

Abstract
Mice with experimental anti-phospholipid syndrome (APS), induced by active immunization with a human anti-cardiolipin MoAb (H-3), were treated with mouse anti-idiotypic MoAb (anti-H3, named S2.9) and with an irrelevant anti-idiotype. The immunized mice produced high titres of mouse anti-cardiolipin antibodies along with clinical manifestations of experimental APS: prolonged activated partial thromboplastin time (aPTT), thrombocytopenia and high rate of fetal loss. Treatment with the specific anti-Id (S2.9) as a whole molecule or F(ab)2 fraction, resulted in a decrease in serum levels of the anti-cardiolipin antibodies, rise in platelet count, shortened aPTT and reduced rate of fetal loss. The anti-Id effect was associated with a rise in the number of IL-2 and interferon-gamma (IFN-γ)-secreting cells (Th1) and reduction in IL-4- and IL-6-secreting cells (Th2). The beneficial effect of the anti-Id treatment in mice with experimental APS induced by active immunization with an idiotype further supports the idiotypic aetiology of experimental APS and points to the role of Th1 cytokines in suppression of its manifestations.
Keywords: anti-phospholipid syndrome, anti-idiotype, Th1 cells, Th2 cells, anti-cardiolipin antibodies
INTRODUCTION
Experimental anti-phospholipid syndrome (APS) may be induced in naive mice by active immunization with human and mouse anti-phospholipid and anti-DNA antibodies [1–5]. Upon stimulation with the autoantibody carrying a specific idiotype (Ab1), naive mice develop anti-antibody (anti-Id = Ab2), and after 2 months an anti-anti-autoantibody (anti-anti-Id = Ab3), that has similar binding characteristics to Ab1, is generated [1]. Mice with experimental APS express the typical serological and clinical manifestations of the APS entailing the presence of anti-cardiolipin (CL) and anti-phosphatidylserine (PS) antibodies, prolonged activated partial thromboplastin time (aPTT), thrombocytopenia and high rate of fetal loss. The availability of this experimental model provided a possibility to evaluate various therapeutic modalities such as the administration of intravenous immunoglobulins (IVIG) [6], recombinant IL-3 [7], anti-CD4 MoAb [8], bromocriptine [9], aspirin [10], low molecular weight heparin [11], and bone marrow transplantation [12].
Since autoimmune diseases were attributed, among other causes, to the disturbance of the idiotypic network, several studies examined the effect of anti-idiotypic modulation on these experimental models [13, 14]. Successful in vitro and in vivo manipulations of autoantibody production by anti-idiotypic (anti-Id) antibodies were previously described in experimental systemic lupus erythematosus (SLE), lupus nephritis, autoimmune tubulointerstitial nephritis, collagen arthritis, experimental myasthenia gravis, autoimmune uveoretinitis and experimental allergic encephalomyelitis [15–21]. On the basis of previous observations and the importance of idiotypes in the pathogenesis of experimental APS we tried to immunomodulate the disease in mice with a specific anti-idiotypic MoAb.
Despite the fact that APS is regarded as an autoantibody-mediated autoimmune condition [22], T cells have been shown to be important in induction and regulation of experimental APS [12, 23]. Two distinct Th cell clones, Th1 and Th2, were identified. The balance between these two Th cell populations and their modulation was shown to play a crucial role in several autoimmune diseases [24]. Indeed, evaluation of the cytokine-secreting splenocytes before and after treatment of experimental APS with the specific anti-Id antibodies pointed to a shift in Th2 to Th1 cytokine pattern.
MATERIALS AND METHODS
Monoclonal antibodies
H-3
Human IgM anti-CL MoAb is generated from a healthy subject immunized with diphtheria and tetanus toxoid [25, 26], and is able to induce experimental APS [1].
S2.9
Mouse IgG anti-idiotypic MoAb is derived from a mouse immunized with H-3 MoAb [26].
Anti-SA-1
A mouse anti-idiotypic directed to anti-DNA antibody was derived from a mouse immunized with SA-1 (an anti-DNA MoAb) [13].
HIgM
Irrelevant human IgM.
Induction of experimental APS in mice
Twelve-week-old BALB/c female mice were immunized intra–dermally with 10 μg of anti-CL MoAb (H-3), emulsified in Freund's complete adjuvant (FCA), in the hind footpads according to previous detailed protocols [1–3]. Three weeks later a boost injection of the H-3 (10 μg) in PBS was given into the same location. The sera of the mice were examined for the presence of anti-CL and anti-PS and anti-ssDNA antibodies every 2 weeks during the period of the treatment. Th1/Th2 cytokine-producing cells (interferon-gamma (IFN-γ), IL-2, and IL-4, IL-6, respectively) were tested monthly, 8 weeks after the boost and at the end of the treatment. Antibody-producing cells were studied at the end of the treatment. The induction of pregnancy (mating procedure) was started 12–14 weeks following the boost immunization. The pregnant mice were killed 15 days after the plug appearance in order to determine the rate of the fetal loss.
