

Abstract
This study investigated the effects of fetal treatment with dexamethasone on ovine fetal cardiovascular defence responses to acute hypoxaemia, occurring either during or 48 h following the period of glucocorticoid exposure. To address the mechanisms underlying these responses, chemoreflex function and plasma concentrations of catecholamines, neuropeptide Y (NPY) and vasopressin were measured. Under general halothane anaesthesia, 26 Welsh Mountain sheep fetuses were surgically prepared for long-term recording at between 117 and 120 days of gestation (dGA; term is ∼145 days) with vascular catheters and a Transonic flow probe around a femoral artery. Following at least 5 days of recovery, fetuses were randomly assigned to one of two experimental groups. After 48 h of baseline recording, at 125 ± 1 dGA, half of the fetuses (n = 13) were continuously infused i.v. with dexamethasone for 48 h at a rate of 2.06 ± 0.13 μg kg−1 h−1. The remaining 13 fetuses were infused with heparinized saline at the same rate (controls). At 127 ± 1 dGA, 2 days from the onset of infusions, seven fetuses from each group were subjected to 1 h of acute hypoxaemia. At 129 ± 1 dGA, 2 days after the end of infusions, six fetuses from each group were subjected to 1 h of acute hypoxaemia. Similar reductions in fetal partial pressure of arterial oxygen occurred in control and dexamethasone-treated fetuses during the acute hypoxaemia protocols. In control fetuses, acute hypoxaemia led to transient bradycardia, femoral vasoconstriction and significant increases in plasma concentrations of catecholamines, vasopressin and NPY. In fetuses subjected to acute hypoxaemia during dexamethasone treatment, the increase in plasma NPY was enhanced, the bradycardic response was prolonged, and the plasma catecholamine and vasopressin responses were diminished. In fetuses subjected to acute hypoxaemia 48 h following dexamethasone treatment, femoral vasoconstriction and plasma catecholamine and vasopressin responses were enhanced, whilst the prolonged bradycardia and augmented plasma NPY responses persisted. These data show that fetal treatment with dexamethasone modifies the pattern and magnitude of fetal cardiovascular responses to acute oxygen deprivation. Modifications to different mechanisms mediating the fetal defence responses to acute hypoxaemia that occur during dexamethasone treatment may reverse, persist or even become enhanced by 48 h following the treatment period.
In the mammalian fetus, episodes of acute hypoxaemia elicit integrated cardiovascular responses that facilitate fetal survival during the period of reduced oxygen availability (Giussani et al. 1994a). The ovine fetal cardiovascular responses to a 1 h episode of acute hypoxaemia have been well characterized at 0.8–0.9 of gestation and include transient bradycardia, an increase in arterial blood pressure (Boddy et al. 1974; Giussani et al. 1993) and redistribution of the combined ventricular output in favour of the adrenal, myocardial and cerebral circulations at the expense of perfusion of peripheral vascular beds (Cohn et al. 1974). The initial bradycardia and increase in peripheral vascular resistance involve activation of the parasympathetic and sympathetic nervous systems, which is triggered by carotid, but not aortic, chemoreflexes (Bartelds et al. 1993; Giussani et al. 1993). Early in hypoxaemia, activation of the parasympathetic nervous system leads to an increase in vagal tone that reduces the fetal heart rate (Parer, 1984; Giussani et al. 1993). Activation of the sympathetic nervous system results in increased peripheral vascular tone, including femoral vasoconstriction, which is mediated by α-adrenergic efferents (Reuss et al. 1982) and is used as a good index of the overall redistribution of blood flow away from the peripheral vascular beds (Giussani et al. 1996). Once initiated, neurally mediated fetal peripheral vasoconstriction is maintained during hypoxaemia by increased release into the fetal circulation of vasoconstrictor agents such as catecholamines, neuropeptide Y (NPY) and arginine vasopressin (AVP; Alexander et al. 1974; Jones et al. 1988; Fletcher et al. 2000a). The increased concentrations of catecholamines in fetal plasma also lead to increased activation of cardiac β-adrenoceptors, opposing the enhanced vagal tone and returning the fetal heart rate back to normoxic levels by the end of the hypoxaemic challenge (Court et al. 1984).
Synthetic glucocorticoids, such as dexamethasone and betamethasone, are used in human clinical practice to treat pregnancies at risk of preterm delivery, maturing the fetus and reducing the incidence of neonatal respiratory distress syndrome, intraventricular haemorrhage and necrotising enterocolitis (Ballard & Ballard, 1995). Recently, it has been shown that fetal treatment with dexamethasone, producing plasma concentrations in the sheep fetus that are approximately one-fifth of the concentration achieved clinically in human infants, modifies basal cardiovascular physiology in the fetus and alters fetal cardiac baroreflex function (Fletcher et al. 2002). However, to date, no study has investigated the effects of fetal exposure to glucocorticoids on chemoreflex function or the fetal cardiovascular defence to acute hypoxaemia. This is important because episodes of acute hypoxaemia are common in utero, particularly during labour and delivery (Huch et al. 1977). In clinical practice, glucocorticoids are administered to pregnant women who are at threat of preterm delivery (Ballard & Ballard, 1995), and since glucocorticoids pass freely through the placenta into the fetal circulation (Bayard et al. 1972), it is very likely that episodes of fetal hypoxaemia will occur during, or shortly following, fetal exposure to synthetic glucocorticoids.
In human obstetric practice, clinical dosing regimens of glucocorticoids involve maternal intramuscular injections with either dexamethasone or betamethasone (NIH Consensus Development Conference, 1994) that expose the human fetus to initially high, but then rapidly decreasing concentrations of synthetic steroid (Ballard & Ballard, 1995). Recently, an experimental model has been developed in which fetal sheep are exposed directly to continuous intravenous infusions of dexamethasone during a 48 h treatment period (Derks et al. 1997; Fletcher et al. 2000b). This model allows assessment of the direct effects of sustained exposure to synthetic glucocorticoid on the fetus without additional confounding influences of glucocorticoid-induced changes in maternal and placental physiology (Wintour et al. 1994; Bennet et al. 1999).
Therefore, the aim of the current study was to determine, in sheep, the effects of direct fetal treatment with dexamethasone on fetal cardiovascular responses to an episode of acute hypoxaemia occurring either during or 48 h following the period of glucocorticoid exposure. To address the effects of glucocorticoids on the neural and endocrine mechanisms mediating the fetal cardiovascular responses to acute hypoxaemia, the effects of dexamethasone on fetal chemoreflex function and plasma concentrations of catecholamines, NPY and vasopressin were also determined.
Methods
Surgical preparation and postsurgical management
All surgical and experimental procedures were performed under the UK Animals (Scientific Procedures) Act 1986. In brief, between 117 and 120 days of gestation (dGA; term is ∼145 days), 26 Welsh Mountain sheep fetuses and their mothers were surgically prepared for long-term recording under general anaesthesia, as described previously in detail (Fletcher et al. 2000a). Food, but not water, was withheld from the ewes for 24 h prior to surgery. Following induction with 20 mg kg−1i.v. sodium thiopentone (Intraval Sodium; Rhone Mérieux, Dublin, Ireland), general anaesthesia (1.5–2.0 % halothane in 50:50 oxygen/nitrous oxide) was maintained using positive-pressure ventilation. A Teflon catheter was inserted into a maternal femoral artery and advanced into the maternal caudal aorta. A lower abdominal midline incision was made and the gravid uterus was exposed. Fetal instrumentation was achieved in two stages. The first uterine incision exposed the fetal head, and translucent PVC catheters (i.d. 0.58 or 0.86 mm; o.d. 0.96 or 1.52 mm, respectively; Critchly Electrical Products, NSW, Australia) were inserted into a fetal carotid artery and jugular vein. The second uterine incision exposed the fetal hindlimbs, and catheters were inserted 4–5 cm into a femoral artery and a femoral vein. An ultrasonic flow transducer (2R or 3S; Transonic Systems, Ithaca, NY, USA) was positioned around the contralateral fetal femoral artery. Another catheter was anchored to the hindlimb for measurement of amniotic cavity pressure. The uterine incisions were closed in layers, catheters were filled with heparinized saline (80 i.u. heparin ml−1 in 0.9 % NaCl) and all catheters and flow probe leads were exteriorized via a small incision in the maternal flank.
Ewes were housed in individual pens, had free access to water and hay, were fed concentrates twice daily (100 g; Sheep Nuts #6; H&C Beart, Kings Lynn, UK), and generally resumed normal feeding patterns within 24 h of surgery. The ewes received 2 days of postoperative analgesia (3 g daily oral phenylbutazone; Equipalozone Paste E-pp, Arnolds Veterinary Products, Shropshire, UK) if required. Antibiotics were administered daily to the ewe (0.20–0.25 mg kg−1i.m. Depocillin; Mycofarm, Cambridge, UK), to the fetus (150 mg kg−1i.v. ampicillin, Penbritin; SmithKline Beecham Animal Health, Surrey, UK) and into the amniotic cavity (300 mg Penbritin). Daily maternal aortic and fetal carotid blood samples (0.4 ml) were taken for analyses of blood gas and acid/base status. Vascular catheters were maintained patent by a slow continuous infusion (0.1 ml h−1) of heparinized saline.
Pressure transducers (COBE; Argon, Texas, USA) were attached to the fetal femoral artery and amniotic cavity catheters. Femoral blood flow (FBF) was monitored via a Transonic T201/T206 monitor (Transonic Systems, Ithaca, NY, USA). A fetal heart-rate meter was triggered from the arterial pressure or FBF pulse.
Treatment groups
Following at least 5 days of postoperative recovery, fetuses were randomly assigned to one of two experimental groups. After 48 h of baseline recording, at 125 ± 1 days, 13 fetuses (six males and seven females) were continuously infused i.v. with dexamethasone (dexamethasone sodium phosphate; Merck, Sharp, Dohme, Herts, UK) for 48 h at a rate of 2.06 ± 0.13 μg kg−1 h−1 (corrected retrospectively for fetal body weight obtained at post-mortem). The remaining fetuses (n = 13; seven males and six females) were infused with heparinized saline at the same rate (0.5 ml h−1) in order to act as age- and sex-matched controls. Maternal aortic and fetal carotid blood samples were taken daily for analysis of plasma dexamethasone concentrations during the experimental period. Samples were collected daily at 10.00 h on the two days before the infusion began (days −1 and 0), on the 2 days of infusion (days 1 and 2) and on the two days after infusion (days 3 and 4). At 127 ± 1 dGA, 45 h from the onset of infusions, seven fetuses from each group (saline: four males, three females; dexamethasone: three males, four females) were subjected to the acute hypoxaemia protocol. At 129 ± 1 dGA, 48 h after the end of the infusion period, six fetuses in each group (saline: three males, three females; dexamethasone: three males, three females) were subjected to the acute hypoxaemia protocol. The study was conducted over two experimental seasons. The experiments involving acute hypoxaemia occurring during the period of infusions were conducted in the first year, whilst those involving acute hypoxaemia following infusions were conducted in the second year.
Acute hypoxaemia protocol
Fetal hypoxaemia was induced by means of reducing the maternal inspired oxygen fraction. The challenge was based on a 3 h protocol: 1 h of normoxia; 1 h of hypoxia; 1 h of recovery (Giussani et al. 1993). At the start of the first hour, a large transparent, respiratory hood was placed over the ewe's head and air was passed through it at a rate of ∼40 l min−1. Following this control period of normoxia, fetal hypoxaemia was induced for 1 h by switching the gas mixture breathed by the ewe to 9 % oxygen in nitrogen (18 l min−1 air; 22 l min−1 nitrogen) with small amounts of carbon dioxide (1–2 l min−1) added to the inspirate in order to maintain fetal Pa,CO2 constant. This gas mixture was designed to reduce fetal Pa,O2 to within 12–15 mmHg. Following the 1 h period of hypoxaemia, the hood was removed and the ewe was allowed to breathe room air for the recovery period.
