

Abstract
The present study was performed to evaluate the presence and the physiological consequences of butyrylcholinesterase (BChE) inhibition on isolated phrenic-hemidiaphragm preparations from normal mice expressing acetylcholinesterase (AChE) and BChE, and from AChE-knockout mice (AChE−/−) expressing only BChE.
Histochemical and enzymatic assays revealed abundance of AChE and BChE in normal mature neuromuscular junctions (NMJs).
In normal NMJs, in which release was reduced by low Ca2+/high Mg2+ medium BChE inhibition with tetraisopropylpyrophosphoramide (iso-OMPA) or bambuterol decreased (∼50%) evoked quantal release, while inhibition of AChE with fasciculin-1, galanthamine (10, 20 μM) or neostigmine (0.1–1 μM) increased (50–80%) evoked quantal release. Inhibition of both AChE and BChE with galanthamine (80 μM), neostigmine (3–10 μM), O-ethylS-2-(diisopropylamino)ethyl-methylphosphono-thioate (MTP) or phospholine decreased evoked transmitter release (20–50%).
In AChE−/− NMJs, iso-OMPA pre-treatment decreased evoked release.
Muscarinic toxin-3 decreased evoked release in both AChE−/− and normal NMJs treated with low concentrations of neostigmine, galanthamine or fasciculin-1, but had no effect in normal NMJs pretreated with iso-OMPA, bambuterol, MTP and phospholine.
In normal and AChE−/− NMJs pretreatment with iso-OMPA failed to affect the time course of miniature endplate potentials and full-sized endplate potentials.
Overall, our results suggest that inhibition or absence of AChE increases evoked quantal release by involving muscarinic receptors (mAChRs), while BChE inhibition decreases release through direct or indirect mechanisms not involving mAChRs. BChE apparently is not implicated in limiting the duration of acetylcholine action on postsynaptic receptors, but is involved in a presynaptic modulatory step of the release process.
Keywords: Butyrylcholinesterase, acetylcholinesterase, acetylcholinesterase knockout mice; muscarinic receptors; cholinesterase inhibitors; acetylcholine release
Introduction
At cholinergic synapses, two closely related enzymes can hydrolyse acetylcholine (ACh): acetylcholinesterase (AChE; EC 3.1.1.7.) and butyrylcholinesterase (BChE; EC 3.1.1.8.). Although the classical function of AChE at cholinergic synapses in regulating the duration of ACh action is well established, the physiological role of BChE has not been systematically examined. This enzyme has been considered physiologically irrelevant since in man the lack of BChE activity does not lead to any abnormality (Primo-Parmo et al., 1996). None the less BChE is found in many animal species so it is likely to have some physiological importance. BChE may co-regulate the degradation of ACh in the human airways tract (Norel et al., 1993), canine tracheal smooth muscle (Adler & Filbert, 1990), and may participate in smooth muscle contraction (Walch et al., 1997). The higher abundance of BChE than AChE in vertebrate embryos (Layer & Willbold, 1995), and during early post-natal development (Silman et al., 1979; Berman et al., 1987; Li et al., 2000) suggested its possible involvement in cell differentiation and development (Chatonnet & Lockridge, 1989; Taylor, 1991; Massoulié et al., 1993; Layer & Willbold, 1995).
The physiological relevance of BChE has also been suggested in gene-targeted mice lacking AChE (Xie et al., 2000), and in mice deficient in the collagen Q (ColQ), via which asymmetric forms of AChE are associated to the synaptic basal lamina (Feng et al., 1999). Mice lacking AChE activity at the NMJ are suspected to survive because BChE, or other esterase-like enzymes, permit(s) a minimal level of cholinergic function. Moreover, a mutation in ache gene in zebrafish, which lacks the bche gene, causes severe morphological defects in embryos, and young larva are paralyzed and die (Behra et al., 2002). This further suggested that in mammals other esterases, such as BChE, may compensate for the absence of AChE activity.
Pioneer pharmacological studies proposed that AChE and BChE complemented each other depending on the ACh concentration in the synaptic cleft. AChE being more active at low ACh concentrations, while BChE would act only at high ACh concentrations (Koelle, 1950). Furthermore, it has been recently hypothesized that BChE is not essential for survival, but may play an important role at certain sites and may act as a back-up to AChE, in conditions when AChE activity is depressed or absent (Li et al., 2000).
The current study was designed to examine the presence, and physiological consequences of BChE inhibition at mature mouse skeletal NMJs. For this purpose, we compared the effects of various BChE and AChE inhibitors on quantal transmitter release in normal and AChE-deficient (AChE−/−) NMJs, and examined the possibility that BChE may contribute to the hydrolysis of ACh in the synaptic cleft.
Methods
Mice
The AChE−/− knockout mice originally generated by Xie et al. (2000), were obtained by mating heterozygous male and females, in order to produce wild-type and nullizygote littermates. Original founders were kindly provided by Prof. O. Lockridge (Eppley Institute, Nebraska, U.S.A.), and were maintained in a 129SVJ strain background. The animals were housed in the transgenic facility of the Gif sur Yvette Campus and the Montpellier animal's house, under standard conditions at a constant temperature of 24°C with a 12 : 12 daylight cycle. Food and water were provided ad libitum. Rearing conditions were in keeping with the guidelines of the French Ministry for Research and Industry relating to the use and storage of transgenic animals.
In our first attempt to raise a colony of AChE−/− mice by breeding AChE+/− mice we obtained a similar rate of animal survival as initially reported by Xie et al. (2000) i.e., the majority of AChE−/− mice remained alive at day 12, but most AChE−/− mice died between postnatal days 14–21, and only 1–2% survived to the adult stage. The emaciated appearance, and absence of body fat, suggested that the cause of death of AChE−/− mice might be starvation. Consequently, efforts were made in our laboratories to facilitate their feeding and caloric intake. For this purpose, pregnant and lactating dams were fed with a highly-energetic liquid-food diet supplemented with essential nutrients (Renutryl, Nestlé Clinical Nutrition, France). The isolation and early weaning (on postnatal day 14) of AChE−/− mice, and their feeding with the liquid diet delivered in petri dishes at the bottom of the cage considerably extended the life span of AChE−/− mice. Thus, 33% of our AChE−/− mice survived over 120 days, and 25% over 240 days, but they remained small at all ages compared to their littermates. The increased life span of AChE−/− mice fed with a high-fat liquid food diet supplemented with essential nutrients, similar to that used in the present study, and the detailed phenotype of adult AChE−/− mice have been recently reported and discussed (Duysen et al., 2002). In this work 2–6-month-old AChE−/− mice were used. For each experiment nullizygote mice were identified by PCR using primers that distinguish wild-type from mutant alleles (Xie et al., 2000). In addition the absence of AChE at the NMJ was histochemically probed as described below.
Female Swiss-Webster mice were purchased from IFFA CREDO (Saint Germain sur l'Arbresle, France). Mice were euthanized by dislocation of the cervical vertebrae followed by immediate exsanguination.
Drugs
Neostigmine methylsulphate was purchased from France Biochem (Meudon, France); bambuterol hydrochloride was kindly provided by AstraZeneca R & D, Lund, Sweden; muscarine chloride, (+)-tubocurarine hydrochloride, thiolacetic acid, 5,5′dithiobis(2-nitrobenzoic acid) (DTNB), tetraisopropylpyro-phosphoramide (iso-OMPA), and 1,5-bis(4-allyldimethylammoniumphenyl)pentan-3-one (BW284C51) were obtained from Sigma-Aldrich Chimie (St. Quentin-Fallavier, France); μ-conotoxin GIIIB was obtained from Calbiochem (San Diego, CA, U.S.A.); O-ethylS-2-(diisopropylamino)ethylmethylphosphonothioate (MTP) was kindly provided by Dr F. Leterrier (Vigny et al., 1978); galanthamine was kindly provided by Dr C. Thal (Institut de Chimie des Substances Naturelles, CNRS, Gif-sur-Yvette, France); O,O′-diethyl S-ethyltrimethylamine phosphorotiolate iodide (phospholine) was kindly provided by Dr M. Israël (CNRS, Gif-sur-Yvette, France). Fasciculin-1 and muscarinic toxin-3 (MT-3) were purified and kindly provided by Dr E. Karlsson (Biomedical Center, Uppsala, Sweden).
