

Abstract
Aims
Changes in drug delivery rate may result in clinically important changes in drug effects. For the loop diuretic frusemide, it would be desirable to develop controlled release preparations, that could maintain an effective urinary excretion rate over a prolonged period of time. The aim of this study was to investigate the influence of frusemide formulation on frusemide recovery, diuretic effect and efficiency.
Methods
Twelve subjects were given 60 mg of four different frusemide controlled release formulations in a single-dose, double-blind, randomized 4-way cross-over design. The formulations were three study drugs with different extended dissolution rates (ER1Tab, ER2Tab and ER3Caps) and one reference drug (LR). Urinary volume and contents of frusemide in urine were measured in samples collected over 24 h.
Results
Substantial differences in frusemide recovery and diuretic efficiency were observed between LR and all other formulations. At 24 h, mean total frusemide recoveries of ER1Tab, ER2Tab and ER3Caps were 52%, 36% and 57% lower, respectively, compared with LR (P < 0.01). Also at 24 h, mean total diuretic efficiency for ER1Tab, ER2Tab and ER3Caps was 83%, 31% and 135% higher, respectively, compared to LR. The rapid dissolution and absorption of LR resulted in a high diuretic response from 0 to 3 h after dosing. However, from 0 to 24 h, there were no differences in diuretic response between the formulations.
Conclusions
Controlled release formulations of frusemide with a low and extended rate of dissolution lead to a more prolonged absorption and subsequent diuresis, but still maintain a similar cumulative response, due to their higher diuretic efficiency.
Keywords: efficiency, extended release, frusemide, pharmacodynamics
Introduction
In the past 30 years, drug delivery systems have been developed, to obtain desirable pharmacological effects by controlling the rate of drug release. Changes in drug delivery rate may result in clinically important changes in drug effects [1]. For the loop diuretic frusemide, it has been shown that if identical doses are given, a slow and constant input of the drug into the body induces a higher total effect compared with a more rapid input. This has been explained by investigating the efficiency of the drug [2–8]. Efficiency denotes the ratio between effect and concentration. In the case of frusemide, efficiency is represented by the ratio between the drug-induced diuretic or natriuretic effect and the frusemide excretion rate.
The pharmacodynamic model for efficiency is closely related to the sigmoid Emax model and is dependent on the same parameters. It can be derived from this model by dividing both sides of the equation by the stimulus (C):
 |
1 |
or in the case of frusemide by the urinary excretion rate (ER):
 |
2 |
Effect and efficiency have different shapes when expressed in relation to ER as previously shown [2, 3, 9]. Because the sigmoid Emax model implies the existence of a maximum effect, efficiency will decrease at high concentrations. Efficiency is also characterized by a maximum value, which is a function of EC50 and the slope factor s and can be derived if s>1. At this point, the ratio between effect and stimulus is most advantageous. The efficiency concept has been used to explain that the time course of drug delivery is an important factor for the cumulated pharmacological effect [2–8].
Clinically, a relatively slow input of frusemide is not only important for obtaining an increased efficiency. It may also help to prevent the compensatory and antagonistic renal sodium retention, that may occur not only during the time of diuretic activity, but also well after the drug effect has subsided [10, 11]. Also, especially in outpatients, it is important to avoid an acute increase in diuresis, which can interfere with normal daily activity. This is often observed with plain frusemide tablets, where diuresis may peak to 25 ml min−1 within 2 h after intake[4]. It would therefore be desirable to use a controlled release preparation of frusemide that could maintain an effective urinary excretion rate over a prolonged period of time.
Frusemide is subject to absorption-limited kinetics, which means that the rate of absorption determines the rate of excretion of the drug in urine. Different controlled release preparations may lead to differences in efficiency, due to different release profiles. The aim of this study was to investigate four different extended release formulations of frusemide with respect to frusemide recovery, diuretic effect and efficiency.
Methods
Subjects
The study protocol was approved by the Ethics Committee at Huddinge University Hospital and by the Swedish Medical Products Agency. Twelve subjects participated in the study after having given written informed consent. All subjects were white males, considered to be healthy according to detailed medical history and physical examination, which included an ECG and laboratory investigations. Their mean age was 28 (range 18–43) years and their mean body weight was 72 (range 60–85) kg. None of the subjects smoked or regularly used any medications.