Calibrated mean femoral arterial and amniotic cavity pressures, fetal heart rate and mean FBF were recorded continually at 1 s intervals throughout the acute hypoxaemia protocol using a computerized Data Acquisition System (Cornell University; Ithaca, NY, USA).
Paired maternal aortic and fetal carotid blood samples (5 ml) were taken every 30 min starting from 15 min of normoxia, for measurement of blood gases, pH and plasma hormone concentrations. In addition, a fetal carotid blood sample was taken 5 min after the onset of hypoxaemia to confirm that a rapid and appropriate fall in fetal arterial partial pressure of oxygen (Pa,O2) had occurred. In the fetuses in which acute hypoxaemia was induced at 48 h following the end of infusions, additional femoral arterial blood samples (0.4 ml) were taken every 30 min starting from 15 min of normoxia, for determination of femoral arterial oxygen content, oxygen carrying capacity and oxygen delivery.
At the end of the experimental protocol, ewes and their fetuses were killed with a lethal dose of sodium pentobarbitone (40 mg kg−1i.v. Pentoject; Animalcare, York, UK). Fetuses were delivered via an abdominal midline incision and hysterotomy. Each fetus was weighed and its sex noted.
Measurements and calculations
Values for maternal and fetal arterial pH (pHa), Pa,O2, arterial partial pressure of carbon dioxide (Pa,CO2) and calculated acid/base excess (ABE) were obtained using a blood gas analyser (ABL 5; Radiometer, Copenhagen, Denmark). Measurements in maternal blood were corrected to 38 °C and those in fetal blood to 39.5 °C. Values for blood haemoglobin concentration ([Hb]) and percentage saturation of haemoglobin (Sat.Hb) were determined using a haemoximeter (OSM2; Radiometer) calibrated for ovine fetal blood. Values for haematocrit were obtained using a microhaematocrit centrifuge (Hawksley, UK).
Amniotic cavity pressure was used as the zero pressure reference level. Fetal femoral vascular resistance was calculated by dividing supra-amniotic mean femoral arterial blood pressure by mean fetal FBF (Fletcher et al. 2000a).
Fetal arterial blood oxygen content (O2,cont) and oxygen capacity (O2,cap) during the acute hypoxaemia protocol were calculated using eqns (1) and (2), respectively:
| (1) |
| (2) |
where 1 molecule of haemoglobin (Mr = 64 450) binds 4 molecules of oxygen. The contribution of oxygen dissolved in plasma is regarded as being negligible.
Femoral arterial oxygen delivery (O2,del) during the acute hypoxaemia protocol was calculated using eqn (3):
| (3) |
Maternal and fetal plasma measurements
Maternal and fetal plasma concentrations of dexamethasone, noradrenaline, adrenaline, NPY and AVP were measured in appropriately treated, frozen plasma aliquots within 4 months from plasma collection.
Dexamethasone. Plasma dexamethasone levels were measured after ether extraction using tritium-labelled dexamethasone as a tracer (Fletcher et al. 2000b), and sheep anti-dexamethasone antiserum (Bioclinical Services International, Cardiff, UK). All values were corrected for recovery (86 %). The interassay coefficient of variation (c.v.) for three control plasma pools (1.8, 5.4 and 26.7 nmol l−1) was 14.6, 9.3 and 8.2 %, respectively. The lower detection limit of the assay was 0.2 nmol l−1. The anti-dexamethasone antiserum showed a 1.6 % crossreactivity against cortisol, and crossreactivities of less than 0.5 % against 11-deoxycortisol, corticosterone, testosterone, progesterone and oestriol.
Catecholamines. Fetal plasma noradrenaline and adrenaline concentrations were measured by high-pressure liquid chromatography with electrochemical detection, as described previously (Fletcher et al. 2002). The interassay c.v. for noradrenaline and adrenaline was 6.2 % and 7.3 %, respectively, and the minimum detectable concentration was 10 pg ml−1.
Neuropeptide Y. Fetal plasma NPY concentrations were measured by radioimmunoassay, as described previously (Fletcher et al. 2000a). The assay used rabbit antiserum and 125I-labelled porcine peptide. Antiserum was produced in-house and the animals used were killed humanely at the end of the experiment. The assay was validated for use in ovine plasma using stripped ovine plasma, and could detect less than 1 pmol l−1 (95 % confidence interval). The interassay c.v. was 6.8 %. There was no detectable crossreactivity of the anti-NPY antiserum with peptide YY.
Arginine vasopressin. Fetal plasma AVP concentrations were measured using a commercially available double-antibody radioimmunoassay kit (Nichols Institute Diagnostics, Saffron Walden, Essex, UK) following separation from plasma proteins by methanol extraction and chromatography (Giussani et al. 1994b). The lower detection limit of the assay was 1.3 pg ml−1. The intra-assay c.v. for four control plasma pools (mean concentrations: 3.2, 9.9, 12.2 and 28.9 pg ml−1) was 10.0, 6.7, 3.7 and 4.6 %, respectively. The interassay c.v. was 6.9 % for a mean value of 10.8 pg ml−1. The anti-AVP antiserum (Nichols Institute Diagnostics) showed crossreactivities of less than 0.1 % with lysine vasopressin, oxytocin and vasotocin.
Data and statistical analyses
Values for all variables are expressed as means ± s.e.m. Statistical significance for comparisons between blood gases, acid/base status and hormone concentrations, was assessed using two-way repeated-measures ANOVA and the Tukey post hoc test. Absolute values and absolute changes from mean normoxic baseline for fetal cardiovascular variables during the acute hypoxaemia protocol were analysed by the summary-of-measures method (Matthews et al. 1990) to focus the number of comparisons (Giussani et al. 2001). Comparisons of values for each variable were made within and between treatment groups using two-way repeated-measures ANOVA and the Tukey post hoc test. Functional chemoreflex analysis was performed to assess the effects of fetal treatment with dexamethasone on fetal cardiovascular chemoreflex responses. For this analysis, linear regression lines were plotted for the cardiovascular chemoreflex response of each fetus within the first 15 min of the onset of hypoxia. The individual values for slopes were then compared between treatment groups using one-way ANOVA or Student's t test for unpaired data. The statistical significance of any correlations was assessed using the Pearson product-moment correlation coefficient test, analysis of slopes and Student's t test for unpaired data, as appropriate. In order to control for interseasonal variation, data for each dexamethasone treatment group were compared with data from the corresponding contemporaneous saline-infused control group. For all statistical tests, significance was accepted when P < 0.05.
Results
In control and dexamethasone-treated groups, maternal plasma dexamethasone concentrations were below the detection limit of the assay throughout the experimental protocol. Whilst dexamethasone was not detected in control fetuses throughout the experimental protocol, fetal i.v. treatment with dexamethasone produced a significant elevation in fetal plasma dexamethasone concentration, averaging 2.73 ± 0.38 nmol l−1 over the 48 h of infusion. In the dexamethasone-treated fetuses, plasma dexamethasone was again undetectable by 48 h following the cessation of infusion.
Blood gases and acid/base status during the acute hypoxaemia protocol
Maternal. During normoxia, values for maternal pHa (during saline: 7.47 ± 0.01; during dexamethasone: 7.47 ± 0.01; following saline: 7.48 ± 0.01; following dexamethasone: 7.50 ± 0.01), Pa,CO2 (during saline: 38.1 ± 0.8 mmHg; during dexamethasone: 36.3 ± 0.6 mmHg; following saline: 36.3 ± 0.8 mmHg; following dexamethasone: 35.6 ± 1.5 mmHg), Pa,O2 (during saline: 97.7 ± 3.9 mmHg; during dexamethasone: 99.7 ± 2.9 mmHg; following saline: 97.2 ± 1.5 mmHg; following dexamethasone: 95.8 ± 2.6 mmHg), ABE (during saline: 3.9 ± 0.6 meq l−1; during dexamethasone: 3.2 ± 0.8 meq l−1; following saline: 4.2 ± 0.6 meq l−1; following dexamethasone: 4.8 ± 1.0 meq l−1) and haematocrit (during saline: 26.9 ± 1.2 %; during dexamethasone: 26.6 ± 1.4 %; following saline: 23.7 ± 0.8 %; following dexamethasone: 27.8 ± 1.3 %) were not significantly different between groups (P > 0.05). During hypoxaemia, maternal Pa,O2 levels fell significantly to a similar level in all groups (during saline: 40.7 ± 4.8 mmHg; during dexamethasone: 41.3 ± 5.3 mmHg; following saline: 40.8 ± 2.0 mmHg; following dexamethasone: 32.0 ± 2.1 mmHg; P < 0.05). These changes occurred without any alteration in any other maternal variable from baseline in any group. During recovery, maternal Pa,O2 reverted to normoxic levels in all groups.
Fetal. Arterial blood gases and acid/base status during the acute hypoxaemia for all groups of fetuses are shown in Table 1. Fetal treatment with dexamethasone had no effect on fetal arterial blood gases or acid/base status during normoxia either during or following the infusion period. Fetal treatment with dexamethasone had no effect on fetal femoral arterial blood O2,cont, O2,cap or O2,del during normoxia at 48 h following the end of infusions (Table 1).
Table 1.
