

Abstract
The M-type potassium current (IM) plays a dominant role in regulating membrane excitability and is modulated by many neurotransmitters. However, except in the case of bradykinin, the signal transduction pathways involved in M-channel modulation have not been fully elucidated. The channels underlying IM are produced by the coassembly of KCNQ2 and KCNQ3 channel subunits and can be expressed in heterologous systems where they can be modulated by several neurotransmitter receptors including histamine H1 receptors. In HEK293T cells, histamine acting via transiently expressed H1R produced a strong inhibition of recombinant M-channels but had no overt effects on the voltage dependence or voltage range of IM activation. In addition, the modulation of IM by histamine was not voltage sensitive, whereas channel gating, particularly deactivation, was accelerated by histamine. Non-hydrolysable guanine nucleotide analogues (GDP-β-S and GTP-γ-S) and pertussis toxin (PTX) treatment demonstrated the involvement of a PTX-insensitive G protein in the signal transduction pathway mediating histamine-induced IM modulation. Abrogation of the histamine-induced modulation of IM by expression of a C-terminal construct of phospholipase C (PLC-β1-ct), which buffers activated Gαq/11 subunits, implicates this G protein α subunit in the modulatory pathway. On the other hand, abrogation of the histamine-induced modulation of IM by expression of two constructs which buffer free βγ subunits, transducin (Gαt) and a C-terminal construct of a G protein receptor kinase (MAS-GRK2-ct), implicates βγ dimers in the modulatory pathway. These findings demonstrate that histamine modulates recombinant M-channels in HEK293T cells via a PTX-insensitive G protein, probably Gαq/11, in a similar manner to a number of other G protein-coupled receptors. However, histamine-induced IM modulation in HEK293T cells is novel in that βγ subunits in addition to Gαq/11 subunits appear to be involved in the modulation of KCNQ2/3 channel currents.
The M-type K+ current (IM) is a slowly activating and non-inactivating K+ current that was first identified in bullfrog sympathetic neurons (Brown & Adams, 1980) and subsequently in many neuronal and non-neuronal cells (Constanti & Brown, 1981; Cassell & McLachlan, 1987; Constanti & Sim, 1987; Moore et al. 1988; Clapp et al. 1992; Tokimasa et al. 1993). The current is termed M since it is inhibited by muscarinic acetylcholine receptor (mAChR) agonists. Suppression of IM by acetylcholine as well as other neurotransmitters relieves the negative effect of IM on neuronal excitability and therefore enhances neuronal excitability. In central and peripheral neurons, IM appears to control neuronal excitability by helping set the resting membrane potential, limiting membrane depolarization, accelerating the decay of the synaptic potential and producing hyperpolarization (for review see Brown, 1988). Thus far, it is generally accepted that IM plays a dominant role in regulating membrane excitability, not only because it is one of few sustained time- and voltage-sensitive currents in the range of action potential initiation, but also because of its modulation by many neurontransmitters and putative regulatory pathways (for reviews see Marrion, 1997; Brown & Yu, 2000). Although the detailed mechanism(s) involved in M-channel modulation have not been fully elucidated, a common finding among studies of IM modulation is that many neurotransmitters attenuate IM by activation of Gq/11 protein-coupled receptors (for review see Marrion, 1997). For example, in rat sympathetic neurons, IM is inhibited by activating endogenous M1 mAChR (Marrion et al. 1989), angiotensin II receptors (Shapiro et al. 1994), B2 bradykinin receptors (Jones et al. 1995) and P2Y2 purinergic receptors (Filippov et al. 1998) via pertussis toxin (PTX)-insensitive G proteins that activate phospholipase C (PLC).
Histamine is a widely distributed biogenic amine neurotransmitter which exerts many modulatory functions by activating its four different types of receptors (H1-H4). Among the four different types of receptor, the histamine H1 receptor (H1R) is a Gq/11 protein-coupled receptor. It has been demonstrated that activation of H1R enhances neuronal excitability and causes strong membrane depolarization and/or increases firing frequency (for review see Brown et al. 2001). For example, in cat and guinea-pig lateral geniculate relay neurons, histamine, acting via H1 receptors, evokes a slow depolarization that is associated with a decrease in a potassium current which can be inhibited by activation of muscarinic receptors (McCormick & Williamson, 1991). In human cortical neurons, activation of H1R by histamine also results in a depolarization associated with an increase in input resistance, probably due to block of a K+ conductance which normally contributes to the resting membrane potential (Reiner & Kamondi, 1994; Brown et al. 2001, review). Recently, we reported that histamine, by activating H1R, could be added to the pool of agonists, muscarinic, P2Y2 purinergic, antiogensin II, and bradykinin receptor agonists, which are known to inhibit M-channels (Guo & Schofield, 2002). In our present studies, we have begun to investigate the mechanism underlying the histamine-induced IM modulation.
Activation of a G protein is always the first step involved in Ca2+ channel and M-channel modulation after G protein coupled receptor (GPCR) activation. Moreover, the G protein involved in M-channel modulation is probably PTX-insensitive (Brown et al. 1989; Jones et al. 1995; Villarroel, 1996). The βγ dimer has been demonstrated to mediate voltage-dependent modulation of N-type Ca2+ channels by many neurotransmitters (Herlitze et al. 1996; Ikeda, 1996) and both Gαq/11 subunits and Gβγ dimers are required in a voltage-independent Ca2+ current modulatory pathway evoked by muscarinic agonists in rat superior cervical ganglion (SCG) neurons (Kammermeier et al. 2000). However, to date only the Gα subunit, rather than the Gβγ subunit, has been suggested to participate in M-channel modulation. The involvement of Gα subunits was proposed due to the fact that microinjection of antibodies specific for the α subunit of Gq/11 protein into rat SCG neurons attenuated muscarinic and bradykinin inhibition of IM (Caulfield et al. 1994; Jones et al. 1995). Using Gαq-deficient mice, Haley et al. (2000) further suggested that IM inhibition by activation of M1 mAChR was mediated partly by Gαq and more predominantly by Gα11. On the other hand, several studies have implied that Gβγ subunits are not required in IM modulation. In rat SCG neurons, overexpression of the C terminus of β-adrenergic receptor kinase 1 (βARK1), which binds and sequesters free βγ subunits, did not attenuate muscarinic inhibition of IM at a concentration of the construct sufficient to attenuate the Gβγ-mediated inhibition of N-type Ca2+ channels (Haley et al. 1998). Furthermore, sequestration of free Gβγ subunits by overexpression of different inactive, GDP-bound Gα subunits to buffer Gβγ subunits in rat SCG neurons also failed to prevent IM modulation by muscarine (Jeong & Ikeda, 1999).
In the present studies, we first tested the effects of non-hydrolysable GDP and GTP analogues on IM modulation by histamine to confirm if, like the activation of other GPCR, activation of a G protein is also involved in histamine-induced recombinant M-channel modulation. Overnight treatment with PTX was performed to determine whether the G protein mediating histamine-induced IM inhibition is PTX-insensitive, a property shared with other receptor systems which inhibit IM. To determine which subunits mediate histamine-induced IM modulation, we then cotransfected HEK293T cells with different constructs to bind and buffer Gα or Gβγ subunits, respectively. The present studies show that although activation of a PTX-insensitive G protein is involved in histamine-induced IM modulation, unlike muscarinic- and bradykinin-induced inhibition, not only Gα but also Gβγ subunits appear to be involved in the histamine-induced IM modulation.
Methods
Cell culture and transfection
The method of cell culture and transfection was described previously (Guo & Schofield, 2002). Briefly, HEK293T cells, kindly provided by Dr Jamboor K. Vishwanatha (University of Nebraska Medical Center, Omaha, NE, USA), were cultured in minimum essential medium (MEM) supplemented with 1 % penicillin-streptomycin, 10 % fetal calf serum under an atmosphere containing 5 % CO2 at 37 °C. The cells were transfected with the human KCNQ2 and rat KCNQ3 cDNAs, H1R cDNA and pEGFP-N1 encoding the green fluorescent protein (to facilitate the identification of transfected cells) as follows. A transfection mixture of 0.5 μg each of KCNQ2/pcDNA3.1, KCNQ3/pcDNA3.1, H1R/pcDNA3, pEGFP-N1, and 6 μl of TransIT-LT1 reagent was made in 75 μl of opti-MEM and preincubated for 20 min. The mixture was then applied to cell culture wells containing 1 ml of culture medium with HEK293T cells at ≈90 % confluence. Concentration-response relationships for expression for transducin (Gαt) and phospholipase-C-β1 C-terminal (PLC-β1-ct) constructs on IM inhibition by histamine were obtained by adding 2.5, 25, 250 and 500 ng of the constructs to the above mixture in 7.5 μl TransIT-LT1 reagent. Alternatively, 500 ng of regulator of G protein signalling 2 (RGS2) or 250 ng of a C-terminal G protein-coupled receptor kinase 2 (MAS-GRK2-ct) construct was added into the transfection mixture when required. After 24 h incubation, the cells were plated onto 35 mm culture dishes which served as the electrophysiological recording chamber. All transfected cells were used within 48 h of plating.
