but showed some homology. Patients 1 and 2 were discordant because only the PBMC from patient 2 gave a positive proliferative response. Patient 21 as well as control subject 15 shared the protective haplotype DR15-DQ6 and both exhibited a T cell response to peptide pool 8 (Tables 1 and 2). Because of the heterogeneity of the HLA haplotypes, we could not ascribe the proliferative response in the presence of a given peptide pool to a specific class II allele in either the type 1 diabetic group or the control group.
Binding of GAD65 peptides to the DPC cells
To evaluate the role of peptide presentation in T cell reactivity, we studied peptide binding on intact DPC cells carrying the HLA class II haplotype which is strongly associated with type 1 diabetes, DRB1*0301/DQB1*0302-DRB1*0401/DQB1*0201 [18]. First, we verified that the DPC cells exhibited surface markers characteristic of activated B cells, namely, CD19, CD40, HLA-DR and HLA-DQ (data not shown). Then, the binding of the 93 individual biotinylated GAD peptides to the DPC cells was measured by cytofluorometric analysis. Twelve peptides were able to bind to the DPC cells with a mean fluorescence greater than five times the background fluorescence determined by incubation with the streptavidin conjugate only (percentage of positive cells > 20%). Figure 4 shows the histograms obtained with three different peptides. To assess the specificity of the binding of these peptides to HLA class II molecules, binding assays were performed with class II-deficient RJ 2.2.5 cells. One peptide (GAD65 residues 205–216) out of the 12 selected with the DPC cells showed significant fluorescence on RJ 2.2.5 cells (Fig. 4). Therefore, the binding of this peptide was not considered to be specific for HLA class II, contrary to the 11 other selected peptides (Table 3). These peptides originated from several regions of the GAD65 sequence. However, three of the five peptides showing the highest binding (mean fluorescence between 82.4 and 105.3) belong to the C-terminal region, in agreement with the proliferation assays (Table 3).
Fig. 4.
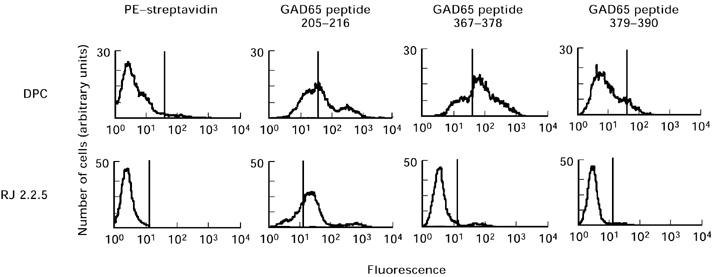
Typical example of flow cytometry histograms of binding of biotinylated GAD65 peptides to DPC cells or HLA class II-deficient RJ 2.2.5 cells.
Table 3.
GAD peptides bound to human HLA class II DRB1*0301/DQB1*0302-DRB1*0401/DQB1*0201

* Mean fluorescence value obtained in a typical experiment. DPC cells were incubated with biotinylated peptides for 18 h at 37°C and then with PE–streptavidin for 30 min at 4°C. Background fluorescence of cells incubated only with PE–streptavidin was subtracted.
