

Abstract
The Directional Atomic Solvation EnergY (DASEY) is an atom-based description of the environment of an amino acid position within a known 3D protein structure. The DASEY has been developed to align and score a probe amino acid sequence to a library of template protein structures for fold assignment. DASEY is computed by summing the atomic solvation parameters of atoms falling within a tetrahedral sector, or petal, extending 16 Å along each of the four bond axes of each α-carbon atom of the protein. The DASEY discriminates between pairs of structurally equivalent positions and random pairs in protein structures sharing a fold but belonging to different superfamilies, unlike some previous descriptors of protein environments, such as buried area. Furthermore, the DASEY values have characteristic patterns of residue replacement, an essential feature of a successful fold assignment method. Benchmarking fold assignment with DASEY achieves coverage of 56% of sequences with 90% accuracy when probe sequences are matched to protein structural templates belonging to the same fold but to a different superfamily, an improvement of greater than 200% over a previous method.
Protein structure provides insight into biochemical function and biological behavior. Because the rate of protein sequence determination continues to outstrip protein structure determination, reliable assignment of sequences to known folds continues to be needed. As of June 2002, the National Center for Biotechnology Information's Non-Redundant GenPept database contained 1,019,589 sequences; the database doubles in size approximately every 18 mo (1). On the same date, the Protein Data Bank contained 16,952 structures; this data bank doubles in size approximately every 3 yr (2). Computational methods that assign a protein's sequence to structure (3–25), including the fold assignment method introduced here, mitigate the disparity between the sizes of the sequence and structure databases.
Two Approaches to Fold Assignment
Computational approaches to assigning sequences to protein folds (Fig. 1) have been of two types: sequence-to-sequence based and sequence-to-structure based. In sequence-to-sequence-based assignments, the probe sequence is matched by dynamic programming (26, 27) to the sequence of a protein with a known fold, and it is inferred that the probe sequence folds in a similar way. In its earliest form, this method was used in the 1960s to assign the sequence of α-lactalbumin to the known structure of hen egg lysozyme (28). Profile methods (29) permitted the information inherent in a whole family of sequences to be used in the fold assignment, and generalizations of the profile methods, including psi-blast (30) and HMMS (11, 13) have greatly expanded the power of sequence-to-sequence fold assignment. Recently, Russell and coworkers (12) have argued that sequence-to-sequence methods of fold assignment are the most sensitive.
Fig 1.

Fold assignment relies on scoring the compatibility of a probe sequence (Top Left) with a known 3D structure (Middle Right). Also available for scoring are the sequence and secondary structure of the protein of known structure (Top Right) and features predicted from the probe sequence (Top Left). The compatibility can be scored by any of the type of functions listed in the Left Middle box. Our method scores the compatibility with DASEY. The DASEY (Inset Bottom Right) describes the hydrophobicity of the environment of a Cα position in a known protein structure. Each of the four dimensions of the DASEY is calculated by summing the Atomic Solvation Parameters (ASPs) of the atoms contained within a tetrahedral sector, or petal, that extends 16 Å along a bond direction from the α-carbon atom of the position. The distance from the bond axis weights down the contribution of each ASP. The four tetrahedral petals are shown (Inset). Two atoms are shown in the petal along the Cα→N direction. The N atom has a negative (polar) ASP; the C atom has a positive (apolar) ASP.
In sequence-to-structure methods (3–10, 15–18, 21, 31), the structure is encoded as a sequence of residue environments. The environment of a residue position has been described in various ways, including the area buried away from solvent in that position or in the distances from that position to surrounding residues. Every type of residue receives a score for occupying a given type of environment, and the sum of all residue scores represents the compatibility of the probe sequence for the structure of the fold.
Directional Atomic Solvation EnergY (DASEY) extends sequence-to-structure methods in two ways. First, the environment of each protein position is encoded as the distribution of nonhydrogen atom types along four tetrahedral directions from the α-carbon of the residue in that position. This distribution is related to the concept of the visible volume proposed by Lo Conte and Smith (32) and may be thought of as extending the residue environments introduced by Bowie et al. (3). We also alter the environmental definition of Bowie et al. by explicitly including a residue's type.
The second extension is in the adoption of the formalism of ref. 16 by computing the preference of a query probe residue and its predicted secondary structure for the template environment to which it is aligned when two proteins having a similar fold and low sequence similarity are superimposed, rather than defining the preference of a residue for an environment in its own structure. The purpose of this is to mimic the fold assignment process in the training method.
Materials and Methods
Domain Databases.
Protein structure files were obtained from the Research Collaboratory for Structural Bioinformatics's Protein Data Bank (2). Structures whose domain boundaries had been specified in the CATH 2.4 (January 2002) database (33) were parsed into their separate domain segments. Discontinuous domains were discarded. For each residue of each domain, we generated additional information, including: solvent-accessible surface area exposed; fraction of area buried by polar atomic groups (3); DASEY secondary structure [using DSSP (34)]; and predicted secondary structure [using PHD (35)].