Treatment of mice with experimental APS
The treatment was started 2 months after the boost injection and given during 8 weeks. The mice were daily injected intra–peritoneally with 200 μg of whole molecule of S2.9, S2.9 F(ab)2, S2.9 Fc fragment, PBS or with irrelevant anti-idiotypic antibody (mouse anti-SA-1, whole molecule and F(ab)2). A group of five mice was killed 1 month after the boost injection in order to determine the number of cytokine-producing cells before the treatment. The number of cytokine-producing cells and the level of cytokines in the serum following treatment was studied in another group of five non-pregnant mice 8 weeks after the treatment was given.
Preparation of F(ab)2 from the S2.9 MoAb
F(ab)2 fragments were prepared from the mouse IgG anti-idiotypic antibody named S2.9 which was dialysed against 100 mm Na-acetate buffer, pH 4.0, and digested with pepsin (2% w/w; Sigma Chemical Co., St Louis, MO) at 37°C for 18 h [27]. Any remaining traces of undigested IgG and Fc fragments were removed by binding to a protein A column (Pharmacia Biotech, Norden AB Sollentuna, Sweden). The efficiency of the S2.9 digestion was confirmed by 12% SDS–PAGE.
Detection of anti-phospholipid antibodies
The titres of anti-phospholipid antibodies in the sera of the immunized mice (anti-CL, anti-PS) were detected by ELISA as previously described [1–3]. Briefly, phospholipid-coated plates were blocked with 5% bovine serum (BS). Mice sera at different dilutions were added, and incubated for 2 h at room temperature. The mice antibodies bound to the solid phase were detected using goat anti-mouse IgG conjugated to alkaline phosphatase (Jackson Immunoresearch Labs Inc., West Grove, PA) and the addition of its substrate p-nitrophenyl phosphate (Sigma). The colour reaction was detected by appropriate substrate and read by employing a Titertek ELISA reader at an optical density (OD) of 405 nm. Each step was followed by extensive washings with PBS.
Detection of anti-ssDNA antibodies
Anti-ssDNA antibodies were detected as previously described [1]. Briefly, polystyrene plates (96 wells; Nunc, Roskilde, Denmark) were coated sequentially with poly-l-lysine (50 μg/ml in water), calf thymus ssDNA (2.5 μg/ml in TBS), and treated with S1-nuclease in nuclease buffer at 37°C and poly-l-glutamate (50 μg/ml).
Detection of lupus anti-coagulant
The presence of lupus anti-coagulant was evaluated by the prolongation of aPTT in a mixing test [1–3], adding one volume of plasma from whole blood mixed with Na-citrate 0.123 mol/l, in a 9 : 1 ratio, to one volume of cephalin, and incubation for 2 min at 37°C. Another volume of 0.02 m CaCl2 was added and the clotting time was recorded in seconds.
Platelet counts
Platelet counts from individual blood samples were quantified in diluted blood using a single optical cytometer (Coulter Counter HC Plus Cell Control; Counter Electronics Ltd, Luton, UK) [1–3].
Evaluation of the pregnancy outcome
The mice vaginal plugs indicating mating were recorded and regarded as day 0 of pregnancy. The mice were killed on day 15 of pregnancy. The percentage index of resorptions of embryos in utero was calculated as follows: %R = R/(R + F): the number of resorbed fetuses (R) divided by resorbed and full term (F) fetuses.
Cytokine-secreting cell ELISPOT assay
Mouse splenocytes (day 15 of pregnancy) were prepared by teasing the spleen and passing through a 45-μm nylon mesh. Following centrifugation at 1400 rev/min, the cells were depleted from macrophages by preincubation on plastic Petri dishes for 1 h at 37°C. The detection of cytokine-secreting cells was determined employing a cytokine-secreting ELISPOT assay as previously described [28]. ELISA plates (Nunc) were coated with capture MoAb 2 μg/ml diluted in PBS (Pharmingen Research Products, Cambridge, UK) and incubated overnight at 4°C. After washing twice with PBS–Tween, the plates were blocked with 200 μg of 25% fetal calf serum (FCS) in PBS for 2 h at 37°C. Splenocytes diluted in complete medium were added and incubated at 37°C in 7% CO2 overnight. Following extensive washings, diluted biotinylated anti-cytokine detecting MoAb (2 μg/ml; Pharmingen) in PBS–Tween containing 1% bovine serum albumin (BSA) was added to detect the secreted cytokine. The plates were incubated at 4°C in a humidified chamber overnight and washed with PBS–Tween. Streptavidin-alkaline phosphatase (Pharmingen) in PBS–Tween 1% FCS was added (100 μg/well) and the plates were incubated for 30 min at 37°C and washed with PBS–Tween. BCIP (50 μg/well; Sigma) (1 mg/ml), dissolved in AMP buffer, warmed to 45°C and mixed with agarose dissolved at 3% in ddH2O, were added. Incubation at 37°C for 8 h resulted in blue spots that were counted under the light microscope.