Fetal arterial blood gas and acid/base status
| Normoxia | Hypoxaemia | Recovery | ||||
|---|---|---|---|---|---|---|
| Variable | N15 | N45 | H15 | H45 | R15 | R45 |
| pHa | ||||||
| During saline | 7.33 ± 0.01 | 7.34 ± 0.01 | 7.32 ± 0.02 | 7.30 ± 0.02 | 7.26 ± 0.02 a | 7.30 ± 0.01 |
| During dexamethasone | 7.36 ± 0.01 | 7.36 ± 0.01 | 7.34 ± 0.02 | 7.27 ± 0.03 a | 7.24 ± 0.04 a | 7.29 ± 0.03 a |
| Following saline | 7.34 ± 0.01 | 7.34 ± 0.01 | 7.31 ± 0.01 | 7.26 ± 0.02 a | 7.26 ± 0.03 a | 7.28 ± 0.02 |
| Following dexamethasone | 7.33 ± 0.02 | 7.32 ± 0.02 | 7.28 ± 0.02 | 7.16 ± 0.04 ab | 7.11 ± 0.04 ab | 7.18 ± 0.04 ab |
| Pa,co2 (mmHg) | ||||||
| During saline | 54.9 ± 0.7 | 54.3 ± 1.2 | 50.1 ± 1.6 a | 49.0 ± 1.4 a | 51.0 ± 0.7 | 52.3 ± 0.7 |
| During dexamethasone | 52.7 ± 0.5 | 52.4 ± 1.1 | 49.1 ± 1.5 | 51.0 ± 1.6 | 49.4 ± 1.5 | 51.3 ± 0.5 |
| Following saline | 53.2 ± 0.8 | 54.0 ± 0.9 | 53.0 ± 0.9 | 52.5 ± 1.0 | 46.3 ± 1.4 a | 50.7 ± 0.7 |
| Following dexamethasone | 51.0 ± 1.7 | 51.4 ± 1.6 | 51.4± 1.2 | 53.0 ± 2.3 | 48.7 ± 1.6 | 50.9 ± 1.0 |
| Pa,o2 (mmHg) | ||||||
| During saline | 22.1 ± 1.3 | 21.4 ± 0.7 | 12.3 ± 0.9 a | 11.6 ± 0.2 a | 22.4 ± 1.1 | 20.4 ± 1.1 |
| During dexamethasone | 21.4 ± 0.7 | 21.7 ± 1.0 | 12.3 ± 0.6 a | 12.4 ± 0.7 a | 22.9 ± 1.1 | 20.4 ± 1.0 |
| Following saline | 22.5 ± 0.5 | 23.0 ± 0.7 | 11.7 ± 0.3 a | 13.2 ± 0.4 a | 23.7 ± 1.5 | 20.7 ± 1.1 |
| Following dexamethasone | 24.0 ± 0.8 | 24.4 ± 1.1 | 12.6 ± 0.4 a | 12.7 ± 0.6 a | 28.1 ± 1.4 ab | 23.0 ± 1.0 |
| ABE (meq l−1) | ||||||
| During saline | 2.0 ± 0.6 | 2.0 ± 0.7 | −0.3 ± 1.2 | −2.9 ± 1.2 a | −3.4 ± 1.8 a | −1.1 ± 1.3 |
| During dexamethasone | 2.8 ± 0.6 | 2.3 ± 0.6 | 0.2 ± 1.1 | −4.3 ± 2.1 a | −6.5 ± 2.6 a | −3.2 ± 1.8 a |
| Following saline | 1.5 ± 1.1 | 1.5 ± 1.1 | −0.3 ± 1.2 | −4.0 ± 1.5 a | −6.8 ± 2.0 a | −3.3 ± 1.6 |
| Following dexamethasone | −0.3 ± 1.4 | −0.6 ± 1.2 | −3.0 ± 1.3 | −10.6 ± 2.2 ab | −14.4 ± 2.0 ab | −9.3 ± 2.2 ab |
| Sat.Hb (%) | ||||||
| During saline | 60.6 ± 2.7 | 59.5 ± 2.6 | 31.6 ± 4.7 a | 32.4 ± 2.9 a | 59.3 ± 4.7 | 55.6 ± 4.8 |
| During dexamethasone | 57.6 ± 2.4 | 58.1 ± 3.3 | 38.0 ± 4.4 a | 30.6 ± 2.4 a | 57.2 ± 4.2 | 51.3 ± 3.6 |
| Following saline | 63.9 ± 2.2 | 64.8 ± 2.1 | 31.9 ± 0.5 a | 32.6 ± 1.8 a | 63.7 ± 3.1 | 54.4 ± 4.4 |
| Following dexamethasone | 63.8 ± 3.9 | 62.9 ± 4.4 | 25.3 ± 3.3 a | 23.8 ± 4.4 a | 52.3 ± 6.6 | 50.2 ± 5.2 |
| [Hb](g dl−1) | ||||||
| During saline | 9.1 ± 0.5 | 8.8 ± 0.4 | 9.6 ± 0.4 a | 10.0 ± 0.5 a | 8.7 ± 0.4 | 8.5 ± 0.4 |
| During dexamethasone | 10.1 ± 0.5 | 9.7 ± 0.5 | 10.3 ± 0.5 | 10.4 ± 0.3 | 9.2 ± 0.3 | 9.2 ± 0.4 |
| Following saline | 8.5 ± 0.6 | 8.4 ± 0.6 | 9.3 ± 0.6 a | 9.0 ± 0.6 a | 7.5 ± 0.6 | 8.0 ± 0.5 |
| Following dexamethasone | 8.1 ± 0.3 | 7.9 ± 0.3 | 8.8 ± 0.5 a | 8.8 ± 0.4 a | 7.6± 0.3 | 7.7 ± 0.4 |
| Hct (%) | ||||||
| During saline | 30.0 ± 2.1 | 29.0 ± 1.2 | 32.9 ± 1.0 | 29.0 ± 1.1 | 29.3 ± 1.1 | 27.2 ± 1.1 |
| During dexamethasone | 32.9 ± 1.8 | 31.7± 1.3 | 33.6 ± 1.3 | 33.1 ± 1.2 | 30.8 ± 1.4 | 30.9 ± 1.2 |
| Following saline | 28.3 ± 1.7 | 28.8 ± 1.5 | 29.5 ± 1.3 | 30.3 ± 1.7 | 26.7 ± 1.9 | 27.0 ± 1.1 |
| Following dexamethasone | 26.9 ± 1.1 | 27.3 ± 0.8 | 28.7 ± 1.4 | 28.7 ± 1.2 | 25.3 ± 1.4 | 24.6 ± 1.1 |
| Femoral arterial O2,cont (μmol ml−1) | ||||||
| During saline | — | — | — | — | — | — |
| During dexamethasone | — | — | — | — | — | — |
| Following saline | 2.8 ± 0.3 | 2.8 ± 0.2 | 1.6 ± 0.2 a | 1.5 ± 0.2 a | 2.6 ± 0.2 | 2.2 ± 0.2 |
| Following dexamethasone | 2.6 ± 0.2 | 2.6 ± 0.3 | 1.1 ± 0.2 a | 1.1 ± 0.3 a | 2.4 ± 0.3 | 2.0 ± 0.3 |
| Femoral arterial O2,cap (μmol ml−1) | ||||||
| During saline | — | — | — | — | — | — |
| During dexamethasone | — | — | — | — | — | — |
| Following saline | 5.4 ± 0.4 | 5.2 ± 0.4 | 5.8 ± 0.4 a | 5.7 ± 0.4 | 5.1 ± 0.3 | 4.9 ± 0.4 |
| Following dexamethasone | 4.8 ± 0.2 | 4.9 ± 0.2 | 5.4 ± 0.2 a | 5.4 ± 0.3 a | 4.7 ± 0.2 | 4.6 ± 0.2 |
| Femoral arterial O2,del (μmol min−1 (kg fetal body weight)−1) | ||||||
| During saline | — | — | — | — | — | — |
| During dexamethasone | — | — | — | — | — | — |
| Following saline | 39.9 ± 3.1 | 36.7 ± 3.6 | 16.7 ± 2.9 a | 14.0 ± 1.9 a | 31.0 ± 6.9 | 34.2 ± 4.4 |
| Following dexamethasone | 37.7 ± 5.9 | 35.9 ± 6.7 | 6.1 ± 1.2 ab | 4.4 ± 1.5ab | 18.7 ± 4.4 a | 25.9 ± 4.9 |
Values are means ± s.e.m. at 15 min (N15) and 45 min (N45) of normoxia, 15 min (H15) and 45 min (H45) of hypoxaemia, and 15 min (R15) and 45 min (R45) of recovery for fetuses exposed to 1 h of hypoxaemia during saline infusion (n = 7), during dexamethasone treatment (n = 7), following saline infusion (n = 6) and following dexamethasone treatment (n = 6). Fetal blood gas values were corrected to 39.5 °C. pHa, arterial pH; Pa,CO2, arterial partial pressure of carbon dioxide; Pa,O2, arterial partial pressure of oxygen; ABE, acid/base excess; Sat.Hb, percentage saturation of haemoglobin; [Hb], blood haemoglobin concentration; Hct, haematocrit; O2,cont, blood oxygen content; O2,cap, blood oxygen carrying capacity; O2,del, oxygen delivery. Data were not available for femoral arterial O2,cont, O2,cap and O2,del during dexamethasone treatment. Significant differences (P < 0.05):
differences by post hoc analysis indicating a significant main effect of time compared with normoxia
differences by post hoc analysis indicating a significant main effect of treatment compared with contemporaneous saline controls (two-way ANOVA + Tukey test).
During hypoxaemia, fetal arterial Pa,O2 and the percentage saturation of haemoglobin fell to similar levels in all groups (Table 1). The reductions in fetal arterial pHa and ABE that occurred during hypoxaemia and recovery were similar in both groups of saline-infused fetuses and in fetuses exposed to hypoxaemia during dexamethasone infusion. However, the magnitude of the acidaemia measured in the arterial blood of fetuses exposed to hypoxaemia following dexamethasone treatment was greater than that observed in the saline-infused fetuses (Table 1). Whilst there was no change from baseline in fetal haematocrit values in any of the treatment groups, fetal blood haemoglobin concentration increased during hypoxaemia in both groups of saline-infused fetuses and in fetuses exposed to hypoxaemia following, but not during, dexamethasone infusion (Table 1). Fetal femoral arterial blood O2,cont decreased, whilst O2,cap increased, during the hypoxaemic challenge in saline-infused fetuses and in fetuses exposed to hypoxaemia following dexamethasone infusion. Whilst femoral arterial blood O2,del fell in saline-infused fetuses and in fetuses exposed to hypoxaemia following dexamethasone infusion, the magnitude of the decline was greater in the dexamethasone-treated fetuses (Table 1).
During recovery, all variables tended to return towards basal values. However, a transient increase in arterial Pa,O2 above normoxic baseline values occurred at 15 min of recovery in fetuses exposed to hypoxaemia following dexamethasone treatment (Table 1). Similarly, in fetuses exposed to hypoxaemia following dexamethasone treatment, fetal pHa, ABE and femoral arterial blood O2,del remained significantly lower than normoxic values and relative to the other groups by the end of the recovery period (Table 1).
Fetal basal cardiovascular variables
Absolute values of fetal cardiovascular variables during normoxic baseline were similar in both groups of saline-infused fetuses (heart rate: 176 ± 3 vs. 161 ± 6 beats min−1; arterial blood pressure: 44.9 ± 2.7 vs. 47.1 ± 1.5 mmHg; FBF: 33.9 ± 2.0 vs. 33.7 ± 3.3 ml min−1; femoral vascular resistance: 1.55 ± 0.13 vs. 1.51 ± 0.13 mmHg (ml min−1)−1; during saline vs. following saline; mean ± s.e.m. for the 60 min of normoxia; P > 0.05; Figs. 1 and 3). During dexamethasone treatment, basal fetal arterial blood pressure (55.4 ± 3.2 vs. 44.9 ± 2.7 mmHg; during dexamethasone vs. during saline; P = 0.01) and femoral vascular resistance (2.94 ± 0.27 vs. 1.55 ± 0.13 mmHg (ml min−1)−1; P < 0.01) were elevated, whilst fetal FBF was reduced (21.9 ± 2.6 vs. 33.9 ± 2.0 ml min−1; P < 0.01) compared with contemporaneous saline-infused controls (Fig. 1). Absolute values of basal fetal heart rate were similar in fetuses during saline infusion and during dexamethasone treatment (182 ± 6 vs. 176 ± 3 beats min−1; during dexamethasone vs. during saline, P > 0.05; Fig. 1).
Figure 1. Fetal cardiovascular variables in the acute hypoxaemia protocol occurring during infusions.
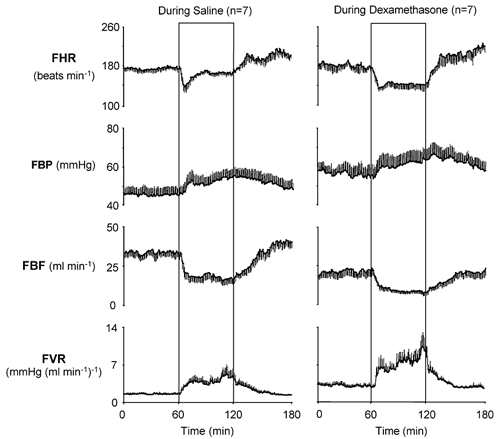
Absolute values of cardiovascular variables for the hypoxaemia protocol during saline infusion (n = 7) and during dexamethasone treatment (n = 7). Values are mean ±s.e.m. for each minute. The boxed area represents the period of hypoxaemia. FHR, fetal heart rate; FBP, fetal arterial blood pressure; FBF, fetal femoral blood flow; FVR, fetal femoral vascular resistance.
Figure 3. Fetal cardiovascular variables in the acute hypoxaemia protocol occurring following infusions.