Measurement of cholinesterase activity
To estimate BChE and AChE activity in muscle, small strips of the innervated region were cut across the hemidiaphragm at either side of the main intramuscular nerve trunk. The sections were then weighed and frozen at −80°C. Frozen tissues were homogenized using a hand-held homogenizer in 10 volumes of ice-cold sodium phosphate buffer (100 mM, pH 7.4) containing 0.5% Tween-20 (see Li et al., 2000). Homogenates were centrifuged at 14,000×g for 10 min at 4°C. To separate globular from asymmetric enzyme molecular forms, sequential extraction was performed in the presence of 1 M NaCl. Supernatants were immediately assayed for cholinesterase by the method of Ellman et al. (1961) using 1 mM acetylthiocholine as substrate, and a temperature-controlled UNIKON 933 Spectrophotometer (Milan, Italy) at 25°C. BChE activity was measured after AChE was inhibited with 0.01 mM BW284C51. AChE activity was measured after BChE activity was inhibited with 0.1 mM iso-OMPA. Cholinesterase activity was expressed in micromoles of substrate hydrolysed min g−1 wet weight of muscle.
Histochemical staining of cholinesterases
Whole-mounts of triangularis sterni muscle were stained for cholinesterase activity by the method of Koelle & Horn (1968) as modified by Gautron (1974), using thiolacetic acid as substrate. Before probing, preparations were incubated for 20 min at room temperature with either 0.01 mM BW284C51 to suppress AChE activity or 0.1 mM iso-OMPA to suppress BChE activity. Control muscles were not pretreated with cholinesterase inhibitors. In order to avoid overstaining, cholinesterase labelling was allowed to proceed for only 12 min at 0°C. At the end of the reaction, preparations were washed and fixed with 4% paraformaldehyde for 20 min. Images of randomly selected endplates were collected using a Zeiss upright microscope equipped with an oil-immersion lens (×20, numerical aperture 0.8) and an extended ISIS CCD, cooled video camera (Photonics Science, U.K.). Digitizing and image analysis were performed with a personal computer using a DT3155 frame grabber (Data Translation MA, U.S.A.) and OPTIMAS software (Media Cybernetics, Silver Spring, U.S.A.).
Electrophysiological recordings on isolated neuromuscular preparations
Electrophysiological recordings were done as previously described (Minic et al., 2002). In brief, left or right hemidiaphragm muscle with their associated phrenic nerves were mounted in Rhodorsil-lined organ baths superfused with an oxygenated standard physiological solution of the following composition (mM): 154 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 5.0 HEPES buffer, pH 7.4 and 11 glucose.
Studies on nerve-evoked quantal transmitter release were performed either in solutions containing low Ca2+ (0.4 mM) and high Mg2+ (6 mM) concentration, or in the standard physiological solution containing 2.2 μM μ-conotoxin GIIIB (to block voltage-depended Na+ channels in muscle fibres), or 3 μM (+)-tubocurarine (to block post-synaptic nicotinic ACh receptor). The motor nerve was stimulated via a suction electrode with current pulses of 0.1 ms duration and supramaximal voltage (typically 3–8 V) at different stimulating frequencies (0.1, 0.3, 10, 20, 40 and 100 Hz). Membrane potentials and synaptic potentials were recorded from endplate regions with intracellular microelectrodes using conventional techniques and an Axoclamp-2A system (Axon Instruments, Union City, CA, U.S.A.). The borosilicate glass microelectrodes were filled with 3 M KCl solution and had 8–12 MΩ resistance. All experiments were performed at 22–24°C.
In experiments with reversible cholinesterase inhibitors (neostigmine, bambuterol, fasciculin-1, galanthamine) preparations were equilibrated for 60 min with each of the inhibitors before recordings, unless otherwise stated. In experiments with irreversible anticholinesterase drugs, preparations were pretreated for 20 min with iso-OMPA, or for 15 min with MTP or phospholine, and then washed out of the inhibitor for 30 min with the physiological medium before recordings.
Data analysis and statistics
The results are presented as the mean of n separate experiments±(s.e.m.). The mean quantal content (mo) of endplate potentials (EPPs), was calculated by the method of failures in which mo=In (N/No), where N, is the number of stimuli; No, is the number of failures i.e. number of stimuli not followed by an EPP.
The amplitudes of full-sized EPPs and MEPPs recorded on junctions treated with μ-conotoxin GIIIB were normalized to a membrane potential of −75 mV. The EPP amplitudes were corrected for non-linear summation (McLachlan & Martin, 1981). The quantal content (m) was calculated at each junction by the direct method (dividing the mean normalized and corrected EPP amplitude by the mean normalized MEPP amplitude). In this last case giant MEPPs were excluded from the amplitude analysis, since they do not enter in the composition of evoked EPPs.
Statistical significance of differences between controls and test values was assessed by Student's t-test (two-tailed), or by the Kolmogorov-Smirnov two-sample test. Data were considered significant at P<0.05.
Results
BChE is present at the neuromuscular junction of adult mice
BChE concentration in chicken and rat skeletal muscle has been reported to fall during muscle maturation (Silman et al., 1979; Berman et al., 1987). Therefore, experiments were first designed to test the presence of BChE at the NMJ of adult mice, and to compare its activity with respect to that of AChE using biochemical and histochemical methods. As shown in Table 1, globular molecular forms of BChE and AChE contributed to about 50% of total cholinesterase activity, whereas AChE asymmetric forms are predominant over BChE asymmetric forms.
Table 1.
AChE and BChE activities in innervated parts of the diaphragm from 2 month-old mice

For histochemical probing BChE and AChE activity whole-mounts of the Triangularis sterni muscle were used (Figure 1). We choose this flat and thin thoracic muscle because it is composed of only a few layers of fibres. As shown in Figure 1(C and D), BChE and AChE staining were detected at motor endplate regions in muscles that were pretreated with either BW284C51 or iso-OMPA to inactivate AChE or BChE respectively (Atack et al., 1989). When muscles were pretreated with both inhibitors no staining was observed (Figure 1B). The enzymatic and histochemical data indicate that both AChE and BChE are located in endplate regions of adult mouse skeletal muscle and exhibit similar levels of activity.
Figure 1.
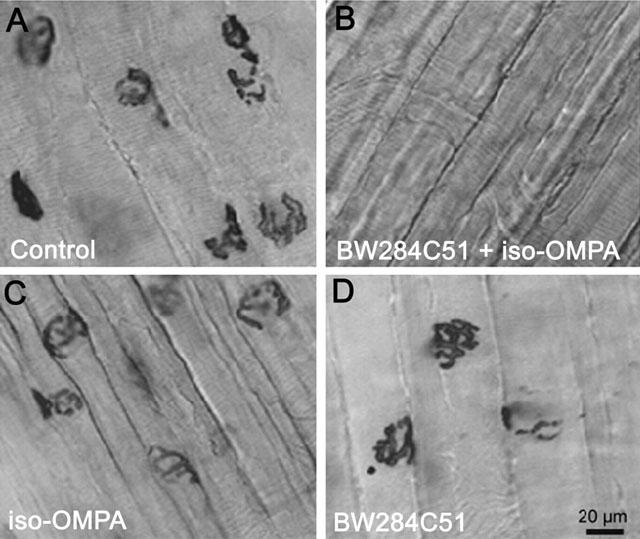
Presence of BChE and AChE activity at the NMJ of mature mouse skeletal muscle fibres. (A) Control untreated Triangularis sterni muscle stained for cholinesterases. (B) Preparation treated successively with 10 μM BW284C51 and 100 μM iso-OMPA to inactivate AChE and BChE. (C) Muscle treated with 100 μM iso-OMPA to reveal AChE. (D) Preparation treated with 10 μM BW284C51 to reveal BChE activity. Scale bar in D (20 μm) applies to all images.