Electrophysiology
HEK293T cells were voltage clamped with the whole-cell perforated-patch configuration of the patch-clamp technique using an Axopatch 1-C or 200A amplifier (Axon Instruments, Foster city, CA, USA). Patch electrodes were fabricated from N51A borosilicate capillary tubing (Garner Glass, Claremont, CA, USA) using a Model P80-PC micropipette puller (Sutter, Novato, CA, USA) and fire-polished to final resistances of < 2 MΩ when filled with the internal solutions. Under nystatin perforated-patch conditions, series resistance was normally less than 15 MΩ; whereas with the open-tip configuration, series resistance was less than 5 MΩ. In both cases, 80 % series resistance compensation was normally applied. Current traces were filtered at 1.0 kHz (-3 dB) using a four-pole low-pass Bessel filter, digitized at 5 kHz with a 12-bit analog-to-digital converter (GW instruments, Somerville, MA, USA) and stored for analysis using a Macintosh Quadra 800 computer. Voltage protocol generation and data acquisition were performed from a 12-bit digital-to-analog converter (GW Instruments) using the S3 data acquisition software (Ikeda SR, NIAAA, Rockville, MD, USA). All recordings were carried out at room temperature (23-25 °C).
Solutions and chemicals
The bath solution contained (mm): NaCl 150, KCl 2, MgCl2 1, CaCl2 2, Hepes 10 and glucose 11, adjusted to pH 7.4 with NaOH. In most experiments, whole-cell IM was recorded by the nystatin perforated-patch technique to prevent rundown which can be prominent under open-tip whole-cell recording conditions. For this purpose, patch pipettes were first tip-filled with an internal solution containing (mm): KCl 55, K2SO4 75, Hepes 10, MgCl2 8, adjusted to pH 7.4 with KOH. Pipettes were then back-filled with the same internal solution containing 240 μg ml−1 nystatin made from a freshly prepared stock solution (60 mg ml−1) of nystatin in dimethyl sulfoxide. In order to apply GTP or related nucleotides to the cytosol of the cells under investigation, IM was recorded in the conventional open-tip whole-cell configuration with an internal solution containing (mm): potassium methylsulfate (KMeSO4) 110, KCl 15, EGTA 0.3, MgCl2 4, Hepes 10, Na2ATP 4, Li3GTP 0.5, adjusted to pH 7.4 with KOH. Where indicated, either 0.5 mm GTPγS or 2 mm GDPβS (lithium salts) were added to the pipette solutions. The osmolalities of the bath and pipette solutions were adjusted with sucrose to 305 and 285 mosmol kg−1, respectively.
Drugs were applied to cells via a custom-built multi-barrelled perfusion system with gravity-fed polyethylene tubes connected to reservoirs containing different solutions. To avoid possible problems due to desensitization, histamine was applied only once for any given cell.
The drugs and chemicals used in these experiments were obtained as follows. KMeSO4 was from ICN (Aurora, OH, USA). Histamine dihydrochloride, Hepes, EGTA, GTP lithium salt, and ATP sodium salt were from Sigma/Aldrich (St Louis, MO, USA). Penicillin-streptomycin, fetal calf serum, Opti-MEM, and MEM were from Gibco/BRL Life Technologies (Gaithersberg, MD, USA). Mirus TransIT-LT1 reagent was from Pan Vera (Madison, WI, USA). PTX, GTPγS lithium salt and GDPβS lithium salt were from Calbiochem (La Jolla, CA, USA).
Data analysis and statistics
Currents were analysed and fitted using Igor Pro software (Wave Metrics, Lake Oswego, OR, USA) on a Macintosh Quadra 800 computer. All data are expressed as means ± s.e.m. ANOVA or Student's t test (paired or unpaired) was used to determine statistical significance. P < 0.05 was considered significant.
Results
Histamine inhibits KCNQ2/3 channel currents
Our previous studies showed that histamine inhibited KCNQ2/3 channel currents via histamine H1 receptors heterologously expressed in HEK293T and HeLa cells (Guo & Schofield, 2002). In cells transfected only with the cDNA encoding the green fluorescent protein, depolarizing pulses elicited a small, voltage-dependent, outward, background current which began to activate positive to -20 mV with a mean amplitude of 390 ± 130 pA (n = 6) at +40 mV.
Figure 1A shows current traces elicited from a HEK293T cell cotransfected with KCNQ2, KCNQ3, H1R cDNAs in the absence and presence of a saturating (10 μm) concentration of histamine. The transfected HEK293T cell was held at -20 mV to avoid contamination with the endogenous voltage-dependent K+ current, and then hyperpolarized to -50 mV to elicit typical inward current relaxation. Histamine (10 μm) produced a significant inhibition of both holding current and inward current relaxation (Fig. 1A). The majority of the cells displayed virtually no leak current and the amplitude of IM was measured as the holding current at -20 mV. In the few cells that displayed a significant leak current, the amplitude of IM was measured from the deactivation tails at -50 mV as the difference between the average of the first 10 ms and last 10 ms of the hyperpolarizing step. The percentage IM inhibition was calculated as:
where Ipre and Ipost are the IM before and after histamine application. The mean inhibition of IM by 10 μm histamine was 78.2 ± 4.3 % (n = 17). IM was usually largely recovered after washout under perforated-patch conditions; in the cell shown in Fig. 1B, the current was totally restored. The histamine-induced IM inhibition was mediated by histamine H1 receptors since pretreatment with the selective H1R antagonist, astemizole, abolished the histamine-induced inhibition (Guo & Schofield, 2002).
Figure 1. Inhibition of KCNQ2/3 channel currents by histamine via histamine H1 receptors.
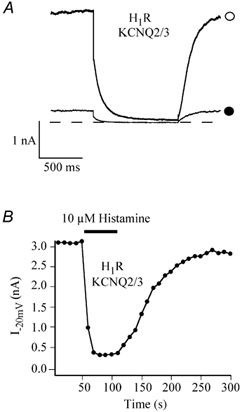
A, superimposed KCNQ2/3 channel currents elicited from a HEK293T cell cotransfected with KCNQ2/3 and H1R cDNAs in the absence (○) and presence (•) of 10 μm histamine, recorded using the nystatin perforated-patch technique. The dashed line represents the zero current level. The cell was held at -20 mV and then hyperpolarized to -50 mV to elicit typical slow inward current relaxation. B, time course of histamine effect in the same cell showing the reversible inhibition of IM.
Voltage-independent M-channel modulation by histamine
Ca2+ channel modulation by activating different GPCRs can be either voltage dependent or voltage independent, whereas it has been reported that muscarinic inhibition of KCNQ2/3 channel current is not voltage dependent (Shapiro et al. 2000). To test whether histamine-induced recombinant M-channel modulation, like that induced by muscarine, was also voltage independent, activation curves in the absence and presence of histamine were constructed as follows: currents were elicited from H1R and KCNQ2/3 cDNA-cotransfected HEK293T cells with 1 s voltage steps in 10 mV increments from -70 to +40 mV from a holding potential of -60 mV, followed by a 500 ms voltage step to -60 mV in the absence and presence of 1 μm histamine (Fig. 2A), a concentration which produces about 75 % maximal inhibition (Guo & Schofield, 2002). Normalized current-voltage (I-V) curves were constructed from peak currents elicited by voltage steps in the absence or presence of 1 μm histamine (Fig. 2C), peak currents were normalized to the peak current generated by a depolarizing step to +40 mV before the application of histamine. M-channels began to activate at around -40 mV both in the absence and presence of histamine. It should be noted that because of the background voltage-dependent K+ channels of HEK293T cells, the magnitude of KCNQ2/KCN3 channel blockade produced by histamine shown in the I-V curve might be underestimated. Since endogenous K+ channels did not produce appreciable tail currents when the cells were hyperpolarized to -60 mV following each voltage step (data not shown), whereas M-channel produced typical inward current relaxation by a hyperpolarizing step (expanded in Fig. 2B), we studied the voltage-independent blockade by measuring the tail currents at -60 mV. Tail current amplitudes were extrapolated from double exponential fits of the current relaxation. The mean percentage inhibitions of tail currents produced by 1 μm histamine were plotted against membrane potential as shown in Fig. 2D. The histamine-induced IM inhibition was not significantly different at test voltages > -30 mV (P > 0.05), demonstrating that the histamine-induced IM modulation is not voltage dependent. At test voltages < -30 mV tail currents were small, particularly after application of histamine which caused large variability of tail current amplitude, and consequently the effects of histamine were not tested at these potentials.
Figure 2. Voltage-independent inhibition of histamine.