Database of Aligned Protein Structures (daps).
A database of aligned protein structures was derived from the domain database. First, all domain pairs were created for each domain-fold family within CATH. Next, we used the Fold classification based on Structure–Structure alignment of Proteins (fssp) database to verify that our domain pairs share a common fold (36). Pairs with dali (37) z scores <2 were discarded. A preliminary structural alignment for each pair was generated by using STAMP (38). Final structural alignments were generated by using COMPARER (39).
Domain Sets for Training, Testing, and Benchmarking.
The domain database was partitioned into five sets. First, training and testing sets were extracted from CATH 1.7 (June, 2000). The training set, used for defining and optimizing the dasey, contained 1,123 domains and their corresponding 9,891 daps-aligned domain pairs for a total of 2,131,404 aligned residue pairs. The test set contained 96,672 residues from 626 domains and their corresponding 4,013 daps aligned domain pairs for a total of 864,758 aligned residue pairs. Plots shown in Fig. 3 were generated by using the test set. Distinct from the training set, two probe sets and one domain template library were created to benchmark the fold assignment performance of dasey. The domain library contained 3,914 domains from CATH 2.0 (January, 2001). The probe set created for BENCHMARK 1 (benchmarking of coverage/accuracy as a function of score) consisted of 1,000 probe domains not classified within CATH 1.7. A second distinct probe set was created for BENCHMARK 2 (benchmarking of coverage as a function of rank) consisting of 500 recent protein chains not classified within CATH 2.0. daps and the domain sets are available from http://fold.doe-mbi.ucla.edu.
Fig 3.

The four dimensions of the DASEY are distributed differently for different probe-to-template position alignments and discriminate structurally similar pairs of positions from random pairs in the test set of 864,758 pairs of aligned residues in similarly folded proteins of different superfamilies. A and B demonstrate the capacity of DASEY to discriminate among different residues for different structural positions in the test set of 864,758 pairs of aligned residues in similarly folded proteins of different superfamilies. A and B show this capacity for one of the four petals. In fact, all four petals contribute to the discrimination and together constitute the environment of a position. The five curves show the different densities P for different residue-secondary structure combinations. Notice the clearly different distributions. C–F plot the value of an environmental descriptor for one residue position of a protein on the x axis, and the descriptor for a structurally equivalent position from a similarly folded protein from a different superfamily on the y axis to demonstrate the degree of conservation of an environmental descriptor; a well-conserved parameter yields a tight line along the diagonal. Note that the DASEY dimensions are better conserved than either Fraction Polar or Area Buried of ref. 3, which are poorly conserved.
Non-dasey Fold Assignment Methods Tested.
In addition to dasey, we also benchmarked the Method of Sequence Derived Properties (sdp) (6), sdp+, an improved version of the Method of Sequence Derived Properties in which secondary structure transition scores are derived from daps, PSI-BLAST (40), and the genthreader server (10). For each probe, PSI-BLAST was executed by first iteratively scanning for homologous proteins within the Non-Redundant GenPept database. We use 0.001 as the threshold for inclusion of a sequence in subsequent iterations as recommended by ref. 41 and used previously by ref. 42. We allow a maximum of six iterations. At each of the six iterations, we save the checkpoint matrix; then the checkpoint matrices are used to search the sequences from the Protein Data Bank.
Evaluating Sequence–Structure Compatibility by Using Structural Environment Dependent Scoring.
We describe the compatibility of sequence for a structure as the odds probability
 |
where items in bold are vector quantities, the superscript “a” denotes “aligned;” the superscript “p” denotes “predicted;” and ∂* indicates the optimal alignment as computed by the Viterbi algorithm on a global–local hidden Markov model similar to those described by ref. 43. The DASEY is defined and described in Results. P(structure|seq) is the normalized probability of an encoded structure given a probe sequence. A structure is encoded as a multidimensional observation vector describing the environment of a structural position as the combination of two discrete parameters, residue and secondary structure, and the continuous, four-dimensional dasey. Transition and emission probabilities, constant throughout the model, are derived directly from daps. The hmm emission probabilities, which are analogous to scores found in a traditional scoring matrix, are computed as the odds ratio describing the preference of a probe residue and its predicted secondary structure to be aligned to a template residue, secondary structure, and DASEY:
 |
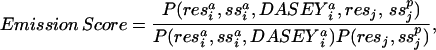 |
where the subscripts i and j denote specific positions of the structure and sequence, respectively. Each of the 3,660 = (60 × 60 + 60) four-dimensional partial probability density functions (PDFs) from which the emission score is computed, P(res ,ss
,ss ,DASEY
,DASEY ,resj,ss
,resj,ss ) and P(res
) and P(res ,ss
,ss ,DASEY
