

Abstract
The pharmacological profiles of presynaptic nociceptin/orphanin FQ (N/OFQ) peptide receptors (NOP) modulating 5-hydroxytryptamine (5-HT) and noradrenaline (NE) release in the rat neocortex were characterized in a preparation of superfused synaptosomes challenged with 10 mM KCl.
N/OFQ concentration-dependently inhibited K+-evoked [3H]-5-HT and [3H]-NE overflow with similar potency (pEC50 ∼7.9 and ∼7.7, respectively) and efficacy (maximal inhibition ∼40%).
N/OFQ (0.1 μM) inhibition of [3H]-5-HT and [3H]-NE overflow was antagonized by selective NOP receptor antagonists of peptide ([Nphe1]N/OFQ(1-13)NH2 and UFP-101; 10 and 1 μM, respectively) and non-peptide (J-113397 and JTC-801; both 0.1 μM) nature. Antagonists were routinely applied 3 min before N/OFQ. However, a 21 min pre-application time was necessary for J-113397 and JTC-801 to prevent N/OFQ inhibition of [3H]-NE overflow.
The NOP receptor ligand [Phe1ψ(CH2-NH)Gly2]N/OFQ(1-13)NH2 ([F/G]N/OFQ(1-13)NH2; 3 μM) did not affect K+-evoked [3H]-NE but inhibited K+-evoked [3H]-5-HT overflow in a UFP-101 sensitive manner. [F/G]N/OFQ(1-13)NH2 antagonized N/OFQ actions on both neurotransmitters.
The time-dependency of JTC-801 action was studied in CHO cells expressing human NOP receptors. N/OFQ inhibited forskolin-stimulated cAMP accumulation and JTC-801, tested at different concentrations (0.1–10 μM) and pre-incubation times (0, 40 and 90 min), antagonized this effect in a time-dependent manner. The Schild-type analysis excluded a competitive type of antagonism.
We conclude that presynaptic NO receptors inhibiting 5-HT and NE release in the rat neocortex have similar pharmacological profiles. Nevertheless, they can be differentiated pharmacologically on the basis of responsiveness to [F/G]N/OFQ(1-13)NH2 and time-dependent sensitivity towards non-peptide antagonists.
Keywords: [F/G]N/OFQ(1-13)NH2, J-113397, JTC-801, 5-hydroxytryptamine, nociceptin/orphanin FQ, noradrenaline, [Nphe1]N/OFQ(1-13NH2, release, synaptosomes, UFP-101
Introduction
The endogenous peptide nociceptin/orphanin FQ (N/OFQ; Meunier et al., 1995; Reinscheid et al., 1995) modulates the function of brain monoaminergic systems via different mechanisms and at different neuroanatomical levels. N/OFQ inhibits the firing of the serotonergic and noradrenergic ascending pathways by acting on cell bodies located in the raphe nuclei (Vaughan & Christie, 1996) and locus coeruleus (Connor et al., 1996; 1999; Okawa et al., 2001) respectively, and modulates 5-hydroxytryptamine (5-HT) and noradrenaline (NE) release by acting on synaptic terminals in receptive areas, in particular the cerebral cortex (reviewed by Schlicker & Morari, 2000). This modulatory action may subserve some of the biological effects of the peptide or its synthetic analogues, such as anxiolysis (Jenck et al., 1997; 2000) and food consumption (Polidori et al., 2000).
Many studies have demonstrated that activation of a specific class of G-protein coupled receptors is responsible for the biological actions of N/OFQ. These receptors share structural and functional homology with classical opioid receptors (e.g. amino acid sequence, transduction mechanisms, cellular effects) but display a distinct pharmacological profile (for recent reviews see Calo et al., 2000; Mogil & Pasternak, 2001). Indeed, N/OFQ actions are typically resistant to the non selective opioid receptor antagonist naloxone but are antagonized by a number of ligands that do not bind to opioid receptors with high affinity. In this respect, the pharmacology of the N/OFQ peptide receptor (henceforth referred to as NOP, according to recent IUPHAR recommendations, Cox et al., 2000) system has been improved over the last few years with the discovery of selective agonists, such as [Nphe1]N/OFQ(1-13)NH2 ([Nphe1]); Calo et al., 2000) and J-113397 (Kawamoto et al., 1999; Ozaki et al., 2000), the first peptide and non-peptide antagonists, respectively, and, more recently, JTC-801 (non-peptide; Shinkai et al., 2000; Yamada et al., 2002) and UFP-101 (peptide, Calo et al., 2002b). The availability of four, chemically distinct NOP receptor selective antagonists is expected to enhance pharmacological characterization of N/OFQ actions and to dissect any possible pharmacological heterogeneity within the NOP receptor system (Burnside et al., 2000; Mogil & Pasternak, 2001). Morphological studies have in fact demonstrated the existence of splice variants of NOP receptors (Mathis et al., 1997; Currò et al., 2001) and NOP receptor heterogeneity has been claimed to interpret variability in binding and distribution of NOP receptor ligands as well as responsiveness to N/OFQ (and partial agonists) observed among different experimental models and/or animal species (Burnside et al., 2000; Mogil & Pasternak, 2001).
Previous work from our laboratories has shown that N/OFQ inhibits K+-evoked [3H]-5-HT and [3H]-NE overflow from rat neocortical synaptosomes in superfusion (Sbrenna et al., 2000; Calo et al., 2002b; Siniscalchi et al., 2002). This is a simple preparation, commonly employed to study the pharmacology of presynaptic receptors (Raiteri & Raiteri, 2000) and particularly useful in the case of N/OFQ which, like opioids, appears to affect neuronal function largely via presynaptic mechanisms (Schlicker & Morari, 2000). N/OFQ inhibition of K+-evoked [3H]-5-HT overflow through presynaptic NOP receptors was insensitive to naloxone and antagonized by [Nphe1] (Sbrenna et al., 2000) and UFP-101 (Calo et al., 2002b). The present study was therefore undertaken to compare the pharmacological profiles of presynaptic NOP receptors inhibiting [3H]-5-HT and [3H]-NE overflows from rat neorocortical synaptosomes. For this purpose, selective NOP receptor antagonists of peptide ([Nphe1] and UFP-101) and non-peptide (J-113397 and JTC-801) nature were employed, together with the NOP receptor ligand [Phe1ψ(CH2-NH)Gly2]N/OFQ(1-13)NH2 ([F/G], Guerrini et al., 1998). Moreover, to provide additional information on the pharmacology of JTC-801 (specifically, to determine its antagonist potency), experiments were performed to examine the ability of JTC-801 to antagonize N/OFQ inhibition of forskolin-stimulated cAMP accumulation in CHO cells expressing recombinant human NOP receptors (CHOhNOP; Okawa et al., 1999).