Inhibition of AChE and BChE activities in muscle homogenates
To determine the specificity of different anticholinesterase drugs for BChE and AChE, an enzymatic assay of cholinesterase activities was performed in muscle homogenates. For this purpose extraction was carried out in Tris-buffer containing 0.5% Tween 20, instead of the more commonly used Triton X-100 which was recently reported to inhibit BChE by binding directly to the enzyme active-site (Li et al., 2000).
As shown in Table 2, the only agent which had no inhibitory effect on BChE, in the range of concentrations tested, was fasciculin-1. All other inhibitors tested affected both AChE and BChE depending on the concentration, but a higher affinity for BChE than for AChE was observed with bambuterol. We further focused our study on the analysis of the physiological effects of various AChE and BChE inhibitors.
Table 2.
Inhibition of total cholinesterase, AChE and BChE in homogenates from mouse diaphragm muscle

Changes in the quantal content of EPPs caused by different inhibitors in normal junctions
To test the presynaptic effects of the chosen inhibitors, the mean quantal content of EPPs was determined in normal NMJs before and after drug application. Experiments were performed in junctions equilibrated for 1 h in physiological solution containing low Ca2+ (0.4 mM)/high Mg2+ (6 mM). Upon nerve stimulation at 0.3 Hz, the quantal content of EPPs was low (0.57±0.15; n=16 fibres from five different muscles) i.e. following some stimuli no quanta were released, while many of the EPPs were generated by a single transmitter quantum.
As shown in Figure 2, inhibition of AChE by fasciculin-1 (70–350 nM) significantly increased mo by about 80% with respect to controls. A similar increase was observed when small doses of either neostigmine (0.1–1 μM), or galanthamine (10 and 20 μM) were used. However, this facilitation of evoked quantal release was reduced and suppressed when the concentration of neostigmine and galanthamine were increased (Figure 2) to levels where they block almost completely both cholinesterases (Table 2).
Figure 2.
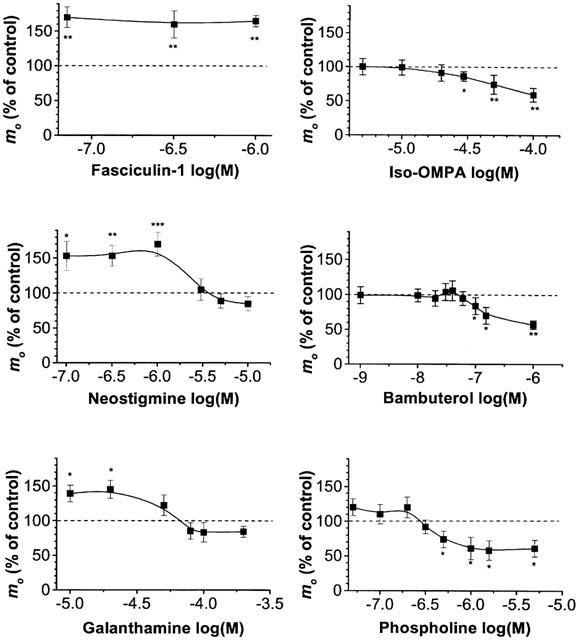
Dose-dependent effect of various cholinesterase inhibitors on the mean quantal content (mo) of EPPs in wild-type NMJs bathed in a physiological solution containing 0.4 mM Ca2+ and 6 mM Mg2+. Effects of inhibitors are given as per cent of the corresponding control mo values (determined before the application of drugs). Each point represents the mean±s.e.mean of n=5–9 separate experiments. Asterisks indicate significant changes in mo (*P<0.05; **P<0.03, ***P<0.001, Student's t-test) versus control values.
When preparations were pretreated with the irreversible BChE inhibitor iso-OMPA (30–100 μM), mo was significantly decreased up to 48% compared to controls (Figure 2). A similar dose-dependent decrease in mo was observed in the presence of 0.1–3 μM bambuterol (Figure 2). Next, we tested if mo changed when both AChE and BChE were irreversibly inhibited. The mo recorded in preparations pretreated with phospholine (0.005–5 μM) decreased depending on the dose used for the pretreatment (Figure 2). Similarly, in preparations pre-incubated with MTP (4 nM) the quantal content of EPP decreased by 51±4% (n=20 fibres from five different muscles) when compared to controls (P<0.01, Student's t-test).
Taken together, these results suggest that inhibition of AChE facilitates evoked quantal transmitter release, while inhibition of BChE alone, or of both AChE and BChE depresses transmitter release.
Effect of iso-OMPA pretreatment on the quantal content of EPPs in AChE−/− NMJs
The effect of BChE inhibition on mo was further tested in AChE−/− NMJs expressing only BChE (Xie et al., 2000; Li et al., 2000). In the low Ca-high Mg solution the mean quantal content of EPPs evoked at 0.3 Hz in AChE−/− NMJs was 1.13±0.42 (n=21 fibres from five different muscles). When AChE−/− NMJs were pretreated with iso-OMPA (100 μM) mo was reduced by 74% with respect to controls (0.29±0.1; n=12 fibres from three different muscles). This result shows that when AChE is absent inhibition of BChE depresses evoked neurotransmitter release and further suggests that presynaptic modulation of quantal transmitter release may depend on the activity of BChE.
Involvement of muscarinic ACh receptors in modulating evoked quantal ACh release
Muscarinic ACh receptors (mAChRs) are known to be involved in the regulation of quantal transmitter release from motor nerve terminals when AChE is inactivated (van der kloot & Molgó, 1994; Minic et al., 2002). Therefore, we tested whether the modulation in the quantal content of EPPs observed after AChE and/or BChE inhibition is mediated by mAChRs. For this purpose, the muscarinic toxin MT-3 was used. This toxin is also known as m-4 toxin, since it shows the highest specificity for M4 mAChR (Caulfield & Birdsall, 1998). However, it can block other mAChR subtypes when applied at higher concentrations. We used this fact and applied MT-3 at concentrations of 2 nM to block M4 mAChRs; 78 nM to block M4 and M1 receptors; and 1.52 μM to block all mAChRs (Jolkkonen et al., 1994).
As shown in Figure 3A, MT-3 had no statistically significant effect per se on the mean quantal content in wild-type NMJs in which both AChE and BChE are functional. However, depending on the concentration MT-3 reduced evoked transmitter release in AChE−/− NMJs (Figure 3B). A similar significant reduction in mo by MT-3 (0.078 and 1.52 μM) was observed in normal junctions treated with neostigmine (1 μM), galanthamine (20 μM) or fasciculin-1 (0.07 μM) (Figure 4A). The significant differences obtained with 0.078 μM MT-3, when compared to 0.002 μM suggests the involvement of M1 mAChR, but not of M4 mAChR, in the enhancement of quantal release when AChE is absent or inactivated.
Figure 3.