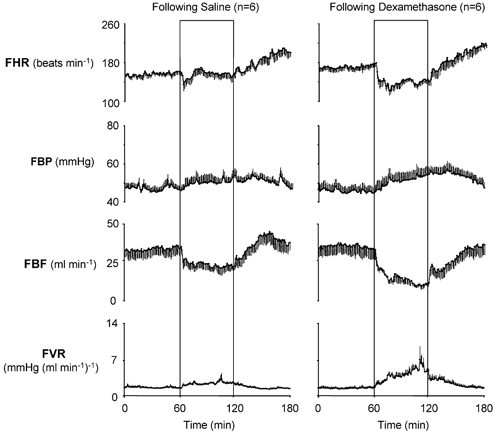
Absolute values of cardiovascular variables for the hypoxaemia protocol following saline infusion (n = 6) and following dexamethasone treatment (n = 6). Values are means ±s.e.m. for each minute. The boxed areas represent the period of hypoxaemia.
Absolute values of fetal cardiovascular variables during normoxic baseline were similar following dexamethasone treatment and following saline infusion (heart rate: 173 ± 6 vs. 161 ± 6 beats min−1; arterial blood pressure: 44.8 ± 2.1 vs. 47.1 ± 1.5 mmHg; FBF: 36.0 ± 4.3 vs. 33.7 ± 3.3 ml min−1; femoral vascular resistance: 1.42 ± 0.19 vs. 1.51 ± 0.13 mmHg (ml min−1)−1; following dexamethasone vs. following saline, P > 0.05; Fig. 3).
Fetal cardiovascular variables during the acute hypoxaemia protocol
Absolute values for fetal cardiovascular variables during the acute hypoxaemia protocol are shown in Figs 1 and 3, and the summary of the statistical analyses for cardiovascular changes from individual normoxic baselines is shown in Figs 2 and 4. During acute hypoxaemia, bradycardia, an increase in arterial blood pressure and a fall in FBF occurred in both groups of saline-infused fetuses. The elevation in fetal arterial blood pressure and fall in FBF resulted in marked increases in calculated femoral vascular resistance values in both groups of saline-infused fetuses (Figs 1–4). In both groups of saline-infused fetuses, whilst the increase in fetal arterial blood pressure and femoral vascular resistance were maintained throughout hypoxaemia, the fall in heart rate was transient, with heart rate returning towards baseline values within 20 min of the onset of the challenge (Figs 1–4).
Figure 2. Statistical summary of changes from baseline for fetal cardiovascular responses to 1 h of hypoxaemia occurring during infusions.
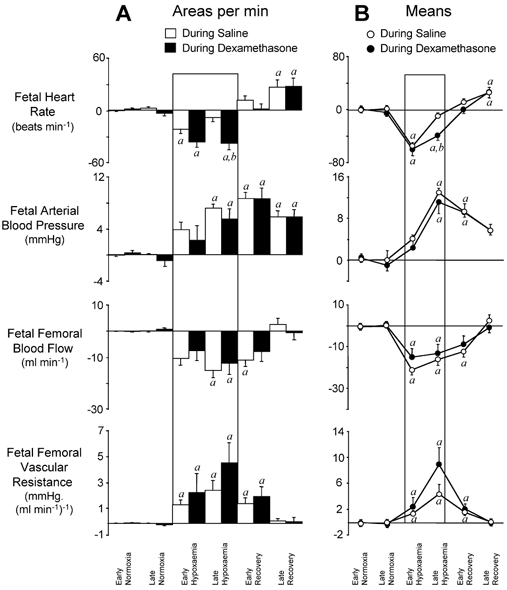
A, mean ±s.e.m. of the areas bounded by the curve for each variable, expressed per minute. B, line graphs representing mean ±s.e.m. of values for changes from individual baselines in fetal heart rate, arterial blood pressure, femoral blood flow and femoral vascular resistance during the following time periods: early (0–45 min) and late (46–60 min) normoxia, early (61–75 min) and late (76–120 min) hypoxaemia, and early (121–135 min) and late (136–180 min) recovery. Fetal cardiovascular responses during saline infusion (○; n = 7) and during dexamethasone treatment (•; n = 7) are represented by open and filled bars, respectively, in A. Significant differences (P < 0.05): a, differences by post hoc analysis indicating a significant main effect of time compared with normoxia; b, differences by post hoc analysis indicating a significant main effect of dexamethasone treatment compared with saline infusion (two-way repeated-measures ANOVA + Tukey test).
Figure 4. Statistical summary of changes from baseline for fetal cardiovascular responses to 1 h of hypoxaemia occurring following infusions.
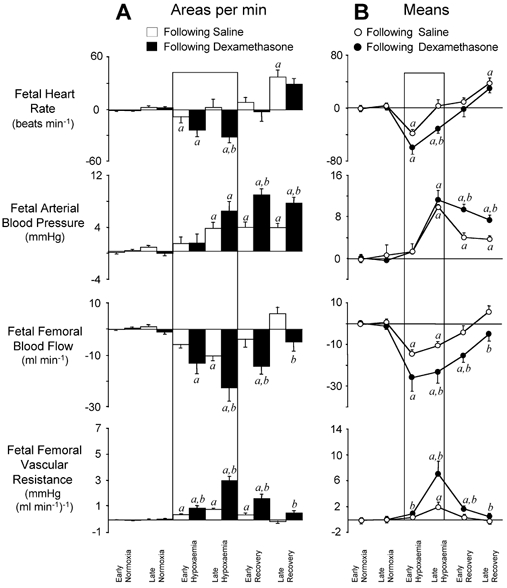
A, mean ±s.e.m. of the areas bounded by the curve, expressed per minute. B, line graphs representing the mean ±s.e.m. of values for changes from individual baselines in fetal heart rate, arterial blood pressure, femoral blood flow and femoral vascular resistance during the following time periods: early (0–45 min) and late (46–60 min) normoxia, early (61–75 min) and late (76–120 min) hypoxaemia, and early (121–135 min) and late (136–180 min) recovery. Fetal cardiovascular responses following saline infusion (○; n = 6) and following dexamethasone treatment (•; n = 6) are represented by open and filled bars, respectively. Significant differences (P < 0.05): a, differences by post hoc analysis indicating a significant main effect of time compared with normoxia; b, differences by post hoc analysis indicating a significant main effect of dexamethasone treatment compared with saline infusion (two-way repeated-measures ANOVA + Tukey test).
In contrast, fetal bradycardia persisted throughout the period of acute hypoxaemia both during and at 48 h following dexamethasone treatment, with fetal heart rate at 45 min of hypoxaemia remaining significantly lower than in saline-infused fetuses (Figs 1–4). Despite the increases in basal fetal arterial blood pressure and femoral vascular resistance and the depressed FBF in normoxia during dexamethasone treatment, further sustained increases in blood pressure and femoral vascular resistance, and a further sustained fall in FBF occurred in response to hypoxaemia during dexamethasone treatment (Figs 1 and 2). In these fetuses, the magnitude of the changes in blood pressure, FBF and femoral vascular resistance from normoxic baseline levels were similar to values observed in saline-infused fetuses (Fig. 2).
Whilst the increase in arterial blood pressure during hypoxaemia was similar, absolute levels of FBF achieved during hypoxaemia were lower in fetuses following dexamethasone treatment compared with saline-infused controls (Figs 3 and 4). This resulted in increases in femoral vascular resistance during hypoxaemia that were significantly greater in fetuses following dexamethasone treatment compared with contemporaneous saline-infused controls (Figs 3 and 4).
During recovery, fetal arterial blood pressure, FBF and femoral vascular resistance returned towards normoxic values in both groups of saline-infused fetuses and in fetuses exposed to hypoxaemia during dexamethasone treatment (Figs 3 and 4). In contrast, in fetuses exposed to hypoxaemia following dexamethasone treatment, fetal arterial blood pressure and femoral vascular resistance remained elevated, whilst FBF remained depressed throughout the recovery period compared with normoxic values (Figs 3 and 4). Tachycardia of similar magnitude developed during the recovery period in all groups (Figs 1–4).
Functional chemoreflex analysis
Correlations between the change from normoxic baseline in fetal Pa,O2 and the changes in fetal heart rate and femoral vascular resistance within the first 15 min of hypoxaemia were made for all groups of fetuses. Analysis of slopes on individual regression lines indicated no significant differences between any of the treatment groups for the change in Pa,O2vs. the change in femoral vascular resistance (during saline: 0.16 ± 0.03 mmHg (ml min−1)−1; during dexamethasone: 0.31 ± 0.26 mmHg (ml min−1)−1; following saline: 0.09 ± 0.04 mmHg (ml min−1)−1; following dexamethasone: 0.15 ± 0.02 mmHg (ml min−1)−1; Δ(femoral vascular resistance)/ΔPa,O2, P > 0.05) or for the change in Pa,O2vs. the change in fetal heart rate (during saline: 2.02 ± 0.84 beats min−1 mmHg−1; during dexamethasone: 4.98 ± 1.76 beats min−1 mmHg−1; following saline: 1.32 ± 0.99 beats min−1 mmHg−1; following dexamethasone: 3.08 ± 0.68 beats min−1 mmHg−1; Δ(fetal heart rate)/ΔPa,O2, P > 0.05).
Endocrine responses during the acute hypoxaemia protocol
Maternal arterial plasma concentrations of catecholamines, NPY and AVP were similar during normoxia and remained unchanged from baseline during the acute hypoxaemia protocol in all treatment groups (Table 2). In contrast, basal fetal plasma noradrenaline concentrations were significantly reduced during, but not following, dexamethasone treatment compared with corresponding saline-infused controls (Fig. 5). Basal fetal plasma concentrations of adrenaline, AVP and NPY were not altered either during or following dexamethasone treatment (Fig. 5).
Table 2.
Maternal plasma hormone concentrations
| Normoxia | Hypoxaemia | Recovery | |||
|---|---|---|---|---|---|
| Variable | N15 | N45 | H15 | H45 | R45 |
| [NA] (pg ml−1) | |||||
| During saline | 498 ± 121 | 664 ± 250 | 671 ± 148 | 543 ± 243 | 635 ± 190 |
| During dexamethasone | 996 ± 282 | 715 ± 279 | 1040 ± 333 | 1361 ± 871 | 634 ± 140 |
| Following saline | 463 ± 128 | 572 ± 187 | 1252 ± 468 | 1219 ± 402 | 737 ± 203 |
| Following dexamethasone | 627 ± 118 | 636 ± 155 | 1220 ± 364 | 1580 ± 862 | 708 ± 252 |
| [ADR] (pg ml−1) | |||||
| During saline | 196 ±80 | 103 ± 5 | 123 ± 62 | 170 ±55 | 177 ± 49 |
| During dexamethasone | 198 ± 81 | 131 ± 54 | 246 ± 81 | 272 ± 54 | 220 ± 52 |
| Following saline | 277 ±169 | 302 ±202 | 272 ± 126 | 337 ± 198 | 299 ± 120 |
| Following dexamethasone | 236 ±81 | 174 ±74 | 262 ± 112 | 420 ± 168 | 286 ±112 |
| [NPY] (pmol l−1) | |||||
| During saline | 9.0 ±1.4 | 9.2 ± 1.6 | 11.5 ±1.2 | 9.0 ±1.5 | 8.5 ± 1.5 |
| During dexamethasone | 10.4 ±1.5 | 10.5 ±1.5 | 12.9 ± 1.1 | 11.5 ±2.2 | 10.1 ± 1.7 |
| Following saline | 3.8 ± 1.0 | 4.9 ± 1.4 | 4.4 ± 1.5 | 5.4 ± 2.7 | 3.9 ± 1.9 |
| Following dexamethasone | 4.4 ±1.4 | 4.8 ± 1.6 | 5.4 ± 1.9 | 13.0 ±4.5 | 5.7 ±2.8 |
| [AVP] (pg ml−1) | |||||
| During saline | 1.8 ± 0.3 | 2.8 ± 0.2 | 4.9 ± 2.8 | 6.1 ± 3.5 | 3.9 ± 1.2 |
| During dexamethasone | 4.0 ±2.1 | 4.0 ± 1.7 | 6.2 ±2.5 | 5.8 ±3.0 | 4.1 ± 1.4 |
| Following saline | 1.7 ±0.4 | 1.7 ±0.4 | 2.7 ±1.1 | 3.2 ± 1.4 | 2.6 ±0.7 |
| Following dexamethasone | 2.4 ± 0.8 | 2.2 ± 0.7 | 2.6 ± 0.7 | 2.8 ± 0.7 | 2.2 ± 0.4 |
Values are means ±s.e.m. at 15 min (N15) and 45 min (N45) of normoxia, 15 min (H15) and 45 min (H45) of hypoxaemia, and 45 min (R45) of recovery for arterial plasma concentrations of noradrenaline ([NA]), adrenaline ([ADR]), neuropeptide Y ([NPY]) and arginine vasopressin ([AVP]) in mothers of fetuses exposed to 1 h of hypoxaemia during saline infusion (n = 7), during dexamethasone treatment (n = 7), following saline infusion (n = 6) and following dexamethasone treatment (n = 6).