Methods
Synaptosome preparation
Male Sprague-Dawley rats (180–240 g), kept under standard conditions (12 h dark/light cycle, free access to food and water), were used. All procedures concerning animal treatment were in accordance with European Communities Council directives (86/609/EEC) and national regulations (D.L. 116/92).
On the morning of the experiment, rats were decapitated under light ether anaesthesia and the fronto-parietal cortex was isolated. Synaptosomes were prepared as previously described (Morari et al., 1998; Sbrenna et al., 2000). Briefly, the cortex was homogenized in ice-cold 0.32 M sucrose buffer at pH 7.4 then centrifuged for 10 min at 1000×gmax (4°C). The supernatant was then centrifuged for 20 min at 12,000×gmax (4°C) with the synaptosomal pellet being resuspended in oxygenated (95% O2, 5% CO2) Krebs solution (mM: NaCl 118.5, KCl 4.7, CaCl2 1.2, MgSO4 1.2, KH2PO4 1.2, NaHCO3 25, glucose 10) containing ascorbic acid (0.05 mM) and disodium EDTA (0.03 mM). Synaptosomes were pre-loaded with [3H]-5-HT or [3H]-NE by incubation (25 min) in medium containing 50 nM [3H]-5-HT or 100 nM [3H]-NE (specific activity of 27.8 and 13.5 Ci mmol−1, respectively, NEN DuPont, Boston, MA, U.S.A.).
One ml aliquots of the suspension (protein concentration of about 0.35 mg protein ml−1) were slowly injected into nylon syringe filters (outer diameter 13 mm, 0.45 μM pore size, internal volume of about 100 μl; Phenomenex, MA, U.S.A.) connected to a peristaltic pump. Filters were maintained at 36.5°C in a thermostatic bath and superfused at a flow rate of 0.4 ml min−1 with a pre-oxygenated Krebs solution. Sample collection (every 3 min) was initiated after a 20 min period of filter washout. K+ stimulation (1 min pulse) was applied at the 38th minute. Under these experimental conditions, the 10 mM K+-evoked [3H]-NE overflow was reduced to 48.4±7.6% and 17.5±6.6% of control (n=7; P<0.05) by perfusion with tetrodotoxin (TTX; 0.5 μM) or omission of Ca2+ from the superfusion medium, respectively. Similar results were obtained for 10 mM K+-evoked [3H]-5HT overflow, which has been previously reported to be largely Ca2+-dependent (90%) and TTX-sensitive (50%; Sbrenna et al., 2000).
N/OFQ was added to the superfusion medium 9 min before the K+ pulse and maintained until the end of the experiment. Antagonists (including [F/G]) were routinely added 3 min before N/OFQ. In a separate set of experiments, to investigate the time-dependency of the action of nonpeptide antagonists, different pre-treatment times were tested (9 and 21 min).
[3H]-5-HT and [3H]-NE analysis
At the end of the experiment, superfusate (3 min samples) and filter retained radioactivity (dissolved with 1 ml of 1 M NaOH followed by 1 ml of 1 M HCl) was determined by liquid scintillation spectrophotometry using a Beckman LS 1800 β-spectrometer and Ultima Gold XR scintillation fluid (Packard Instruments B.V., Groningen, The Netherlands). The chemical nature of the released radioactivity from superfused rat brain synaptosomes was previously investigated by Collard et al. (1981) ([3H]-5-HT) and de langen et al. (1979) ([3H]-NE) who demonstrated that during high K+ stimulation, tritium overflow was almost exclusively due to the unmetabolized neurotransmitters.
cAMP accumulation in CHOhNOP cells
CHOhNOP cells were maintained in DMEM (Dulbecco's Modified Eagle's Medium): Ham F12 (50 : 50) containing 5% FCS (Foetal Calf Serum), 2 mM glutamine, 50 IU ml−1 penicillin (P), 50 μg ml−1 streptomycin (S), 200 μg ml−1 hygromycin B and 200 μg ml−1 G418 as previously described (Okawa et al., 1999). Cell cultures were maintained at 37°C in 5% CO2/humidified air. When confluency was reached (3–4 days), cells were harvested for use by the addition of 0.9% saline containing HEPES (10 mM)/EDTA (0.05%). CHO cell suspensions were washed twice with and resuspended in Krebs/HEPES buffer of the following composition (mM): Na+ 143.3, K+ 4.7, Ca2+ 2.5, Mg2+ 1.2, Cl− 125.6, H2PO42− 1.2, SO42− 1.2, glucose 11.7, and HEPES 10, BSA (Bovine Serum Albumin) 0.5%, pH 7.4 with 10 M NaOH. cAMP formation was measured in 0.3 ml volumes of whole cell suspensions in the presence of isobutylmethylxanthine (IBMX; 1 mM) and forskolin (1 μM). Peptidase inhibitors were not used as they did not influence the response to N/OFQ (unpublished observation). N/OFQ was included in various concentrations in order to obtain a full concentration–response curve both in the presence and absence of JTC-801 (0.1–10 μM). After 15 min incubation at 37°C, reactions were terminated and cAMP was extracted and assayed as previously described (Okawa et al., 1999). To study the time-dependence of JTC-801 action, JTC-801 was either co-applied with N/OFQ or given 40 and 90 min prior to it.
Drugs
N/OFQ, [Nphe1], [F/G] UFP-101 and J-113397 (1-[3R, 4R)-1-cyclooctylmethyl - 3 - hydroxymethyl- 4 -piperidyl]-3-ethyl-1,3-dihydro-2H-benzimidazole-2-one) were prepared as previously described (Calo et al., 1998; Guerrini et al., 2000; de risi et al., 2001). J-11339 used in our experiments is a racemic mixture. JTC-801 (N-(4-amino-2-methylquinolin-6-yl)-2-(4-ethylphenoxymethyl)benzamide hydrochloride) was also synthesized in our laboratories as described in the literature (Shinkai et al., 2000). Naloxone was purchased from Tocris Cookson (Bristol, U.K.) and tetrodotoxin (TTX) from Sigma Chemical Company (St Louis, MO, U.S.A.).
With the exception of JTC-801 (in DMSO) all drugs were dissolved in distilled water (1 mM) and stock solutions were stored at −20°C until use.
Data presentation and statistical analysis
All data are expressed as means±s.e.mean of n experiments. Data from experiments in synaptosomes are calculated as fractional release (FR; i.e. tritium efflux expressed as percentage of the tritium content in the filter at the onset of the corresponding collection period) and expressed as percentage of K+-evoked neurotransmitter overflow. K+-evoked neurotransmitter overflow was calculated by subtracting the estimated spontaneous efflux (obtained by interpolation between the samples preceding and following the stimulation) from the total efflux observed in the stimulated sample.