,DASEY ,gap), is modeled as shown in Eq. 3 as a mixture of multivariate normal density functions:
,gap), is modeled as shown in Eq. 3 as a mixture of multivariate normal density functions:
 |
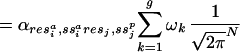 |
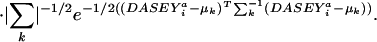 |
These functions are generated as shown schematically in Fig. 2 and described in the figure legend. Each multivariate normal density k within the mixture is defined by ωk, μk and Σk, the mix weight, mean vector, and covariance matrix, respectively. emmix (44) was used to estimate g, the appropriate number of mixture components, and to derive the parameters ωk, μk, and Σk of the PDFs from the populations of DASEYs binned by (res ,ss
,ss ,resj,ss
,resj,ss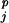 ). The population size of each bin was used to derive
). The population size of each bin was used to derive 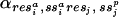 which weights the partial density functions (Eq. 3) and ensures that the complete mixture of partial PDFs integrates to one over all DASEY values for the support set of all resa,ssa,DASEY,res,ssp combinations. For poorly populated bins, we use a PDF uniform with respect to DASEY environments rather than attempting to overfit a complex density function. To test the stability of the generated mixture PDFs and to verify we did not overfit the data, density functions were computed first with all of the training data and a second time with 10% of the aligned domain pairs removed. The two sets of PDFs were indistinguishable, implying that the distributions were not overfit despite the high degree of parameterization. Although it is possible to derive PDFs for P(resa,ssa,DASEYa) directly, we instead compute P(resa,ssa,DASEYa), as shown in Eq. 4, as a sum of the partial densities P(res
which weights the partial density functions (Eq. 3) and ensures that the complete mixture of partial PDFs integrates to one over all DASEY values for the support set of all resa,ssa,DASEY,res,ssp combinations. For poorly populated bins, we use a PDF uniform with respect to DASEY environments rather than attempting to overfit a complex density function. To test the stability of the generated mixture PDFs and to verify we did not overfit the data, density functions were computed first with all of the training data and a second time with 10% of the aligned domain pairs removed. The two sets of PDFs were indistinguishable, implying that the distributions were not overfit despite the high degree of parameterization. Although it is possible to derive PDFs for P(resa,ssa,DASEYa) directly, we instead compute P(resa,ssa,DASEYa), as shown in Eq. 4, as a sum of the partial densities P(res ,ss
,ss ,DASEY
,DASEY ,resj,ss
,resj,ss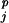 ) with respect to the 60 resj,ss
) with respect to the 60 resj,ss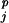 combinations and gap:
combinations and gap:
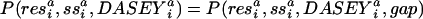 |
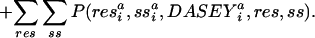 |
Notice that, in addition to the 60 residue and secondary structure classes (20 residue types times 3 secondary structure types: α, β, and coil), we introduce an additional “gap” character to treat insertions and deletions. The PDF describing the likelihood of emitting a gap insertion in the probe is computed similarly to the other residue types. However, daps is used to derive P(gapi resj,ss ), the likelihood of aligning a gap to a resj,ss
), the likelihood of aligning a gap to a resj,ss from the probe sequence(deletion in the probe).
from the probe sequence(deletion in the probe).
Fig 2.

DASEY is used to align and score a probe sequence with a known template structure. This is achieved with PDFs of the form P(res ,ss
,ss ,DASEY
,DASEY ,resj,ss
,resj,ss ) (Eq. 5), that give the likelihood of aligning residue type (res) j of the probe to structure position i of the template. This PDF depends on DASEY, res, and secondary structure type (ss). In the example of a DAPS aligned residue pair shown in Step 2, position i of structure I, which happens to be occupied by a valine in a sheet (E) with a DASEY of [30, 56, 25, 48] and PHD (35) predicted secondary structure of sheet (E) is aligned to position j of structure J, an arginine in a coil (C) with a DASEY of [32, 53, 20, 41] and PHD-predicted secondary structure of helix (H). The DASEY of [32, 53, 20, 41] means that the weighted sum of atomic solvation parameters for atoms within the Cα→N petal is 32, and so forth. Notice that the DASEY vectors from the aligned residue pair contribute to two bins. We place the DASEY vector [30, 56, 25, 48] from position i into the first bin, which we denote “VE RH.” This means this DASEY represents a valine strand aligned to an arginine-predicted helix. Next we place the DASEY vector [32, 53, 20, 41] into a second bin, which we denote “RC VE.” Appropriately binned DASEY vectors from the training set of aligned known structures give density plots as shown on the left in Step 4. The distribution of DASEY values in each of the 3,600 bins (one bin for each combination of residue, predicted secondary structure, aligned residue, aligned secondary structure) is modeled as a mixture of multivariate normal PDFs, as shown by the equation on the right in Step 4. These density functions are the dominant terms of the emission score and describe the likelihood of aligning a residue and predicted secondary structure from a probe sequence of unknown structure with a residue, secondary structure, and DASEY from a template structure.