Determination of cytokine secretion
The measurement of the concentration of IL-2, IFN-γ, IL-4 and IL-6 in the sera of mice was performed by commercial kits (R&D Research and Diagnostic Systems, Minneapolis, MN).
Spot ELISA to determine anti-phospholipid and anti-DNA- secreting cells
Mouse splenocytes (1 × 107 cells/ml) were assayed for their ability to secrete in vitro anti-CL, anti-PS and anti-ssDNA antibodies [15]. The preparation of splenocytes was done by teasing the spleen and passing the splenocytes through 0.45-μm nylon wool. The erythrocytes were lysed with 0.83% Tris-buffered ammonium chloride. The cells were seeded in RPMI 1640 into 24-well tissue culture plates (Nunc) precoated with CL, PS or ssDNA. Anti-mouse polyvalent alkaline phosphatase was added for 2 h at 37°C. Following extensive washings, BCIP (Sigma) was added in 2-aminopropranolol Triton X-405 MgCl2 buffer to 3% agar (type I, low electroendosmosis; Sigma) heated and diluted in BCIP buffer at a 4:1 ratio, resulting in a 0.6% agar solution. Overnight incubation in 37°C resulted in blue spots. The specific ELISPOTS were evaluated in comparison with ELISPOT of total IgG- secreting cells.
RESULTS
The effect of treatment with S2.9 on the manifestations of experimental APS
Treatment of experimental APS with whole molecule of S2.9 resulted in a decrease in the mean s.d. of the serum levels of the anti-CL and anti-PS antibodies (1.491 ± 0.249 OD and 1.678 ± 0.327 OD before treatment and 0.467 ± 0.67 and 0.505 ± 0.99 after treatment, respectively; P < 0.001) (Table 1). Treatment with the whole molecule of S2.9 shortened the aPTT (42 ± 4 s versus 69 ± 4 s in mice treated with PBS; P < 0.001) (Table 1). The platelet count in mice treated with S2.9 whole molecule was significantly higher (989 242 × 103/mm3) than in mice treated with an irrelevant anti-Id (528 ± 176/mm3) (P < 0.001), and the rate of fetal loss was reduced in the treated group (18 ± 2%) in comparison with the mice given an irrelevant anti-Id (63 ± 7%; P < 0.001) (Table 1). The titres of anti-phospholipid antibodies, aPTT, platelet count and the rate of fetal loss in mice treated with F(ab)2 of S2.9 did not differ significantly from those of the mice treated with S2.9 as a whole molecule (Table 1). The application of Fc fragment of S2.9, or an irrelevant MoAb (anti-SA-1) did not alter the clinical or serological findings of experimental APS (Table 1).
Table 1.
Serological and clinical manifestations in mice with experimental anti-phospholipid syndrome (APS), treated with specific (S2.9) and irrelevant mouse anti-SA-1 (MαSa-1) anti-idiotypic antibodies

* Sera were tested 2 months after boost administration (= before treatment) and 8 weeks after treatment, at a dilution of 1 : 200. Values are expresssed as mean ± s.d. OD at 405 nm, the numbers in parentheses demonstrate the values before treatment; each group represents 25 mice.
† aPTT, Activated partial thromboplastin time (n = 20).
Percent fetal loss = % resorption = resorbed fetuses/resorbed + full term fetuses.
‡ Statistical analyses were performed by anova test. P < 0.001 was found in the groups of exp. APS treated with S2.9 whole molecule (WM), S2.9 F(ab)2, in comparison with the values before treatment and after treatment with S2.9 Fc.
P > 0.5 when treatment was carried out with an irrelevant anti-idiotypic antibody (mouse anti-SA-1).
Analysis of the kinetics of the immunosuppressive effect of the treatment showed a decline in the titres of the autoantibodies in sera of mice given the whole molecule or the F(ab)2 fragment of S2.9 during the 8 weeks of administration (Fig. 1). Discontinuation of the injections of the S2.9 was followed by a rise in the titres of anti-phospholipid antibodies, yet 6 weeks after the cessation of the administration these titres were still below pretreatment levels (Fig. 1).
Fig. 1.