The pharmacological terminology adopted in this paper is consistent with IUPHAR recommendations (Jenkinson et al., 1995). Agonist potencies were measured as pEC50, which is the negative logarithm to base 10 of the agonist molar concentration that produces 50% of the maximal possible effect of that agonist. JTC-801 antagonist potency is expressed in terms of pKB: due to the clear non-equilibrium observed in CHOhNOP cells (see results), pKB values were estimated for each concentration of JTC-801 that produced a shift in the response to N/OFQ using the Gaddum Schild equation (pKB=−log((CR-1)/[Antagonist])), assuming a slope equal to unity. Statistical analysis was performed on FR values by the Kruskal–Wallis test (non-parametric ANOVA) followed by the Dunn's test for multiple comparisons.
Results
[3H]-5-HT overflow in synaptosomes
Basal [3H]-5-HT efflux (6.8±0.15 fmol mg prot−1 min−1, n=92) corresponded to a fractional release of 3.94±0.6%. One min pulse of KCl 10 mM evoked a tritium overflow (7.5±0.29 fmol mg prot−1 min−1, n=22) that was inhibited in a concentration–dependent manner by N/OFQ (0.001–3 μM; Figure 1). Analysis of the concentration–response curve of N/OFQ yielded a pEC50 value of 7.87±0.13. Maximal inhibition (42±4%) was observed at 3 μM.
Figure 1.
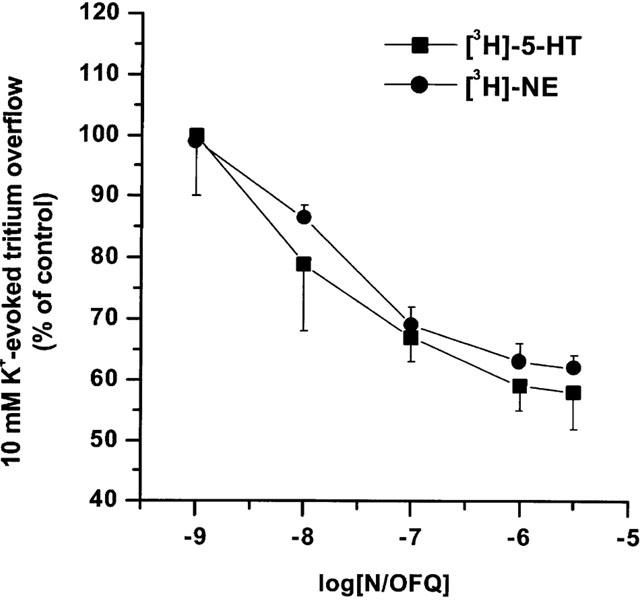
N/OFQ inhibits synaptosomal [3H]-5-HT and [3H]-NE overflow. Concentration–response curves of the inhibitory effects of N/OFQ (0.001–3 μM) on 10 mM K+-evoked [3H]-5-HT and [3H]-NE overflow. Significant inhibition of [3H]-5-HT and [3H]-NE overflow was found at 10 nM N/OFQ. Data are expressed as per cent of the K+ stimulation and are means±s.e.mean of at least eight experiments.
The inhibitory effect of N/OFQ was antagonized by selective NOP receptor peptide and non-peptide antagonists (Figure 2). Antagonist concentrations were chosen on the basis of present and previous experiments on CHOhNOP cells showing that [Nphe1], UFP-101 and J-113397 antagonized N/OFQ inhibition of cAMP accumulation with pA2 values of 6.0 (Calo et al., 2000), 7.11 (Calo et al., 2002b) and 7.52 (Bigoni et al., 2000), respectively. [Nphe1] (10 μM), UFP-101 (1 μM) as well as J-113397 and JTC-801 (both 0.1 μM) antagonised the effect of N/OFQ (0.1 μM; Figure 2). None of the antagonists modified K+-stimulated [3H]-5-HT overflow per se (Table 1).
Figure 2.
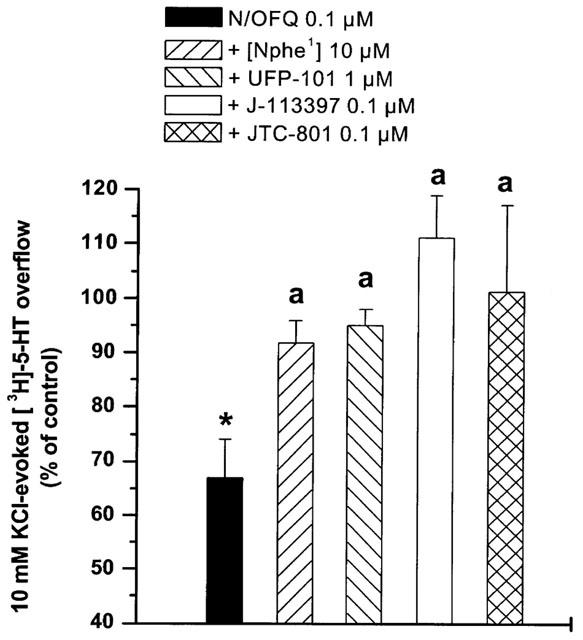
Selective NOP receptor antagonists prevent N/OFQ inhibition of [3H]-5-HT overflow. Effects of [Nphe1], UFP-101, J-113397 and JTC-801 on the inhibition of K+-evoked [3H]-5-HT overflow induced by N/OFQ (0.1 μM). Antagonists were perfused 3 min before N/OFQ and maintained until the end of experiment. Data are means±s.e.mean of at least eight experiments. *P<0.05; different from control. aP<0.05; different from N/OFQ.
Table 1.
Selective peptide and non-peptide NOP receptor antagonists did not alter 10 mM K+-evoked [3H]-5-HT and [3H]-NE overflow

[3H]-5-HT overflow evoked by 10 mM K+ was also inhibited (18±0.8%) by a supramaximal concentration of the NOP receptor ligand [F/G] (3 μM; Sbrenna et al., 2000; Figure 3). Since it has been previously shown that [F/G] inhibited K+-evoked [3H]-5-HT release from neocortical synaptosomes also via opioid (possibly MOP) receptors (Sbrenna et al., 2000) experiments were performed in the presence of naloxone (3 μM), which also did not affect K+-evoked stimulation per se (Sbrenna et al., 2000). The inhibitory effect of [F/G] was prevented by UFP-101 (1 μM). As previously reported (Sbrenna et al., 2000), [F/G] (3 μM) also antagonized the effect of N/OFQ 0.1 μM. Indeed the N/OFQ effect in the presence of [F/G] was reduced compared to that elicited by [F/G] alone (Figure 3).
Figure 3.