Figure 5. Fetal arterial plasma hormone concentrations during the acute hypoxaemia protocol.
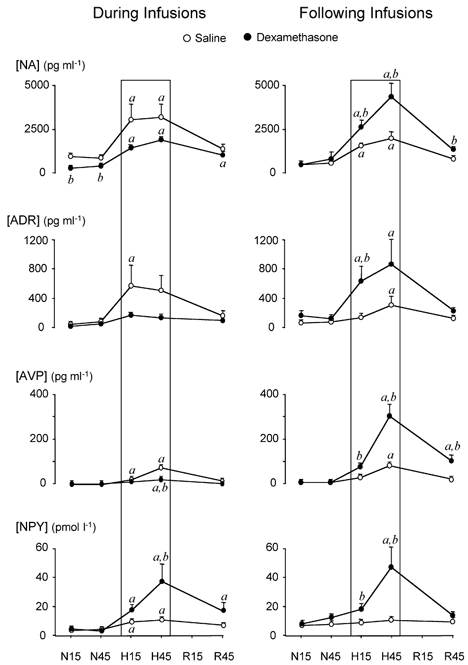
Arterial plasma concentrations of noradrenaline (NA), adrenaline (ADR), neuropeptide Y (NPY) and arginine vasopressin (AVP) in fetuses exposed to 1 h of hypoxaemia during saline infusion (n = 7), during dexamethasone treatment (n = 7), following saline treatment (n = 6) and following dexamethasone treatment (n = 6). Values are means ±s.e.m. at 15 min (N15) and 45 min (N45) of normoxia, 15 min (H15) and 45 min (H45) of hypoxaemia, and 45 min (R45) of recovery. The boxed area represents the period of hypoxaemia. Significant differences (P < 0.05): a, differences by post hoc analysis indicating a significant main effect of time compared with normoxic baseline; b, differences by post hoc analysis indicating a significant main effect of dexamethasone treatment compared with saline infusion (two-way repeated-measures ANOVA + Tukey test).
During acute hypoxaemia, significant increases in arterial plasma concentrations of noradrenaline, adrenaline, NPY and AVP occurred in both groups of saline-infused fetuses, and responses were not different between these two groups of control fetuses (Fig. 5). In fetuses exposed to hypoxaemia during dexamethasone treatment, the magnitude of the plasma noradrenaline response was maintained, the adrenaline and AVP responses were attenuated or abolished, and the plasma NPY response was enhanced compared with contemporaneous saline-infused controls (Fig. 5). In fetuses exposed to hypoxaemia following dexamethasone treatment, fetal arterial plasma noradrenaline, adrenaline, AVP and NPY responses were enhanced compared with corresponding saline-infused fetuses (Fig. 5). When data for 15 and 45 min of normoxia and 15 and 45 min of hypoxaemia were combined, irrespective of fetal treatment group, a highly significant positive correlation was found between values for femoral vascular resistance during the acute hypoxaemia protocol and the corresponding fetal plasma NPY concentrations (y = 0.13x + 1.86, r = 0.769; P < 0.001), but not plasma noradrenaline, adrenaline or AVP concentrations.
Discussion
The data from the present study demonstrate that direct fetal treatment with dexamethasone, producing circulating dexamethasone concentrations in the sheep fetus approximating one-fifth of the levels measured in human umbilical blood following maternal antenatal glucocorticoid therapy (Kream et al. 1983), modifies the pattern and magnitude of the fetal cardiovascular responses to acute oxygen deprivation. The data show that the modifications to different aspects of the fetal defence responses to acute hypoxaemia that occur during dexamethasone treatment may reverse, persist or even become enhanced by 48 h following the treatment period.
Dexamethasone dose and route of administration
Fetal plasma dexamethasone concentrations during infusion in the present study (2.73 ± 0.38 nmol l−1; mean ± s.e.m. during the 48 h infusion regimen for all treated fetuses) were approximately one-eighth of the mean value produced by Derks and colleagues (1997) in fetal sheep (20 nmol l−1) and were one-fifth of the mean value measured in umbilical arterial blood samples taken from human infants at caesarean section 12 h after the completion of a course of maternal antenatal glucocorticoid treatment (5 mg dexamethasone intramuscularly every 12 h for 48 h; Kream et al. 1983). Clinical dosing regimens involve maternal intramuscular injections (NIH Consensus Development Conference, 1994) that will expose the human fetus to initially high, but then rapidly decreasing concentrations of synthetic steroid (Ballard & Ballard, 1995). In the present studies, the sheep fetuses were exposed to continuous intravenous infusions of dexamethasone during the 48 h treatment period. In this way, intrafetal administration of dexamethasone allowed assessment of the direct effects of the synthetic glucocorticoid on fetal cardiovascular, endocrine and metabolic variables. This minimized possible confounding influences of dexamethasone-induced changes in maternal homeostasis, such as the maternal hyperglycaemia that results from intramuscular injection of dexamethasone in pregnant ewes (Bennet et al. 1999), and may also have avoided some of the modifications in placental morphology and function associated with maternal glucocorticoid administration (Wintour et al. 1994; Tangalakis et al. 1995). Direct intrafetal infusions also permit relatively controllable exposure of the fetuses to dexamethasone and avoid further confounding influences of differences in transplacental passage between individual animals and between sheep and primate placentae. In addition, some studies have demonstrated differential effects of maternal vs. fetal administration of synthetic glucocorticoids on the sheep fetus. For example, Bennet et al. (1999) observed a reduction in fetal Pa,O2 over the first few hours following a maternal intramuscular injection of 12 mg of dexamethasone in pregnant ewes, whereas direct intrafetal infusion has been shown to have no effect on fetal Pa,O2 (Derks et al. 1997; Fletcher et al. 2002).
Fetal heart rate response
The mechanisms accounting for the persistence of bradycardia during acute hypoxaemia in dexamethasone-treated fetuses may include enhanced negative chronotropic effects and/or depressed positive chronotropic influences on the fetal heart. Furthermore, the relative importance of these factors in contributing to persistence of fetal bradycardia in response to acute hypoxaemia may be different during and following the period of dexamethasone treatment.
In previous studies, the absolute change in fetal heart rate per unit change in the percentage saturation of fetal haemoglobin has been used as an indirect measure of the gain of the fetal cardiac chemoreflex pathway (e.g. Baan et al. 1993). However, as the carotid body senses changes in Pa,O2 and not changes in the oxygen saturation of fetal haemoglobin (see Blanco et al. 1984; Giussani et al. 1994a), in the present study chemoreflex function was assessed by determining the absolute changes in fetal heart rate and femoral vascular resistance vs. changes in fetal Pa,O2 within the first 15 min of the onset of hypoxaemia. The initial bradycardia and the initial increase in femoral vascular resistance within this time period are well-established carotid chemoreflexes (Bartelds et al. 1993; Giussani et al. 1993). Data from the current study suggest that whilst dexamethasone treatment produced persistent resetting of fetal baroreflex function (Fletcher et al. 2002), the fetal cardiac chemoreflex was not significantly altered either during or following the period of dexamethasone treatment. This suggests that the mechanisms mediating increased negative chronotropic drive during acute hypoxaemia in dexamethasone-treated fetuses do not include enhanced activity and/or gain of the fetal chemoreflex. However, these findings do not exclude the persistent predominance of negative chronotropic chemoreflex activity into the period beyond the first 15 min of hypoxaemia.
Enhanced vagal and/or depressed sympathetic signal transduction in the efferent arm of the fetal chemoreflex pathway may also account for the persistence of bradycardia throughout the hypoxaemia induced during and following the period of dexamethasone treatment. For example, endogenous glucocorticoids play an important role in increasing myocardial muscarinic acetylcholine receptor affinity both in vivo (Jacobsson et al. 1983) and in vitro (Ransnas et al. 1987) in the rat. Also, a reduction in sensitivity to isoprenaline was observed postnatally in rats when dexamethasone was administered either prenatally to the mother (Hou & Slotkin, 1989) or postnatally to the offspring (Lau & Slotkin, 1981). This effect was attributed to a reduction in β-adrenoceptor coupling, since the results of binding assays indicate that receptor expression remains unaltered. Bian and colleagues (1990) reported a dose-dependent effect of maternal dexamethasone treatment on coupling of β-adrenoceptor activation to intracellular cAMP production in the neonatal rat kidney and heart. Although ‘low’ doses of dexamethasone (0.2 mg kg−1s.c. in maternal rats on 17, 18 and 19 dGA) led to an enhancement of β-adrenoceptor transduction in the heart, higher doses (0.8 mg kg−1), above the threshold for promoting maturation of lung function in the rat fetus (Kudlacz et al. 1989; Bian et al. 1990), had a suppressive effect on myocardial sensitivity to β-adrenergic stimulation. Combined, the past and present data therefore suggest that persistence of the bradycardic response to acute hypoxaemia during and following dexamethasone treatment of the ovine fetus is due to increased myocardial sensitivity to vagal stimulation and/or depressed myocardial sensitivity to β-adrenergic stimulation.
Peripheral vasoconstrictor response
Despite the elevation in basal fetal femoral vascular resistance during dexamethasone treatment, the additional increment in femoral vascular resistance that was observed during acute hypoxaemia was at least as large in dexamethasone-treated fetuses compared with corresponding saline-infused controls. However, by 48 h after the end of the period of dexamethasone treatment, the magnitude of the increment in femoral vascular resistance during hypoxaemia was greater in the fetuses exposed to dexamethasone compared with contemporaneous control fetuses. Data presented in the current study show that dexamethasone does not affect the magnitude or gain of the femoral vasoconstrictor chemoreflex response to hypoxaemia, and that dexamethasone attenuates the plasma catecholamine and vasopressin responses, but enhances the plasma NPY responses to hypoxaemia during the period of treatment. The data also show that dexamethasone augments the plasma NPY, catecholamine and vasopressin responses to acute hypoxaemia following the period of treatment. Hence, the findings of this study suggest that enhancement of the femoral vasoconstrictor response to acute hypoxaemia during dexamethasone treatment cannot be explained by enhancement of the chemoreflex and/or plasma catecholamine and vasopressin responses to acute hypoxaemia. The data indicate that increased NPY-ergic activity may contribute, at least in part, to the enhanced vasoconstrictor response to acute hypoxaemia in dexamethasone-treated fetuses. Indeed, in this study, when data were pooled regardless of treatment group, values of fetal femoral vascular resistance were found to correlate positively with fetal plasma NPY concentrations, but not with fetal plasma catecholamine or AVP levels.