) (Eq. 5), that give the likelihood of aligning residue type (res) j of the probe to structure position i of the template. This PDF depends on DASEY, res, and secondary structure type (ss). In the example of a DAPS aligned residue pair shown in Step 2, position i of structure I, which happens to be occupied by a valine in a sheet (E) with a DASEY of [30, 56, 25, 48] and PHD (35) predicted secondary structure of sheet (E) is aligned to position j of structure J, an arginine in a coil (C) with a DASEY of [32, 53, 20, 41] and PHD-predicted secondary structure of helix (H). The DASEY of [32, 53, 20, 41] means that the weighted sum of atomic solvation parameters for atoms within the Cα→N petal is 32, and so forth. Notice that the DASEY vectors from the aligned residue pair contribute to two bins. We place the DASEY vector [30, 56, 25, 48] from position i into the first bin, which we denote “VE RH.” This means this DASEY represents a valine strand aligned to an arginine-predicted helix. Next we place the DASEY vector [32, 53, 20, 41] into a second bin, which we denote “RC VE.” Appropriately binned DASEY vectors from the training set of aligned known structures give density plots as shown on the left in Step 4. The distribution of DASEY values in each of the 3,600 bins (one bin for each combination of residue, predicted secondary structure, aligned residue, aligned secondary structure) is modeled as a mixture of multivariate normal PDFs, as shown by the equation on the right in Step 4. These density functions are the dominant terms of the emission score and describe the likelihood of aligning a residue and predicted secondary structure from a probe sequence of unknown structure with a residue, secondary structure, and DASEY from a template structure.
Once the 3,660 PDFs were derived, each describing the likelihood that a probe residue and its predicted secondary structure are aligned to a (DASEY, template residue, template secondary structure) set, we were able to compute emission probabilities and use them in a hidden Markov model to generate sequence–structure compatibility scores between a probe sequence and template structures.
Results
The DASEY.
The DASEY, shown in Fig. 1 Lower Right Inset, describes the chemical environment around a residue position in a known structure in terms of the distribution of hydrophobic and hydrophilic atomic groupings. The sequence of these distributions is thought to be characteristic of a protein fold (45). Around each Cα, we compute the weighted sum of the atomic solvation parameters of the atoms contained in four tetrahedral sectors, or petals, whose major axes lie along a bond direction and extend 16 Å. The contribution of an atom to the sum is Gaussian weighted by distance from the bond axis so that less than 5% of the volume overlaps between directions. Values of atomic solvation parameters were taken from ref. 46. Atomic solvation parameters are derived from measured energies of transfer of atomic groups from a polar solvent to a nonpolar solvent. Consequently, the DASEY can be thought of as a description of the hydrophobicity of the environment in which a residue is situated. Large DASEY values imply that a residue is in a hydrophobic environment. Small DASEY values imply that a residue is in a hydrophilic environment. As shown in Table 3, which is published as supporting information on the PNAS web site, www.pnas.org, DASEY can be defined in numerous ways. Permutations of the listed parameters were tested within the training set to determine the parameter set that best discriminates structurally equivalent positions from random positions and best separates residue distributions, as explained in the following.
Properties of the DASEY.
DASEY is useful for fold assignment in part because each distinct set of summed atomic solvation parameters prefers particular residues. That is, the DASEY discriminates residue substitution preferences. Fig. 3 A and B shows two of the four one-dimensional projections (one for each petal) of sample DASEY distributions for five of the total of 3,660 distributions for the variables (residue, ss, residue, ssp). Notice that each distribution is multimodal. The multimodality of the distributions is informative, revealing the preferences that residues have for different structural environments. For example, “AH AH” substitutions appear favorable in both hydrophobic and hydrophilic environments, whereas “AH EH” substitutions are most favored in hydrophilic environments. Note that each distribution is distinctive, demonstrating each residue's environment-dependent substitution preferences.
The DASEY is better able to discriminate structurally equivalent positions from random positions than either “area buried” or “fraction polar” the environmental descriptors used by Bowie et al. (3). If a continuous environmental descriptor were perfectly conserved at structurally equivalent positions, a scatter plot of that descriptor's value in each of the structurally equivalent positions would populate the diagonal. Conversely, if the parameter were poorly conserved such a scatter plot would be diffuse. Fig. 3 C and D show log cell-plots of the values of two of the tetrahedral directions of the DASEY at structurally equivalent positions. Fig. 3 E and F show log cell-plots plots of area buried and fraction polar respectively for structurally equivalent positions within the daps test set. Notice the diffuse character of the graphs shown in Fig. 3 E and F as compared with Fig. 3 C and D where a trend toward the diagonal is evident. The other two DASEY directions are also better conserved than area buried and fraction polar (data not shown). Fig. 3 C–F reveal the dasey is highly conserved at structurally equivalent positions, whereas “area buried” and “fraction polar” are not. Furthermore, the dasey is not conserved at random pairs of positions (data not shown) implying that the DASEY discriminates structurally equivalent positions from random positions.
Performance in Fold Assignment Accuracy and Coverage of DASEY Relative to the Earlier Methods sdp and sdp+.