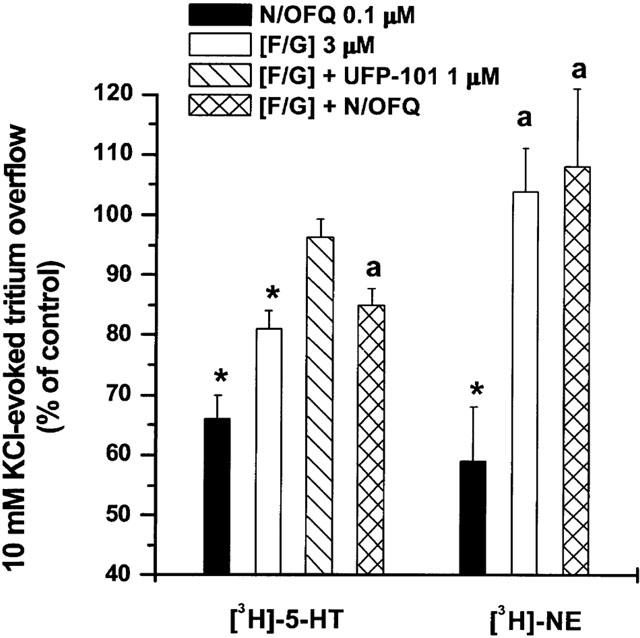
The NOP receptor ligand [F/G] prevents N/OFQ inhibition of [3H]-5-HT and [3H]-NE overflow. Effect of [F/G] (3 μM) on the inhibition of [3H]-5-HT and [3H]-NE overflow induced by N/OFQ (0.1 μM). [F/G] was applied (in the presence of naloxone 3 μM) 3 min before N/OFQ and maintained until the end of experiment. Data are means±s.e.mean of at least eight experiments. *P<0.05; different from control. aP<0.05; different from N/OFQ.
[3H]-NE overflow in synaptosomes
Basal [3H]-NE efflux (8.0±0.2 fmol mg prot−1 min−1, n=92) corresponded to a fractional release of 7.73±0.9%. One min pulse of KCl 10 mM evoked a tritium overflow (7.4±0.6 fmol mg prot−1 min−1, n=22) that was inhibited in a concentration-dependent manner by N/OFQ (0.001–3 μM; Figure 1). Analysis of the concentration-response curve of N/OFQ yielded a pEC50 value of 7.72±0.03. Maximal inhibition (38±2%) was observed at 3 μM.
As shown for [3H]-5-HT, the effect of N/OFQ was antagonized by [Nphe1] (10 μM) and UFP-101 (1 μM; Figure 4). Surprisingly, however, the non-peptide antagonists J-113397 and JTC-801 (both 0.1 μM) were unable to antagonise the action of N/OFQ when added to the perfusion medium 3 and 9 min before the peptide (Figure 5). A pre-application time of 21 min was necessary for the antagonist effect to develop. None of the antagonists modified K+-stimulated [3H]-NE efflux per se when applied 3 min before N/OFQ (Table 1) or longer (data not shown).
Figure 4.
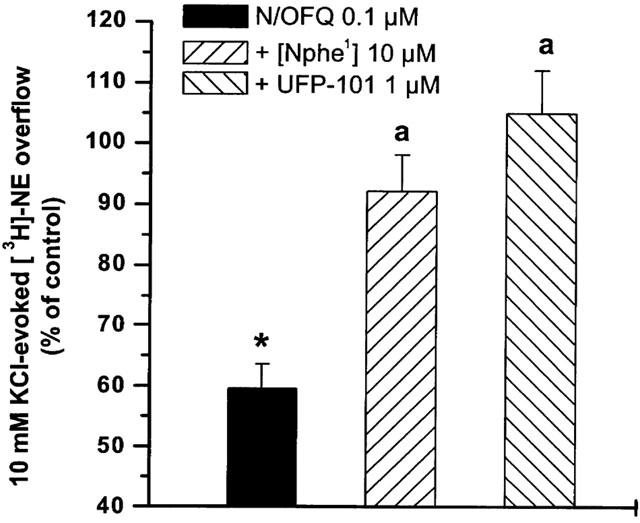
Selective peptide NOP receptor antagonists prevent N/OFQ inhibition of [3H]-NE overflow. Effect of [Nphe1] and UFP-101 on the inhibition of [3H]-NE overflow induced by N/OFQ (0.1 μM). Antagonists were perfused 3 min before N/OFQ and maintained until the end of experiment. Data are means±s.e.mean of at least six experiments. *P<0.05; different from control. aP<0.05; different from N/OFQ.
Figure 5.
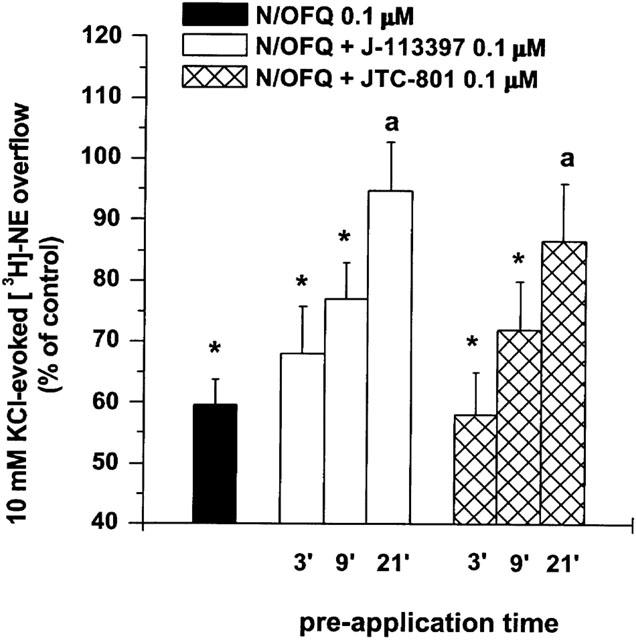
Selective non-peptide NOP receptor antagonists prevent N/OFQ inhibition of [3H]-NE overflow. Effect of J-113397 and JTC-801 on the inhibition of [3H]-NE overflow induced by N/OFQ (0.1 μM). Antagonists were applied at different times (3, 9 and 21 min) before N/OFQ and maintained until the end of experiment. Data are means±s.e.mean of at least six experiments. *P<0.05; different from control. aP<0.05; different from N/OFQ.
[F/G] (3 μM, applied 3 min prior to N/OFQ) also antagonized the effect of N/OFQ. At variance with data for [3H]-5-HT, however, [F/G] alone did not significantly affect K+-evoked [3H]-NE overflow at this concentration (Figure 3).
cAMP accumulation in CHOhNOP cells
N/OFQ produced a concentration dependent and saturable inhibition of the forskolin (1 μM)-stimulated cAMP formation in CHOhNOP cells (Figure 6). JTC-801 (0.1–10 μM), did not affect per se forskolin-stimulated cAMP formation (data not shown) but prevented the effect of N/OFQ. Its action was time- and concentration-dependent. Indeed, when co-applied with N/OFQ (Figure 6A), only JTC-801 10 μM displaced the concentration response curve of the natural peptide to the right, the curves being parallel and reaching similar maximal effects (pKB ∼7.0). However when JTC-801 was pre-incubated for 40 min (Figure 6B) and 90 min (Figure 6C) there was a time dependent increase in pKB estimated using the highest concentration of JTC-801. More importantly an inhibition at the lower (1 μM) concentration was uncovered which yielded time dependent increases in pKB values. For all the three sets of data, the slope of the Schild regression lines were significantly higher than 1 (data not shown) excluding a competititve type of antagonism. The effects of pre-incubation are summarized in Table 2. In all three sets of experiments N/OFQ displayed similar potency (pEC50 ∼9.73–10.05).