NPY is known to be colocalized with noradrenaline in postganglionic sympathetic nerve terminals innervating the peripheral vasculature (Lundberg et al. 1983; Ekblad et al. 1984), and is also present in high concentrations in the adrenal medulla (Allen et al. 1983). Despite the relatively high adrenal medullary NPY activity, adrenalectomy in adult rats had no effect on basal NPY or on the increment in plasma NPY in response to acute psychological stress (Mormede et al. 1990). In addition, experiments in intact calves (Bloom et al. 1988) and adrenalectomized, weaned lambs (Bloom et al. 1989) have provided evidence for a negligible role of the adrenal medulla in contributing to the increase in circulating NPY concentrations during splanchnic nerve stimulation. Taken together, these data suggest that adrenal medullary NPY contributes little to the increment in plasma NPY observed during splanchnic nerve stimulation and during acute stress, and that overspill from perivascular sympathetic nerve terminals is the most likely source of plasma NPY levels under these conditions. Furthermore, in contrast to noradrenaline, NPY lacks synaptic reuptake mechanisms (Lundberg, 1996). Therefore, in the current study, changes in circulating NPY may provide an indirect measure of peripheral sympathetic nerve activity and, if so, the data suggest that fetal exposure to dexamethasone increases the activity of the sympathetic nervous system. Indeed, dexamethasone treatment has been shown to enhance tyrosine hydroxylase activity by 50 % in the superior cervical ganglion of adult rats, indicating increased pre-ganglionic activity (Sze & Hedrick, 1983). Furthermore, antenatal dexamethasone treatment has been shown to accelerate the postnatal maturation of central nervous system catecholaminergic signalling in rats (Slotkin et al. 1992), and to enhance renal sympathetic nervous activity in lambs at preterm delivery (Segar et al. 1998). The increase in plasma NPY concentrations may also be an index of sympathetic nervous system discharge frequency, since nerve stimulation at high frequencies results in NPY overspill, which is proportionately greater than that of noradrenaline (Lundberg, 1996). Hence, the increased plasma NPY responses to acute hypoxaemia during and following dexamethasone treatment may indicate modification to sympathetic nervous system discharge patterns and enhancement of discharge frequency, as well as a possible shift in the dependence of signal transmission from noradrenergic to peptidergic mechanisms. Although the origin of circulating plasma noradrenaline concentrations in the adult is also primarily of spillover from sympathetic nerve terminals, the origin of circulating noradrenaline in the fetus has been shown to be largely a result of increased adrenal output of noradrenaline (Cohen et al. 1984, 1991), since adrenalectomy abolished not only the fetal plasma adrenaline response, but also 90 % of the rise in plasma noradrenaline concentrations during acute hypoxaemia (Jones et al. 1988). Consequently in the fetal period, the circulating level of noradrenaline is not a representative marker of general sympathetic nervous system activity.
The endocrine data from the current study clearly demonstrate differential effects of the treatment protocol on different hormonal axes at different times relative to the period of dexamethasone infusion. The increases in circulating concentrations of adrenaline and AVP measured in this study were attenuated during, but enhanced following, the period of dexamethasone treatment. The latter findings are similar to those of Ervin et al. (2000), where pre-exposure to a single intrafetal injection of betamethasone at 126–127 dGA enhanced the premature newborn lamb plasma AVP and noradrenaline responses to 20 min of acute hypoxaemia following delivery 24 h later. Other studies have reported that fetal exposure to glucocorticoids suppresses plasma catecholamine concentrations under basal conditions (Derks et al. 1997), and that it significantly attenuates the rise in plasma catecholamine concentrations normally seen at birth in preterm lambs (Padbury et al. 1995) and in human infants (Kallio et al. 1998). Particularly pertinent to the findings of the current study is the observation that NPY administration to adult rats inhibits both the release of noradrenaline from sympathetic nerve terminals and the secretion of adrenaline from the adrenal medulla that is evoked by electrical stimulation of preganglionic sympathetic nerves (Linton-Dahlof, 1989). Thus, the suppression of the fetal plasma adrenaline response to acute hypoxaemia during dexamethasone exposure may not only be a result of a direct inhibitory effect of the glucocorticoid, but may also be due to indirect effects mediated by enhanced NPY activity.
In this study, the attenuation of the fetal plasma AVP response to acute hypoxaemia during dexamethasone treatment contrasts with the lack of effect of cortisol infusion on the ovine fetal plasma AVP response to acute hypoxaemia reported by Akagi et al. (1990). However, the findings are in accordance with those obtained in conscious adult dogs, where cortisol infusion suppressed basal and hypotension-stimulated plasma AVP concentrations (Raff et al. 1990; Papanek & Raff, 1994). Similarly, single or repeated antenatal betamethasone administration attenuates the normal increases in plasma AVP following preterm delivery (Ervin et al. 1996, 1997). Indeed, dexamethasone treatment in vitro has been shown to inhibit AVP release from hypothalamic cells of juvenile male rats (Hellbach et al. 1998). Furthermore, in fetal sheep between 109 and 125 dGA, AVP and corticotrophin releasing hormone mRNA abundance in the parvocellular region of the paraventricular nucleus is augmented by adrenalectomy, with cortisol replacement inhibiting this increase, supporting an inhibitory action of endogenous glucocorticoids on hypothalamic AVP production (Unno et al. 1998).
The data in the current study also indicate differential endocrine responses to the hypoxaemic challenge between mothers and fetuses. Whilst the hypoxaemic challenge elicited increases in fetal plasma noradrenaline, adrenaline, NPY and AVP concentrations, no changes were detected in maternal plasma in any of the study groups. These observations may partially reflect the well-established differences in set-points and sensitivities of the fetal compared with the postnatal/adult chemoreflex (Blanco et al. 1984) for endocrine axes known to have a neural influence, such as the hypothalamopituitary-adrenal axis (Giussani et al. 1994c). Similarly, the differences between maternal and fetal endocrine responses may result from variations in the set-point and sensitivity of the endocrine organs themselves to reductions in Pa,O2 and oxygen saturation. Further studies are required to address more fully the mechanisms and implications of the differences between the maternal and fetal responses to acute hypoxaemia.
The mechanisms accounting for the enhancement of the fetal plasma noradrenaline, adrenaline and vasopressin responses to acute hypoxaemia following dexamethasone treatment remain unknown, but glucocorticoid-mediated upregulation of adrenal phenylethanolamine N-methyltransferase (PNMT) activity may be a contributing factor to the enhanced adrenergic response. For example, Kennedy & Ziegler (2000) have shown that treatment of rat fetuses with dexamethasone for the major part of gestation increases adrenal PNMT activity. Similarly, in adult rats, dexamethasone treatment (1 mg kg−1s.c. for 12 days) enhances PNMT expression and activity in skeletal muscle, and these changes persist 6 days after the cessation of glucocorticoid treatment (Kennedy et al. 1993). However, this effect on PNMT may be glucocorticoid and dose specific, since cortisol (2.5–3.0 mg day−1) infused into fetal sheep for 7 days between 109 and 116 dGA suppressed adrenal PNMT mRNA expression (Adams et al. 1999). Fetal exposure to dexamethasone may also lead to precocious glucocorticoid-dependent maturation of the fetal plasma AVP and catecholamine responses to acute hypoxaemia, since several investigators have reported that the magnitude of these fetal endocrine responses to acute hypoxaemia increase with advancing gestational age in the sheep fetus (Cohn et al. 1982; Stark et al. 1982; Jones & Wei, 1985; Iwamoto, 1989; Cheung, 1990; Raff & Wood, 1992) and with incubation time in the chicken embryo (Mulder et al. 2000). Possible glucocorticoid-dependent maturational effects on fetal endocrine axes may be masked by glucocorticoid negative feedback if axes activation occurs during the period of dexamethasone treatment. Therefore, glucocorticoid-dependent maturation of endocrine axes responsiveness may only become unmasked once the steroid has cleared from the fetal circulation. Our previous studies have shown similar effects of glucocorticoids on the fetal ACTH and cortisol response to hypoxaemia during (Fletcher et al. 2000b) and following (Fletcher et al. 2002) dexamethasone treatment. Alternatively, the augmented endocrine responses to acute hypoxaemia following dexamethasone treatment may be partly driven by the increased degree of fetal acidaemia that develops following exposure to the glucocorticoid. Clinically, it has been shown that the catecholamine concentrations in human umbilical cord blood samples correlate both to the degree of hypoxaemia and the degree of acidosis at delivery (Newnham et al. 1984), and experimentally, Gardner et al. (2002) have reported greater plasma catecholaminergic and vasopressinergic responses to acute hypoxaemia in acidaemic fetuses.
Although the maturational effects of glucocorticoids on sympathetic nervous activity and on endocrine vasoconstrictor hormone responses may explain in part the enhanced fetal peripheral vasoconstrictor response to acute hypoxaemia in dexamethasone-treated fetuses in the present study, they are not the only explanation. Glucocorticoid treatment may induce structural and functional changes at the level of vascular smooth muscle and connective tissue, thereby modifying basal vascular tone and the gain of superimposed vasoconstrictor stimuli (Altura & Altura, 1974; Leitman et al. 1984; Pierce et al. 1995). Treatment of adult rabbits with dexamethasone increases transmembrane Ca2+ flux in aortic smooth muscle cells (Kornel et al. 1995) and induces expression of transport proteins for transmembrane sodium and calcium influx (Kornel et al. 1993), augmenting the intracellular signal for vascular smooth muscle cell contraction. In addition, treatment of cultured rat vascular smooth muscle cells with dexamethasone (10−7m) for 48 h enhances the production of inositol trisphosphate in response to α-adrenoceptor stimulation with noradrenaline (Liu et al. 1992), and fetal treatment with betamethasone for 48 h has been shown to enhance the maximum vasoconstrictor tension generated by fetal femoral arterial segments in response to depolarizing potassium solutions (Anwar et al. 1999). Other studies have demonstrated dose-, time- and agent-specific effects of glucocorticoids on vascular sensitivity to endocrine and paracrine vasoactive factors (Walker & Williams, 1992). Furthermore, recent studies using small-vessel wire myography have shown that in vivo treatment of fetal sheep with glucocorticoids enhances the in vitro responses of peripheral arteries both to vasoconstrictors, such as noradrenaline and endothelin, and to endothelium-dependent vasodilators, such as acetylcholine, whilst reducing sensitivity to the endothelium-independent vasodilator effects of bradykinin and forskolin, and blunting endothelin-induced nitric oxide synthesis in peripheral resistance arteries (Anwar et al. 1999; Docherty et al. 2001a,b; Molnar et al. 2002).
Acknowledgments
This work was funded by Tommy's - The Baby Charity, UK. The authors would like to thank Mr Malcolm Bloomfield and Mr Paul Hughes for excellent technical assistance, and Mrs Sue Nicholls and Miss Vicky Johnson for the care of the animals used in this study. A.J.W.F. was supported by the Foster Studentship, Department of Physiology, University of Cambridge, UK. D.A.G. is a Fellow of the Lister Institute for Preventive Medicine, UK.