DASEY displays greater coverage at several accuracies than either sdp (6) or sdp+. In our first benchmark, a sequence–structure compatibility score was generated for each member of a set of 1,000 probe sequences whose true folds have been classified in CATH 2.0 with each member of a structural template library whose folds had also been classified in CATH 2.0. Resulting scores are partitioned into three bins depending on the classified fold and superfamily of the probe and template. If the probe and template have different folds [different class, architecture, and topology (C.A.T.), as defined in CATH], we call the match incorrect and the sequence–structure compatibility score for that match contributes to the probability of false alarm. If the probe and template belong to the same fold and to the same homologous superfamily (same C.A.T.H as defined in CATH), the match is discarded as being appropriate for what could be termed remote homology recognition rather than for fold assignment. If the probe and template belong to the same fold and to different homologous superfamilies (same C.A.T., different H), the match is retained, denoted correct, and contributes to the probability of detection. Consequently, only fold level matches were considered. The average sequence identity between pairs in our set of true-positive matches was 7%.
Three fold-assignment methods were tested. The first method was Fischer and Eisenberg's (6) Method of Sequence Derived Properties (sdp). The second method was an extension to sdp denoted sdp+. In sdp+, the score for the match or mismatch of the secondary structure element comes from a log-odds table derived from daps, rather than from an identity matrix, enhancing the fold recognition capacity of sdp slightly. Finally, the third method uses DASEY structural environment-dependent scoring. The method of Bowie et al. (3) was not tested because sdp was previously found to be more effective (47).
We used sdp, sdp+, and dasey to generate sequence–structure compatibility scores between 1,000 probe sequences and 3,914 domain templates; generating ≈4,000,000 scores for each method. From the sets of correct and incorrect scores, probabilities of detection and false alarm were computed and used to generate the Receiver Operator Characteristic plot shown in Fig. 4. dasey presents a clear improvement over sdp and sdp+. As shown in Table 1, with error rates of 5 or 10%, the dasey-based method achieves greater than twice the coverage of the sdp methods.
Fig 4.

BENCHMARK 1 assesses the accuracy and coverage of the 3-fold assignment methods, DASEY, SDP (6), and SDP+. For the 1,000 protein sequences of the probe set and the 3,914 structures of the template library, the fraction of correct fold assignments is given. The coverage or probability of detection describes the method's sensitivity or true positive fraction. The probability of false alarm describes the method's false positive (error) fraction, (1-accuracy). These two quantities are plotted on a Receiver Operator Characteristic plot (48). Perfect performance on a Receiver Operator Characteristic plot is a horizontal line along the top of the graph representing 100% coverage and 0% error. The DASEY method has nearly double the coverage of the SDP methods at low error rates. The vertical bars refer to thresholds in Table 1.
Table 1.
Performance of 5-fold assignment methods in two challenging benchmarks
| Method
|
Benchmark 1 | Benchmark 2 | |||
|---|---|---|---|---|---|
| Coverage, %
|
Accuracy, %
|
No. (%) of 500 probes correctly identified fold in top | |||
| 1 | 5 | 10 | |||
| psi-blast (30) | NA | NA | 30 (6) | 35 (7) | 43 (9) |
| sdp (6) | 12 | 95 | 45 (9) | 64 (13) | 87 (17) |
| 21 | 90 | ||||
| sdp+ | 14 | 95 | 52 (10) | 95 (19) | 138 (28) |
| 26 | 90 | ||||
| genthreader (10) | NA | NA | 83 (17) | 110 (22) | 152 (30) |
| dasey | 34 | 95 | 124 (25) | 192 (38) | 239 (48) |
| 56 | 90 | ||||
Two distinct benchmarks of dasey's fold assignment performance were executed: one looks at dasey scores, the other looks at template rankings. For both benchmarks, we denote a match to be “correct” only if the two proteins share a fold but are members of different superfamilies. In Benchmark 1 (see Materials and Methods), we compare the coverage and accuracy of dasey scores relative to methods for fold assignment on challenging targets: 1,000 probe sequences of known structures, distinct from the training set, were compared with 3,914 template domains. In Benchmark 2 (see Results), 500 probe sequences not in cath 2.0 were submitted to the five methods. Note that dasey correctly identifies more fold matches at higher ranks than other methods.
Fold Assignment Rank Performance of dasey Relative to psi-blast (30), genthreader (10), sdp (6), and sdp+.
As an additional benchmark, we selected 500 proteins from the Protein Data Bank not classified in CATH 2.0. These proteins were not members of either our training set, or fold library (which exclusively contain proteins from CATH 2.0 or before). These proteins were also not members of genthreader's fold library. The sequences were submitted to sdp, sdp+, dasey, psi-blast and genthreader. As the genthreader server returns only its top 10 matches, we cannot construct the Receiver Operator Characteristic plots shown above and can ask only the question, “For what number of the probes was a correct structural template assigned within the top scoring (1, 5, or 10) templates, excluding templates belonging to the same homologous family as the probe sequence?” For this benchmark, we do not consider sequence–structure compatibility scores. Instead, we focus solely on the existence of a correct fold within the top scoring 1, 5, or 10 template assignments. The performance of psi-blast on this benchmark is discussed below. As before, we consider only fold level matches; superfamily matches are discarded. Benchmark results are shown in Table 1. dasey displays a significantly greater coverage than other methods.