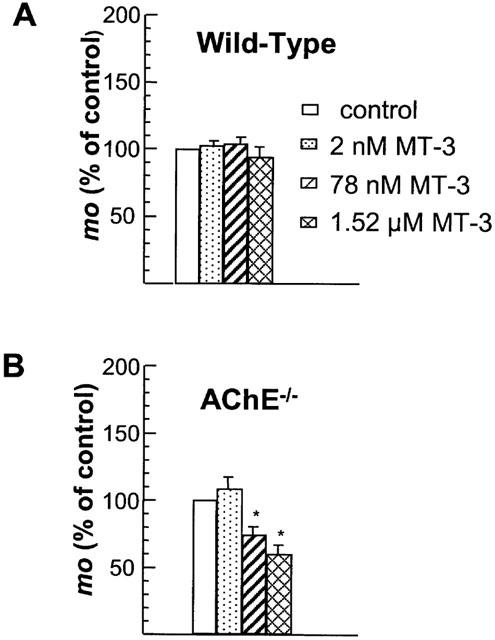
Effect of different concentrations of MT-3 on the mean quantal content (mo) of EPPs, in wild-type (A), and AChE−/− (B) NMJs bathed in a physiological solution containing 0.4 mM Ca2+ and 6 mM Mg2+. Note that MT-3 had a significant effect only in AChE−/− NMJs. Values are expressed as a percentage of the respective controls, and they represent the mean±s.e.mean, calculated from 4–9 separate experiments. Asterisks denote values significantly different from control values (*P<0.05, Student's t-test).
Figure 4.
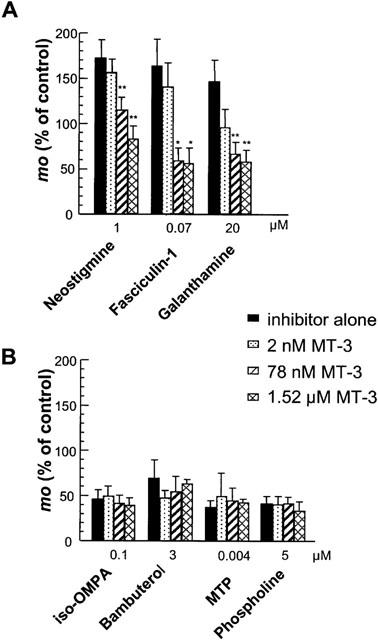
Effect of various concentrations of MT-3 on the mean quantal content (mo) of EPPs recorded in wild-type NMJs treated, or pretreated with various cholinesterase inhibitors (see Methods). Experiments were performed in standard saline containing 0.4 mM Ca2+ and 6 mM Mg2+. Values are expressed as percentage of the respective controls (determined before drug application). Note the absence of MT-3 effect on mo when BChE, and both BChE and AChE are inactivated. Each column represents the mean±s.e.mean, calculated from 4–9 separate experiments. Asterisks indicate significant changes in mo (*P<0.05; **P<0.03, Student's t-test) with respect to values obtained with the corresponding inhibitor alone.
MT-3 had no effect on the quantal content of EPPs in junctions in which BChE was inhibited by iso-OMPA or bambuterol (Figure 4B). Similarly MT-3 was also ineffective after both AChE and BChE were inhibited with the organophosphate compounds MTP and phospholine (Figure 4B). Thus, the modulation of quantal transmitter release by mAChRs appears to require depressed activity of AChE and/or BChE.
Effect of BChE inhibition on the time course and quantal content of full-sized EPPs in AChE−/− and normal NMJs
To determine whether BChE contributed to the hydrolysis of ACh in AChE−/− NMJs, experiments were performed to analyse the effect of iso-OMPA (100 μM) pretreatment on the time course of evoked EPPs (0.3 Hz) recorded in standard physiological solution containing 2.2 μM μ-conotoxin GIIIB. For comparison, parallel experiments were carried out in wild-type NMJs. As illustrated in Figure 5(A,B), EPPs recorded in AChE−/− NMJs exhibit a prolonged decay, as compared to EPPs recorded in wild-type junctions at similar resting membrane potentials. The time constant of decay (determined from 90–10% of EPP amplitude) had a mean value of 19.4±2.9 ms in AChE−/− NMJs (n=11 fibres from three muscles; resting membrane potential range −60 to −67 mV) and 2.6±0.7 ms in wild-type NMJs (n=21 fibres from five muscles; membrane potential range −64 to −72 mV). Although the time course of the EPP reflects the time constant of the muscle membrane, it is likely that the prolonged decay detected in AChE−/− NMJs is due to the lack of AChE which allows ACh to persist in the synaptic cleft, and to activate ACh receptors repetitively, as proposed by Katz & Miledi (1973). However, when BChE was inactivated by iso-OMPA the decay time constant of EPPs was not modified in both AChE−/− and wild-type NMJs. These results suggest that inactivation of BChE by iso-OMPA does not sustain the elevated concentration of ACh in the synaptic cleft, as the absence, or inhibition of AChE does.
Figure 5.
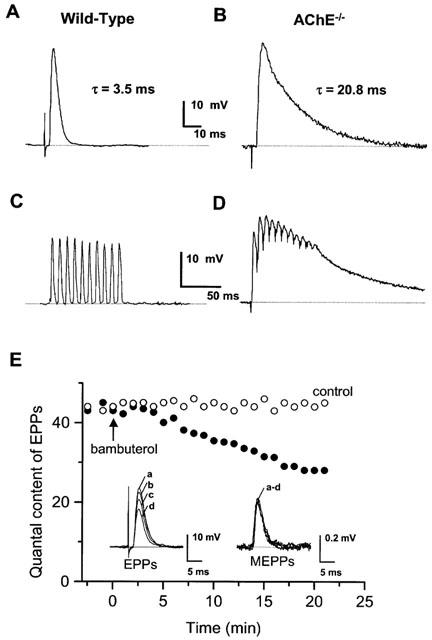
Examples of EPPs recorded in wild-type (A,C) and AChE−/− (B,D) NMJs bathed in normal physiological saline containing 2.2 μM μ-conotoxin GIIIB. EPPs evoked by single nerve stimulation (A,B), and during a train of 100 ms duration at 100 Hz (C,D). The decay time constant (τ) of EPPs is indicated for comparison in A and B. Data in A,C and B,D are from the same NMJs. The resting membrane potential during recordings were −65 mV (A,C) and −67 mV (B,D). (E) Quantal content of full-sized EPPs evoked at 0.1 Hz and recorded in a wild-type junction bathed in standard physiological solution containing μ-conotoxin GIIIB (2.2 μM) before, and after the addition (arrow) of 150 nM bambuterol to the medium (filled circles). For comparison data obtained in another junction during the same time period under control conditions (open circles) are also shown. Each circle represents the data computed during 1 min. The insets show averaged traces of EPPs and MEPPs recorded in the same junction before (a), and 5 (b), 10 (c), and 20 (d) min after the addition of bambuterol.
To test whether inhibition of BChE modifies the quantal content (m) of full-sized EPPs evoked at 0.3 Hz in the presence of 2.2 μM μ-conotoxin GIIIB, the amplitude of EPPs and MEPPs was determined in AChE−/− and wild-type NMJs before, and after pretreatment with 100 μM iso-OMPA. In AChE−/− NMJs such pretreatment significantly decreased m values from 48.0±3.2 to 36.4±2.5 (n=11 fibres, from three muscles; P<0.05, Student's t-test). Equivalent results were obtained in wild-type NMJs pretreated with iso-OMPA in which m decreased from 43.5±2.8 to 34.2±2.7 (n=21 fibres, from five muscles; P<0.05, Student's t-test). A similar decrease in m was observed in wild-type NMJs treated with 150 nM bambuterol, as shown in Figure 5E. Under these conditions, bambuterol decreased EPP amplitudes without affecting that of MEPPs (Figure 5E, insets).