Study design
The study had a single dose double-blind randomized 4-way cross-over design balanced for sequence. The washout-period between the 4 study days was at least 1 week. On the study days, each subject was given 60 mg of one of four oral frusemide preparations with different in vitro dissolution rates: two exploratory extended release compositions based on a tablet matrix of insoluble cellulose polymer containing frusemide (ER1Tab and ER2Tab), one exploratory capsule containing pellets of frusemide coated with insoluble cellulose polymer (ER3Caps) and the reference product Lasix® Retard® (LR) consisting of enteric coated granules containing frusemide. The different in vitro dissolution profiles of the respective compositions are shown in Figure 1. The preparations were given with 150 ml of water. Standardized meals were provided the day before and during each study day, containing 159 mmol sodium and 81 mmol potassium per day. Caffeinated drinks were not allowed the day before and during the study day. Urine was collected for 24 h before the study day to assess adherence to the diet and to estimate basal diuresis. The subjects were asked to fast for 8 h before each drug administration and the study started in the morning at the closure of the 24 h urine collection. Food and fluid intake were strictly standardized for the first 10 h of each study day. Breakfast was served at 3 h after dosing, lunch after 5 h, fruit after 6.5 h and dinner after 9 h. With each meal, except with the fruit, the subjects drank 150 ml water. Fluid intake between the meals was 100 ml every hour of an oral rehydration fluid for 10 h. The fluid contained 12.7 mmol l−1 sodium (292 mg l−1), 2.1 mmol l−1 potassium (82 mg l−1), 7.4 mmol l−1 chloride (261 mg l−1), 1.4 mmol l−1 phosphate (136 mg l−1) and 83.3 mmol l−1 glucose (15 gm l−1). The total fluid intake during the first 10 h was 1600 ml. After 10 h, the intake of water was ad libitum. During each study day, urine was collected at the following intervals: just before drug administration (at 0 h), and from 0 to 0.5, 0.5–1, 1–1.5, 1.5–2, 2–2.5, 2.5–3, 3–3.5, 3.5–4, 4–4.5, 4.5–5, 5–6, 6–7, 7–8, 8–9, 9–10, and 10–24 h after drug administration. The urine volumes were weighed and aliquots were carefully protected from light and frozen at −70° C until assayed for frusemide.
Figure 1.

Mean (s.d.) in vitro dissolution profiles of ER1Tab (•), ER2Tab (▪), ER3Caps (○) and LR (▵).
In vitro dissolution
The in vitro dissolution profiles of the four frusemide formulations (Figure 1) were generated using the Paddle method (USPII) at a stirring speed of 100 rotations min−1. The dissolution fluid was a phosphate buffer of pH 6.8 according to USP and the experiments were performed at a temperature of 37° C. The content of frusemide was analysed on-line with a spectrophotometer at a wavelength of 277 nm. Three (LR) and six to 12 tablets or capsules (ER1Tab, ER2Tab or ER3Caps) containing 60 mg frusemide each were run in parallel. Eighty percent of frusemide was dissolved after 20, 120, 240 and 540 min for LR, ER2Tab, ER1Tab and ER3Caps, respectively.
Analytical methods
Frusemide was analysed using a previously described method [12]. The LLQ was 0.5 μg ml−1 based on accuracy (mean deviation less than 20%) and precision (CV less than 20%) on the back calculated data. The intra-assay CV ranged from 0.4 to 4.1% at 2.6 μg ml−1, from 0.6 to 3.7% at 10.8 μg ml−1 and from 2 to 3.5% at 27.1 μg ml−1. The interassay CV values were 3.4%, 2.5% and 2.8%, respectively.