Kinetics of mouse anti-cardiolipin titres in the sera of mice with experimental anti-phospholipid syndrome (APS) following treatment with a specific (S2.9) and irrelevant mouse anti-SA-1 (MαSA-1) anti-idiotypic antibodies, whole molecule (WM), F(ab)2 and Fc fragments (n = 5).
Effect of treatment with S2.9 on cytokine-secreting cells
One month after the boost injection with H-3 antibody the number of IL-2-secreting cells was higher by 8.9, IFN-γ by 7.5, IL-4 by 1.8 and IL-6 by 2.1 than the numbers in the non-immunized mice (P < 0.001) (Fig. 2). No significant difference was found between the number of cytokine-producing cells 1 month following boost injection of H-3 antibody and human IgM. Two months following the boost injection the mice were treated with S2.9 whole molecule, F(ab)2, Fc fragments, PBS and irrelevant anti-Id (anti-SA-1). The profile of Th1/Th2 cytokine-producing cells was studied at the end of the treatment. The treatment was given during 8 weeks and five mice from each group were killed in order to evaluate the number of cytokine- and antibody-producing cells. Treatment with whole molecule S2.9 and F(ab)2 fragment S2.9 reduced the number of Th2 IL-4-secreting cells to 45% of its pretreatment value, as well as significantly lowering the number of IL-6-secreting cells (to 31% and 59% of the pretreatment values, respectively) (Fig. 3). The application of anti-SA-1 antibody or the Fc fragment of the S2.9 antibody did not affect the number of various cells secreting cytokines.
Fig. 2.

Representative profile of cytokine spot-forming cells 1 month after boost injection (n = 5 for each group). Data are expressed as mean ± s.d. for each mouse.
Fig. 3.

The number of IL-4- and IL-6-secreting splenocytes from mice with experimental anti-phospholipid syndrome (APS) 8 weeks after treatment with specific (S2.9) and irrelevant mouse anti-SA-1 (MαSA-1) anti-idiotypic antibodies, whole molecule (WM), F(ab)2 and Fc fragments (n = 5).
Figure 4 demonstrates that the treatment with S2.9 whole molecule and S2.9 F(ab)2 fragment resulted in an increase in the number of Th1 IL-2-producing cells by 1.7 from the number counted before treatment. Production of IFN-γ increased by 1.8 following treatment with S2.9.
Fig. 4.

The number of IL-2- and IFN-γ-secreting splenocytes in mice with experimental anti-phospholipid syndrome (APS) following treatment with specific (S2.9) anti-idiotypic antibody and irrelevant mouse anti-SA-1 (MαSA-1) anti-idiotypic antibodies, whole molecule (WM), F(ab)2 and Fc fragments.
The effect of treatment with S2.9 on the concentrations of cytokines in the sera of APS mice
The treatment with S2.9 resulted in a rise in the concentrations of IL-2 and IFN-γ and decrease in the concentrations of IL-4 and IL-6 (P < 0.001; Fig. 5). Treatment with an irrelevant anti-Id (anti-SA-1) did not alter significantly the concentrations of the cytokines in the mice sera.
Fig. 5.

The concentrations of Th1/Th2 cytokines in the sera of anti-phospholipid syndrome (APS) mice, following treatment with specific (S2.9) and irrelevant mouse anti-SA-1 (MαSA-1) anti-idiotypic antibodies, whole molecule (WM), F(ab)2 and Fc fragments (n = 10).
The effect of treatment with S2.9 on specific autoantibody- producing cells
The treatment with whole molecule S2.9 or the F(ab)2 fragment resulted in a reduction in the number of anti-CL antibody forming cells by 3.4 and 2.7, respectively (Fig. 6). The number of anti-PS antibody-secreting cells decreased by 6.7 and 7.8 after treatment with S2.9 whole molecule and with the F(ab)2 fragment. Treatment with the Fc fragment of S2.9 or with the anti-SA-1 irrelevant anti-Id did not affect significantly the number of anti-CL or anti-PS forming cells (Fig. 6)
Fig. 6.
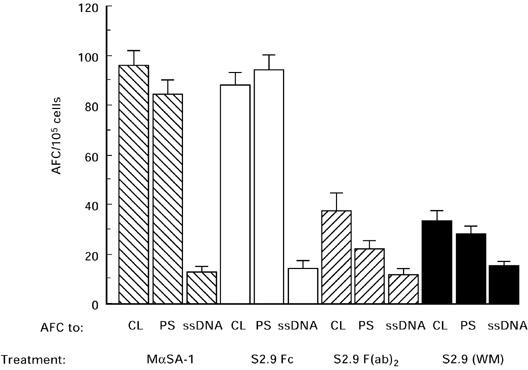
Anti-cardiolipin (CL), anti-phosphatidylserine (PS) and anti-ssDNA antibody-forming cells (AFC) in mice with experimental anti-phospholipid syndrome (APS) following treatment with specific (S2.9) and irrelevant mouse anti-SA-1 (MαSA-1) anti-idiotypic antibodies, whole molecule (WM), F(ab)2 and Fc fragments.
