

Abstract
The natural history of arrhythmogenic right ventricular cardiomyopathy is determined by the electrical instability of the dystrophic myocardium, which can precipitate arrhythmic cardiac arrest any time during the course of the disease and by the progressive myocardial loss that results in ventricular dysfunction and heart failure. Sudden death accounts for the majority of the fatal events but its occurrence is mostly unpredictable. There are no prospective and controlled studies assessing clinical markers that can predict the occurrence of life‐threatening ventricular arrhythmias. However, the noninvasive risk profile, which emerges from retrospective analysis of clinical and pathologic series, is characterized by history of syncope, physical exercise, spontaneous ventricular tachycardia or ventricular fibrillation, right ventricular dysfunction, left ventricular involvement, right precordial negative T wave, right bundle branch block, QT‐QRS dispersion, right precordial ST‐segment elevation and late potentials. At present only QRS dispersion, history of syncope and right and/or left ventricular abnormalities at radionuclide angiography proved to be independent noninvasive predictors of sudden death.
Keywords: arrhythmias, ECG, right ventricular cardiomyopathy, risk stratification
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is a primary myocardial disease, often familial, that is characterized by structural and functional abnormalities of the right ventricle due to replacement of the myocardium by fatty and fibrous tissue. 1 , 2 Arrhythmias of right ventricular origin are the main clinical manifestations. 3 They range from isolated premature ventricular beats to sustained ventricular tachycardia (VT) or ventricular fibrillation (VF). Other clinical characteristics include ECG depolarization/repolarization changes in right precordial leads as well as global or regional dysfunction and structural alterations of the right ventricle. Right or biventricular heart failure that mimics dilated cardiomyopathy may appear during the clinical course. 4 , 5
Along the natural history, ARVC may manifest with the following clinical phases: concealed period, overt electrical disorder, right ventricular failure, and biventricular failure. 6 , 7 The “concealed” phase is characterized by subtle right ventricular changes, with or without ventricular arrhythmias. The pathologic process may be segmental and involve only one region of the so‐called triangle of dysplasia: the subtricuspidal region, the apex, and the outflow tract. In this phase patients are usually asymptomatic, present with or without non‐complex ventricular arrhythmias, and nevertheless are at risk of sudden death. The “overt” clinical phase is typical of symptomatic arrhythmic patients, who show overt right ventricular functional and structural abnormalities, in the setting of an increasing risk of sudden death. Right ventricular failure is caused by massive involvement of the right ventricle, determining a global dysfunction with a relatively preserved left ventricular function. Biventricular failure is the final stage of ARVC, explained by pronounced left ventricular involvement and often associated with atrial fibrillation and thromboembolic events.
The natural history of ARVC is determined by the electrical instability of dystrophic myocardium, which can precipitate sudden arrhythmic cardiac arrest any time during the course of the disease and by the progressive myocardial loss that results in ventricular dysfunction and heart failure. Sudden death accounts for the majority of the fatal events and usually occurs in adolescents and young adults. Conversely heart failure may complicate the follow‐up of ARVC and in pathologic series may give reason for 20% of death. 4 While ventricular dysfunction is progressive up to heart failure, the occurrence of fatal arrhythmic events is mainly unpredictable. 5 , 8