DISCUSSION
This study examined human T cell proliferative responses to peptides from the GAD65 protein, one of the dominant antigens involved in type 1 diabetes. T cell reactivity of freshly isolated PBMC was tested in patients with very recent-onset diabetes. The experiments performed using pooled GAD peptides in the present study represent a first step in localizing the immunogenic regions of GAD65 associated with type 1 diabetes. The sensitivity of the PBMC proliferation assay may be somewhat limited by the low frequency of antigen-specific T cells among the PBMC and by possible competition between peptides for binding to HLA molecules. Consequently, cell proliferation expressed in terms of an SI was taken as a qualitative rather than a quantitative criterion and was considered positive when > 3. Under these conditions, we found that 66.6% of patients at diabetes onset showed a positive T cell proliferative response to at least one GAD peptide pool. This frequency is close to that found in previous studies of T cell proliferation with recombinant human GAD65 [6, 23]. However, a high frequency (39.1%) of positive responses was also found with PBMC from control subjects. This finding is also consistent with previous studies demonstrating frequent recognition by control subjects of GAD65 peptides sharing homology with coxsackie virus protein [22], and of several GAD65 and GAD67 peptides [24, 25]. In addition, our findings demonstrate that the nine GAD65 regions defined by the peptide pools were able to elicit a proliferative response of PBMC from type 1 diabetic patients as well as from control subjects, albeit to a variable extent. The N-terminal region (GAD residues 1–66) seemed to be weakly immunogenic, since it was found to be reactive in only two patients. In our study, PBMC from type 1 diabetic patients were more reactive than those from controls to peptides from the C-terminal region (residues 379–585) of GAD65, which includes the region identified in a similar study by Lohmann et al. [25]. The highest proportion of patients with a positive proliferative response was observed in experiments with peptides from the GAD65 region 379–450 (pool 7). This finding was especially noteworthy, since no control subject exhibited reactivity to this peptide pool. Moreover, individual analysis of the peptides from pool 7 showed that peptide 379–390 elicited PBMC proliferation in four out of the five selected diabetic patients with pool 7 PBMC reactivity and did not stimulate the PBMC from the control subjects. In this way, GAD region 379–450, different from the one determined in the study of Lohmann et al. [25], and peptide 379–390 could constitute a novel GAD65 region implicated in type 1 diabetes with a new antigenic motif.
Different type 1 diabetes susceptible or protective haplotypes have been defined [10, 11, 26, 27]. In accordance with these data, a large majority (85.7%) of type 1 diabetic patients included in this study shared at least one HLA DR-DQ haplotype (DR4-DQB1*0201 or DR3-DQB1*0302) which has consistently been observed in association with type 1 diabetes. Analysis of all the complete class II haplotypes showed multiple allele combinations (Table 1), which precluded direct correlation with the observed proliferative responses. Despite this apparent heterogeneity of HLA class II haplotypes in the groups studied, some trends in T cell reactivities were noted. Indeed, 57% (12/21) of the responses of type 1 diabetic patients who showed a significant proliferation were obtained with peptides from the C-terminal region of GAD65. These results might reflect immunodominance of the C-terminal region of the GAD65. We propose the following hypotheses to explain the discrepancy in the T cell reactivity between HLA-identical or semi-identical individuals. First, the lack of sensitivity of the proliferation assay is a possible explanation, since specific T lymphocytes may be present at a very low level in the peripheral circulation. Second, different longitudinal studies have reported a decrease of the T cell response a few months after diabetes onset [5, 13]. It is thus easy to imagine deviation of GAD immunity between the first asymptomatic autoimmune reactivity and the time of clinical diabetes (or of diabetes of long standing). Finally, absence of a proliferative response in the same HLA class II context might be explained by the fact that there is no T cell capable of being activated. This latter situation is particularly highlighted by the similar distribution of the HLA class II alleles among the type 1 diabetic and the control groups.
By flow cytometry, we tried to establish the entire peptide binding capacity of DPC cells expressing two highly disease-susceptible haplotypes DRB1*0301/DQB1*0302 and DRB1*0401/DQB1*0201 in comparison with class II-deficient cells. Since the synthetic GAD65 peptides were blocked at their NH2- (biotinylation) and COOH-termini (carboxamidation), which both represent positions strongly fixed to the edges of the groove of MHC class I molecules [28], we assumed that the selected GAD65 peptides could only bind to the class II molecules on DPC cells. Peptides bound to DPC belong to several GAD regions (Table 3). These results are partially in accordance with previous studies using different approaches to identify GAD65 T cell epitopes in the context of HLA-DR [14, 29, 30]. These latter studies also identified several immunogenic regions in the GAD65 sequence in the context of the DRB1*0401 molecule. Among the 11 peptides we identified in this study only one (GAD65 residues 265–276) corresponds to a GAD sequence with the four putative anchor positions P1, P4, P6 and P9 for the DRB1*0401 molecule: M267, L270, R272 and A275 [28]. Eight out of the 10 other bound peptides contained at least three residues which could represent anchor positions, mostly for DRB1*0401 molecule, but also for DRB1*0301 molecule. For some of these peptides, several combinations of P1, P4, P6 and P9 anchor positions could be determined. In contrast to studies in myasthenia gravis and malaria, which reported a good correlation between the binding observed by FACS analysis and PBMC proliferative responses [31, 32], our study showed little concordance between the results of this binding assay and the proliferation assay. Indeed, DPC cells share independently the same HLA-DQB1 or HLA-DRB1 allele with some patients who showed a proliferative response to regions 1–66, 259–324 and 445–522. However, none of the peptides defined in the proliferation experiments with individual peptides from pool 7 were shown to bind to DPC cells, not even the peptide 379–390 which contained the binding motif of the DRB1*0401 molecule (L381, V384, R386, S389) [28]. Therefore, the ability to screen for potential pathogenic epitopes by measuring the direct binding of biotinylated peptides to diabetic patients' cells as a complementary approach to proliferation assays must be further assessed.