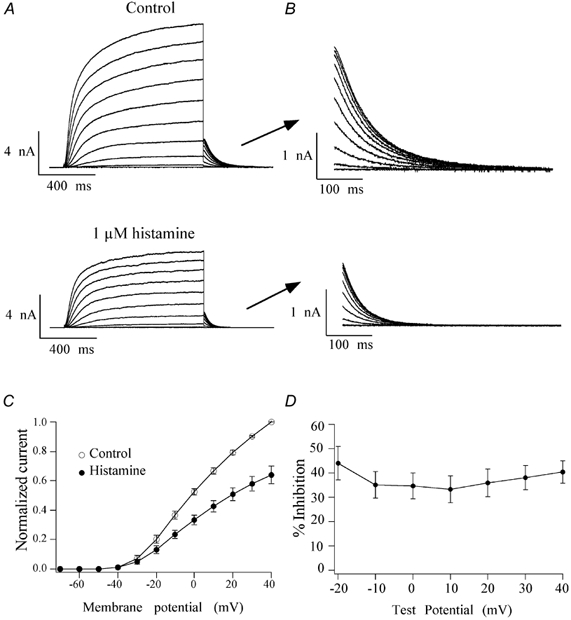
A, families of currents elicited by depolarizing steps from -70 mV to +40 mV from a holding potential of -60 mV in the absence and presence of 1 μm histamine, recorded using the nystatin perforated-patch technique. B, tail currents shown on a faster time scale. C, normalized current-voltage relationship of M-channels in the absence (○) and presence (•) of histamine. The peak current at each voltage step was normalized to the peak current elicited by the depolarizing step to +40 mV in the absence of histamine. D, the mean percentage inhibition of tail currents was plotted against each voltage step to demonstrate the voltage-independent inhibition of IM by histamine. Error bars indicate the s.e.m.
Histamine-induced IM inhibition does not shift voltage dependence of channel activation
A shift of the Ca2+ channel activation curve is involved in Ca2+ channel modulation by many neurotransmitters (Herlitze et al. 1996), whereas muscarinic inhibition of KCNQ2/3 channels is not associated with changes in channel voltage dependence (Shapiro et al. 2000). We tested whether there was a shift of recombinant M-channel voltage dependence in the histamine-induced IM modulation using a similar protocol to that described in Fig. 2. Tail current amplitude at -60 mV resulting from various test pulses (Fig. 2B) was well described by a modified Boltzmann equation:
where I is the tail current amplitude at the test potential V, Imax is the fitted maximum tail current, V1/2 is the half-activation voltage, and k the slope factor of activation. The M-channel activation curves before and after application of 1 μm histamine are shown in Fig. 3. Like muscarinic inhibition, histamine did not significantly change the V1/2 (-11.9 ± 1.7 mV vs. -14.0 ± 1.6 mV before and after application of histamine, n = 7; P > 0.05), however, the slope of the activation was slightly but significantly increased by histamine (13.1 ± 1.1 mV vs. 9.9 ± 0.8 mV before and after application of histamine, n = 7; P < 0.05). This slight change in activation slope might be due to M-current rundown and/or histamine receptor desensitization.
Figure 3. Activation curves in the absence (○) and presence (•) of 1 μm histamine.
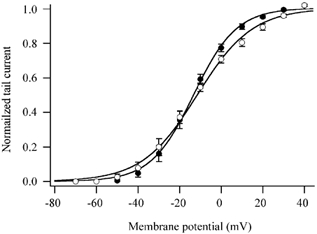
Curves were obtained from tail currents recorded at -60 mV following voltage steps from -70 mV to +40 mV as shown in Fig. 2B. The activation curves were fitted with a modified Boltzmann equation (see text). Error bars are the s.e.m.
Effect of histamine on IM gating
The activation phase of IM was somewhat variable. Usually IM activated with a sigmoid time course which displayed two components and was well fitted by the following equation:
where Imax,f and Imax,s are the amplitudes of the fast and slow activation phases and τf and τs are the fast and slow time constants. The remaining M-currents were fitted by a single exponential function raised to the second power. Figure 4 shows examples of the rising phase of the M-current elicited by a 1 s pulse to -20 mV from a holding potential of -60 mV in the absence and presence of 10 μm histamine. The current records shown are plotted at 1/50th of the original point density in order to more clearly reveal the superimposed fit. The continuous line is a fit of the data to the above equation. The residuals, calculated from the data and fitted line, are shown on an expanded scale below each record. Both the fast and slow activation time constants showed a tendency to be reduced by histamine but these changes did not reach statistical significance. The fast time constant was 73.1 ± 17.6 ms and 59.7 ± 5.7 ms (n = 6) in the absence and presence of histamine, respectively. The slow activation time constant was 599.3 ± 168.9 ms (n = 6) and 263.4 ± 56.1 ms (n = 5) in the absence and presence of histamine, respectively.
Figure 4. Effects of histamine on IM activation.
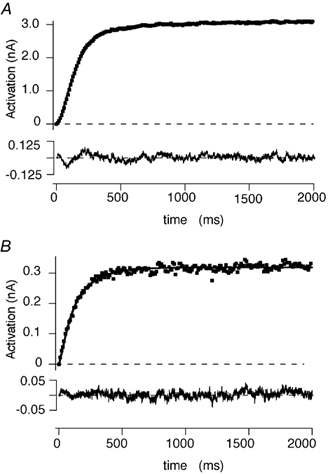
A and B, the activation phase of IM elicited by a 2 s depolarizing step from -60 mV to -20 mV in the absence and presence of 10 μm histamine, respectively. Currents were recorded using the nystatin perforated-patch technique. The currents are plotted at 1/50th of the original density. The continuous lines are fits to a double exponential function (see text). The dashed lines represent the zero current level. The residuals calculated from the difference between the original record and the fitted line are plotted below each record.
The deactivation process of IM was well fitted with a double exponential equation both in the absence and presence of histamine. Figure 5 shows examples of the current relaxation phase of the M-current elicited by repolarizing the membrane to -50 mV after a 1 s pulse to -20 mV in the absence and presence of 10 μm histamine. The current records shown are plotted at 1/50th of the original point density. The continuous line is a fit of the data to a double exponential equation:
where Imax,f and Imax,s are the amplitudes of the fast and slow deactivation phases and τf and τs are the fast and slow time constants. The residuals, calculated from the data and fitted line, are shown on an expanded scale below each record. Histamine decreased both the fast and slow deactivation time constants of IM. The bar graphs of Fig. 5C and D show the fast and slow time constants, in the absence and presence of 10 μm histamine, respectively. The fast time constant decreased from 69.3 ± 5.6 ms in the absence of histamine to 43.7 ± 6.7 ms in the presence of histamine (n = 10, P < 0.05). The slow time constant decreased from 316.0 ± 58.0 ms in the absence of histamine to 170.0 ± 30.4 ms in the presence of histamine (n = 9, P < 0.05). The effects of histamine on IM deactivation were examined at two other potentials, -35 mV and -65 mV (data not shown). Under control conditions, stepping from -20 mV to -35 mV also resulted in a double exponential relaxation of IM but both τf and τs were significantly greater than at -50 mV, and histamine (10 μm) significantly decreased both time constants. At -65 mV however, IM deactivation in the absence and presence of 10 μm histamine appeared to be dominated by the fast time constant such that the currents tended to decay as a single exponential process. Histamine had no significant effect on the proportion of the two decay components at the potentials tested.
Figure 5. Effects of histamine on IM deactivation.
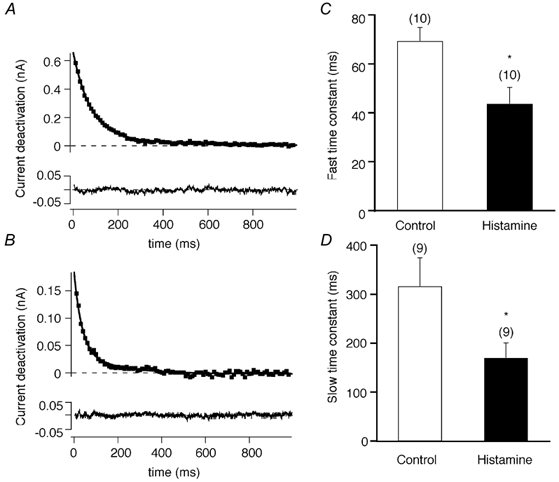
A and B, the deactivation tail current of IM elicited by repolarizing the membrane to -50 mV after a 2 s depolarizing step to -20 mV from -60 mV in the absence and presence of 10 μm histamine, respectively. Currents were recorded using the nystatin perforated-patch technique. The currents are plotted at 1/50th of the original density. The continuous lines are fits to a double exponential function (see text). The dashed lines represent the zero current level. The residuals calculated from the difference between the original record and the fitted line are plotted below each record. C and D, the effects of histamine on the fast and slow time constants, respectively. The numbers in parentheses represents the number of cells tested, *P < 0.05 compared with control. Error bars are the s.e.m.