Calculations
The primary objective of the study was to compare the four extended release formulations of frusemide with regard to the time profiles of diuresis and efficiency. The desired property of the formulation was to produce a gradual onset and extended diuretic pattern. The first 3 h after dose intake were regarded as the initial time interval for the drug effect. Since the intake of food and fluid were standardized for the first 10 h, this interval was also of interest. The total frusemide-induced diuresis from 0–3 h, 0–10 h and 0–24 h was calculated by subtracting the basal diuresis from the total diuresis within each time period. Basal diuresis was calculated for each subject individually, as the mean value from the four 24 h urine collections on the day before each separate study day. The values for basal diuresis ranged from 0.55 to 1.69 ml min−1 between the subjects. The total diuretic efficiency from 0–3 h, 0–10 h and 0–24 h was calculated as the ratio between the drug-induced diuresis and the frusemide urinary recovery within that time period as follows:
 |
3 |
where i represents 3, 10 or 24 h, respectively, and Ae is the total amount of frusemide excreted within the respective time period.
Differences between the formulations in total induced diuresis, total frusemide recovery and total diuretic efficiency from 0–3 h, 0–10 h and 0–24 h were analysed by using an analysis of variance model including factors for subject, treatment, period and carry-over. The significance level in this study was 0.05. However, P values were adjusted using Bonferroni correction.
Results
Total frusemide recovery, total induced diuretic response and total diuretic efficiency for all subjects and treatments are shown in Table 1 and 2. Substantial differences in frusemide recovery were observed between LR and all other formulations. At the end of the study day at 24 h, the mean total frusemide recoveries of ER1Tab, ER2Tab and ER3Caps were 52%, 36% and 57% lower, respectively, compared to LR (P < 0.01). There were no significant differences in frusemide recovery between ER1Tab, ER2Tab and ER3Caps. The rapid dissolution and absorption of the LR formulation resulted in a powerful diuretic response during the first 3 h after dosing. In this period, diuresis was significantly higher, about 600 ml, compared to ER1Tab, ER2Tab and ER3Caps (P < 0.01). However, over a longer period of time, from 0–10 h and from 0–24 h, there were no statistically significant differences in total diuretic response between the four formulations (Table 1 and 2). Mean values for all subjects, for frusemide excretion rate, diuretic response and diuretic efficiency, are shown in Figure 2. During most of the study day, mean diuretic efficiency of LR was lower compared with all other formulations, especially compared with ER3Caps, which had the slowest dissolution rate. Over the entire study day (the 0–24 h time period) total diuretic efficiency for ER1Tab, ER2Tab and ER3Caps was 83%, 31% and 135% higher, respectively, compared with LR (P < 0.05, NS, P < 0.01). For ER2Tab, diuretic efficiency was also significantly lower (P < 0.01) compared with ER3Caps for the 0–10 h and 0–24 h time periods.
Table 1.
Urinary recovery of frusemide, total induced diuretic response and total diuretic efficiency from 0–3 h, 0–10 h and 0–24 h. Mean (s.d.) n =12.
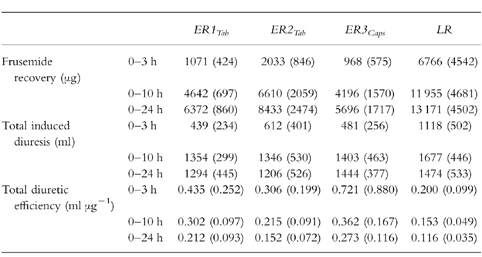
Table 2.
Adjusted means and 95% confidence intervals for the different between the formulations in urinary recovery of frusemide, total induced diuretic response and total diuretic efficiency from 0-3 h, 0-10 h and 0-24 h (n =12).

Figure 2.

Mean frusemide excretion rate (a), induced diuresis (b) and diuretic efficiency (c) vs time following the administration of ER1Tab (•), ER2Tab (▪), ER3Caps (○) and LR (▵).