The frequency of sudden death varies in the studies on the long‐term follow‐up. Sudden death occurred in 2 of 15 patients reported by Blomström‐Lundqvist et al. 9 (mean follow‐up 8.8 years), in no subjects of the original series of Marcus et al. 10 (mean follow‐up 8.4 years), in 1 of 7 patients of the series of Reiter et al. 11 (mean follow‐up 5.1 years), in 1 of 58 individuals of the series of Leclercq and Coumel 12 (mean follow‐up 8.8 years), in 2 of 17 adolescent subjects of Daliento et al. 13 (mean follow‐up 7 years), in 14 of 106 subjects in the series of Le Guludec et al. 14 (mean follow‐up 4 years), and in 1 of 151 familial cases of Nava et al. 15 (mean follow‐up of 8.5 years). About half of these subjects who died suddenly were not protected by antiarrhythmic therapy. ARVC seems to have a favorable outcome when recognized and treated. Fontaine et al. 16 estimated a rate of arrhythmic death of 2% per year, despite various forms of pharmacological and nonpharmacological treatments. Canu et al. 17 calculated a 10‐year actuarial survival rate of 75% for patients with VT, treated medically. In patients with severe arrhythmias, defined as sustained VT or symptomatic nonsustained VT, the frequency of sudden death during a mean follow‐up of about 4 years reached 13.2% (3.4% per year). 14 Familial cases exhibited a mortality rate of 0.08 patient per year. 15
This relatively low incidence of sudden death makes it difficult to assess the arrhythmic risk in the patients predisposed to such an ominous event. There are no prospective and controlled studies assessing clinical markers, which can predict the occurrence of life‐threatening ventricular arrhythmias. The noninvasive risk profile, which emerges from retrospective analysis of clinical and pathologic series is characterized by the history of syncope, physical exercise, spontaneous VT and VF, right ventricular dysfunction, left ventricular involvement, right precordial negative T wave, right bundle branch block, QT‐QRS dispersion, right precordial ST‐segment elevation, and late potentials.
SYNCOPE
Marcus et al. 10 analyzed syncope as a prognostic factor for arrhythmic death in their series of 33 patients. They suggested a more favorable long‐term prognosis in those patients who did not have a history of syncope. Turrini et al. 18 found syncope to be an independent predictor of sudden death with a sensitivity of 40% and a specificity of 90% (Table 1). Also, when the course of ventricular arrhythmias seems to be unpredictable in individual patients, syncope appears to be a prognostic factor. Among the 15 patients of Blomström‐Lundqvist et al. 9 a history of syncope was present in all the 3 patients who died and in only 2 of the 12 patients who survived. Syncope is more frequent in young than in adult patients 13 and it is the most important warning symptom preceding sudden death in the familial ARVC patients. Nava et al. 15 demonstrated that syncope was the only significant clinical variable that distinguished the clinical picture of 19 probands who died suddenly from 132 living affected patients.
Table 1.
Comparison of Clinical and ECG Data in Three ARVC Groups (from Turrini et al., 18 modified)
| Group I (20 pts) | Group II (20 pts) | Group III (20 pts) | |
|---|---|---|---|
| Age (yrs) | 24.8 ± 6.5 | 26.4 ± 6.2 | 22.4 ± 7.4 |
| Sex | M = 17, F = 3 | M = 18, F = 2 | M = 16, F = 4 |
| Complete RBBB | 4 (20%) | 3 (15%) | 1 (5%) |
| ST‐segment elevation on right precordial leads | 8 (40%) | 8 (40%) | 3 (15%) |
| Epsilon wave | 7 (35%) | 5 (25%) | 6 (30%) |
| Negative T wave beyond V1 | 17 (85%)a | 14 (70%) | 9 (45%) |
| Syncope | 8 (40%)b | 4 (20%) | 0 |
F = female; Group I = patients who died suddenly; Group II = patients with sustained ventricular tachycardia; Group III = patients with no sustained ventricular tachycardia; M = male; aP = 0.02 versus Group II and III; bP = 0.007 versus Group II and III; pts = patients; RBBB = right bundle‐branch block; yrs = years.