Effect of non-hydrolysable GTP and GDP analogues on IM inhibition by histamine
Neuronal function is widely regulated by activation of GPCRs in both the peripheral and central nervous systems. It is well known that in rat sympathetic neurons, the initial step in the neurotransmitter-mediated inhibition of IM upon receptor activation (e.g. M1 mAChR and B2 bradykinin receptor) is the activation of a G protein. Since the release of intracellular Ca2+ caused by H1 receptor activation occurs through activation of Gq/11, we sought to determine if histamine inhibition of IM also occurred through a G protein. Using the open-tip configuration, HEK293T cells cotransfected with H1R and KCNQ2/3 cDNAs were dialysed with GTP (0.5 mm) alone, GTP (0.5 mm) and GTPγS (0.5 mm), or GTP (0.5 mm) and GDPβS (2 mm). GTP was not omitted from the pipette solution when GTPγS or GDPβS was included because inclusion of GTP helps prevent significant current rundown in the open-tip configuration (data not shown). When the pipette solution contained 0.5 mm GTP, 10 μm histamine significantly inhibited IM as, under nystatin-perforated patch conditions, the average inhibition by 10 μm histamine was 88.6 ± 3.7 % (n = 5); the recovery from histamine inhibition was partially reversible under the open-tip configuration (Fig. 6A).
Figure 6. Effects of intracellular GTP and GDP analogues on the histamine-induced inhibition of KCNQ2/3 currents recorded using the open-tip configuration.
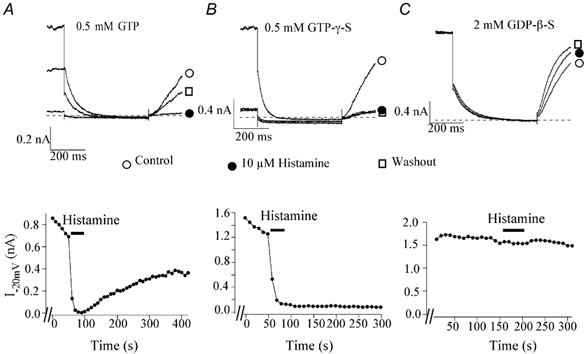
A-C, KCNQ2/3 currents before (○), during (•) and after (□) 10 μm histamine application with 0.5 mm GTP (A), 0.5 mm GTPγS(B) or 2 mm GDPβS (C) added to the intracellular solution. The current traces were elicited by a 1 s step to -50 mV from a holding potential of -20 mV. The dashed lines represent the zero current level. The lower panel shows the time course of the histamine effect on M-currents elicited from the corresponding cells.
The non-hydrolysable GTP analogue, GTPγS, binds to the α subunits of G proteins, thus trapping the G proteins in an active conformation and conferring irreversible G protein activation. Inclusion of 0.5 mm GTPγS in the pipette solution was performed to test whether it rendered the histamine-induced inhibition irreversible. After 2-4 min dialysis with GTPγS, 10 μm histamine produced an irreversible inhibition of IM of 97.0 ± 1.0 % (n = 6; Fig. 6B). In some experiments, very brief application of histamine (< 20 s) was used, to avoid reaching the maximum inhibition. Under this condition, IM remained attenuated after washout reaching a steady state residual level, whereas when only GTP was included in the pipette solution, IM attenuation was terminated by washout and IM began to return to the pre-drug level (data not shown) as in Fig. 6A.
GDPβS is a non-hydrolysable GDP analogue that is poorly phosphorylated and has been shown to prevent the G protein-mediated effects by trapping the G protein in an inactive conformation. Inclusion of 2 mm GDPβS in the pipette solution was performed to test whether histamine-induced IM modulation could be prevented. After a 10 min dialysis to achieve a stable concentration of GDPβS, histamine-induced IM inhibition was significantly attenuated (Fig. 6C), the mean inhibition was 16.3 ± 8.5 % (n = 6; P < 0.05vs. control). Inclusion of GDPβS itself did not significantly change IM except for the current rundown seen in all open-tip recording.
The inclusion of guanine nucleotide analogues in the pipette solution resulted in the addition of up to 6 mm Li+ to the internal solutions. Since it is possible that Li+ could modify some of the intracellular signal transduction pathways (Drummond, 1987) and may therefore interfere with IM modulation, we tested the direct effects of Li+ by adding 6 mm LiCl to the pipette solution. Under these conditions, IM was not altered during dialysis for 2-15 min and 10 μm histamine still produced strong (80.3 ± 5.9 %, n = 6) IM inhibition (data not shown).
A PTX-insensitive G protein is involved in IM inhibition by histamine
In sympathetic neurons, inhibition of M-current by muscarinic and bradykinin receptors has been shown to be mediated by the α subunit of the Gq/11 heterotrimer which is not sensitive to PTX (Jones et al. 1995; Haley et al. 1998). To determine if the IM inhibition by histamine was also PTX-insensitive, we pretreated the cells with 500 ng ml−1 PTX. In our experiments, overnight pretreatment with PTX did not prevent histamine-induced IM inhibition (Fig. 7A). The histamine-induced IM inhibition after PTX treatment (56.8 ± 7.4 %, n = 7) was not significantly different from the control group (76.9 ± 6.2 %, n = 9; P > 0.05; Fig. 7B). Since PTX treatment resulted in additional time between transfecting and measurement of the histamine-induced IM modulation, a separate control group was utilized in this series of experiments. In rat SCG neurons, noradrenaline (NA; norepinephrine) inhibition of N-type Ca2+ channels is partially PTX-sensitive (Schofield, 1990). To ensure that PTX was effective, we used the NA-induced N-type Ca2+ channel modulation as a positive control. Overnight treatment of rat SCG neurons with 500 ng ml−1 PTX significantly prevented 10 μm NA-induced inhibition of N-type Ca2+ currents (data not shown).
Figure 7. PTX does not prevent KCNQ2/3 channel inhibition by histamine.
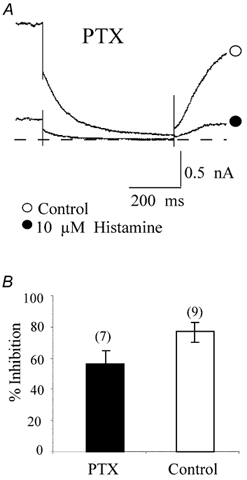
A, IM channel currents in the absence (○) and presence (•) of 10 μm histamine elicited from a HEK293T cell pretreated with PTX, recorded using the nystatin perforated-patch technique. The current traces were elicited by a 1 s step to -50 mV from a holding potential of -20 mV. The dashed line represents the zero current level. B, mean inhibition of IM by 10 μm histamine with and without PTX treatment. The numbers in parentheses represent the number of cells tested. Error bars are the s.e.m.
Effect of Gα buffer on histamine-induced IM inhibition
As described in the introduction, the muscarinic and bradykinin inhibition of IM is mediated by the α subunit rather than the βγ subunits of the Gq/11 heterotrimer in sympathetic neurons. To determine whether the α subunit or the βγ dimer of the activated G protein acts as the primary mediator in histamine-induced IM inhibition, we expressed a GFP-tagged, C terminal fusion construct of phospholipase C-β1 (PLC-β1-ct). This construct encodes a PLC-β1 C-terminal protein and prevents downstream effector activation since it selectively binds activated GTP-bound Gαq/11 but lacks PLC-β1 enzymatic activity (Wu et al. 1993; Kammermeier & Ikeda, 1999). Microinjection of this construct into rat SCG neurons prevented Ca2+ channel and M-channel modulation by heterologously expressed metabotropic glutamate receptor 1a (mGluR 1a; Kammermeier & Ikeda, 1999). Expression of PLC-β1-ct also selectively attenuated voltage-independent muscarinic inhibition of Ca2+current in rat SCG neurons (Kammermeier et al. 2000). If histamine-induced M-channel inhibition is mediated by the Gαq/11 subunit, one should expect the expression of PLC-β1-ct to prevent histamine-induced IM inhibition. In our studies, cotransfection of PLC-β1-ct significantly attenuated the histamine-induced inhibition of IM, indicating that Gαq/11 is an intermediary of IM modulation by histamine (Fig. 8A and B). The effect of expression of PLC-β1-ct at different transfection concentrations (e.g. the amount of cDNA added in a transfection mixture) on histamine-induced IM inhibition is shown in Fig. 8C.
Figure 8. Expression of PLC-β1-ct reduces the histamine modulation of KCNQ2/3 channels in HEK 293T cells.
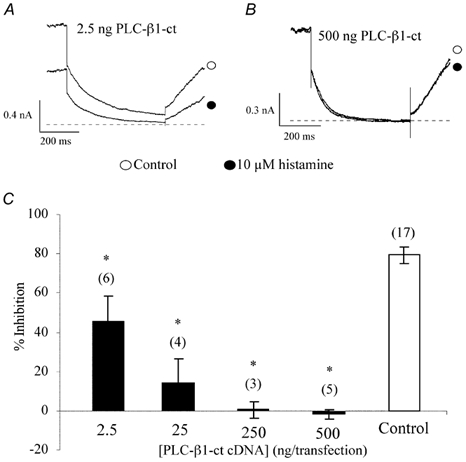
A and B, IM in the absence (○) and presence (•) of 10 μm histamine showing the effect of PLC-β1-ct (2.5 ng and 500 ng transfection concentration, respectively) on histamine-induced inhibition. The current traces were elicited by a 1 s step to -50 mV from a holding potential of -20 mV and recorded using the nystatin perforated-patch technique. The dashed lines represent the zero current level. C, concentration-response relationship of PLC-β1-ct effect on histamine-induced IM inhibition. The numbers in parentheses represent the number of cells tested, *P < 0.05 compared with control. Error bars are the s.e.m.