References
- Adams MB, Ross JT, Butler TG, McMillen IC. Glucocorticoids decrease phenylethanolamine N-methyltransferase mRNA expression in the immature foetal sheep adrenal. J Neuroendocrinol. 1999;11:569–575. doi: 10.1046/j.1365-2826.1999.00359.x. [DOI] [PubMed] [Google Scholar]
- Akagi K, Berdusco ET, Challis JR. Cortisol inhibits ACTH but not the AVP response to hypoxaemia in fetal lambs at days 123–128 of gestation. J Dev Physiol. 1990;14:319–324. [PubMed] [Google Scholar]
- Alexander DP, Bashore RA, Britton HG, Forsling ML. Maternal and fetal arginine vasopressin in the chronically catheterised sheep. Biol Neonate. 1974;25:242–248. doi: 10.1159/000240696. [DOI] [PubMed] [Google Scholar]
- Allen JM, Adrian TE, Polak JM, Bloom SR. Neuropeptide Y (NPY) in the adrenal gland. J Auton Nerv Syst. 1983;9:559–563. doi: 10.1016/0165-1838(83)90013-9. [DOI] [PubMed] [Google Scholar]
- Altura BM, Altura BT. Peripheral vascular actions of glucocorticoids and their relationship to protection in circulatory shock. J Pharmacol Exp Ther. 1974;190:300–315. [PubMed] [Google Scholar]
- Anwar MA, Schwab M, Poston L, Nathanielsz PW. Betamethasone-mediated vascular dysfunction and changes in hematological profile in the ovine fetus. Am J Physiol. 1999;276:H1137–1143. doi: 10.1152/ajpheart.1999.276.4.H1137. [DOI] [PubMed] [Google Scholar]
- Baan J, Jr, Boekkooi PF, Teitel DF, Rudolph AM. Heart rate fall during acute hypoxemia: a measure of chemoreceptor response in fetal sheep. J Dev Physiol. 1993;19:105–111. [PubMed] [Google Scholar]
- Ballard PL, Ballard RA. Scientific basis and therapeutic regimens for use of antenatal glucocorticoids. Am J Obstet Gynecol. 1995;173:254–262. doi: 10.1016/0002-9378(95)90210-4. [DOI] [PubMed] [Google Scholar]
- Bartelds B, Van Bel F, Teitel DF, Rudolph AM. Carotid, not aortic, chemoreceptors mediate the fetal cardiovascular response to acute hypoxemia in lambs. Pediatr Res. 1993;34:51–55. doi: 10.1203/00006450-199307000-00013. [DOI] [PubMed] [Google Scholar]
- Bayard F, Louvet JP, Ruckebusch Y, Boulard C. Transplacental passage of dexamethasone in sheep. J Endocrinol. 1972;54:349–350. doi: 10.1677/joe.0.0540349. [DOI] [PubMed] [Google Scholar]
- Bennet L, Kozuma S, McGarrigle HH, Hanson MA. Temporal changes in fetal cardiovascular, behavioural, metabolic and endocrine responses to maternally administered dexamethasone in the late gestation fetal sheep. Br J Obstet Gynaecol. 1999;106:331–339. doi: 10.1111/j.1471-0528.1999.tb08270.x. [DOI] [PubMed] [Google Scholar]
- Bian XP, Seidler FJ, Bartolome J, Kavlock RJ, Bartolome M, Slotkin TA. Dose-dependent effect of prenatal dexamethasone treatment on beta-adrenergic receptor coupling to ornithine decarboxylase and cyclic AMP. J Dev Physiol. 1990;14:125–130. [PubMed] [Google Scholar]
- Blanco CE, Dawes GS, Hanson MA, McCooke HB. The response to hypoxia of arterial chemoreceptors in fetal sheep and new-born lambs. J Physiol. 1984;351:25–37. doi: 10.1113/jphysiol.1984.sp015229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom SR, Edwards AV, Jones CT. The adrenal contribution to the neuroendocrine responses to splanchnic nerve stimulation in conscious calves. J Physiol. 1988;397:513–526. doi: 10.1113/jphysiol.1988.sp017016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom SR, Edwards AV, Jones CT. Neuroendocrine responses to stimulation of the splanchnic nerves in bursts in conscious, adrenalectomized, weaned lambs. J Physiol. 1989;417:79–89. doi: 10.1113/jphysiol.1989.sp017791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boddy K, Dawes GS, Fisher R, Pinter S, Robinson JS. Foetal respiratory movements, electrocortical and cardiovascular responses to hypoxemia and hypercapnia in sheep. J Physiol. 1974;243:599–618. doi: 10.1113/jphysiol.1974.sp010768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung CY. Fetal adrenal medulla catecholamine response to hypoxia-direct and neural components. Am J Physiol. 1990;258:R1340–1346. doi: 10.1152/ajpregu.1990.258.6.R1340. [DOI] [PubMed] [Google Scholar]
- Cohen WR, Piasecki GJ, Cohn HE, Susa JB, Jackson BT. Sympathoadrenal responses during hypoglycemia, hyperinsulinemia, and hypoxemia in the ovine fetus. Am J Physiol. 1991;261:E95–102. doi: 10.1152/ajpendo.1991.261.1.E95. [DOI] [PubMed] [Google Scholar]
- Cohen WR, Piasecki GJ, Cohn HE, Young JB, Jackson BT. Adrenal secretion of catecholamines during hypoxemia in fetal lambs. Endocrinology. 1984;114:383–390. doi: 10.1210/endo-114-2-383. [DOI] [PubMed] [Google Scholar]
- Cohn HE, Piasecki GJ, Jackson BT. The effect of beta-adrenergic stimulation on fetal cardiovascular function during hypoxemia. Am J Obstet Gynecol. 1982;144:810–816. doi: 10.1016/0002-9378(82)90357-x. [DOI] [PubMed] [Google Scholar]
- Cohn HE, Sacks EJ, Heymann MA, Rudolph AM. Cardiovascular responses to hypoxemia and acidemia in fetal lambs. Am J Obstet Gynecol. 1974;120:817–824. doi: 10.1016/0002-9378(74)90587-0. [DOI] [PubMed] [Google Scholar]
- Court DJ, Parer JT, Block BS, Llanos AJ. Effects of beta-adrenergic blockade on blood flow distribution during hypoxaemia in fetal sheep. J Dev Physiol. 1984;6:349–358. [PubMed] [Google Scholar]
- Derks JB, Giussani DA, Jenkins SL, Wentworth RA, Visser GH, Padbury JF, Nathanielsz PW. A comparative study of cardiovascular, endocrine and behavioural effects of betamethasone and dexamethasone administration to fetal sheep. J Physiol. 1997;499:217–226. doi: 10.1113/jphysiol.1997.sp021922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Docherty CC, Kalmar-Nagy J, Engelen M, Koenen SV, Nijland M, Kuc RE, Davenport AP, Nathanielsz PW. Effect of in vivo fetal infusion of dexamethasone at 0. 75 GA on fetal ovine resistance artery responses to ET-17. Am J Physiol. 2001a;281:R261–268. doi: 10.1152/ajpregu.2001.281.1.R261. [DOI] [PubMed] [Google Scholar]
- Docherty CC, Kalmar-Nagy J, Engelen M, Nathanielsz PW. Development of fetal vascular responses to endothelin-1 and acetylcholine in the sheep. Am J Physiol. 2001b;280:R554–562. doi: 10.1152/ajpregu.2001.280.2.R554. [DOI] [PubMed] [Google Scholar]
- Ekblad E, Edvinsson L, Wahlestedt C, Uddman R, Hakanson R, Sundler F. Neuropeptide Y co-exists and co-operates with noradrenaline in perivascular nerve fibers. Regul Pept. 1984;8:225–235. doi: 10.1016/0167-0115(84)90064-8. [DOI] [PubMed] [Google Scholar]
- Ervin MG, Berry LM, Ikegami M, Jobe AH, Padbury JF, Polk DH. Single dose fetal betamethasone administration stabilizes postnatal glomerular filtration rate and alters endocrine function in premature lambs. Pediatr Res. 1996;40:645–651. doi: 10.1203/00006450-199611000-00001. [DOI] [PubMed] [Google Scholar]
- Ervin MG, Ikegami M, Jobe AH, Polk DH, Newnham J. Multiple maternal glucocorticoid exposures alter preterm newborn endocrine responses. J Soc Gynecol Invest. 1997;4:81A. [Google Scholar]
- Ervin MG, Padbury JF, Polk DH, Ikegami M, Berry LM, Jobe AH. Antenatal glucocorticoids alter premature newborn lamb neuroendocrine and endocrine responses to hypoxia. Am J Physiol. 2000;279:R830–838. doi: 10.1152/ajpregu.2000.279.3.R830. [DOI] [PubMed] [Google Scholar]
- Fletcher AJW, Edwards CMB, Gardner DS, Fowden AL, Giussani DA. Neuropeptide Y in the sheep fetus: Effects of acute hypoxemia and dexamethasone during late gestation. Endocrinology. 2000a;141:3976–3982. doi: 10.1210/endo.141.11.7770. [DOI] [PubMed] [Google Scholar]
- Fletcher AJW, Goodfellow MR, Forhead AJ, Gardner DS, McGarrigle HHG, Fowden AL, Giussani DA. Low doses of dexamethasone suppress pituitary-adrenal function but augment the glycemic response to acute hypoxemia in fetal sheep during late gestation. Pediatr Res. 2000b;47:684–691. doi: 10.1203/00006450-200005000-00021. [DOI] [PubMed] [Google Scholar]
- Fletcher AJW, McGarrigle HHG, Edwards CMB, Fowden AL, Giussani DA. Effects of low dose dexamethasone treatment on basal cardiovascular and endocrine function in fetal sheep during late gestation. J Physiol. 2002;545:649–660. doi: 10.1113/jphysiol.2001.015693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner DS, Fletcher AJW, Bloomfield MR, Fowden AL, Giussani DA. Effects of prevailing hypoxaemia, acidaemia or hypoglycaemia upon the cardiovascular, endocrine and metabolic responses to acute hypoxaemia in the ovine fetus. J Physiol. 2002;540:351–366. doi: 10.1113/jphysiol.2001.013434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giussani DA, Gardner DS, Cox DR, Fletcher AJ. Purinergic contribution to the circulatory, metabolic, and adrenergic responses to acute hypoxemia in fetal sheep during late gestation. Am J Physiol. 2001;280:R678–685. doi: 10.1152/ajpregu.2001.280.3.R678. [DOI] [PubMed] [Google Scholar]
- Giussani DA, McGarrigle HHG, Moore PJ, Bennet L, Spencer JAD, Hanson MA. Carotid sinus nerve section and the increase in plasma cortisol during acute hypoxia in fetal sheep. J Physiol. 1994c;477:75–80. doi: 10.1113/jphysiol.1994.sp020172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giussani DA, McGarrigle HHG, Spencer JAD, Moore PJ, Bennet L, Hanson MA. Effect of carotid denervation on plasma vasopressin levels during acute hypoxia in the late-gestation sheep fetus. J Physiol. 1994b;477:81–87. doi: 10.1113/jphysiol.1994.sp020173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giussani DA, Riquelme RA, Moraga FA, McGarrigle HH, Gaete CR, Sanhueza EM, Hanson MA, Llanos AJ. Chemoreflex and endocrine components of cardiovascular responses to acute hypoxemia in the llama fetus. Am J Physiol. 1996;271:R73–83. doi: 10.1152/ajpregu.1996.271.1.R73. [DOI] [PubMed] [Google Scholar]
- Giussani DA, Spencer JA, Hanson MA. Fetal cardiovascular reflex responses to hypoxaemia. Fetal Matern Med Rev. 1994a;6:17–37. [Google Scholar]
- Giussani DA, Spencer JA, Moore PJ, Bennet L, Hanson MA. Afferent and efferent components of the cardiovascular reflex responses to acute hypoxia in term fetal sheep. J Physiol. 1993;461:431–449. doi: 10.1113/jphysiol.1993.sp019521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou QC, Slotkin TA. Effects of prenatal dexamethasone or terbutaline exposure on development of neural and intrinsic control of heart rate. Pediatr Res. 1989;26:554–557. doi: 10.1203/00006450-198912000-00005. [DOI] [PubMed] [Google Scholar]