Discussion
Frusemide is a widely prescribed drug and different formulations for oral use have become available for therapeutic administration[13]. The aim of this study was to determine which type of controlled release characteristics of frusemide would lead to a mild but extended diuretic response with a high diuretic efficiency. From the four formulations studied, the Lasix® Retard® formulation had the most rapid dissolution profile, successively followed by the two matrix tablets, ER2Tab and ER1Tab. The ER3Caps capsule formulation had the most extended dissolution profile. Similar patterns were seen in vivo, as shown by the frusemide excretion rate vs time profiles and the differences in frusemide recovery between the formulations from 0 to 3 h, 0–10 h and 0–24 h. This may indicate that the in vitro dissolution rate profiles generated at the described conditions may be predictive of the in vivo behaviour of the studied formulations. Several studies have attempted to correlate the in vitro dissolution rate with measurements of bioavailability of frusemide, however, with conflicting results[14–16]. Differences in dissolution methodology between the studies and intersubject and intrasubject variability in frusemide absorption are possible explanations.
The rapid dissolution and absorption profile of the LR formulation found in our study seems remarkably different from earlier observations, where the administration of similar 60 mg oral doses of Lasix® Retard® to 26 healthy and fasting subjects resulted in peak frusemide excretion rates of only 30 (s.d. 20) μg min−1 [4]. This discrepancy may be explained by an extreme variability in the absorption of Lasix® Retard®. Intra- and intersubject variability in the rate and extent of frusemide absorption have been reported to be large[13, 17]. It may also be due to the high variability in dissolution rate for this particular formulation. The mean in vitro dissolution profiles of 15 different batches of Lasix® Retard® generated under similar conditions to those described earlier are shown in Figure 3. Alternatively, the dissolution characteristics of Lasix® Retard® may have changed since 1992.
Figure 3.

Mean in vitro dissolution profiles of 15 batches of Lasix® Retard®.
The high frusemide excretion rates observed for LR were associated with marked inital diuretic response. On average, 76% of the total diuresis induced from 0–24 h was produced within the first 3 h after dosing, leading to significant differences between the formulations for the 0–3 h time period. However, for the 0–10 h time period (where food and fluid intake were standardized) and for the entire study day, there were no differences in response between the formulations. This was caused by the higher diuretic efficiency of the ER1Tab, ER2Tab and ER3Caps formulations compared with LR. Diuretic efficiency was highest for ER3Caps, followed by the ER1Tab formulation.
The results of our study confirm the efficiency concept and stress the importance of the time course of the frusemide excretion rate for the cumulative diuretic response. It also implies that pharmacokinetic bioequivalence is relatively unimportant for frusemide. Formulations with a lower rate and extent of absorption may lead to a more constant diuresis while still resulting in a similar cumulative response, due to their higher diuretic efficiency. There is a need for studies of the use of frusemide controlled release formulations in patients. Such studies should preferably focus not only on bioavailability or response, but also on intra- and interpatient variability, compliance and patient outcome.
Acknowledgments
The technical and analytical assistance of Joacim Lindström, Bengt Lundberg, Malin Löfqvist, Monja Zrimsek (Astra Arcus, Sweden) and Christina Alm, Kerstin Burman and Eva Götharson (Division of Clinical Pharmacology, Huddinge University Hospital) is gratefully acknowledged. Jonas Román (Hässle Läkemedel, Sweden) is thanked for statistical advice. This study was supported by the Swedish MRC (nr 12590) and by funds of Karolinska Institutet.