The effects reported indicate that inhibition of BChE leads to a decrease in the mean number of quanta entering in the composition of full-sized EPPs.
Effect of BChE inhibition on EPPs evoked byhigh-frequency nerve stimulation
Further experiments using high-frequency nerve stimulation were carried out to examine whether BChE inhibition by iso-OMPA pretreatment affected synaptic transmission in AChE−/− and normal NMJs. For this, preparations were equilibrated for 1 h in standard solution containing either μ-conotoxin GIIIB, or (+)-tubocurarine and EPPs were evoked by trains of 10–100 Hz.
In AChE−/− neuromuscular preparations bathed in standard solution containing μ-conotoxin GIIIB (2.2 μM), repetitive phrenic nerve stimulation at frequencies higher than 20 Hz resulted in a staircase phenomenon. This was characterized by a decline in successive EPP peak-amplitudes and by a residual sustained depolarization of the junctional membrane that could attain about 10–15 mV at 100 Hz of nerve stimulation. The endplate depolarization was reversed upon cessation of nerve stimulation (Figure 5D), but remained unaffected by iso-OMPA pretreatment (data not shown).
In wild-type junctions, neither residual endplate depolarization nor the staircase phenomenon were observed during nerve stimulation at 100 Hz (Figure 5C). Furthermore, after iso-OMPA pretreatment (100 μM), no residual endplate depolarization was detected when the motor nerve was stimulated at 10–100 Hz, and the staircase phenomenon was not observed (data not shown).
In (+)-tubocurarine-treated AChE−/− junctions bathed with standard medium, EPPs exhibited a similar decline in amplitude upon nerve stimulation at 10, 20 and 40 Hz, as observed in normal NMJs under the same conditions (Figure 6). However, AChE−/− junctions were unable to maintain an effective transmission during a 1 s train of stimuli at 100 Hz, as illustrated in Figure 6A. Furthermore, the fall in EPP amplitudes was coincident with the ‘build-up' of a junctional depolarization of about 1–2 mV. The preincubation of wild-type or AChE−/− NMJs with iso-OMPA (100 μM) did not cause an additional EPP depression at all stimulation frequencies investigated (Figure 6A,B). These findings suggest that during high-frequency trains of nerve stimulation, the depression of EPP amplitudes is little affected by BChE activity.
Figure 6.
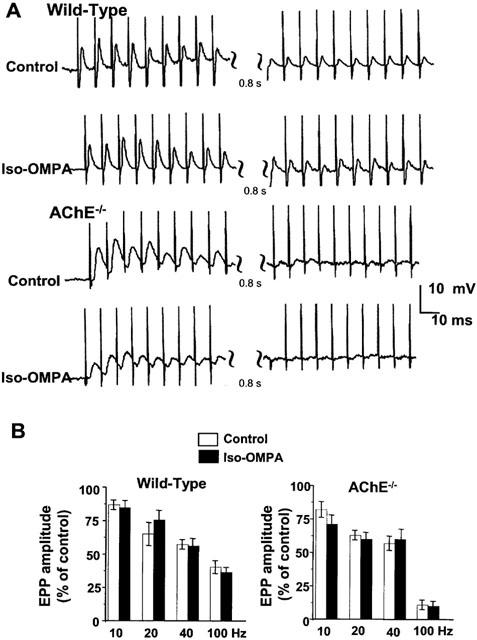
(A) Representative repetitive EPPs evoked during 1 s nerve stimulation at 100 Hz and recorded in wild type and AChE−/− NMJs bathed in normal physiological saline containing 3 μM (+)-tubocurarine. EPPs are shown at the beginning (left) and at the end of the train (right). Note the slight enhancement of EPP amplitudes at the beginning of stimulation and their reduction at the end of the train, and the fact that this pattern was unaffected by pretreatment with iso-OMPA (100 μM). Also note the inability of AChE−/− NMJs to sustain evoked EPPs at the end of the train. (B) Changes in EPP amplitudes during nerve stimulation, under control conditions (open bars) and after pretreatment with 100 μM iso-OMPA (filled bars) in wild-type and AChE−/− NMJs. Nerve stimuli were delivered at 10, 20 and 40 Hz for 10 s, and at 100 Hz for 1 s. For each set of data, the first and last EPPs in each train were computed, and results are expressed as per cent of the 1st EPP. Note that the depression of EPPs was unaffected by iso-OMPA pretreatment. Bars represent the mean±s.e.mean. (n=3–9).
Effects of iso-OMPA on MEPPs in normal andAChE−/− NMJs
Inactivation of AChE by inhibitors is known to change MEPPs (Bowman et al., 1986). Therefore, the effects of the inactivation of BChE and/or absence of AChE were examined by measuring the frequency, amplitude, and time course of MEPPs in hemidiaphragm preparations pretreated with iso-OMPA (100 μM). Analysis on MEPPs were performed in normal and AChE−/− NMJs bathed in standard physiological solution.
When MEPPs were recorded in AChE−/− NMJs, a striking feature was the presence of a high proportion of events with amplitudes 2–10 times higher than the modal value of MEPPs. In 25 junctions investigated, from five different muscles, the proportion of these giant MEPPs (G-MEPPs) varied from 7 to 16% of the total population. Spontaneous potentials recorded in AChE−/− NMJ are shown in Figure 7A to illustrate G-MEPPs.
Figure 7.
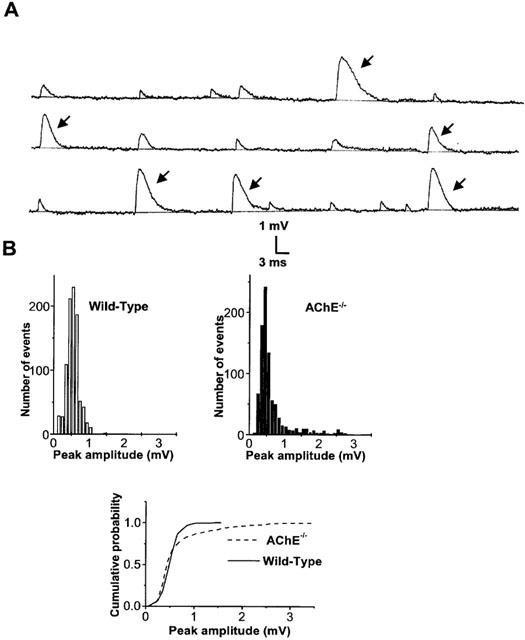
(A) Representative spontaneous MEPPs and G-MEPPs (arrows) recorded in an AChE−/− NMJ bathed in standard physiological solution. (B) Amplitude-distribution histograms and a cumulative plot of spontaneous events recorded in wild-type and AChE−/− NMJs. Note the different cumulative distribution of events in AChE−/− when compared to wild-type NMJs (P<0.001, Kolmogorov-Smirnov test) which is due to the presence of G-MEPPs.
Analysis of the amplitude distribution of MEPPs recorded in normal junctions revealed the presence of only a small proportion of G-MEPPs (1–3%) with respect to the total number of events recorded. Figure 7B shows peak-amplitude histograms and the cumulative probability distribution of MEPPs recorded in AChE−/− and wild-type junctions. Pretreatment of hemidiaphragm preparations with iso-OMPA (100 μM) failed to affect significantly the proportion of G-MEPPs either in wild-type (2.2±1.6% vs 1.5±0.6 for controls, n=4 different muscles, P>0.1) or in AChE−/− NMJs (10.5±3.6% vs 9.5±0.7 for controls, n=5 different muscles, P>0.1). In addition, when G-MEPPs were excluded from the analysis, MEPP amplitudes and the half-decay time of MEPPs were not significantly affected by iso-OMPA pretreatment either in wild-type, or in AChE−/− NMJs (Figure 8A,B).
Figure 8.