Comparison of the different studies on T cell reactivity to GAD65 suffers from variations in the T cell assays and the purity of the recombinant antigen (First International Workshop on Autoreactive T Cells, Canberra, 1996). The use of peptides could enhance the reproducibility of such studies, since their purity and chemical structure can be easily checked. The few studies, including our own, using GAD65 peptides to identify T cell epitopes in human diabetes [13, 14, 23–25] reported relatively discordant results regardless of the HLA restriction. However, our findings demonstrate that the GAD65 protein exhibits several immunogenic determinants in its entire sequence, but only those from the C-terminal region seem to be associated with type 1 diabetes.
Acknowledgments
We thank the Departments of Paediatric and Adult Endocrinology in Lyon and Saint-Etienne for recruitment of patients, the Blood Transfusion Centre of Lyon for HLA typing and the Blood Transfusion Centre of Saint-Etienne, especially Dr L. Absi, for the recruitment of the bone marrow donor subjects and for HLA typing. We thank A. Durand, S. Monbeig and A. Stefanutti for excellent technical assistance. We are indebted to Professor R. Sabatier for helpful statistical analysis and to S.L. Salhi for critical reading of the manuscript.
REFERENCES
- 1.Baekkeskov S, Aanstoot HJ, Christgau S, et al. Identification of the 64K autoantigen in insulin-dependent diabetes as the GABA-synthesizing enzyme glutamic acid decarboxylase. Nature. 1990;347:151–6. doi: 10.1038/347151a0. [DOI] [PubMed] [Google Scholar]
- 2.Nepom GT. Glutamic acid decarboxylase and other autoantigens in IDDM. Curr Opin Immunol. 1995;7:825–30. doi: 10.1016/0952-7915(95)80055-7. [DOI] [PubMed] [Google Scholar]
- 3.Payton MA, Hawkes CJ, Christie MR. Relationship of the 37,000- and 40,000–Mr tryptic fragments of islet antigens in IDDM to the protein tyrosine phosphatase-like molecule IA-2 (ICA512) J Clin Invest. 1995;96:1506–11. doi: 10.1172/JCI118188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brooks-Worrell BM, Starkebaum GA, Greenbaum C, Palmer JP. Peripheral blood mononuclear cells of insulin-dependent diabetic patients respond to multiple islet cell proteins. J Immunol. 1996;157:5668–74. [PubMed] [Google Scholar]
- 5.Durinovic-Bello I, Hummel M, Ziegler A-G. Cellular immune response to diverse islet cell antigens in IDDM. Diabetes. 1996;45:795–800. doi: 10.2337/diab.45.6.795. [DOI] [PubMed] [Google Scholar]
- 6.Roep BO. T-cell responses to autoantigens in IDDM. The search for the holy Grail. Diabetes. 1996;45:1147–56. doi: 10.2337/diab.45.9.1147. [DOI] [PubMed] [Google Scholar]
- 7.Honeyman MC, Stone N, De Aizpurua H, Rowley MJ, Harrison LC. High T cell responses to glutamic acid decarboxylase (GAD) isoform 67 reflect a hyperimmune state that precedes the onset of insulin-dependent diabetes. J Autoimmun. 1997;10:165–73. doi: 10.1006/jaut.1996.0124. [DOI] [PubMed] [Google Scholar]