DISCUSSION
In the present study experimental APS was induced in mice by active immunization with human anti-CL MoAb (H3). The mice were treated intraperitoneally with a specific mouse anti-Id antibody (S2.9). Treatment with anti-Id antibody as a whole molecule or as a F(ab)2 fragment resulted in a decrease in the serum level of the anti-phospholipid antibodies and a subsidence of the clinical signs of APS (prolonged aPTT, thrombocytopenia and fetal resorptions). These findings support the potential for idiotypic manipulation in APS. A recent study in which an experimental APS was successfully treated with high-dose IVIG [6] provides additional support to the potency of anti-Id treatment in this condition, since IVIG efficacy in autoimmune states is attributed mainly to its content of a panoply of anti-Id antibodies [29].
In order to evaluate further possible mechanisms by which the suppressive effect of anti-Id antibody on experimental APS is achieved, we examined the number of Th1 and Th2 cells and the concentration of the cytokines secreted by these cells: IL-2, IFN-γ and IL-4, IL-6, respectively. Treatment with anti-Id S2.9 resulted in a rise in the number of IL-2- and IFN-γ-secreting cells (Th1) as well as elevation of the concentrations of these cytokines in the sera of the mice and reduction in the number of IL-4- and IL-6-producing cells (Th2), along with a decrease in the concentrations of the latter lymphokines in the sera of APS mice. This phenomenon was accompanied by a decrease in the number of specific anti-CL and anti-PS antibody-forming cells.
In previous studies we have pointed to the importance of T cells in the induction of experimental SLE and APS. The induction of experimental SLE and APS was achieved by syngeneic bone marrow transplantation [12, 30]. Experimental APS may be induced in mice by infusion of Th2clone specific for anti-PS (unpublished data). The results of the present study suggest that Th2 cells have the important role in the development of APS in mice, while a switch of Th2 to Th1 pattern is associated with induction of remission. Various diseases have been shown to be associated with predominance of either Th1 or Th2 cell type [24, 31]. Dominance of Th1 cells was found in patients and in animal models of organ-specific autoimmune states such as Grave's disease [32], rheumatoid arthritis [33], experimental autoimmune encephalomyelitis [34] and insulin-dependent diabetes mellitus (IDDM) in non-obese mice [35].
The situation seems different in systemic autoimmune disorders. For instance, it was shown recently that autoimmune vasculitis in Brown Norway rats induced by injection of gold or mercuric chloride (HgCl2) is associated with reduction in Th1-like activity and increase in production of IL-4, which is secreted by Th2 cells [36]. Several observations support the role of Th2 cells in the immunopathogenesis of SLE in humans and in animal models. Prevalent Th2 activity was found in experimental SLE induced by allogeneic stimulation or chemicals [37], and in murine lupus [38]. In humans, high expression of IL-4, IL-6, IL-10 and IFN-γ (Th2 profile) was found in SLE patients compared with healthy subjects [39], and elevated levels of CD30, a known marker of Th2 cells, were detected in patients with active lupus [40].
Despite the above, the interaction between Th1 and Th2 cells in SLE is probably more complex. For example, IFN-γ, although secreted mainly by Th1 cells, was found to be essential for the pathogenesis of SLE in the murine model (NZB/W mice) [41] and in human disease [42]. High concentrations of this cytokine probably augment subsequent Th2 activity and aggravate the severity of disease progression. A recent study in which SLE in mice was induced by active immunization with human anti-DNA MoAbs showed that the induction and development of experimental SLE were dependent on two stages of T cell activation and cytokine secretion. First, the Th1-type represented by high IL-2 and IFN-γ production, followed later by the Th2-type cytokine secretion (high IL-4 and IL-10 production) [43].
APS is closely related to SLE. In humans, APS may develop as a secondary disorder in the course of SLE. Mice with experimental SLE induced by active immunization with human anti-DNA antibody exhibit, along with elevation in the titre of anti-DNA antibodies and clinical signs of SLE, high levels of anti- phospholipid antibodies and clinical manifestations of APS [3]. The results of the present study suggest that there is a Th2predominance in the course of experimental APS in mice, similar to their prevalence in SLE in the NZB/W model. It is still not clear whether the Th1and Th2 profile noted in APS is primary or secondary to the underlying immunopathogenesis.