- Hellbach S, Gartner P, Deicke J, Fischer D, Hassan AH, Almeida OF. Inherent glucocorticoid response potential of isolated hypothalamic neuroendocrine neurons. FASEB J. 1998;12:199–207. doi: 10.1096/fasebj.12.2.199. [DOI] [PubMed] [Google Scholar]
- Huch A, Huch R, Schneider H, Roth G. Continuous transcutaneous monitoring of fetal oxygen tension during labour. Br J Obstet Gynaecol. 1977;84:1–39. doi: 10.1111/j.1471-0528.1977.tb16231.x. [DOI] [PubMed] [Google Scholar]
- Iwamoto HS. Cardiovascular responses to reduced oxygen delivery: studies in fetal sheep at 0. 55–0.7 gestation. In: Gluckman PD, Johnston BM, Nathanielsz PW, editors. Advances in Fetal Physiology: Reviews in Honor of G. C. Liggins. Ithaca, New York: Perinatology Press; 1989. pp. 43–54. [Google Scholar]
- Jacobsson BA, Bergh CH, Hjalmarson A. Corticosteroid modulation of muscarinic receptors in rat myocardial membranes. Biochim Biophys Acta. 1983;760:77–83. doi: 10.1016/0304-4165(83)90126-5. [DOI] [PubMed] [Google Scholar]
- Jones CT, Roebuck MM, Walker DW, Johnston BM. The role of the adrenal medulla and peripheral sympathetic nerves in the physiological responses of the fetal sheep to hypoxia. J Dev Physiol. 1988;10:17–36. [PubMed] [Google Scholar]
- Jones CT, Wei G. Adrenal-medullary activity and cardiovascular control in the fetal sheep. In: Kunzel W, editor. Fetal Heart Rate Monitoring. Berlin: Springer-Verlag; 1985. pp. 127–135. [Google Scholar]
- Kallio J, Karlsson R, Toppari J, Helminen T, Scheinin M, Kero P. Antenatal dexamethasone treatment decreases plasma catecholamine levels in preterm infants. Pediatr Res. 1998;43:801–807. doi: 10.1203/00006450-199806000-00014. [DOI] [PubMed] [Google Scholar]
- Kennedy B, Elayan H, Ziegler MG. Glucocorticoid induction of epinephrine synthesizing enzyme in rat skeletal muscle and insulin resistance. J Clin Invest. 1993;92:303–307. doi: 10.1172/JCI116567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy B, Ziegler MG. Ontogeny of epinephrine metabolic pathways in the rat: role of glucocorticoids. Int J Dev Neurosci. 2000;18:53–59. doi: 10.1016/s0736-5748(99)00106-9. [DOI] [PubMed] [Google Scholar]
- Kornel L, Nelson WA, Manisundaram B, Chigurupati R, Hayashi T. Mechanism of the effects of glucocorticoids and mineralocorticoids on vascular smooth muscle contractility. Steroids. 1993;58:580–587. doi: 10.1016/0039-128x(93)90099-9. [DOI] [PubMed] [Google Scholar]
- Kornel L, Prancan AV, Kanamarlapudi N, Hynes J, Kuzianik E. Study on the mechanisms of glucocorticoid-induced hypertension: glucocorticoids increase transmembrane Ca2+ influx in vascular smooth muscle in vivo. Endocrinol Res. 1995;21:203–210. doi: 10.3109/07435809509030436. [DOI] [PubMed] [Google Scholar]
- Kream J, Mulay S, Fukushima DK, Solomon S. Determination of plasma dexamethasone in the mother and the newborn after administration of the hormone in a clinical trial. J Clin Endocrinol Metab. 1983;56:127–133. doi: 10.1210/jcem-56-1-127. [DOI] [PubMed] [Google Scholar]
- Kudlacz EM, Navarro HA, Slotkin TA. Phosphatidic acid phosphatase in neonatal rat lung: effects of prenatal dexamethasone or terbutaline treatment on basal activity and on responsiveness to beta adrenergic stimulation. J Pharmacol Exp Ther. 1989;250:236–240. [PubMed] [Google Scholar]
- Lau C, Slotkin TA. Maturation of sympathetic neurotransmission in the rat heart. VII. Suppression of sympathetic responses by dexamethasone. J Pharmacol Exp Ther. 1981;216:6–11. [PubMed] [Google Scholar]
- Leitman DC, Benson SC, Johnson LK. Glucocorticoids stimulate collagen and noncollagen protein synthesis in cultured vascular smooth muscle cells. J Cell Biol. 1984;98:541–549. doi: 10.1083/jcb.98.2.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linton-Dahlof P. Modulatory interactions of neuropeptide Y (NPY) on sympathetic neurotransmission. Acta Physiol Scand Suppl. 1989;586:1–85. [PubMed] [Google Scholar]
- Liu J, Haigh RM, Jones CT. Enhancement of noradrenaline-induced inositol polyphosphate formation by glucocorticoids in rat vascular smooth muscle cells. J Endocrinol. 1992;133:405–411. doi: 10.1677/joe.0.1330405. [DOI] [PubMed] [Google Scholar]
- Lundberg JM. Pharmacol of cotransmission in the autonomic nervous system: integrative aspects on amines, neuropeptides, adenosine triphosphate, amino acids and nitric oxide. Pharmacol Rev. 1996;48:113–178. [PubMed] [Google Scholar]
- Lundberg JM, Terenius L, Hokfelt T, Goldstein M. High levels of neuropeptide Y in peripheral noradrenergic neurons in various mammals including man. Neurosci Lett. 1983;42:167–172. doi: 10.1016/0304-3940(83)90401-9. [DOI] [PubMed] [Google Scholar]
- Matthews JNS, Altman DG, Campbell MJ, Royston P. Analysis of serial measurements in medical research. Br Med J. 1990;300:230–235. doi: 10.1136/bmj.300.6719.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molnar J, Nijland M, Howe D, Nathanielsz PW. Evidence for microvascular dysfunction after prenatal dexamethasone at 0. 7, 0.75 and 0.8 gestation in sheep. Am J Physiol. 2002;283:R561–567. doi: 10.1152/ajpregu.00031.2002. [DOI] [PubMed] [Google Scholar]
- Mormede P, Castagne V, Rivet JM, Gaillard R, Corder R. Involvement of neuropeptide Y in neuroendocrine stress responses. Central and peripheral studies. J Neural Transm Suppl. 1990;29:65–75. doi: 10.1007/978-3-7091-9050-0_8. [DOI] [PubMed] [Google Scholar]
- Mulder AL, Golde JM, Goor AA, Giussani DA, Blanco CE. Developmental changes in plasma catecholamine concentrations during normoxia and acute hypoxia in the chick embryo. J Physiol. 2000;527:593–599. doi: 10.1111/j.1469-7793.2000.00593.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newnham JP, Marshall CL, Padbury JF, Lam RW, Hobel CJ, Fisher DA. Fetal catecholamine release with preterm delivery. Am J Obstet Gynecol. 1984;149:888–893. doi: 10.1016/0002-9378(84)90610-0. [DOI] [PubMed] [Google Scholar]
- NIH Consensus Development Conference. National Institutes of Health Consensus Development Conference statement: effect of corticosteroids for fetal maturation on perinatal outcomes. Am J Obstet Gynecol. 1994;173:246–253. [Google Scholar]
- Padbury JF, Polk DH, Ervin MG, Berry LM, Ikegami M, Jobe AH. Postnatal cardiovascular and metabolic responses to a single intramuscular dose of betamethasone in fetal sheep born prematurely by cesarean section. Pediatr Res. 1995;38:709–715. doi: 10.1203/00006450-199511000-00013. [DOI] [PubMed] [Google Scholar]
- Papanek PE, Raff H. Physiological increases in cortisol inhibit basal vasopressin release in conscious dogs. Am J Physiol. 1994;266:R1744–1751. doi: 10.1152/ajpregu.1994.266.6.R1744. [DOI] [PubMed] [Google Scholar]
- Parer JT. The effect of atropine on heart rate and oxygen consumption of the hypoxic fetus. Am J Obstet Gynecol. 1984;148:1118–1122. doi: 10.1016/0002-9378(84)90638-0. [DOI] [PubMed] [Google Scholar]
- Pierce RA, Mariencheck WI, Sandefur S, Crouch EC, Parks WC. Glucocorticoids upregulate tropoelastin expression during late stages of fetal lung development. Am J Physiol. 1995;268:L491–500. doi: 10.1152/ajplung.1995.268.3.L491. [DOI] [PubMed] [Google Scholar]
- Raff H, Skelton MM, Cowley AW., Jr Cortisol inhibition of vasopressin and ACTH responses to arterial hypotension in conscious dogs. Am J Physiol. 1990;258:R64–69. doi: 10.1152/ajpregu.1990.258.1.R64. [DOI] [PubMed] [Google Scholar]
- Raff H, Wood CE. Effect of age and blood pressure on the heart rate, vasopressin, and renin response to hypoxia in fetal sheep. Am J Physiol. 1992;263:R880–884. doi: 10.1152/ajpregu.1992.263.4.R880. [DOI] [PubMed] [Google Scholar]
- Ransnas L, Hjalmarson A, Sabler E, Jacobsson B. Effect of corticosteroids on muscarinic receptors on cultured myocardial cells. Pharmacol Toxicol. 1987;61:107–110. doi: 10.1111/j.1600-0773.1987.tb01785.x. [DOI] [PubMed] [Google Scholar]
- Reuss ML, Parer JT, Harris JL, Krueger TR. Hemodynamic effects of alpha-adrenergic blockade during hypoxia in fetal sheep. Am J Obstet Gynecol. 1982;142:410–415. doi: 10.1016/s0002-9378(16)32381-x. [DOI] [PubMed] [Google Scholar]
- Segar JL, Lumbers ER, Nuyt AM, Smith OJ, Robillard JE. Effect of antenatal glucocorticoids on sympathetic nerve activity at birth in preterm sheep. Am J Physiol. 1998;274:R160–167. doi: 10.1152/ajpregu.1998.274.1.R160. [DOI] [PubMed] [Google Scholar]
- Slotkin TA, Lappi SE, McCook EC, Tayyeb MI, Eylers JP, Seidler FJ. Glucocorticoids and the development of neuronal function: effects of prenatal dexamethasone exposure on central noradrenergic activity. Biol Neonate. 1992;61:326–336. doi: 10.1159/000243761. [DOI] [PubMed] [Google Scholar]
- Stark RI, Wardlaw SL, Daniel SS, Husain MK, Sanocka UM, James LS, Vande Wiele RL. Vasopressin secretion induced by hypoxia in sheep: developmental changes and relationship to beta-endorphin release. Am J Obstet Gynecol. 1982;143:204–215. doi: 10.1016/0002-9378(82)90656-1. [DOI] [PubMed] [Google Scholar]
- Sze PY, Hedrick BJ. Effects of dexamethasone and other glucocorticoid steroids on tyrosine hydroxylase activity in the superior cervical ganglion. Brain Res. 1983;265:81–86. doi: 10.1016/0006-8993(83)91336-7. [DOI] [PubMed] [Google Scholar]
- Tangalakis K, Moritz K, Shandley L, Wintour EM. Effect of maternal glucocorticoid treatment on ovine fetal fluids at 0. 6 gestation. Reprod Fertil Dev. 1995;7:1595–1598. doi: 10.1071/rd9951595. [DOI] [PubMed] [Google Scholar]
- Unno N, Wu WX, Ding XY, Li C, Hing WK, Nathanielsz PW. The effects of fetal adrenalectomy at 110 days gestational age on AVP and CRH mRNA expression in the hypothalamic paraventricular nucleus of the ovine fetus. Dev Brain Res. 1998;106:119–128. doi: 10.1016/s0165-3806(97)00203-4. [DOI] [PubMed] [Google Scholar]
- Walker BR, Williams BC. Corticosteroids and vascular tone: mapping the messenger maze. Clin Sci. 1992;82:597–605. doi: 10.1042/cs0820597. [DOI] [PubMed] [Google Scholar]
- Wintour EM, Alcorn D, McFarlane A, Moritz K, Potocnik SJ, Tangalakis K. Effect of maternal glucocorticoid treatment on fetal fluids in sheep at 0. 4 gestation. Am J Physiol. 1994;266:R1174–1181. doi: 10.1152/ajpregu.1994.266.4.R1174. [DOI] [PubMed] [Google Scholar]