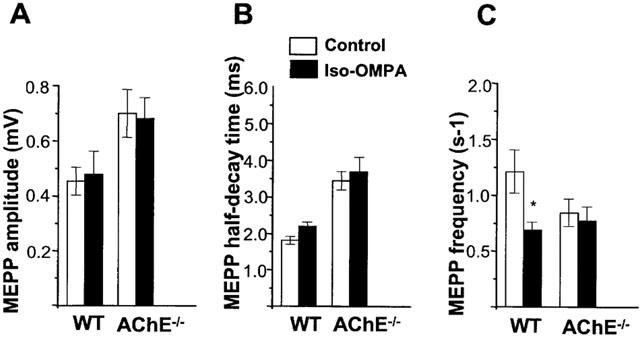
Effect of pretreatment with iso-OMPA (100 μM) on amplitude (A), half-decay time (B) and frequency (C) of MEPPs recorded in wild-type (WT) and AChE−/− NMJs. G-MEPPs were excluded for the calculation of the three parameters. Data were obtained from 21 fibres (six different WT muscles) and from 11 fibres (four different AChE−/− muscles) and represent the mean±s.e.mean (*P<0.05, Student's t-test). Resting membrane potential of the fibres was comprised between −70 and −67 mV for WT and AChE−/− NMJs.
In wild-type NMJs pretreatment with 100 μM iso-OMPA significantly decreased the frequency of MEPPs with respect to controls (Figure 8C). However, in AChE−/− NMJs the decrease in MEPP frequency was not significant after iso-OMPA pretreatment (Figure 8C).
Discussion
The aim of this study was to compare the effects of specific AChE and BChE inhibitors on quantal ACh release at normal and AChE−/− junctions in order to investigate the physiological role of BChE at the skeletal NMJ.
Although our histochemical and biochemical experiments provide evidence that both enzymes can hydrolyse ACh, and both are expressed at mature motor-endplates, their inhibition affects quantal transmitter release differently. We used various cholinesterase inhibitors, which have different molecular composition (organophosphates, carbamates, alkaloid, polypeptide), since most of the inhibitors are not rigorously specific for cholinesterases but also affect other targets. The results show that the molecular type of the inhibitors does not matter, it is their targets that matter. In low Ca2+/high Mg2+-blocked NMJs, inhibition of BChE by iso-OMPA or bambuterol reduced quantal ACh release. In contrast, inhibition of AChE by low concentrations of fasciculin-1, neostigmine or galanthamine enhanced release. It has been reported that neostigmine blocks K+ channels in motor nerve terminals (Braga et al., 1993), and galanthamine is known to allostericaly potentiate neuronal nicotinic ACh receptors in mammalian central nervous system (Santos et al., 2002), and these actions may contribute to facilitate ACh release. However, in higher concentrations, neostigmine and galanthamine also inhibited BChE and decreased quantal ACh release. A similar decrease was observed with MTP and phospholine which irreversibly inhibited both AChE and BChE. Our results show that when only a few ACh quanta are released following nerve stimulation all inhibitors of BChE tested reduced evoked ACh release which suggests the involvement of BChE in a presynaptic modulatory step of the release process.
In AChE−/− NMJs, the absence of AChE enhanced the quantal content of EPPs when compared to wild-type NMJs bathed in the same low Ca2+/high Mg2+ medium and the inhibition of BChE by iso-OMPA strongly reduced mo values. A similar increase in mo values has been previously reported in NMJs which lack AChE from ColQ-deficient mice (Minic et al., 2002).
When the probability of quantal release was high, as in junctions bathed in standard medium containing μ-conotoxin GIIIB (to record full-sized EPPs without being disturbed by muscle action potentials and contraction), inactivation of BChE decreased m values in both wild-type and AChE−/− NMJs.
Experiments performed in standard medium withμ-conotoxin GIIIB, revealed that inhibition of BChE did not affect the time course of EPPs in both wild-type and AChE−/− NMJs. These findings suggest that BChE inhibition does not lead to increased ACh levels in the synaptic cleft. Furthermore, during high-frequency nerve stimulation, inhibition of BChE failed to cause the so-called staircase phenomenon that was evident in AChE−/− NMJs, and that has also been reported for acute organophosphate poisoning (Maselli & Soliven, 1991). The staircase phenomenon observed in AChE−/− NMJ was markedly reduced by (+)-tubocurarine, which indicates that in the absence of AChE activity this effect was caused by constant stimulation of post-synaptic nicotinic ACh receptors.
It has been proposed that during high frequency nerve stimulation the diminution of EPP amplitude (or run-down) becomes enhanced in the presence of (+)-tubocurarine due to its inhibitory action on presynaptic nicotinic receptors (Tian et al., 1994). In (+)-tubocurarine treated AChE−/− NMJs, the decrease in EPP amplitudes at high frequency (100 Hz) nerve stimulation was still more marked than in wild-type NMJs. This enhancement in run-down may be caused by negative feedback action of ACh that accumulated in the synaptic cleft (Chang et al., 1990). It is also possible that high ACh concentration may modulate postsynaptic nicotinic ACh receptor responses, as observed for agonist induced potentiation and desensitisation, after inactivation of AChE (Feltz & Trautmann, 1980). However, the run-down of EPPs was little affected after BChE inhibition in both wild-type and AChE−/− NMJs, suggesting that in (+)-tubocurarine-treated junctions BChE inactivity did not additionally increase ACh concentration in the synaptic cleft. Moreover, BChE inhibition did not affect the amplitude and the time course of spontaneous MEPPs in wild-type and AChE−/− NMJs. These results strongly suggest that BChE inhibition does not prolong the ACh action on endplate receptors. It should be noted that in AChE−/− mice the activity of BChE at the NMJ is not significantly different from that found in wild-type junctions (Li et al., 2000).
Cholinesterase inhibitors are known to increase the proportion of G-MEPPs in normal NMJs (Molgó & Thesleff, 1982; Thesleff et al., 1990; Maselli & Soliven, 1991). This is in agreement with a high proportion of G-MEPPs observed in AChE−/− NMJs. In contrast, iso-OMPA did not enhance the proportion of G-MEPPs. The only effect on spontaneous events detected at normal NMJs after BChE inhibition was a reduction in MEPP frequency. A similar reduction in the frequency of MEPPs with iso-OMPA has been previously reported in frog NMJs (Duncan & Publicover, 1979). However, in AChE−/− NMJs the frequency of MEPPs remained unaffected by iso-OMPA pretreatment.
Although AChE has a central role in cholinergic transmission, its absence in AChE−/− mice resulted in a very mild phenotype (Xie et al., 2000; Duysen et al., 2002). Some of the animal characteristics, such as muscle weakness, have also been reported in endplate AChE deficient humans (Ohno et al., 2000), in ColQ−/− mice (Feng et al., 1999; and results unpublished), and are typically observed during poisoning by AChE inhibitors. The enhancement in the quantal content of EPPs in NMJs of adult AChE−/− mice may contribute to the animal's adaptation to the loss of AChE activity and to the survival process. However, complementary studies (that will be published elsewhere) indicate that there is an important remodelling of the pre- and post-synaptic constitutive elements of the NMJ, which either adapt or compensate for the absence of AChE catalytic activity.