Figure 6.
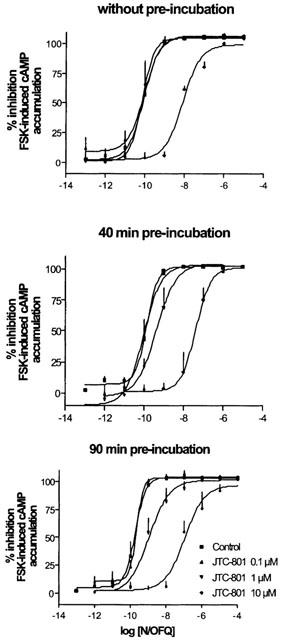
N/OFQ inhibits cAMP accumulation in CHOhNOP cells. The inhibitory effect of N/OFQ (0.001–1 μM) on forskolin-stimulated cyclic AMP accumulation in CHO cells expressing human recombinant NOP receptors was measured in the absence or the presence of the non-peptide NOP receptor antagonist JTC-801 (0.1–10 μM) at different pre-incubation times (0, 40 and 90 min; panel A, B and C, respectively). Data are means±s.e.mean of ⩾3 experiments.
Table 2.
Effects of incubation time on JTC-801 antagonist properties

Discussion
The pharmacological profiles of presynaptic NOP receptors modulating monoamine release from rat neocortical synaptosomes were studied and compared using four different selective NOP receptor antagonists and the NOP receptor ligand [F/G]. The results indicate that NOP receptors located on serotonergic and noradrenergic terminals are equally sensitive to both peptide ([Nphe1] and UFP-101) and non-peptide (J-113397 and JTC-801) antagonists, indicating that these populations of NOP receptors have similar pharmacological profiles. However, the peculiar time-dependence of action of J-113397 and JTC-801 together with the lack of an agonist effect of [F/G] on noradrenergic terminals suggests that the two native NOP receptor populations can be pharmacologically differentiated.
This conclusion is further substantiated by considering the similarities between the neurosecretory efficiency at noradrenergic and serotonergic terminals. Indeed, a 1 min pulse of KCl 10 mM evoked quantitatively a similar overflow of [3H]-5-HT and [3H]-NE (see Results). Moreover, both [3H]-5-HT and [3H]-NE overflow was inhibited to the same extent by omission of extracellular calcium and TTX application (for data on 5-HT see Sbrenna et al., 2000), suggesting a similar involvement of exocytotic and sodium dependent (i.e. quasi physiological) mechanisms. Finally, N/OFQ concentration response curves for K+-evoked [3H]-5-HT and [3H]-NE overflow were essentially identical (see pEC50 and Emax values), suggesting no preferential action of the endogenous peptide on the two populations of nerve endings.
Pharmacological studies of the N/OFQ system can now take advantage of different putative NOP receptor selective antagonists of peptide and non-peptide nature. The first peptide antagonist, [Nphe1] (reviewed by Calo et al., 2000; 2002a), has been consistently proven to be competitive, selective for NOP receptors and devoid of residual agonist activity, although characterized by low potency and poor metabolic stability. These problems have been overcome by the discovery of UFP-101 (Calo et al., 2002b), that competitively antagonized N/OFQ inhibition in peripheral tissues in vitro (pA2 7.1–7.3) and N/OFQ-induced hyperalgesia and hypolocomotion in mice in vivo (Calo et al., 2002b), displaying a potency one order of magnitude higher than [Nphe1] and a selectivity over classical opioid receptors of about three orders of magnitude. Accordingly, UFP-101 prevented in a competitive way N/OFQ inhibition of synaptosomal [3H]-5-HT release with a pA2 of 7.66 without exerting primary agonist activity (Calo et al., 2002b). Since, in the same preparation, a pA2 of 6.7 was found for [Nphe1] (Sbrenna et al., 2000), it is likely that pharmacology of presynaptic NOP receptors on serotonergic terminals is similar to that reported for recombinant human NOP receptors expressed in CHOhNOP cells (pA2 of 7.11 for UFP-101 and 6.0 for [Nphe1]). NOP receptors on noradrenergic terminals also display sensitivity towards [Nphe1] and UFP-101. This is in keeping with previous observations that [Nphe1] antagonized N/OFQ inhibition of NE release from neocortical slices (Okawa et al., 2001; Siniscalchi et al., 2002) and, when co-injected with N/OFQ into the locus coeruleus in vivo, prevented inhibition of neocortical NE release induced by the natural peptide (Okawa et al., 2001).
N/OFQ actions on synaptosomal 5-HT and NE release were also antagonized by submicromolar (i.e. 0.1 μM) concentrations of the NOP receptor selective non-peptide antagonist J-113397. This is in line with a recent study in rat neocortical slices where J-113397 antagonized N/OFQ inhibition of [3H]-NE release with a pA2 of 8.47 (Rominger et al., 2002) and, in general, with previous in vitro and in vivo observations showing that J-113397 is a pure (i.e. devoid of primary activity), selective (>300 fold over opioid receptors; Ozaki et al., 2000) and potent (pA2 in the 7.4–8.2 range) NOP receptor antagonist (reviewed by Calo et al., 2002a).
In contrast, to our knowledge only two studies (Shinkai et al., 2000; Yamada et al., 2002), have so far reported the antagonist properties of JTC-801 at NOP receptors, showing, in particular, that JTC-801 10 μM (co-applied with N/OFQ) antagonized both N/OFQ binding to recombinant human NOP receptors and N/OFQ inhibition of forskolin-stimulated cAMP accumulation in HeLa cells. We also showed that JTC-801, when co-applied with N/OFQ, prevents N/OFQ action on cAMP formation in CHOhNOP cells at 10 μM, being ineffective at lower concentrations. These results which are indeed very similar to those recently published by the Japanese group (Yamada et al., 2002), strongly suggest a non equilibrium condition. This was confirmed by the finding that following pre-incubation (40 min) the antagonist potency of JTC-801 increased (from inactivity to a pKB of 6.46 for the 1 μM and from 6.97 to 7.49 for the 10 μM concentration). Although it is evident that under the present experimental conditions pKB values have a pure descriptive value, this finding indicates that JTC-801 potency is time-dependent. Indeed, evidence has been presented that binding kinetics of non-peptide antagonists are in some case slower compared to peptide antagonists (for NK2 tachykinin receptors see Edmonds-Alts et al., 1992, and for B2 bradykinin receptors see Camarda et al., 2002). The selective NOP receptor non-peptide agonists Ro 64-6198 also displays different kinetics of action (slowly developing and pseudoirreversible effect) compared to N/OFQ (Rizzi et al., 2001). Quite surprisingly, however, the pKB values at 1 and 10 μM JTC-801 were also different after 90 min pre-incubation. This unexpected finding may be related to the mixed competitive/non-competitive antagonist nature of the compound (Yamada et al., 2002). The high receptor reserve of this preparation is probably the cause of the underestimation of the JTC-801 antagonist potency when low concentrations of antagonist are used (for a detailed discussion of this topic see Kenakin, 1993).