The regulators of G protein signalling (RGS) molecules are a new family of proteins that participate in the G protein cycle by acting as GTPase-activating proteins. They limit the lifetime of active, GTP-bound Gα subunits by accelerating the intrinsic GTPase activity of the Gα subunit and thus attenuate responses or hasten the signal termination (Doupnik et al. 1997; Chuang et al. 1998; Chen et al. 2000). Moreover, binding of RGS proteins to active GTP-bound Gα subunits also interferes with the interaction between Gα-GTP and effector proteins and thus blocks effector activation and downstream signalling (Hepler et al. 1997). Among many identified RGS proteins, RGS2 has been shown to interact selectively with Gαq/11 (Heximer et al. 1999). Like PLC-β1-ct, microinjection of the RGS2 encoding construct at the same concentration as PLC-β1-ct into mGluR1a-expressing rat SCG neurons significantly attenuated Ca2+ channel and M-channel inhibition by glutamate (Kammermeier & Ikeda, 1999). Therefore, to parallel the experiments with PLC-β1-ct, we cotransfected HEK293T cells with the RGS2 encoding construct to test whether cotransfection of RGS2 could prevent histamine-induced IM inhibition, as was observed for the expression of PLC-β1-ct. Unexpectedly, expression of RGS2 at a concentration of 500 ng per transfection, the maximum transfection concentration used for PLC-β1-ct and the other constructs (see below), did not significantly attenuate IM inhibition by histamine. In cells expressing RGS2, 10 μm histamine still displayed IM inhibition (73.0 ± 7.7 %, n = 11; Fig. 9), which was not significantly different from that in non-RGS-expressing cells (P > 0.05).
Figure 9. Expression of RGS2 fails to prevent histamine modulation of KCNQ2/3 channels.
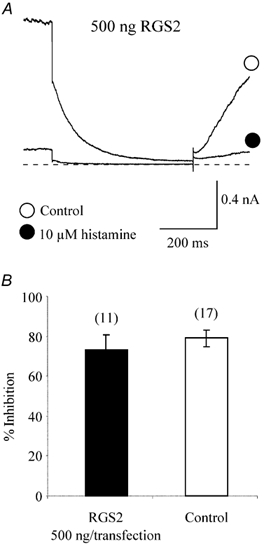
A, IM elicited from a RGS2-expressing HEK293T cell in the absence (○) and presence (•) of histamine. The current traces were elicited by a 1 s step to -50 mV from a holding potential of -20 mV using the nystatin perforated-patch technique. The dashed line represents the zero current level. B, mean inhibition of KCNQ2/3 currents by 10 μm histamine in control and RGS2-expressing cells. The numbers in parentheses represent the number of cells tested. Error bars are the s.e.m.
Histamine-induced M-channel modulation is inhibited by Gβγ buffering
Gαt is a Gα subunit which attains predominantly the GDP-bound state when overexpressed in SCG neurons and exhibits a high affinity for Gβγ (Kammermeier & Ikeda, 1999). Overexpression of heterologous Gαt buffers free Gβγ subunits released after G protein activation and thereby prevents interaction of Gβγ subunits with their effector proteins. In mGluR1a-expressing rat SCG neurons, expression of Gαt significantly attenuated voltage-dependent Ca2+ channel inhibition by glutamate, but did not affect glutamate-induced IM modulation (Kammermeier & Ikeda, 1999). To determine if Gβγ subunits are involved in KCNQ2/3 channel inhibition by histamine, we cotransfected a construct encoding Gαt along with H1R and KCNQ2/3 cDNAs. If, like muscarinic and glutamatergic inhibition of M-channels, Gβγ is not involved in histamine-induced IM inhibition, expression of Gαt should have no effect on histamine-induced inhibition. In our studies, at lower transfection concentrations, expression of Gαt did not significantly attenuate histamine-induced IM modulation (83.8 ± 9.4 %, n = 4; 62.9 ± 13.5 %, n = 4 at 2.5 ng and 25 ng per transfection, respectively; P > 0.05; Fig. 10A and C). However, interestingly, at higher transfection concentrations, expression of Gαt significantly attenuated IM inhibition by histamine (38.5 ± 7.1 %, n = 5 at 250 ng, P < 0.05; 16.9 ± 6.2 %, n = 4 at 500 ng, P < 0.05, Fig. 10B and C). These data implicate Gβγ subunits in histamine-induced IM modulation.
Figure 10. Expression of Gαt reduces histamine-induced modulation of KCNQ2/3 channels.
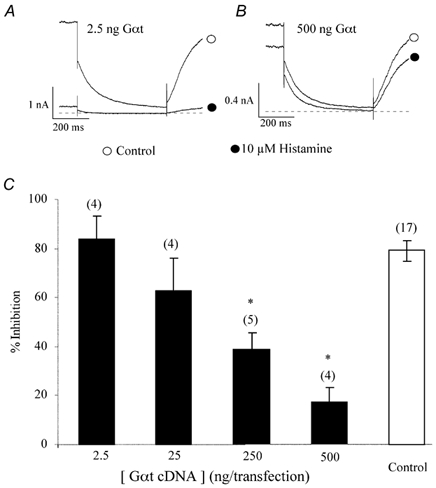
A and B, IM in the absence (○) and presence (•) of 10 μm histamine showing the effect of Gαt on histamine-induced inhibition at two different concentrations (2.5 ng and 500 ng transfection concentration, respectively). The current traces were elicited by a 1 s step to -50 mV from a holding potential of -20 mV using the nystatin perforated-patch technique. The dashed lines represent the zero current level. C, concentration-response relationship of Gαt on histamine-induced IM inhibition. The numbers in parentheses represents the number of cells tested, *P < 0.05 compared with control. Error bars are the s.e.m.
MAS-GRK2-ct is a C-terminal construct derived from G protein-coupled receptor kinase 2 (GRK2) with a myristic acid attachment signal at the N-terminus of the constructs (Kammermeier & Ikeda, 1999). Since this construct does not have kinase activity but retains the ability to bind free Gβγ dimers, MAS-GRK2-ct, like Gαt, has been used as a Gβγ buffer. In rat sympathetic neurons heterologously expressing mGLuR1a, expression of MAS-GRK2-ct significantly reduced mGLuR1a-mediated Ca2+ channel inhibition, but did not affect IM modulation by glutamate, findings similar to those from coexpression of mGLuR1a and Gαt (Kammermeier & Ikeda, 1999). As parallel experiments to further study the involvement of Gβγ in histamine-induced IM modulation, MAS-GRK2-ct was cotransfected with KCNQ2/3 subunit cDNAs into HEK293T cells. Similarly, expression of MAS-GRK2-ct at a concentration of 250 ng per transfection also significantly attenuated histamine-induced IM inhibition (33.7 ± 10.9 %, n = 5; P < 0.05, Fig. 11), further implicating Gβγ involvement in the signal transduction pathway mediating histamine-induced IM inhibition.
Figure 11. Expression of MAS-GRK2-ct attenuates histamine-induced modulation of KCNQ2/3 channels.
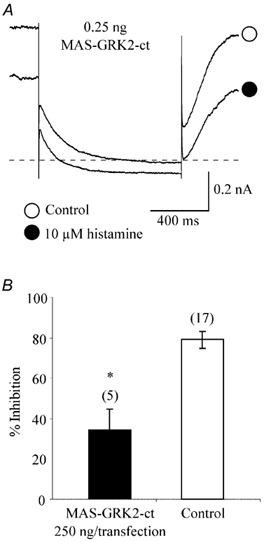
A, IM elicited from a MAS-GRK2-ct -expressing HEK293T cell in the absence (○) and presence (•) of histamine. The current traces were elicited by a 1 s step to -50 mV from a holding potential of -20 mV. The dashed line represents the zero current level. B, mean inhibition of KCNQ2/3 currents by histamine in control and MAS-GRK2-ct-expressing cells. The numbers in parentheses represent the number of cells tested, *P < 0.05 compared with control. Error bars are the s.e.m.
Discussion
Our studies show that, like muscarinic and bradykinin receptor-mediated inhibition of M-current, activation of a PTX-insensitive G protein is involved in histamine-induced modulation of recombinant M-channels expressed in HEK293T cells. However, unlike muscarinic and bradykinin inhibition where only Gαq/11 subunits appear to participate in the modulation, not only Gαq/11 but also Gβγ subunits are involved in this histamine-induced M-channel modulation.