Alteration of the cytokine profile in autoimmune conditions has been utilized to modulate their natural course. For instance, antibodies to IL-12, a major Th1stimulant, inhibited the development of IDDM in non-obese diabetic (NOD) mice [44]. Treatment with an anti-IL-10 MoAb delayed the onset of autoimmune manifestations of lupus-prone NZB/W mice, whereas administration of IL-10, a Th2 cytokine, resulted in exacerbation of the disease [45].
In our study the switch from Th1 to Th2 cells in mice with experimental APS was induced by treatment with a specific anti-Id antibody. The fact that F(ab)2 fragment was as efficient as the whole molecule suggests that the anti-Id caused neutralization of the circulating Id in the serum. The effect of the administration of the anti-Id antibody was transient: discontinuation of the treatment resulted in a rise of the titres of the anti-phospholipid antibodies. This may be explained by the fact that the cytokine secretion profile following treatment was similar to the profile recorded 1 month after the boost injection, when the serological and clinical manifestations were not yet evident, but the cascade of Th cell activation had been already initiated. Further studies are needed in order to evaluate the effect of pretreatment of mice with experimental APS before or immediately after induction of the disease.
It seems that experimental APS in mice is a Th2 predominant state. A shift toward Th1 predominance brings about subsidence of clinical and serological manifestations of the disease, and this shift may be promoted by treatment with a specific anti-Id antibody.
Acknowledgments
This study was supported by the Japanese-Israeli Binational Grant for Research, no. 6113, Israeli Ministry of Science.
REFERENCES
- 1.Bakimer R, Fishman P, Blank M, et al. Induction of primary anti-phospholipid syndrome in mice by immunization with a human monoclonal anti-cardiolipin antibody (H-3) J Clin Invest. 1995;89:1558–63. doi: 10.1172/JCI115749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blank M, Tincani A, Shoenfeld Y. Induction of experimental anti-phospholipid syndrome in naive mice with purified IgG anti- phosphatidylserine antibodies. J Rheumatol. 1994;21:100–4. [PubMed] [Google Scholar]
- 3.Blank M, Krause I, Ben-Bassat M, et al. Induction of experimental anti-phospholipid syndrome associated with SLE following immunization with human monoclonal pathogenic anti-DNA idiotype. J Autoimmun. 1992;5:495–509. doi: 10.1016/0896-8411(92)90008-e. [DOI] [PubMed] [Google Scholar]
- 4.Pierangeli SS, Harris EN. Induction of phospholipid-binding antibodies in mice and rabbits by immunization with human β2 glycoprotein 1 or anti-cardiolipin antibodies alone. Clin Exp Immunol. 1993;93:269–72. doi: 10.1111/j.1365-2249.1993.tb07978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Branch DW, Dudley DJ, Mitchell DM, et al. Immunoglobulin G fractions from patients with anti-phospholipid antibodies cause fetal death in BALB/c mice: a model for autoimmune fetal loss. Am J Obstet Gynecol. 1990;164:210–6. doi: 10.1016/s0002-9378(11)90700-5. [DOI] [PubMed] [Google Scholar]
- 6.Krause I, Blank M, Kopolovic J, et al. Abrogation of experimental systemic lupus erythematosus and primary antiphospholipid syndrome with intravenous gamma globulin. J Rheumatol. 1995;22:1068–74. [PubMed] [Google Scholar]
- 7.Fishman P, Falash Vaknine E, Zigelman R, et al. Prevention of fetal loss in experimental anti-phospholipid syndrome by in vivo administration of recombinant interleukin-3. J Clin Invest. 1993;91:1834–7. doi: 10.1172/JCI116396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tomer Y, Blank M, Shoenfeld Y. Suppression of experimental antiphospholipid syndrome and systemic lupus erythematosus in mice by anti-CD4 monoclonal antibodies. Arthritis Rheum. 1994;37:1236–44. doi: 10.1002/art.1780370819. [DOI] [PubMed] [Google Scholar]
- 9.Blank M, Krause I, Buskila D, et al. Bromocriptine immunomodulation of experimental SLE and primary anti-phospholipid syndrome via induction of nonspecific T suppressor cells. Cell Immunol. 1995;162:114–22. doi: 10.1006/cimm.1995.1058. [DOI] [PubMed] [Google Scholar]