The finding that JTC-801 (and also J-113397) did not prevent N/OFQ action on [3H]-NE overflow when applied 3 or 9 min before the peptide is in line with data obtained in CHOhNOP cells. However, it remains unclear and somewhat intriguing why only a 3 min pre-treatment time was sufficient for JTC-801 and J-113397 to block N/OFQ effects on [3H]-5-HT overflow. It is possible that non-peptide antagonists bind with slower kinetics to NOP receptors located on noradrenergic compared to serotonergic nerve terminals. In this case, the expression of different subtypes of NOP receptors, or modulatory proteins affecting receptor properties, on the two populations of nerve terminals should be considered (see Introduction). Nevertheless, conclusive functional evidence for NOP receptor heterogeneity is still lacking (Mogil & Pasternak, 2001) and the present data support the view that pharmacological differences between NOP receptors expressed on noradrenergic and serotonergic neurones, if any, are minimal. Alternatively, the time-dependence of J-11397 and JTC-801 antagonism may be explained considering that a higher number of NOP receptors needs to be occupied on noradrenergic compared to serotonergic terminals before antagonism can be detected. The finding that [F/G] acts as partial agonist on serotonergic terminals and pure antagonist on noradrenergic terminals, however, may contradict this view. Indeed, the activity of [F/G] at NOP receptors has been shown to vary (from almost pure antagonist to full agonist; Calo et al., 2000) with the expression level (or stimulus-response coupling efficiency) of NOP receptors in different experimental models (Burnside et al., 2000). Thus the differential behaviour of [F/G], also observed in rat neocortical slices (Siniscalchi et al., 1999; 2002), may be related to a lower density (or a less efficient receptor coupling) of presynaptic NOP receptors on noradrenergic rather than serotonergic terminals.
Whatever the neurobiological mechanism, the finding that [F/G] inhibits synaptosomal 5-HT but not NE release differently from the endogenous peptide N/OFQ, suggests that this compound may be employed to selectively modulate neurosecretion at serotonergic terminals. More generally, we envisage that partial agonists may selectively affect neuronal function and represent a possible new approach for targeting subpopulations of NOP receptors regulating specific brain functions. This hypothesis is supported by the finding that i.v. injection of the NOP receptor partial agonists [F/G], Ac-RYYRIK-NH2 and Ac-RYYRWK-NH2 produced a selective free-water diuresis without altering cardiovascular functions which was different from the full agonist N/OFQ (Kapusta et al., 2002).
In conclusion, pharmacological analysis of presynaptic NOP receptors localized on serotonergic and noradrenergic terminals in the rat neocortex shows subtle differences with respect to time-dependent sensitivity towards non-peptide receptor antagonists and responsiveness to [F/G]. Although the explanation(s) for this behaviour are presently under investigation, these results indicate that the two receptor systems can be differentiated pharmacologically and possibly targeted by specific therapeutic agents.
Acknowledgments
This work was supported by a cofin 99 grant from the Italian Ministry of University and British Journal of Anaesthesia (Leicester studies).
Abbreviations
- CHO
Chinese hamster ovary
- [F/G]
[Phe1ψ(CH2-NH)Gly2]nociceptin/orphanin FQ(1-13)NH2
- JTC-801
N-(4-amino-2-methylquinolin-6-yl)-2-(4-ethylphenoxymethyl)benzamide hydrochloride
- J-113397
(±)trans-1-[1-cyclooctylmethyl-3-hydroxymethyl-4-piperidyl]-3-ethyl-1,3-dihydro-2H-benzimidazol-2-one
- N/OFQ
nociceptin/orphanin FQ
- [Nphe1]
[Nphe1]nociceptin/orphanin FQ(1-13)NH2
- TTX
tetrodotoxin
- UFP-101
[Nphe1,Arg14,Lys15]N/OFQ-NH2
References
- BIGONI R., CALO G., RIZZI A., GUERRINI R., DE RISI C., HASHIMOTO Y., HASHIBA E., LAMBERT D.G., REGOLI D. In vitro characterization of J-113397, a non-peptide nociceptin/orphanin FQ receptor antagonist. Naunyn Schmiedebergs Arch. Pharmacol. 2000;361:565–568. doi: 10.1007/s002100000220. [DOI] [PubMed] [Google Scholar]
- BURNSIDE J.L., RODRIGUEZ L., TOLL L. Species differences in the efficacy of compounds at the nociceptin receptor (ORL1) Peptides. 2000;21:1147–1154. doi: 10.1016/s0196-9781(00)00253-9. [DOI] [PubMed] [Google Scholar]
- CALO G., GUERRINI R., BIGONI R., RIZZI A., BIANCHI C., REGOLI D., SALVADORI S. Structure-activity study of the nociceptin(1-13)-NH2 N-terminal tetrapeptide and discovery of a nociceptin receptor antagonist. J. Med. Chem. 1998;41:3360–3366. doi: 10.1021/jm970805q. [DOI] [PubMed] [Google Scholar]
- CALO G., GUERRINI R., RIZZI A., SALVADORI S., REGOLI D. Pharmacology of nociceptin and its receptor: a novel therapeutic target. Br. J. Pharmacol. 2000;129:1261–1283. doi: 10.1038/sj.bjp.0703219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CALO G., RIZZI A., BIGONI R., GUERRINI R., SALVADORI S., REGOLI D. Pharmacological profile of nociceptin/orphanin FQ receptors. Clin. Exp. Pharmacol. Physiol. 2002a;29:223–228. doi: 10.1046/j.1440-1681.2002.03633.x. [DOI] [PubMed] [Google Scholar]