Histamine inhibits recombinant M-channels by activating histamine H1 receptors
Brain histamine is involved in many central nervous system functions by causing arousal, increasing anxiety, controlling feeding behaviour, facilitating learning and memory processes and enhancing synaptic plasticity, etc. (for review see Brown, 2001). Like bradykinin, which is an important mediator of inflammation and hyperalgesia (Jones et al. 1995), histamine in the central nervous system plays an important role in antinociception. The central histamine system also controls the cardiovascular system by activating the sympathetic nervous system and increasing the concentration of plasma catecholamines (for review see Brown et al. 2001). Among the four types of histamine receptor, H1 receptors are found especially in the hypothalamus and other limbic regions where they are mainly located in the postsynaptic membrane and are associated with Gq/11 protein. H1R stimulation activates PLC, which leads to the hydrolysis of phosphatidyl-4,5-bisphosphate and the generation of inositol trisphosphate and diacylglycerol. Although activation of H1R has been reported to cause a hyperpolarization by activating Ca2+-dependent K+ channels in C6 glial cells via release of Ca2+ from intracellular Ca2+ stores (Weiger et al. 1997), in many neurons, it has been shown that activation of H1 receptors causes strong membrane depolarizations by blocking a potassium conductance, activating a non-specific cation channel or a sodium-calcium exchanger (for review see Brown et al. 2001).
Previously we have shown that histamine inhibits recombinant M-channels expressed in HEK293T and HeLa cells cotransfected with H1R, KCNQ2 and KCNQ3 cDNAs, suggesting that activation of H1R by histamine could modulate neuronal excitability by suppressing M-channels provided the H1R and native M-channels colocalize in the same neurons. During the course of this study, Wallace et al. (2002) showed that histamine, probably acting via histamine H1 receptors, caused a membrane depolarization associated with an increase in membrane resistance in bovine adrenal chromaffin cells induced by inhibiting an M-current. Our present studies further show that the histamine-induced IM inhibition is not voltage dependent and histamine does not shift the M-channel activation voltage dependence, effects which are shared with muscarinic inhibition of native and recombinant M-channels (Shapiro et al. 2000).
Effects of histamine on M-channel gating
The rising phase of IM activation has been described as either a single or double exponential function without the delay displayed by most other voltage-gated potassium channels. Thus, it has been suggested that M-channels do not gate through consecutive closed states as do Shaker potassium channels (Selyanko & Brown, 1999). Our data, on the other hand, clearly show a delay before current activation and generally the currents were well fitted by a double exponential function. However, to accommodate the delay prior to activation it was necessary to raise the fast exponential component to the second power. Although the origin of the delay in activation is not the focus of this paper, it is clearly not due to series resistance artifacts resulting from the nystatin perforated-patch conditions since no such delay was observed in the deactivation phase. Moreover, in open-tip recordings, where the series resistance was lower, a similar delay was observed before current activation. The deactivation kinetics of the currents shown here were similar to those described for IM recorded using the amphotericin B-perforated patch technique applied to rat SCG neurons. Both the fast and slow time constants of deactivation were similar to those described for IM deactivation in rat SCG neurons (Selyanko & Brown, 1999).
Several studies have shown that neurotransmitters that inhibit IM have no effect on the kinetic components of the current (Boehm, 1998). Our records on the other hand clearly show that histamine decreased both the fast and slow time constants of IM deactivation. The changes in deactivation time constants suggest that histamine decreases the channel open times or shifts the channel gating into a short open-time mode as has been shown for muscarinic inhibition of IM in bullfrog sympathetic neurons (Marrion, 1993). However, recent single channel studies of M-channel gating indicate that modal gating does not take place in rat sympathetic neurons (Selyanko & Brown, 1999). Further single channel studies will be required to determine if recombinant M-channels, consisting of KCNQ2 and KCNQ3 subunits, gate in a modal fashion in HEK293T cells and whether consecutive closed states of those channels explain the delay in activation of macroscopic IM in those cells.
Coupling of the H1R to a recombinant M-channel via a PTX-insensitive G protein
It has been well documented that receptor-mediated suppression of the M-current acts through a heterotrimeric G protein, most likely a member of the Gq/11 class of G proteins. The present studies provide strong evidence that histamine inhibition of IM by activation of H1R is also mediated by activation of a G protein as indicated by the effects of non-hydrolysable GTP and GDP analogues where inclusion of GDPβS abolished the IM inhibition by histamine, whereas inclusion of GTPγS rendered the histamine-induced inhibition irreversible.
A PTX-insensitive G protein has been implicated in the mechanism of muscarine, brandykinin, angiotensin II, and uridine triphosphate (UTP)-induced inhibition of the M-channel in rat sympathetic neurons. PTX blocks the signal transduction pathway involved in Gi/o-, but not Gq/11-mediated IM inhibition. Since other H1R-mediated effects are associated with the Gq/11 protein, it is not surprising that overnight pretreatment with PTX did not prevent histamine-induced inhibition of IM.
Both Gα and βγ subunits are involved in M-channel modulation
Many neurotransmitters can modulate both Ca2+ channels and M-channels. Although the involvement of Gβγ subunits in voltage-dependent inhibition of Ca2+ channels is well demonstrated, thus far only Gα subunits have been implicated in IM modulation. Caulfield et al. (1994), using antibodies against the C-terminal domain of different Gα subunits, first proposed that only Gq/11 protein α subunits were involved in M1 mAChR-mediated inhibition of IM in rat SCG neurons. However, the C-terminus of the Gα subunit is thought to be the site where GDP-bound Gα subunits interact with receptors and where GTP-bound Gα subunits interact with PLC-β1, therefore it is possible that the antibodies may prevent the interaction between the receptor and G protein and thus disable both branches of the bifurcating signalling pathway, i.e. the activation of GTP-bound Gα subunits and dissociated or free Gβγ dimers (Haley et al. 1998; Kammermeier & Ikeda, 1999). On the other hand, Haley et al. (1998) reported that intranuclear microinjection of a βARK1 construct (200 and 400 μg ml−1) to buffer free βγ did show some attenuation of muscarinic inhibition of IM. Although the attenuation of muscarinic inhibition by expression of the βARK1 construct at a low concentration (200 μg ml−1) did not achieve significance, injection of a high concentration (400 μg ml−1) of the βARK1 construct significantly attenuated the muscarinic inhibition. The authors attributed this attenuation to a non-specific effect of the construct.
In our experiments, the attenuation of histamine-induced suppression by buffering free Gβγ after expression of Gαt or MAS-GRK2-ct strongly suggests that the Gβγ dimer, in addition to the Gα subunit, also participates in this histamine-induced modulation. The effect of buffering Gβγ dimers is unlikely to be a non-specific effect since the expression of two different constructs produced similar effects in regard to the histamine-induced modulation. However, several questions remain. Firstly, is this βγ involvement expression system specific? Thus far all studies concerning G protein subunit involvement in M-channel modulation have been performed in native (rat or mouse) SCG neurons heterologously expressing different constructs or antibodies. It is possible that the involvement of the Gβγ subunit in histamine-induced IM modulation is a peculiarity of the HEK293T expression environment. Secondly, does Gβγ participate only in recombinant M-channel modulation? To date few papers have been published to address the question of the modulation of recombinant M-channels by activation of GPCR. Moreover, we are not aware of any studies that address the effect of particular G protein subunits in heterologously expressed recombinant M-channel modulation. Whether modulation of native and recombinant M-channels is mediated by the same modulatory pathway(s) has not been elucidated. Finally, does Gβγ participate only in histamine-induced IM modulation? The histamine H1 receptor is a new member of the receptor pool which can modulate M-channels. At the present time it is not known whether the signal transduction pathway involved in IM modulation by histamine is unique.
The attenuation of histamine-induced M-channel modulation after expressing PLC-β1-ct strongly supports the notion that Gαq/11 is involved. That observation seems to be contradicted by the lack of effect of RGS2 expression. However, there are some differences between the two constructs. The PLC-β1-ct construct is targeted to the membrane by residues in the C-terminus (Wu et al. 1993). Although the RGS2 construct possesses an N-terminal domain capable of targeting the molecule to the plasma membrane, several studies indicate that the RGS2 construct is predominantly targeted to the nucleus (Chatterjee & Fisher, 2000; Heximer et al. 2001; Song et al. 2001). For example, in COS-7 cells, GTP-fusion RGS2 is localized exclusively in the nucleus and no accumulation of RGS2 proteins at the plasma membrane is observed (Chatterjee & Fisher, 2000). In human astrocytoma 1321N1 cells and neuroblastoma SH-SY5Y cells, endogenous and transfected RGS2 is predominantly, although not entirely, localized in the nucleus (Song et al. 2001; Zmijewski et al. 2001). Similarly, RGS2 is massively localized in the nucleus in HEK293 cells, although it can translocate to the cytoplasm and plasma membrane. These studies suggest that RGS2 localization is regulated in a cell-type-specific manner (Heximer et al. 2001). Although there may be an activity-dependent translocation of the construct out of the nucleus, for instance, in HEK293 cells, expression of activated Gq increased RGS2 association with the plasma membrane and decreased accumulation in the nucleus (Heximer et al. 2001). However, RGS2 may have to be massively overexpressed to get significant concentrations to the point of interest. Even RGS2 which leaks from the nucleus may not be targeted to the membrane - thus the effective concentration of PLC-β1-ct may be higher by several orders of magnitude than that of RGS2. The concentration of RGS2 cDNA used in the present studies was the maximum concentration used for the other constructs, but even at this concentration, expression of RGS2 did not attenuate histamine-induced inhibition of IM. Higher concentrations of RGS2 were not used since although increasing the RGS2 concentration may enhance expression by leakage from the nucleus and translocation to the cell membrane, they may also increase the risk of non-specific effects.