Discussion
Strengths and Weaknesses of dasey in Relation to Other Fold-Assignment Methods.
Any new approach to fold assignment must be compared with the powerful sequence-profile-based program psi-blast (30), which is incorporated in several approaches to fold assignment (10, 12). In the benchmark described above with 500 probe sequences, psi-blast was unable to assign folds, either correctly or incorrectly, for 455 of the 500 probe sequences. These are proteins with fold level but not sequence level similarity to the probe. For 225 of the 455 proteins that psi-blast was unable to assign, dasey is able to rank a correct, structural homolog in the top 10 scoring folds. Table 2 gives 10 examples of protein chains from the Protein Data Bank that were part of our second benchmark; these were correctly assigned to a fold with rank one and high confidence by dasey and could not be assigned to a structure, either correctly or incorrectly, by using psi-blast.
Table 2.
A sample of protein sequences from Benchmark 2 confidently assigned to folds by dasey but not by psi-blast
| Probe ID | Probe protein | Template ID | Template protein | P | RMSD | % ID |
|---|---|---|---|---|---|---|
| 1du3L | Death Receptor 5 | 1gbg | Glucanase | 0.99 | 3.8 | 2 |
| 1eygD | Ssb-C | 1bcpD | Pertussis toxin | 0.95 | 3.1 | 4 |
| 1fewA | Smac | 1vls | Aspartate receptor | 0.99 | 3.6 | 4 |
| 1dysB | Glucanase Cel6B | 1cz1A | Exo-B-(1,3)-glucanase | 0.99 | 4.2 | 5 |
| 1gh8A | Translation EF 1B | 1psdA | D-3-P dehydrogenase | 0.95 | 3.1 | 6 |
| 1ge8A | Pcna | 1b77A | Sliding clamp | 0.99 | 3.1 | 11 |
| 1dr9A | B7-1 (Cd80) | 1rlw | Phospholipase A2 | 0.99 | 3.1 | 12 |
| 1gcjB | Importin-Beta | 1b3uA | Pp2A | 0.99 | 3.7 | 12 |
| 1erjC | Tup1 | 1qfmA | Prolyl oligopeptidase | 0.99 | 3.7 | 13 |
| 1e20A | Hal3 | 1bn6A | Haloalkane dehalogenase | 0.99 | 4.2 | 14 |
Of the 500 probe protein chains from Benchmark 2, psi-blast assigned a fold for only 45, and 10 of these assignments were incorrect. Of the remaining 455 proteins from Benchmark 2, we select 10 protein sequences that dasey confidently assigned to folds at rank one. For these 10, columns 1 and 3 give the PDB accession no. of the probe sequence and template structure, respectively. Columns 2 and 4 give the functional names of the probe and template proteins, respectively. Column 5 lists the probability value assigned by dasey to the match. Column 6 lists the rms deviation (RMSD) of the dali structural alignment between the probe's actual structure and the template's structure. Column 7 lists the percent identity of dali structural alignment between the probe and template structures. Each protein belongs to a different fold family. Note that dasey confidently (i.e. large P) assigns distant sequences [small percentage identical residue (% ID)] to similar folds (small RMSD).
Conversely, by using the June 2002 sequence databases, psi-blast was able to correctly assign folds to 29 of the 261 proteins dasey was unable to assign correctly. psi-blast is powerful when a probe protein has numerous diverse homologs within sequence databases (e.g., GenPept). In contrast, dasey relies solely on the comparison of a single probe sequence to a library of template structures. Consequently, its performance is not related to the number or diversity of homologs a probe sequence might have. The 29 proteins predicted preferentially by psi-blast each had a large diverse collection of homologs within GenPept. If superfamily matches are included, psi-blast's performance increases dramatically. Instead of assigning only 43 proteins to correct folds, psi-blast is able to assign 176. On the other hand, dasey's improvement is less marked, assigning 307 correctly, whereas with only fold matches it was able to assign 239.
genthreader utilizes psi-blast, but appears to yield greater coverage and accuracy than psi-blast alone: genthreader is able to assign 20 proteins that dasey is unable to. On the other hand, we find that dasey is able to assign 105 proteins that genthreader is unable to, an improvement of 17%.
dasey's Performance Gain Is Not Just from Improved Handling of Secondary Structure Information.
dasey scoring differs from the sdp and Bowie methods in two major respects: improved handling of secondary structure information and more elaborate descriptions of structural environments. It might be argued that the improved performance of dasey is primarily due to a better use of secondary structure and that the environmental component is irrelevant. However, a comparison of the difference in performance of sdp+ and dasey reveals improved performance of dasey is mainly from the environmental-dependent scoring, because the scoring tables used in sdp+, were derived from daps. Although sdp+ is able to assign 23 proteins dasey is unable to, dasey is able to assign 130 proteins sdp+ is unable to.