- 10.Krause I, Blank M, Gilbrut B, Shoenfeld Y. The effect of aspirin on recurrent fetal loss in experimental antiphospholipid syndrome. Am J Reprod Immunol. 1993;29:155–61. doi: 10.1111/j.1600-0897.1993.tb00581.x. [DOI] [PubMed] [Google Scholar]
- 11.Inbar O, Blank M, Faden D, et al. Prevention of fetal loss in experimental antiphospholipid syndrome by low-molecular-weight heparin. Am J Obstet Gynecol. 1993;169:423–6. doi: 10.1016/0002-9378(93)90100-w. [DOI] [PubMed] [Google Scholar]
- 12.Blank M, Tomer Y, Slavin S, Shoenfeld Y. Induction of tolerance to experimental anti phospholipid syndrome (APS) by syngeneic bone marrow transplantation. Scand J Immunol. 1995;42:226–34. doi: 10.1111/j.1365-3083.1995.tb03649.x. [DOI] [PubMed] [Google Scholar]
- 13.Shoenfeld Y, Isenberg D. The mosaic of autoimmunity. Amsterdam: Elsevier; 1989. Idiotype network; pp. 103–25. [Google Scholar]
- 14.Shoenfeld Y. Idiotypic induction of autoimmunity: a new aspect of the idiotypic network. FASEB. 1994;8:1297–301. doi: 10.1096/fasebj.8.15.8001742. [DOI] [PubMed] [Google Scholar]
- 15.Blank M, Manosroi J, Tomer Y, et al. Suppression of experimental systemic lupus erythematosus (SLE) with specific anti-idiotypic antibody–saporin conjugate. Clin Exp Immunol. 1994;98:434–41. doi: 10.1111/j.1365-2249.1994.tb05509.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hahn BH, Ebling FM. Suppression of murine lupus nephritis by administration of anti-idiotypic antibody to anti-DNA. J Immunol. 1984;132:187–90. [PubMed] [Google Scholar]
- 17.Zanetti M, Mampaso F, Wilson CB. Anti idiotype as a probe in the analysis of autoimmune tubulointerstitial nephritis in Brown Norway rat. J Immunol. 1984;131:1268–74. [PubMed] [Google Scholar]
- 18.Nordling C, Holmdahl R, Klareskog L. Down-regulation of collagen arthritis after in vivo treatment with a syngenic monoclonal anti-idiotypic antibody to a cross-reactive idiotype on collagen II autoantobodies. Immunol. 1991;72:486–90. [PMC free article] [PubMed] [Google Scholar]
- 19.Dwyer DA, Schonbeck S. Anti-idiotypic regulation of the immune response against the acetylcholine receptor. In: Golstein G, Bach JF, Wigzell H, editors. Immune regulation by characterized polypeptides. New York: Alan R Liss; 1987. pp. 607–18. [Google Scholar]
- 20.Koazky DE, Mirshahi M. Experimental autoimmune uveoretinitis: idiotypic regulation and disease suppression. Int Ophthalmol. 1990;14:43–47. doi: 10.1007/BF00131168. [DOI] [PubMed] [Google Scholar]
- 21.Zou SR, Whitaker JN. Specific modulation of T cells and murine experimental allergic encephalomyelitis by monoclonal anti idiotypic antibodies. J Immunol. 1993;150:1629–42. [PubMed] [Google Scholar]
- 22.Blank M, Cohen J, Toder V, Shoenfeld Y. Induction of primary anti-phospholipid syndrome by anti-cardiolipin antibodies. Proc Natl Acad Sci USA. 1991;88:3069–73. doi: 10.1073/pnas.88.8.3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fishman P, Bakimer R, Blank M, et al. The putative role of cytokines in the induction of primary anti-phospholipid syndrome in mice. Clin Exp Immunol. 1992;70:266–70. doi: 10.1111/j.1365-2249.1992.tb07940.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mosmann TR, Sad S. The expanding universe of T-cell subsets: Th1, Th2 and more. Immunol Today. 1996;17:138–46. doi: 10.1016/0167-5699(96)80606-2. [DOI] [PubMed] [Google Scholar]
- 25.Hohman A, Comacchio R, Boswarva V, Sutjita M, Bradley J. The H-3 anti-phospholipid idiotype is found in patients with SLE but not in patients with syphilis. Clin Exp Immunol. 1991;86:207–11. doi: 10.1111/j.1365-2249.1991.tb05797.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sutjita M, Hohmann A, Comacchio R, et al. A common anti-cardiolipin antibody idiotype in autoimmune disease: identification using a mouse monoclonal antibody directed against a naturally-occurring anti- phospholipid antibody. Clin Exp Immunol. 1989;75:211–6. [PMC free article] [PubMed] [Google Scholar]
- 27.Parham P. On the fragmentation of monoclonal IgG1, IgG2a, and IgG2b from BALB/c mice. J Immunol. 1983;131:2895–902. [PubMed] [Google Scholar]