During inhibition of AChE, the enhancement or depression of quantal ACh release has been attributed to the activation of presynaptic receptors during the delayed diffusion of ACh molecules in the synaptic cleft (see reviews by Bowman et al., 1990; van der kloot & Molgó, 1994). We recently reported the involvement of M1 mAChR in the enhancement of quantal release in ColQ-deficient NMJs, and in wild-type NMJs during inhibition of AChE by fasciculin-2 (Minic et al., 2001; 2002). In the present study we observed that enhancement of quantal release during AChE inhibition by low concentrations of neostigmine, fasciculin-1 and galanthamine was antagonized by MT-3, which by itself had no effect when AChE is active. MT-3 also decreased the quantal content in AChE−/− NMJs. These results suggest the implication of mAChRs in the facilitatory effect detected during the inhibition or absence of AChE. Interestingly, MT-3 failed to antagonize the decrease in the mean quantal content of EPPs caused by iso-OMPA, bambuterol, MTP and phospholine. This raises the question why after the action of those inhibitors there is no regulation of evoked transmitter release by mAChRs? One possibility is that mAChRs are inactivated due to a direct interaction with the inhibitors, so that they can no longer respond to MT-3. Another possibility is that organophosphate or carbamate inhibitors decrease the number of mAChRs available in the membrane, as it has been recently reported for the M2 and M4 mAChRs in rat striatal neurones following treatment with various cholinesterase inhibitors (Liste et al., 2002).
In conclusion, our results indicate that the different effects of cholinesterase inhibitors on quantal transmitter release are related to their specificity for AChE and BChE. BChE is apparently not involved in regulating the duration of ACh action at the post-synaptic membrane which suggests that it is unlikely that BChE acts simply as a backup for AChE in hydrolysing ACh in either normal or AChE−/− isolated neuromuscular preparations. However, inhibitors of BChE decreased evoked quantal transmitter release, suggesting that BChE is involved in a presynaptic modulatory step of the release process and can play a constitutive physiological role at mature NMJs.
Acknowledgments
We thank Drs E. Karlsson, M. Israël, C. Thal and F. Leterrier for providing some of the drugs used and Drs W. Van der Kloot and J. Massoulié for critical reading of the manuscript. We thank Prof. O. Lockridge for the gift of the knockout AChE founders and constant support. We are grateful to Miss L. Faille for histochemical assistance, and Miss P. Villeneuve and Miss L. Lepourry for taking care of the animals. This research was supported in part by the Association Française contre les Myopathies and by C.N.R.S. J. Minic was supported by fellowships from the Association Française contre les Myopathies and the Fondation pour la Recherche Médicale.
Abbreviations
- ACh
acetylcholine
- AChE
acetylcholinesterase
- BChE
butyrylcholinesterase
- BW284C51
1,5-bis(4-allyldimethylammoniumphenyl) pentan-3-one
- DTNB
5,5′-dithiobis(2-nitrobenzoic acid)
- EPP
endplate potential
- G-MEPP
giant miniature endplate potential
- iso-OMPA
tetraisopropylpyrophosphoramide
- m
quantal content of endplate potentials calculated by the direct method
- MEPP
miniature endplate potential
- mo
mean quantal content of endplate potentials calculated by the method of failures
- MT-3
muscarinic toxin-3
- MTP
O-ethylS-2-(diisopropylamino)ethylmethylphos-phonothioate
- NMJ
neuromuscular junction
References
- ADLER M., FILBERT M.G. Role of butyrylcholinesterase in canine tracheal smooth muscle function. FEBS Lett. 1990;267:107–110. doi: 10.1016/0014-5793(90)80300-8. [DOI] [PubMed] [Google Scholar]
- ATACK J.R., YU Q.S., SONCRANT T.T., BROSSI A., RAPOPORT S.I. Comparative inhibitory effects of various physostigmine analogs against acetyl- and butyrylcholinesterases. J. Pharmacol. Exp. Ther. 1989;249:194–202. [PubMed] [Google Scholar]
- BEHRA M., COUSIN X., BERTRAND C., VONESCH J.L., BIELLMANN D., CHATONNET A., STRÄHLE U. Acetylcholinesterase is required for neuronal and muscular development in the zebrafish embryo. Nat. Neurosci. 2002;5:111–118. doi: 10.1038/nn788. [DOI] [PubMed] [Google Scholar]
- BOWMAN W.C., GIBB A.J., HARVEY A.L., MARSHALL I.G.Prejunctional actions of cholinoceptor agonist and antagonist and of anticholinesterase drugs Handbook of Experimental Pharmacology, Vol. 79, New neuromuscular Blocking Agents 1986Berlin: Springer-Verlag; 141–170.ed. Kharkevich, D. [Google Scholar]
- BOWMAN W.C., PRIOR C., MARSHALL I.G. Presynaptic receptors in the neuromuscular junction. Ann. N.Y. Acad. Sci. 1990;604:69–81. doi: 10.1111/j.1749-6632.1990.tb31983.x. [DOI] [PubMed] [Google Scholar]
- BRAGA M.F., ROWAN E.G., HARVEY A.L., BOWMAN W.C. Prejunctional action of neostigmine on mouse neuromuscular preparations. Br. J. Anaesth. 1993;70:405–410. doi: 10.1093/bja/70.4.405. [DOI] [PubMed] [Google Scholar]
- BERMAN H.A., DECKER M.M., JO S. Reciprocal regulation of acetylcholinesterase and butyrylcholinesterase in mammalian skeletal muscle. Dev. Biol. 1987;120:154–161. doi: 10.1016/0012-1606(87)90113-8. [DOI] [PubMed] [Google Scholar]
- CAULFIELD M.P., BIRDSALL N.M.J. Classification of muscarine acetylcholine receptors. Pharmacol. Rev. 1998;50:279–290. [PubMed] [Google Scholar]
- CHANG C.C., HUANG C.Y., HONG S.J. Organic calcium channel antagonists provoke acetylcholine receptor autodesensitization on train stimulation of motor nerve. Neuroscience. 1990;38:731–742. doi: 10.1016/0306-4522(90)90066-d. [DOI] [PubMed] [Google Scholar]
- CHATONNET A., LOCKRIDGE O. Comparison of butyrylcholinesterase and acetylcholinesterase. Biochem. J. 1989;260:625–634. doi: 10.1042/bj2600625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DUNCAN C.J., PUBLICOVER S.J. Inhibitory effects of cholinergic agents on the release of transmitter at the frog neuromuscular junction. J. Physiol. 1979;294:91–103. doi: 10.1113/jphysiol.1979.sp012917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DUYSEN E.G., STRIBLEY J.A., FRY D.L., HINRICHS S.H., LOCKRIDGE O. Rescue of the acetylcholinesterase knockout mouse by feeding a liquid diet; phenotype of the adult acetylcholinesterase deficient mouse. Dev. Brain Res. 2002;137:43–54. doi: 10.1016/s0165-3806(02)00367-x. [DOI] [PubMed] [Google Scholar]
- ELLMAN G.L., COURTNEY D.K., ANDRES V., FEATHERSTONE R.M. A new and rapid colorimentric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961;7:88–95. doi: 10.1016/0006-2952(61)90145-9. [DOI] [PubMed] [Google Scholar]