- CALO G., RIZZI A., RIZZI D., BIGONI R., GUERRINI R., MARZOLA G., MARTI M., MCDONALD J., MORARI M., LAMBERT D.G., SALVADORI S., REGOLI D. [Nphe1, Arg14, Lys15]Nociceptin-NH2, a novel potent and selective antagonist of the nociceptin/orphanin FQ receptor. Br. J. Pharmacol. 2002b;136:303–311. doi: 10.1038/sj.bjp.0704706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CAMARDA V., RIZZI A., CALO' G., WIRTH K., REGOLI D. Pharmacological characterization of novel kinin B2 receptor ligands. Can. J. Physiol. Pharmacol. 2002;80:281–286. doi: 10.1139/y02-037. [DOI] [PubMed] [Google Scholar]
- COLLARD K.J., CASSIDY D.M., PYE M.A., TAYLOR R.M. The stimulus-induced release of unmetabolized 5-hydroxytryptamine from superfused rat brain synaptosomes. J. Neurosci. Meth. 1981;4:163–179. doi: 10.1016/0165-0270(81)90051-0. [DOI] [PubMed] [Google Scholar]
- CONNOR M., VAUGHAN C.W., CHIENG B., CHRISTIE M.J. Nociceptin receptor coupling to a potassium conductance in rat locus coeruleus neurons in vitro. Br. J. Pharmacol. 1996;119:1614–1618. doi: 10.1111/j.1476-5381.1996.tb16080.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CONNOR M., VAUGHAN C.W., JENNINGS E.A., ALLEN G.A., CHRISTIE M.J. Nociceptin, Phe1ψ-nociceptin1-13, nocistatin and prepronociceptin154-181 effects on calcium channel currents and a potassium current in rat locus coeruleus in vitro. Br. J. Pharmacol. 1999;119:1614–1618. doi: 10.1038/sj.bjp.0702971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COX B.M., CHAVKIN C., CHRISTIE M.J., CIVELLI O., EVANS C., HAMON M.D., HOELLT V., KIEFFER B., KITCHEN I., MCKNIGHT A.T., MEUNIER J.C., PORTOGHESE P.S.Opioid receptors The IUPHAR Compendium of Receptor Characterization and Classification 2000London: IUPHAR Media Ltd; Girdlestone, D. ed [Google Scholar]
- CURRÒ D., YOO Y.H., ANDERSON M., SONG I., DEL VALLE J., OWYANG C. Molecular cloning of the orphanin FQ receptor gene and differential tissue expression of splice variants in the rat. Gene. 2001;266:139–145. doi: 10.1016/s0378-1119(00)00553-9. [DOI] [PubMed] [Google Scholar]
- DE LANGEN C.D.J., HOGENBOOM F., MULDER A.H. Presynaptic noradrenergic α-receptors and modulation of 3H-noradrenaline release from rat brain synaptosomes. Eur. J. Pharmacol. 1979;60:79–89. doi: 10.1016/0014-2999(79)90054-2. [DOI] [PubMed] [Google Scholar]
- DE RISI C., POLLINI P., TRAPELLA C., PERETTO I., RONZONI S., GIARDINA G.A. A new synthetic approach to 1-[3R, 4R)-1-cyclooctylmethyl-3-hydroxymethyl-4-piperidyl]-3-ethyl-1,3-dihydro-2H-benzimidazol-2-one (J-113397), the first non-peptide ORL-1 receptor antagonist. Bioorg. Med. Chem. 2001;9:1871–1877. doi: 10.1016/s0968-0896(01)00085-2. [DOI] [PubMed] [Google Scholar]
- EDMONDS-ALT X.P., VILAIN P., GOULAOUIC P., PROIETTO V., VAN BROECK D., ADVENIER C., NALINE E., NELIAT G., LE FUR G., BRELIERE J.C. A potent and selective nonpeptide antagonist of the neurokinin A (NK2) receptor. Life. Sci. Pharmacol. Lett. 1992;50:PL101. doi: 10.1016/0024-3205(92)90352-p. [DOI] [PubMed] [Google Scholar]
- GUERRINI R., CALO G., BIGONI R., RIZZI A., VARANI K., TOTH G., GESSI S., HASHIBA E., HASHIMOTO Y., LAMBERT D.G., BOREA P.A., TOMATIS R., SALVADORI S., REGOLI D. Further studies on nociceptin-related peptides: discovery of a new chemical template with antagonist activity on the nociceptin receptor. J. Med. Chem. 2000;43:2805–2813. doi: 10.1021/jm990075h. [DOI] [PubMed] [Google Scholar]
- GUERRINI R., CALO G., RIZZI A., BIGONI R., BIANCHI C., SALVADORI S., REGOLI D. A new selective antagonist of the nociceptin receptor. Br. J. Pharmacol. 1998;123:163–165. doi: 10.1038/sj.bjp.0701640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JENCK F., MOREAU J.-L., MARTIN J.R., KILPATRICK G., REINSCHEID R.K., MONSMA F.J., NOTHAKER H.-P., CIVELLI O. Orphanin FQ acts as an anxiolytic to attenuate behavioural responses to stress. Proc. Natl. Acad. Sci. 1997;94:14854–14858. doi: 10.1073/pnas.94.26.14854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JENCK F., WICHMANN J., DAUTZENBERG F.M., MOREAU J.-L., OUAGAZZAL A.M., MARTIN J.R., LUNDSTROM K., CESURA A., POLI S.M., ROEVER S., KOLCZEWSI S., ADAM G., KILPATRICK G. A synthetic agonist at the orphanin FQ/nociceptin receptor ORL1: anxiolytic profile in the rat. Proc. Natl. Acad. Sci. 2000;97:4938–4943. doi: 10.1073/pnas.090514397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JENKINSON D.H., BARNARD E.A., HOYER D., HUMPREY P.P.A., LEFF P., SHANKLEY N.P. International Union of Pharmacology Committee on receptor nomenclature and drug classification XI Recommendations on terms and symbols in quantitative pharmacology. Pharmacol. Rev. 1995;47:255–266. [PubMed] [Google Scholar]
- KAPUSTA D.R., CALO' G., KENIGS V.A. Experimental Biology. New Orleans, USA; 2002. Peripheral administration of partial agonists of the nociceptin/orphanin FQ peptide (NOP) receptor produce water diuresis in conscious rats. [Google Scholar]
- KAWAMOTO H., OZAKI S., ITOH Y., MIYAJI M., ARAI S., NAKASHIMA H., KATO OHTA H., IWASAWA Y. Discovery of the first potent and selective small molecule opioid receptor-like (ORL1) antagonist: 1-[3R, 4R)-1-cyclooctylmethyl-3 - hydroxymethyl -4-piperidyl]-3-ethyl-1,3-dihydro-2H-benzimidazol-2-one (J-113397) J. Med. Chem. 1999;42:5061–5063. doi: 10.1021/jm990517p. [DOI] [PubMed] [Google Scholar]
- KENAKIN T. Raven Press, New York, USA; 1993. Pharmacological analysis of drug-receptor interactions; pp. 323–343. [Google Scholar]