- 8.Kaufman DL, Clare-Salzler M, Tian J, et al. Spontaneous loss of T-cell tolerance to glutamic acid decarboxylase in murine insulin-dependent diabetes. Nature. 1993;366:69–72. doi: 10.1038/366069a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tish R, Yang XD, Singer SM, Liblau RS, Fugger L, McDevitt HO. Immune response to glutamic acid decarboxylase correlates with insulitis in non-obese diabetic mice. Nature. 1993;366:72–75. doi: 10.1038/366072a0. [DOI] [PubMed] [Google Scholar]
- 10.She J-X. Susceptibility to type 1 diabetes: HLA-DQ and DR revisited. Immunol Today. 1996;17:323–9. doi: 10.1016/0167-5699(96)10014-1. [DOI] [PubMed] [Google Scholar]
- 11.Boitard C, Caillat-Zucman S, Timsit J. Insulin-dependent diabetes and human leucocyte antigens. Diabetes Metabol. 1997;23:22–28. [PubMed] [Google Scholar]
- 12.Walden P. T-cell epitope determination. Curr Opin Immunol. 1996;8:68–74. doi: 10.1016/s0952-7915(96)80107-5. [DOI] [PubMed] [Google Scholar]
- 13.Bach JM, Otto H, Nepom G, Jung G, Cohen H, Timsit J, Boitard C, Van Endert PM. High affinity presentation of an autoantigenic peptide in type I diabetes by an HLA class II protein encoded in a haplotype protecting from disease. J Autoimmun. 1997;10:375–86. doi: 10.1006/jaut.1997.0143. [DOI] [PubMed] [Google Scholar]
- 14.Endl J, Otto H, Jung G, et al. Identification of naturally processed T cell epitopes from glutamic acid decarboxylase presented in the context of HLA-DR alleles by T lymphocytes of recent onset IDDM patients. J Clin Invest. 1997;99:2405–15. doi: 10.1172/JCI119423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rharbaoui F, Granier C, Kellou M, Mani J-C, van Endert P, Madec A-M, Boitard C, Bouanani M. Peptide specificity of high-titer anti-glutamic acid decarboxylase (GAD) 65 autoantibodies. Immunol Letters. 1998;62:123–30. doi: 10.1016/s0165-2478(98)00036-4. [DOI] [PubMed] [Google Scholar]
- 16.Petersen JS, Hejnaes KR, Moody A, et al. Detection of GAD65 antibodies in diabetes and other autoimmune diseases using a simple radioligand assay. Diabetes. 1994;43:459–67. doi: 10.2337/diab.43.3.459. [DOI] [PubMed] [Google Scholar]
- 17.Schmidli RS, Colman PG, Bonifacio E participating laboratories. Disease sensitivity and specificity of 52 assays for glutamic acid decarboxylase antibodies. The second international GADAb Workshop. Diabetes. 1995;44:636–40. doi: 10.2337/diab.44.6.636. [DOI] [PubMed] [Google Scholar]
- 18.Madec A-M, Rousset F, Ho S, Robert F, Thivolet C, Orgiazzi J, Lebecque S. Four IgG anti-islet human monoclonal antibodies isolated from a type 1 diabetes patient recognize distinct epitopes of glutamic acid decarboxylase 65 and are somatically mutated. J Immunol. 1996;156:3541–9. [PubMed] [Google Scholar]
- 19.Accolla RS. Human B cell variants immunoselected against a single Ia antigen subset have lost expression of several Ia antigen subsets. J Exp Med. 1983;157:1053–8. doi: 10.1084/jem.157.3.1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bodmer JG, Marsh SG, Albert ED, et al. Nomenclature for factors of the HLA system, 1995. Tissue Antigens. 1995;46:1–18. doi: 10.1111/j.1399-0039.1995.tb02470.x. [DOI] [PubMed] [Google Scholar]