Accessibility of dasey and Avenues for Improvement.
Although dasey performs better than some earlier procedures, there is room for improvement, as shown by Fig. 4. We use the area underneath Receiver Operator Characteristic curves to measure of dasey's ability to use a given template structure, or fold class, to discriminate sequences sharing fold-level similarity from sequences not sharing fold level similarity. With this measure, we observe that dasey is least able to assign sequences belonging to templates of small proteins and of proteins containing a large percentage of coil. For example, protein templates belonging to CATH class four, protein domains which have low secondary structure content, were less well assigned than protein domains whose structures belong to classes one, two, or three. Furthermore protein domains such as 3ldh01 and 1c0dA0, which contain extended coil regions, are poorly assigned despite belonging to the Rossmann and TIM Barrel folds, respectively, which overall are among the best-assigned fold classes. We also observe that short proteins will often preferentially match substructures that resemble their native fold within larger domains of a different fold type. For example, a sequence whose true fold is a helix-turn-helix might on occasion preferentially match a helix-turn-helix region in a large mixed α-β fold.
Probe sequences may be submitted to dasey for assignment to known folds at the University of California Los Angeles Department of Energy Fold Server, http://fold.doe-mbi.ucla.edu.
Supplementary Material
Acknowledgments
We thank Rob Grothe, Charlotte Deane, and Lukasz Salwinski for extensive discussions, technical advice, and suggestions. The work of P.M. was funded in part by the National Science Foundation. We also thank BER–Department of Energy, National Institutes of Health, and Howard Hughes Medical Institute for support.
Abbreviations
daps, Database of Aligned Pairs of Structures
dasey, Directional Atomic Solvation EnergY
PDF, Probability Density Function
sdp, Sequence Derived Properties
References
- 1.Benson D. A., Karsch-Mizrachi, I., Lipman, D. J., Ostell, J., Rapp, B. A. & Wheeler, D. L. (2002) Nucleic Acids Res. 30, 17-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Berman H. M., Battistuz, T., Bhat, T. N., Bluhm, W. F., Bourne, P. E., Burkhardt, K., Feng, Z., Gilliland, G. L., Iype, L., Jain, S., et al. (2002) Acta Crystallogr. D 58, 899-907. [DOI] [PubMed] [Google Scholar]
- 3.Bowie J. U., Lüthy, R. & Eisenberg, D. (1991) Science 253, 164-170. [DOI] [PubMed] [Google Scholar]
- 4.Bryant S. H. & Lawrence, C. E. (1993) Proteins 16, 92-112. [DOI] [PubMed] [Google Scholar]
- 5.Defay T. R. & Cohen, F. E. (1996) J. Mol. Biol. 262, 314-323. [DOI] [PubMed] [Google Scholar]
- 6.Fischer D. & Eisenberg, D. (1996) Protein Sci. 5, 947-955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Godzik A. & Skolnick, J. (1992) Proc. Natl. Acad. Sci. USA 89, 12098-12102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hendlich M., Lackner, P., Weitckus, S., Floeckner, H., Froschauer, R., Gottsbacher, K., Casari, G. & Sippl, M. J. (1990) J. Mol. Biol. 216, 167-180. [DOI] [PubMed] [Google Scholar]
- 9.Huang E. S., Subbiah, S., Tsai, J. & Levitt, M. (1996) J. Mol. Biol. 257, 716-725. [DOI] [PubMed] [Google Scholar]
- 10.Jones D. T. (1999) J. Mol. Biol. 287, 797-815. [DOI] [PubMed] [Google Scholar]
- 11.Karplus K., Barrett, C., Cline, M., Diekhans, M., Grate, L. & Hughey, R. (1999) Proteins Suppl, 121-125. [DOI] [PubMed] [Google Scholar]
- 12.Koretke K. K., Russell, R. B. & Lupas, A. N. (2002) Protein Sci. 11, 1575-1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krogh A., Brown, M., Mian, I. S., Sjolander, K. & Haussler, D. (1994) J. Mol. Biol. 235, 1501-1531. [DOI] [PubMed] [Google Scholar]
- 14.Lundstrom J., Rychlewski, L., Bujnicki, J. & Elofsson, A. (2001) Protein Sci. 10, 2354-2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.MacCallum R. M., Kelley, L. A. & Sternberg, M. J. (2000) Bioinformatics 16, 125-129. [DOI] [PubMed] [Google Scholar]