- MATHIS J.P., RYAN MORO J., CHANG A., HOM J.S.H., SCHEINBERG D.A., PASTERNAK G.W. Biochemical evidence for orphanin FQ/Nociceptin receptor heterogeneity in mouse brain. Biochem. Biophys. Res. Comm. 1997;230:462–465. doi: 10.1006/bbrc.1996.5867. [DOI] [PubMed] [Google Scholar]
- MEUNIER J.C., MOLLEREAU C., TOLL L., SUAUDEAU C., MOISAND C., ALVINERIE P., BUTOUR J.L., GUILLEMOT J.C., FERRARA P., MONSARRAT B., MAZARGUIL H., VASSART G., PARMENTIER M., COSTENTIN J. Isolation and structure of the endogenous agonist of opioid receptor-like ORL1 receptor. Nature. 1995;377:532–535. doi: 10.1038/377532a0. [DOI] [PubMed] [Google Scholar]
- MOGIL J.S., PASTERNAK G.W. The molecular and behavioral pharmacology of the orphanin FQ/Nociceptin peptide and receptor family. Pharmacol. Rev. 2001;53:381–415. [PubMed] [Google Scholar]
- MORARI M., SBRENNA S., MARTI M., CALIARI C., BIANCHI C., BEANI L. NMDA and non-NMDA ionotropic glutamate receptors modulate striatal acetylcholine release via pre- and postsynaptic mechanisms. J. Neurochem. 1998;71:2006–2017. doi: 10.1046/j.1471-4159.1998.71052006.x. [DOI] [PubMed] [Google Scholar]
- OKAWA H., KUDO M., KUDO T., GUERRINI R., LAMBERT D.G., KUSHIKATA T., YOSHIDA H., MATSUKI A. Effects of nociceptinNH2 and [Nphe1]nociceptin(1-13)NH2 on rat brain noradrenaline release in vivo and in vitro. Neurosci. Lett. 2001;303:173–176. doi: 10.1016/s0304-3940(01)01721-9. [DOI] [PubMed] [Google Scholar]
- OKAWA H., NICOL B., BIGONI R., HIRST R.A., CALO G., GUERRINI R., ROWBOTHAM D.J., SMART D., MCKNIGHT A.T., LAMBERT D.G. Comparison of the effects of [Phe1ψ(CH2-NH)Gly2]nociceptin-(1-13)-NH2 in rat brain, rat vas deferens, and CHO cells expressing recombinant human nociceptin receptors. Br. J. Pharmacol. 1999;127:123–130. doi: 10.1038/sj.bjp.0702539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OZAKI S., KAWAMOTO H., ITOH Y., MIYAJI M., AZUMA T., ICHIKAWA D., NAMBU H., IGUCHI T., IWASAWA Y., OTA H. In vitro and in vivo pharmacological characterization of J-113397, a potent and selective non-peptidyl ORL1 receptor antagonist. Eur. J. Pharmacol. 2000;402:45–53. doi: 10.1016/s0014-2999(00)00520-3. [DOI] [PubMed] [Google Scholar]
- POLIDORI C., CALO G., CICCOCIOPPO R., GUERRINI R., REGOLI D., MASSI M. Pharmacological characterization of the nociceptin receptor mediating hyperphagia: identification of a selective antagonist. Psychopharmacology. 2000;148:430–437. doi: 10.1007/s002130050073. [DOI] [PubMed] [Google Scholar]
- RAITERI L., RAITERI M. Synaptosomes still viable after 25 years of superfusion. Neurochem. Res. 2000;25:1265–1274. doi: 10.1023/a:1007648229795. [DOI] [PubMed] [Google Scholar]
- REINSCHEID R.K., NOTHACKER H.P., BOURSON A., ARDATI A., HENNINGSEN R.A., BUNZOW J.R., GRANDY D.K., LANGEN H., MONSMA F.J., JR, CIVELLI O. Orphanin FQ: a neuropeptide that activates an opioid like G protein-coupled receptor. Science. 1995;270:792–794. doi: 10.1126/science.270.5237.792. [DOI] [PubMed] [Google Scholar]
- RIZZI D., BIGONI R., RIZZI A., JENCK F., WICHMANN J., GUERRINI R., REGOLI D., CALO G. Effects of Ro 64-6198 in nociceptin/orphanin FQ-sensitive isolated tissues. Naunyn Schmiedebergs Arch. Pharmacol. 2001;363:551–555. doi: 10.1007/s002100100399. [DOI] [PubMed] [Google Scholar]
- ROMINGER A., FORSTER S., ZENTNER J., DOOLEY D.J., MCKNIGHT A.T., FEUERSTEIN T.J., JACKISCH R., VLASKOVSKA M. Comparison of the ORL1 receptor-mediated inhibition of noradrenaline release in human and rat neocortical slices. Br. J. Pharmacol. 2002;135:800–806. doi: 10.1038/sj.bjp.0704523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SBRENNA S., MARTI M., MORARI M., CALO G., GUERRINI R., BEANI L., BIANCHI C. Modulation of 5-hydroxytryptamine efflux from rat cortical synaptosomes by opioids and nociceptin. Br. J. Pharmacol. 2000;130:425–433. doi: 10.1038/sj.bjp.0703321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHLICKER E., MORARI M. Nociceptin/orphanin FQ and neurotransmitter release in the central nervous system. Peptides. 2000;21:1023–1029. doi: 10.1016/s0196-9781(00)00233-3. [DOI] [PubMed] [Google Scholar]
- SHINKAI H., ITO T., IIDA T., KITAO Y., YAMADA H., UCHIDA I. 4-Aminoquinolines: novel nociceptin antagonists with analgesic activity. J. Med. Chem. 2000;43:4667–4677. doi: 10.1021/jm0002073. [DOI] [PubMed] [Google Scholar]
- SINISCALCHI A., RODI D., BEANI L., BIANCHI C. Inhibitory effect of nociceptin on [3H]-5HT release from rat cerebral cortex slices. Br. J. Pharmacol. 1999;128:119–123. doi: 10.1038/sj.bjp.0702793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SINISCALCHI A., RODI D., MORARI M., MARTI M., CAVALLINI S., MARINO S., BEANI L., BIANCHI C. Direct and indirect inhibition by nociceptin/orphanin FQ on noradrenaline release from rodent cerebral cortex in vitro. Br. J. Pharmacol. 2002;136:1178–1184. doi: 10.1038/sj.bjp.0704841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VAUGHAN C.W., CHRISTIE M.J. Increase by the ORL1 (opioid receptor-like 1) ligand, nociceptin, of inwardly rectifying K conductance in dorsal raphe nucleus neurones. Br. J. Pharmacol. 1996;117:1609–1611. doi: 10.1111/j.1476-5381.1996.tb15329.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YAMADA H., NAKAMOTO H., SUZUKI Y., ITO T., AISAKA K. Pharmacological profiles of a novel opioid receptor-like 1 (ORL1) receptor antagonist, JTC-801. Br. J. Pharmacol. 2002;135:323–332. doi: 10.1038/sj.bjp.0704478. [DOI] [PMC free article] [PubMed] [Google Scholar]