References
- 1.Banerjee PS, Robinson JR. Novel drug delivery systems: an overview of their impact on clinical pharmacokinetic studies. Clin Pharmacokinet. 1991;20:1–14. doi: 10.2165/00003088-199120010-00001. [DOI] [PubMed] [Google Scholar]
- 2.Kaojarern S, Day B, Brater DC. The time course of delivery of furosemide into urine: an independent determinant of overall response. Kidney Int. 1982;22:69–74. doi: 10.1038/ki.1982.134. [DOI] [PubMed] [Google Scholar]
- 3.Alván G, Helleday L, Lindholm A, Sanz E, Villén T. Diuretic effect and diuretic efficiency after intravenous dosage of frusemide. Br J Clin Pharmacol. 1990;29:215–219. doi: 10.1111/j.1365-2125.1990.tb03622.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alván G, Paintaud G, Eckernäs S-Å, Grahnén A. Discrepancy between bioavailability as estimated from urinary recovery of frusemide and total diuretic effect. Br J Clin Pharmacol. 1992;34:47–52. doi: 10.1111/j.1365-2125.1992.tb04106.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van Meyel JJ, Smits P, Russel FG, Gerlag PG, Tan Y, Gribnau FW. Diuretic efficiency of furosemide during continuous administration versus bolus injection in healthy volunteers. Clin Pharmacol Ther. 1992;51:440–444. doi: 10.1038/clpt.1992.44. [DOI] [PubMed] [Google Scholar]
- 6.Paintaud G, Alván G, Eckernäs S-Å, Wakelkamp M, Grahnén A. The influence of food intake on the effect of two controlled release formulations of furosemide. Biopharm Drug Dispos. 1995;16:221–232. doi: 10.1002/bdd.2510160307. [DOI] [PubMed] [Google Scholar]
- 7.Dormans TPJ, van Meyel JJM, Gerlag PGG, Tan Y, Russel FGM, Smits P. Diuretic efficiency of high dose furosemide in severe heart failure: bolus injection versus continuous infusion. J Am Coll Cardiol. 1996;28:376–382. doi: 10.1016/0735-1097(96)00161-1. [DOI] [PubMed] [Google Scholar]
- 8.Wakelkamp M, Alván G, Paintaud G. Natriuretic efficiency of frusemide as a consequence of drug input rate. Br J Clin Pharmacol. 1997;43:481–491. doi: 10.1046/j.1365-2125.1997.00592.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hammarlund MM, Odlind B, Paalzow LK. Acute tolerance to furosemide diuresis in humans. Pharmacokinetic-pharmacodynamic modeling. J Pharmacol Exp Ther. 1985;233:447–453. [PubMed] [Google Scholar]
- 10.Wilcox CS, Mitch WE, Kelly RA, et al. Response of the kidney to furosemide I. Effects of salt intake and renal compensation. J Lab Clin Med. 1983;102:450–458. [PubMed] [Google Scholar]
- 11.Ferguson JA, Sundblad KJ, Becker PK, Gorski JC, Rudy DW, Brater DC. Role of duration of diuretic effect in preventing sodium retention. Clin Pharmacol Ther. 1997;62:203–208. doi: 10.1016/S0009-9236(97)90069-2. [DOI] [PubMed] [Google Scholar]
- 12.Wakelkamp M, Alván G, Paintaud G. The time of maximum diuretic effect for model selection in pharmacokinetic-pharmacodynamic analysis of furosemide. Br J Clin Pharmacol. 1998;45:63–70. doi: 10.1046/j.1365-2125.1998.00637.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Murray MD, Haag KM, Black PK, Hall SD, Brater DC. Variable furosemide absorption and poor predictability of response in elderly patients. Pharmacotherapy. 1997;17:98–106. [PubMed] [Google Scholar]
- 14.Waller ES, Crismon ML, Smith RV, Bauza MT, Doluisio JT. Comparative bioavailability of furosemide from solution and 40 mg tablets with different dissolution characteristics following oral administration in normal men. Biopharm Drug Dispos. 1988;9:211–218. doi: 10.1002/bod.2510090209. [DOI] [PubMed] [Google Scholar]
- 15.McNamara PJ, Foster TS, Digenis GA, et al. Influence of tablet dissolution on furosemide bioavailability: a bioequivalence study. Pharm Res. 1987;4:151–153. doi: 10.1023/a:1016427321532. [DOI] [PubMed] [Google Scholar]
- 16.Kingsford M, Eggers NJ, Soteros G, Maling TJB, Shirkey RJ. An in-vivo-in-vitro correlation for the biovailability of frusemide tablets. J Pharm Pharmacol. 1984;36:536–538. doi: 10.1111/j.2042-7158.1984.tb04446.x. [DOI] [PubMed] [Google Scholar]
- 17.Grahnén A, Hammarlund M, Lundqvist T. Implications of intraindividual variability in bioavailability studies of furosemide. Eur J Clin Pharmacol. 1984;27:595–602. doi: 10.1007/BF00556898. [DOI] [PubMed] [Google Scholar]