- 28.Taguchi T, Jr, McGhee RL, Coffman KW. Detection of individual splenic T cells producing IFNγ and IL-5 using the enzyme linked immunospot (ELISPOT) assay. J Immunol Methods. 1990;128:65–73. doi: 10.1016/0022-1759(90)90464-7. [DOI] [PubMed] [Google Scholar]
- 29.Lacriox-Desmazes S, Mouthon L, Spalter SH. Immunoglobulins and the regulation of autoimmunity through the immune network. Clin Exp Rheumatol. 1996;14(Suppl.):S9–S15. [PubMed] [Google Scholar]
- 30.Blank M, Ben-Bassat M, Shoenfeld Y. Modulation of SLE induction in naive mice by specific T cells suppressor activity to pathogenic anti-DNA idiotype. Cell Immunol. 1991;137:1–13. doi: 10.1016/0008-8749(91)90095-s. [DOI] [PubMed] [Google Scholar]
- 31.McFarland HF. Complexities in the treatment of autoimmune disease. Science. 1996;274:2037–8. doi: 10.1126/science.274.5295.2037. [DOI] [PubMed] [Google Scholar]
- 32.De Carli MD, D'Elios MM, Mariotti S. Cytolytic T cells with Th1 like cytokine profile predominate in retro orbital lymphocytic infiltrates of Grave's disease. J Clin Endocrinol Metab. 1993;77:1120–4. doi: 10.1210/jcem.77.5.8077301. [DOI] [PubMed] [Google Scholar]
- 33.Dolhain RJEM, Van der Heiden AN, Ter Haar NT. Shift toward T lymphocytes with a T helper 1 cytokine-secretion profile in the joints of patients with rheumatoid arthritis. Arthritis Rheum. 1996;39:1961–9. doi: 10.1002/art.1780391204. [DOI] [PubMed] [Google Scholar]
- 34.Baron JL, Madri JA, Ruddle NH. Surface expression of 4 integrin by CD4+ T cells is required for their entry into brain parenchyma. J Exp Med. 1993;177:57–68. doi: 10.1084/jem.177.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Katz JD, Bnoist C, Mathis D. T helper cell subsets in insulin- dependent-diabetes. Science. 1995;268:1185–8. doi: 10.1126/science.7761837. [DOI] [PubMed] [Google Scholar]
- 36.Qasim FJ, Thiru S, Gillespie K. Gold and d-penicillamine induce vasculitis and up-regulate mRNA for IL-4 in the Brown Norway rat: support for a role for Th2 cell activity. Clin Exp Immunol. 1997;108:438–45. doi: 10.1046/j.1365-2249.1997.2971296.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goldman M, Druet P, Gleichman E. Th2 cells in systemic autoimmunity: insights from allogeneic diseases and chemically-induced autoimmunity. Immunol Today. 1992;12:223–7. doi: 10.1016/0167-5699(91)90034-Q. [DOI] [PubMed] [Google Scholar]
- 38.Nakajima A, Azuma M, Kodera S. Preferential dependence of autoantibody production in murine lupus on CD86 costimulatory molecule. Eur J Immunol. 1995;25:3060–9. doi: 10.1002/eji.1830251112. [DOI] [PubMed] [Google Scholar]
- 39.Richaud-Patin Y, Alcocer-Vaela J, Llorente L. High levels of Th2 cytokine gene expression in SLE. Rev Invest Clin. 1995;47:267–72. [PubMed] [Google Scholar]
- 40.Caligaris-Cappio F, Bertero MT, Converso M. Circulating levels of soluble CD30 a marker of cell producing Th2-type cytokines, are increased in patients with systemic lupus erythematosus and correlate with disease activity. Clin Exp Immunol. 1995;13:339–43. [PubMed] [Google Scholar]
- 41.Jacob CH, Van der MeidePH, Mcdevitt HO. In vivo treatment of (NZBXNZW) F1 lupus-like nephritis with monoclonal antibody to gamma interferon. J Exp Med. 1987;166:798–803. doi: 10.1084/jem.166.3.798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hagivara E, Gourley MF, Lee S, Klinman DK. Disease severity in patients with systemic lupus erythematosus correlates with an increased ratio of interleukin-10: interferon-gamma-secreting cells in the peripheral blood. Arthritis Rheum. 1996;39:379–85. doi: 10.1002/art.1780390305. [DOI] [PubMed] [Google Scholar]
- 43.Segal R, Bermas BL, Dayan M, et al. Kinetics of cytokine production in experimental systemic lupus erythematosus. J Immunol. 1997;158:3009–16. [PubMed] [Google Scholar]
- 44.Trebleau S, Germann T, Gately MK, et al. The role of IL-12 in the induction of organ-specific autoimmune diseases. Immunol Today. 1995;16:383–6. doi: 10.1016/0167-5699(95)80006-9. [DOI] [PubMed] [Google Scholar]
- 45.Ishida H, Muchamuel T, Sakguchi S. Onset of autoimmunity in NZB/WF1 mice. J Exp Med. 1994;179:305–10. doi: 10.1084/jem.179.1.305. [DOI] [PMC free article] [PubMed] [Google Scholar]