- 16.Rice D. W. & Eisenberg, D. (1997) J. Mol. Biol. 267, 1026-1038. [DOI] [PubMed] [Google Scholar]
- 17.Ouzounis C., Sander, C., Scharf, M. & Schneider, R. (1993) J. Mol. Biol. 232, 805-825. [DOI] [PubMed] [Google Scholar]
- 18.Rost B., Schneider, R. & Sander, C. (1997) J. Mol. Biol. 270, 471-480. [DOI] [PubMed] [Google Scholar]
- 19.Russell R. B., Copley, R. R. & Barton, G. J. (1996) J. Mol. Biol. 259, 349-365. [DOI] [PubMed] [Google Scholar]
- 20.Salwinski L. & Eisenberg, D. (2001) Protein Sci. 10, 2460-2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sippl M. J. & Weitckus, S. (1992) Proteins 13, 258-271. [DOI] [PubMed] [Google Scholar]
- 22.Vila J., Williams, R. L., Vasquez, M. & Scheraga, H. A. (1991) Proteins 10, 199-218. [DOI] [PubMed] [Google Scholar]
- 23.Wilmanns M. & Eisenberg, D. (1993) Proc. Natl. Acad. Sci. USA 90, 1379-1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rychlewski L., Jaroszewski, L., Li, W. & Godzik, A. (2000) Protein Sci. 9, 232-241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kelley L. A., MacCallum, R. M. & Sternberg, M. J. (2000) J. Mol. Biol. 299, 499-520. [DOI] [PubMed] [Google Scholar]
- 26.Smith T. F. & Waterman, M. S. (1981) J. Mol. Biol. 147, 195-197. [DOI] [PubMed] [Google Scholar]
- 27.Needleman S. B. & Wunsch, C. D. (1970) J. Mol. Biol. 48, 443-453. [DOI] [PubMed] [Google Scholar]
- 28.Browne W. J., North, A. C., Phillips, D. C., Brew, K., Vanaman, T. C. & Hill, R. L. (1969) J. Mol. Biol. 42, 65-86. [DOI] [PubMed] [Google Scholar]
- 29.Gribskov M., McLachlan, A. D. & Eisenberg, D. (1987) Proc. Natl. Acad. Sci. USA 84, 4355-4358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Altschul S. F., Madden, T. L., Schäffer, A. A., Zhang, J., Zhang, Z., Miller, W. & Lipman, D. J. (1997) Nucleic Acids Res. 25, 3389-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jones D. T., Taylor, W. R. & Thornton, J. M. (1992) Nature 358, 86-89. [DOI] [PubMed] [Google Scholar]
- 32.Lo Conte L. & Smith, T. F. (1997) J. Mol. Biol. 273, 338-348. [DOI] [PubMed] [Google Scholar]
- 33.Orengo C. A., Michie, A. D., Jones, S., Jones, D. T., Swindells, M. B. & Thornton, J. M. (1997) Structure (Cambridge, U.K.) 5, 1093-1108. [DOI] [PubMed] [Google Scholar]
- 34.Kabsch W. & Sander, C. (1983) Biopolymers 22, 2577-2637. [DOI] [PubMed] [Google Scholar]
- 35.Rost B. (1996) Methods Enzymol. 266, 525-539. [DOI] [PubMed] [Google Scholar]
- 36.Holm L. & Sander, C. (1997) Nucleic Acids Res. 25, 231-234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Holm L. & Sander, C. (1995) Trends Biochem. Sci. 20, 478-480. [DOI] [PubMed] [Google Scholar]
- 38.Russell R. B. & Barton, G. J. (1992) Proteins 14, 309-323. [DOI] [PubMed] [Google Scholar]
- 39.Sali A. & Blundell, T. L. (1990) J. Mol. Biol. 212, 403-428. [DOI] [PubMed] [Google Scholar]
- 40.Altschul S. F. & Koonin, E. V. (1998) Trends Biochem. Sci. 23, 444-447. [DOI] [PubMed] [Google Scholar]
- 41.Jones D. T. & Swindells, M. B. (2002) Trends Biochem. Sci. 27, 161-164. [DOI] [PubMed] [Google Scholar]
- 42.Huynen M., Doerks, T., Eisenhaber, F., Orengo, C., Sunyaev, S., Yuan, Y. & Bork, P. (1998) J. Mol. Biol. 280, 323-326. [DOI] [PubMed] [Google Scholar]
- 43.Hughey R. & Krogh, A. (1996) Comput. Appl. Biosci. 12, 95-107. [DOI] [PubMed] [Google Scholar]
- 44.McLachlan, G. J., Peel, D., Basford, K. E. & Adams, P. (1999) J. Stat. Software 4.
- 45.Sweet R. M. & Eisenberg, D. (1983) J. Mol. Biol. 171, 479-488. [DOI] [PubMed] [Google Scholar]
- 46.Eisenberg D. & McLachlan, A. D. (1986) Nature 319, 199-203. [DOI] [PubMed] [Google Scholar]
- 47.Fischer, D., Elofsson, A., Rice, D. & Eisenberg, D. (1996) Pac. Symp. Biocomput. 300–318. [PubMed]
- 48.Scharf L. L., (1991) Statistical Signal Processing: Detection, Estimation and Time Series Analysis (Addison–Wesley, Menlo Park, CA).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.