Functional implications
Recently it has been shown that histamine inhibits M-channels of bovine chromaffin cells (probably via histamine H1 receptors), leading to enhanced membrane excitability (Wallace et al. 2002). Although histamine-induced M-channel modulation has not been demonstrated in neurons, there are several neuronal tissues where histamine-induced inhibition of IM might occur. For example, it has been shown that in guinea-pig myenteric neurons, histamine produces slow depolarization associated with increased input resistance, enhances excitability, and generates repetitive spike discharge (Nemeth et al. 1984; Tamura & Wood, 1992). Histamine, acting via H1 receptors, also excites guinea-pig SCG neurons by producing transient membrane depolarization, increasing input resistance and transiently blocking a long-duration component of the spike after-hyperpolarization (Christian et al. 1989). Christian & Weinreich (1992), correlated the effects of 0.1-100 μm histamine with the effects of antigenic challenge on excitatory postsynaptic potentials of guinea-pig SCG neurons. The authors concluded that endogenous histamine released during an immunological response can activate histamine H1 and H3 receptors to modulate synaptic efficacy in sympathetic ganglia. The concentrations of histamine used in this study are physiological since they fall in the range used by Christian & Weinreich (1992).
The experiments with non-hydrolysable guanine nucleotide analogues and PTX provide strong evidence that a PTX-insensitive G protein is involved in histamine-induced IM modulation, an effect shared with all other receptors known to modulate IM. The second messenger signalling pathway involved in this histamine-induced IM modulation remains to be elucidated. It remains to be seen whether histamine exerts its effects on IM directly via the release of intracellular Ca2+, as is the case for bradykinin and UTP. Alternatively, histamine may utilize the depletion of phosphatidylinositol-4,5-bisphosphate for signal transduction as has recently been suggested for the activation of other Gq/11-coupled receptors (Stemkowski, et al. 2002; Suh & Hille, 2002).
Acknowledgments
We wish to thank Dr Stephen Ikeda, NIH/NIAAA/DICBR/LMP for KCNQ2 and KCNQ3 cDNA and for the Gαt, PLC-β1-ct, RGS2 and MAS-GRK2-ct constructs. We thank Dr Grace Athas, LSU Health Sciences Center, New Orleans, LA, for the cDNA encoding the histamine H1 receptor.
References
- Boehm S. Selective inhibition of M-type potassium channels in rat sympathetic neurons by uridine nucleotide preferring receptors. British Journal of Pharmacology. 1998;124:1261–1269. doi: 10.1038/sj.bjp.0701956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown BS, Yu SP. Modulation and genetic identification of the M channel. Progress in Biophysics and Molecular Biology. 2000;73:135–166. doi: 10.1016/s0079-6107(00)00004-3. [DOI] [PubMed] [Google Scholar]
- Brown DA. M currents. Ion Channels. 1988;1:55–94. doi: 10.1007/978-1-4615-7302-9_2. [DOI] [PubMed] [Google Scholar]
- Brown DA, Adams PR. Muscarinic suppression of a novel voltage-sensitive K+ current in a vertebrate neurone. Nature. 1980;283:673–676. doi: 10.1038/283673a0. [DOI] [PubMed] [Google Scholar]
- Brown DA, Marrion NV, Smart TG. On the transduction mechanism for muscarine-induced inhibition of M-current in cultured rat sympathetic neurones. Journal of Physiology. 1989;413:469–488. doi: 10.1113/jphysiol.1989.sp017664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown RE, Stevens DR, Haas HL. The physiology of brain histamine. Progress in Neurobiology. 2001;63:637–672. doi: 10.1016/s0301-0082(00)00039-3. [DOI] [PubMed] [Google Scholar]
- Cassell JF, McLachlan EM. Muscarinic agonists block five different potassium conductances in guinea-pig sympathetic neurons. British Journal of Pharmacology. 1987;91:259–261. doi: 10.1111/j.1476-5381.1987.tb10279.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caulfield MP, Jones S, Vallis Y, Buckley NJ, Kim GD, Milligan G, Brown DA. Muscarinic M-current inhibition via Gαq/11 and α-adrenoceptor inhibition of Ca2+ current via Gαo in rat sympathetic neurones. Journal of Physiology. 1994;477:415–422. doi: 10.1113/jphysiol.1994.sp020203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee TK, Fisher RA. Cytoplasmic, nuclear, and golgi localization of RGS proteins. Evidence for N-terminal and RGS domain sequences as intracellular targeting motifs. Journal of Biological Chemistry. 2000;275:24013–24021. doi: 10.1074/jbc.M002082200. [DOI] [PubMed] [Google Scholar]
- Chen CK, Burns ME, He W, Wensel TG, Baylor DA, Simon MI. Slowed recovery of rod photoresponse in mice lacking the GTPase accelerating protein RGS9–1. Nature. 2000;403:557–560. doi: 10.1038/35000601. [DOI] [PubMed] [Google Scholar]
- Christian EP, Undem BJ, Weinreich D. Endogenous histamine excites neurones in the guinea-pig superior cervical ganglion in vitro. Journal of Physiology. 1989;409:297–312. doi: 10.1113/jphysiol.1989.sp017498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian EP, Weinreich D. Presynaptic histamine H1 and H3 receptors modulate sympathetic ganglionic synaptic transmission in the guinea-pig. Journal of Physiology. 1992;457:407–430. doi: 10.1113/jphysiol.1992.sp019385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang HH, Yu M, Jan N, Jan LY. Evidence that the nucleotide exchange and hydrolysis cycle of G proteins causes acute desensitization of G-protein gated inward rectifier K+ channels. Proceedings of the National Academy of Sciences of the USA. 1998;95:11727–11732. doi: 10.1073/pnas.95.20.11727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapp LH, Sims SM, Singer JJ, Walsh JV., Jr Role for diacylglycerol in mediating the actions of ACh on M-current in gastric smooth muscle cells. American Journal of Physiology. 1992;263:C1274–1281. doi: 10.1152/ajpcell.1992.263.6.C1274. [DOI] [PubMed] [Google Scholar]
- Constanti A, Brown DA. M-currents in voltage-clamped mammalian sympathetic neurones. Neuroscience Letters. 1981;24:289–294. doi: 10.1016/0304-3940(81)90173-7. [DOI] [PubMed] [Google Scholar]
- Constanti A, Sim JA. Muscarinic receptors mediating suppression of the M-current in guinea-pig olfactory cortex neurones may be of the M2-subtype. British Journal of Pharmacology. 1987;90:3–5. doi: 10.1111/j.1476-5381.1987.tb16818.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doupnik CA, Davidson N, Lester HA, Kofuji P. RGS proteins reconstitute the rapid gating kinetics of Gbetagamma-activated inwardly rectifying K+ channels. Proceedings of the National Academy of Sciences of the USA. 1997;94:10461–10466. doi: 10.1073/pnas.94.19.10461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond AH, Joels LA, Hughes PJ. The interaction of lithium ions with inositol lipid signalling systems. Biochemical Society Transactions. 1987;15:32–35. doi: 10.1042/bst0150032. [DOI] [PubMed] [Google Scholar]
- Filippov AK, Webb TE, Barnard EA, Brown DA. P2Y2 nucleotide receptors expressed heterologously in sympathetic neurons inhibit both N-type Ca2+ and M-type K+ currents. Journal of Neuroscience. 1998;18:5170–5179. doi: 10.1523/JNEUROSCI.18-14-05170.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J, Schofield GG. Histamine inhibits KCNQ2/KCN3 channel current via recombinant histamine H1 receptors. Neuroscience Letters. 2002;328:285–288. doi: 10.1016/s0304-3940(02)00484-6. [DOI] [PubMed] [Google Scholar]