- 21.Busch R, Strang G, Howland K, Rothbard JB. Degenerate binding of immunogenic peptides to HLA-DR proteins on B cell surfaces. Int Immunol. 1990;2:443–51. doi: 10.1093/intimm/2.5.443. [DOI] [PubMed] [Google Scholar]
- 22.Schloot NC, Roep BO, Wegmann DR, Yu L, Wang TB, Eisenbarth GS. T-cell reactivity to GAD65 peptide sequences shared with coxsackie virus protein in recent-onset IDDM, post-onset IDDM patients and control subjects. Diabetologia. 1997;40:332–8. doi: 10.1007/s001250050683. [DOI] [PubMed] [Google Scholar]
- 23.Hummel M, Durinovic-Bello I, Ziegler A-G. Relation between cellular and humoral immunity to islet cell antigens in type 1 diabetes. J Autoimmun. 1996;9:427–30. doi: 10.1006/jaut.1996.0059. [DOI] [PubMed] [Google Scholar]
- 24.Atkinson MA, Bowman MA, Campbell L, Darrow BL, Kaufman DL, Maclaren NK. Cellular immunity to a determinant common to glutamic acid decarboxylase and coxsackie virus in insulin dependent diabetes. J Clin Invest. 1994;94:2125–9. doi: 10.1172/JCI117567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lohmann T, Leslie RDG, Hawa M, Geysen M, Rodda S, Londei M. Immunodominant epitopes of glutamic acid decarboxylase 65 and 67 in insulin-dependent diabetes mellitus. Lancet. 1994;343:1607–8. doi: 10.1016/s0140-6736(94)93061-9. [DOI] [PubMed] [Google Scholar]
- 26.Van der Auwera B, Van Waeyenberge C, Schuit F, Heimberg H, Vandewalle C, Gorus F, Flament J Belgian Diabetes Registry. DRB1*0403 protects against IDDM in caucasians with the high-risk heterozygous DQA1*0301-DQB1*0302/DQA1*0501-DQB1*0201 genotype. Diabetes. 1995;44:527–30. doi: 10.2337/diab.44.5.527. [DOI] [PubMed] [Google Scholar]
- 27.Undlien DE, Friede T, Rammensee H-G, et al. HLA-encoded genetic predisposition in IDDM. Diabetes. 1997;46:143–9. doi: 10.2337/diab.46.1.143. [DOI] [PubMed] [Google Scholar]
- 28.Rammensee H-G. Chemistry of peptides associated with MHC class I and II molecules. Curr Opin Immunol. 1995;7:85–96. doi: 10.1016/0952-7915(95)80033-6. [DOI] [PubMed] [Google Scholar]
- 29.Wicker LS, Chen SL, Nepom GT, et al. Naturally processed T cell epitopes from human glutamic acid decarboxylase identified using mice transgenic for the type 1 diabetes-associated human MHC class II allele, DRB1*0401. J Clin Invest. 1996;98:2597–603. doi: 10.1172/JCI119079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Patel SD, Cope AP, Congia M, Chen TT, Kim E, Fugger L, Wherrett D, Sonderstrup-McDevitt G. Identification of immunodominant T cell epitopes of human glutamic acid decarboxylase 65 by using HLA-DR (A1*0101, B1*0401) transgenic mice. Proc Natl Acad Sci USA. 1997;94:8082–7. doi: 10.1073/pnas.94.15.8082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zisman E, Brautbar C, Sela M, et al. Binding of peptides of the human acetylcholine receptor æ-subunit to HLA class II of patients with myasthenia gravis. Human Immunol. 1995;44:121–30. doi: 10.1016/0198-8859(95)00094-1. [DOI] [PubMed] [Google Scholar]
- 32.Calvo-Calle JM, Hammer J, Sinigaglia F, Clavijo P, Moya-Castro ZR, Nardin EH. Binding of malaria T cell epitopes to DR and DQ molecules in vitro correlates with immunogenicity in vivo. J Immunol. 1997;159:1362–73. [PubMed] [Google Scholar]