- FELTZ A., TRAUTMANN A. Interaction between nerve-related acetylcholine and bath applied agonists at the frog end-plate. J. Physiol. 1980;299:533–552. doi: 10.1113/jphysiol.1980.sp013141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FENG G., KREJCI E., MOLGÓ J., CUNNINGHAM J.M., MASSOULIÉ J., SANES J.R. Genetic analysis of collagen Q: roles in acetylcholinesterase and butyrylcholinesterase assembly and in synaptic structure and function. J. Cell Biol. 1999;144:1349–1360. doi: 10.1083/jcb.144.6.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GAUTRON J. Cytochimie ultrastructurale des acétyl-cholinestérases. J. Microscopie. 1974;21:259–264. [Google Scholar]
- JOLKKONEN M., VAN GIERSBERGEN P.L., HELLMAN U., WERNSTEDT C., KARLSSON E. A toxin from the green mamba Dendroaspis angusticeps: amino acid sequence and selectivity for muscarinic m4 receptors. FEBS Lett. 1994;352:91–94. doi: 10.1016/0014-5793(94)00933-3. [DOI] [PubMed] [Google Scholar]
- KATZ B., MILEDI R. Binding of acetylcholine to receptors and its removal from the synaptic cleft. J. Physiol. 1973;231:549–574. doi: 10.1113/jphysiol.1973.sp010248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KOELLE G.B. The histochemical differentiation of types of cholinesterases and their localization in tissues of the cat. J. Pharmacol. Exp. Ther. 1950;100:158–179. [PubMed] [Google Scholar]
- KOELLE G.B., HORN R.S. Acetyl disulfide, (CH3COS)2, a major active component in the thiolacetic acid histochemical method for acetylcholinesterase. J. Histochem. Cytochem. 1968;16:743–753. doi: 10.1177/16.12.743. [DOI] [PubMed] [Google Scholar]
- LAYER P.G., WILLBOLD E. Novel functions of cholinesterases in development, physiology and disease. Prog. Histochem. Cytochem. 1995;29:1–94. doi: 10.1016/s0079-6336(11)80046-x. [DOI] [PubMed] [Google Scholar]
- LI B., STRIBLEY J.A., TICU A., XIE W., SCHOPFER L.M., HAMMOND P., BRIMIJOIN S., HINRICHS S.H., LOCKRIDGE O. Abundant tissue butyrylcholinesterase and its possible function in the acetylcholinesterase knockout mouse. J. Neurochem. 2000;75:1320–1331. doi: 10.1046/j.1471-4159.2000.751320.x. [DOI] [PubMed] [Google Scholar]
- LISTE I., BERNARD V., BLOCK B. Acute and chronic acetylcholinesterase inhibition regulates in vivo the localization and abundance of muscarinic receptors m2 and m4 at the cell surface and in the cytoplasm of striatal neurons. Mol. Cell. Neurosci. 2002;20:244–256. doi: 10.1006/mcne.2001.1083. [DOI] [PubMed] [Google Scholar]
- MASELLI R.A., SOLIVEN B.C. Analysis of the organophosphate-induced electromyographic response to repetitive nerve stimulation; paradoxical response to edrophonium and D-tubocurarine. Muscle Nerve. 1991;14:1182–1188. doi: 10.1002/mus.880141207. [DOI] [PubMed] [Google Scholar]
- MASSOULIÉ J., PEZZEMENTI L., BON S., KREJCI E., VALLETTE F.M. Molecular and cellular biology of cholinesterases. Prog. Neurobiol. 1993;41:31–91. doi: 10.1016/0301-0082(93)90040-y. [DOI] [PubMed] [Google Scholar]
- MCLACHLAN E.M., MARTIN A.R. Non-linear summation of endplate potentials in the frog and mouse. J. Physiol. 1981;311:307–324. doi: 10.1113/jphysiol.1981.sp013586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MINIC J., MOLGÓ J., KARLSSON E., KREJCI E. Muscarinic receptors and the regulation of acetylcholine release at the mouse neuromuscular junction. J. Physiol. 2001;536 Suppl.:P125. doi: 10.1046/j.0953-816x.2001.01875.x. [DOI] [PubMed] [Google Scholar]
- MINIC J., MOLGÓ J., KARLSSON E., KREJCI E. Regulation of acetylcholine release by muscarinic receptors at the mouse neuromuscular junction depends on the activity of acetylcholinesterase. Eur. J. Neurosci. 2002;15:439–448. doi: 10.1046/j.0953-816x.2001.01875.x. [DOI] [PubMed] [Google Scholar]
- MOLGÓ J., THESLEFF S. 4-aminoquinoline-induced ‘giant' miniature endplate potentials at mammalian neuromuscular junctions. Proc. R. Soc. Lond. B Biol. Sci. 1982;214:229–244. doi: 10.1098/rspb.1982.0006. [DOI] [PubMed] [Google Scholar]
- NOREL X., ANGRISANI M., LABAT C., GORENNE I., DULMET E., ROSSI F., BRINK C. Degradation of acetylcholine in human airways: role of butyrylcholinesterase. Br. J. Pharmacol. 1993;108:914–919. doi: 10.1111/j.1476-5381.1993.tb13486.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OHNO K., ENGEL A.G., BRENGMAN J.M., SHEN X.M., HEIDENREICH F., VINCENT A., MILONE M., TAN E., DEMIRICI M., WALSH P., NAKANO S., AKIGUCHI I. The spectrum of mutations causing end-plate acetylcholinesterase deficiency. Ann. Neurol. 2000;47:162–170. [PubMed] [Google Scholar]
- PRIMO-PARMO S.L., BARTELS C.F., WIERSEMA B., VAN DER SPEK A.F., INNIS J.W., LA DU, B.N. Characterization of 12 silent alleles of the human butyrylcholinesterase (BCHE) gene. Am. J. Hum. Genet. 1996;58:52–64. [PMC free article] [PubMed] [Google Scholar]
- SANTOS M.D., ALKONDON M., PEREIRA E.F., ARACAVA Y., EISENBERG H.M., MAELICKE A., ALBUQUERQUE E.X. The nicotinic allosteric potentiating ligand galantamine facilitates synaptic transmission in the mammalian central nervous system. Mol. Pharmacol. 2002;61:1222–1234. doi: 10.1124/mol.61.5.1222. [DOI] [PubMed] [Google Scholar]
- SILMAN I., DI GIAMBERERDINO L., LYLES L., COURAUD E.A. Parallel regulation of acetylcholinesterase and pseudocholinesterase in normal, denervated and dystrophic chicken skeletal muscle. Nature. 1979;280:160–162. doi: 10.1038/280160a0. [DOI] [PubMed] [Google Scholar]
- TAYLOR P. The cholinesterases. J. Biol. Chem. 1991;266:4025–4028. [PubMed] [Google Scholar]
- TIAN L., PRIOR C., DEMPSTER J., MARSHALL I.G. Nicotinic antagonist-produced frequency-dependent changes in acetylcholine release from rat motor nerve terminals. J. Physiol. 1994;476:517–529. doi: 10.1113/jphysiol.1994.sp020151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- THESLEFF S., SELLIN L.C., TAGERUD S. Tetrahydroaminoacridine (tacrine) stimulates neurosection at mammalian motor endplates. Br. J. Pharmacol. 1990;100:487–490. doi: 10.1111/j.1476-5381.1990.tb15834.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VAN DER KLOOT W., MOLGÓ J. Quantal acetylcholine release at the vertebrate neuromuscular junction. Physiol. Rev. 1994;74:899–991. doi: 10.1152/physrev.1994.74.4.899. [DOI] [PubMed] [Google Scholar]
- VIGNY M., BON S., MASSOULIÉ J., LETERRIER F. Active-site catalytic efficiency of acetylcholinesterase molecular forms in Electrophorus, torpedo, rat and chicken. Eur. J. Biochem. 1978;85:317–323. doi: 10.1111/j.1432-1033.1978.tb12241.x. [DOI] [PubMed] [Google Scholar]
- WALCH L., TAISNER C., GASCARD J.P., NASHASHIBI N., BRINK C., NOREL X. Cholinesterase activity in human pulmonary arteries and veins. Br. J. Pharmacol. 1997;121:986–990. doi: 10.1038/sj.bjp.0700158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- XIE W., STRIBLEY J.A., CHATONNET A., WILDER P.J., RIZZINO A., MCCOMB R.D., TAYLOR P., HINRICHS S.H., LOCKRIDGE O. Postnatal developmental delay and supersensitivity to organophosphate in gene-targeted mice lacking acetylcholinesterase. J. Pharmacol. Exp. Ther. 2000;293:896–902. [PubMed] [Google Scholar]