- Haley JE, Abogadie FC, Delmas P, Dayrell M, Vallis Y, Milligan G, Caulfield MP, Brown DA, Buckley NJ. The alpha subunit of Gq contributes to muscarinic inhibition of the M-type potassium current in sympathetic neurons. Journal of Neuroscience. 1998;18:4521–4531. doi: 10.1523/JNEUROSCI.18-12-04521.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haley JE, Delmas P, Offermanns S, Abogadie FC, Simon MI, Buckley NJ, Brown DA. Muscarinic inhibition of calcium current and M current in Galpha q-deficient mice. Journal of Neuroscience. 2000;20:3973–3979. doi: 10.1523/JNEUROSCI.20-11-03973.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hepler JR, Berman DM, Gilman AG, Kozasa T. RGS4 and GAIP are GTPase-activating proteins for Gq alpha and block activation of phospholipase C beta by gamma-thio-GTP-Gq alpha. Proceedings of the National Academy of Sciences of the USA. 1997;94:428–432. doi: 10.1073/pnas.94.2.428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herlitze S, Garcia DE, Mackie K, Hille B, Scheuer T, Catterall WA. Modulation of Ca2+ channels by G-protein beta gamma subunits. Nature. 1996;380:258–262. doi: 10.1038/380258a0. [DOI] [PubMed] [Google Scholar]
- Heximer SP, Lim H, Bernard JL, Blumer KJ. Mechanisms governing subcellular localization and function of human RGS2. Journal of Biological Chemistry. 2001;276:14195–14203. doi: 10.1074/jbc.M009942200. [DOI] [PubMed] [Google Scholar]
- Heximer SP, Srinivasa SP, Bernstein LS, Bernard JL, Linder ME, Hepler JR, Blumer KJ. G protein selectivity is a determinant of RGS2 function. Journal of Biological Chemistry. 1999;274:34253–34259. doi: 10.1074/jbc.274.48.34253. [DOI] [PubMed] [Google Scholar]
- Ikeda SR. Voltage-dependent modulation of N-type calcium channels by G-protein beta gamma subunits. Nature. 1996;380:255–258. doi: 10.1038/380255a0. [DOI] [PubMed] [Google Scholar]
- Jeong SW, Ikeda SR. Sequestration of G-protein beta gamma subunits by different G-protein alpha subunits blocks voltage-dependent modulation of Ca2+ channels in rat sympathetic neurons. Journal of Neuroscience. 1999;19:4755–4761. doi: 10.1523/JNEUROSCI.19-12-04755.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S, Brown DA, Milligan G, Willer E, Buckley NJ, Caulfield MP. Bradykinin excites rat sympathetic neurons by inhibition of M current through a mechanism involving B2 receptors and G alpha q/11. Neuron. 1995;14:399–405. doi: 10.1016/0896-6273(95)90295-3. [DOI] [PubMed] [Google Scholar]
- Kammermeier PJ, Ikeda SR. Expression of RGS2 alters the coupling of metabotropic glutamate receptor 1a to M-type K+ and N-type Ca2+ channels. Neuron. 1999;22:819–829. doi: 10.1016/s0896-6273(00)80740-0. [DOI] [PubMed] [Google Scholar]
- Kammermeier PJ, Ruiz-Velasco V, Ikeda SR. A voltage-independent calcium current inhibitory pathway activated by muscarinic agonists in rat sympathetic neurons requires both Galpha q/11 and Gbeta gamma. Journal of Neuroscience. 2000;20:5623–5629. doi: 10.1523/JNEUROSCI.20-15-05623.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick DA, Williamson A. Modulation of neuronal firing mode in cat and guinea pig LGNd by histamine: possible cellular mechanisms of histaminergic control of arousal. Journal of Neuroscience. 1991;11:3188–3199. doi: 10.1523/JNEUROSCI.11-10-03188.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrion NV. Selective reduction of one mode of M-channel gating by muscarine in sympathetic neurons. Neuron. 1993;11:77–84. doi: 10.1016/0896-6273(93)90272-s. [DOI] [PubMed] [Google Scholar]
- Marrion NV. Control of M-current. Annual Review of Physiology. 1997;59:483–504. doi: 10.1146/annurev.physiol.59.1.483. [DOI] [PubMed] [Google Scholar]
- Marrion NV, Smart TG, Marsh SJ, Brown DA. Muscarinic suppression of the M-current in the rat sympathetic ganglion is mediated by receptors of the M1-subtype. British Journal of Pharmacology. 1989;98:557–573. doi: 10.1111/j.1476-5381.1989.tb12630.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore SD, Madamba SG, Joels M, Siggins GR. Somatostatin augments the M-current in hippocampal neurons. Science. 1988;239:278–280. doi: 10.1126/science.2892268. [DOI] [PubMed] [Google Scholar]
- Nemeth PR, Ort CA, Wood JD. Intracellular study of effects of histamine on electrical behaviour of myenteric neurones in guinea-pig small intestine. Journal of Physiology. 1984;355:411–425. doi: 10.1113/jphysiol.1984.sp015427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner PB, Kamondi A. Mechanisms of antihistamine-induced sedation in the human brain: H1 receptor activation reduces a background leakage potassium current. Neuroscience. 1994;59:579–588. doi: 10.1016/0306-4522(94)90178-3. [DOI] [PubMed] [Google Scholar]
- Schofield GG. Norepinephrine blocks a calcium current of adult rat sympathetic neurons via an alpha 2-adrenoceptor. European Journal of Pharmacology. 1990;180:37–47. doi: 10.1016/0014-2999(90)90590-3. [DOI] [PubMed] [Google Scholar]
- Selyanko AA, Brown DA. M-channel gating and simulation. Biophysical Journal. 1999;77:701–713. doi: 10.1016/S0006-3495(99)76925-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro MS, Roche JP, Kaftan EJ, Cruzblanca H, Mackie K, Hille B. Reconstitution of muscarinic modulation of the KCNQ2/KCNQ3 K+ channels that underlie the neuronal M current. Journal of Neuroscience. 2000;20:1710–1721. doi: 10.1523/JNEUROSCI.20-05-01710.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro MS, Wollmuth LP, Hille B. Angiotensin II inhibits calcium and M current channels in rat sympathetic neurons via G proteins. Neuron. 1994;12:1319–1329. doi: 10.1016/0896-6273(94)90447-2. [DOI] [PubMed] [Google Scholar]
- Song L, Zmijewski JW, Jope RS. RGS2: regulation of expression and nuclear localization. Biochemical and Biophysical Research Communications. 2001;283:102–106. doi: 10.1006/bbrc.2001.4742. [DOI] [PubMed] [Google Scholar]
- Stemkowski PL, Tse FW, Peuckmann V, Ford CP, Colmers WF, Smith PA. ATP-inhibition of M current in frog sympathetic neurons involves phospholipase C but not Ins P3, Ca2+, PKC, or Ras. Journal of Neurophysiology. 2002;88:277–288. doi: 10.1152/jn.2002.88.1.277. [DOI] [PubMed] [Google Scholar]
- Suh B, Hille B. Recovery from muscarinic modulation of m current channels requires phosphatidylinositol 4, 5-bisphosphate synthesis. Neuron. 2002;35:507–520. doi: 10.1016/s0896-6273(02)00790-0. [DOI] [PubMed] [Google Scholar]
- Tamura K, Wood JD. Effects of prolonged exposure to histamine on guinea pig intestinal neurons. Digestive Diseases and Sciences. 1992;37:1084–1088. doi: 10.1007/BF01300291. [DOI] [PubMed] [Google Scholar]
- Tokimasa T, Tsurusaki M, Akasu T. Chemosensitivity of C-cells in bullfrog dorsal root ganglia to substance P and adenosine 5′-triphosphate. Neuroscience Letters. 1993;163:169–172. doi: 10.1016/0304-3940(93)90374-t. [DOI] [PubMed] [Google Scholar]
- Villarroel A. M-current suppression in PC12 cells by bradykinin is mediated by a pertussis toxin-insensitive G-protein and modulated by intracellular calcium. Brain Research. 1996;740:227–233. doi: 10.1016/s0006-8993(96)00870-0. [DOI] [PubMed] [Google Scholar]
- Wallace DJ, Chen C, Marley PD. Histamine promotes excitability in bovine adrenal chromaffin cells by inhibiting an M-current. Journal of Physiology. 2002;540:921–939. doi: 10.1113/jphysiol.2001.013370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiger T, Stevens DR, Wunder L, Haas HL. Histamine H1 receptors in C6 glial cells are coupled to calcium-dependent potassium channels via release of calcium from internal stores. Naunyn-Schmiedeberg's Archives of Pharmacology. 1997;355:559–565. doi: 10.1007/pl00004983. [DOI] [PubMed] [Google Scholar]
- Wu D, Jiang H, Katz A, Simon MI. Identification of critical regions on phospholipase C-beta 1 required for activation by G-proteins. Journal of Biological Chemistry. 1993;268:3704–3709. [PubMed] [Google Scholar]
- Zmijewski JW, Song L, Harkins L, Cobbs CS, Jope RS. Second messengers regulate RGS2 expression which is targeted to the nucleus. Biochimica et Biophysica Acta. 2001;1541:201–211. doi: 10.1016/s0167-4889(01)00144-6. [DOI] [PubMed] [Google Scholar]