PHYSICAL EXERCISE
Effort is a significant precipitating factor of sudden death in patients with ARVC. In the study of Thiene et al. 1 among 60 sudden unexplained deaths under the age of 35 years, 10 out of 12 individuals (87%) with ARVC died during exertion. In 5 cases, sudden death was the first sign of the disease. Furthermore Corrado et al. 19 showed that ARVC was the leading cause of sudden death among young competitive athletes. An imbalanced autonomic input in the heart is likely to intervene in the functional chain of events culminating in sudden death. Leclercq et al. 20 reported a change in the vagosympathetic balance with an increased sympathetic tone in patients with spontaneous VT at Holter monitoring. Haissaguerre et al. 21 demonstrated that high dose of isoproterenol, mimicking catecholamines effect, induced VT in the great majority of the patients. Wichter et al. 22 found an increased vulnerability to the cathecolamines effects in patients with regional abnormalities of sympathetic innervation at 131 I‐MIBG imaging. The myocardium deprived of its nerve supply responds abnormally to catecholamine exposure, a phenomenon called denervation supersensitivity. Genetic predisposition may enhance the effect of catecholamines. In the subgroup of patients with a peculiar familial form of ARVD2, mapping to chromosome 1q42‐43, which recently was ascribed to defective gene encoding ryanodine receptor 2, adrenergic stimulation induced life‐threatening ventricular arrhythmias. 23 , 24 These patients had right ventricular apical aneurisms and exercise‐induced polymorphic VT associated with a high risk of sudden death during effort. Premature ventricular beats showed a long coupling interval and a polymorphic QRS morphology, often featuring bidirectional VT.
SPONTANEOUS VT AND VF
In ARVC monomorphic VT and VF are two unrelated arrhythmic manifestations. Usually VT does not precede VF. In the series of Nimkhedkar et al. 25 concerning 10 patients undergoing surgery for life‐threatening drug‐refractory ventricular arrhythmias, none had a history of VF. In the series of Leclercq and Coumel 12 only two out of 50 patients with VT also had a history of VF. No episodes of VF were recorded in patients who have had insertion of an implantable cardioverter‐defibrillator, after a mean follow‐up of 11 months. 26 Conversely, cardiac arrest may be the first manifestation of the disease and it is most likely precipitated by abrupt VF. Aouate et al. 27 described a primary VF recorded on Holter monitoring, in a sudden death case which was found at postmortem to have ARVC.
Frequent VT requiring hospital admission has a less favorable long‐term outcome, even though the VT appeared to be controlled by antiarrhythmic drugs. 17 Recurrences of VT cannot be predicted by noninvasive test such as Holter monitoring or exercise testing and by programmed ventricular stimulation. 28 Also, resuscitated VF is a poor prognostic factor. Among the 20 patients of Canu et al., 17 a history of VF was present in two of the three who died suddenly, despite pharmacological and/or ablative therapy.
In patients with right ventricular outflow tachycardia (RVOT), accurate investigation of underlying abnormalities of right ventricular structure and function is required. The diagnosis of idiopathic RVOT, which is defined as a primary electrical disease in the absence of structural heart disease, is one of exclusion. Although multiple mechanisms have been implicated with the genesis of idiopathic RVOT, the most common is triggered activity from delayed afterdepolarizations, which are cAMP dependent and related to intracellular calcium overload. These forms of VT are facilitated by catecholamines and characteristically terminate with adenosine as well as vagal maneuvers, beta‐blockers, and verapamil. In the idiopathic RVOT, conventional imaging modalities, including echocardiography and right cine angiography, do not demonstrate remarkable structural abnormalities. In contrast, magnetic resonance imaging (MRI) consistently reveals focal thinning, fatty infiltrations, and wall motion abnormalities of the right ventricle in 95% of the patients. 29 The functional significance of these anatomic abnormalities is uncertain. In the series of Markowitz et al. 30 there was a poor correlation between the site of MRI abnormalities and the origin of VT. Whether idiopathic RVOT is a concealed form of ARVC needs to be clarified. Recently, body surface mapping proved to be useful in differentiating patients with ARVC from patients with idiopathic RVOT, especially in the early diagnostic evaluation, or in asymptomatic family members, or when the results of other diagnostic techniques are inconclusive. 31 , 32
The good clinical course of idiopathic RVOT explains the importance of an accurate diagnosis. In the report of Pietras et al. 33 sudden death occurred in 2 of the 15 patients with right ventricular abnormalities not diagnosed with ARVC, while no death occurred in 23 patients with completely normal hearts.
RIGHT VENTRICULAR DYSFUNCTION
Right ventricular enlargement has been observed in ARVC patients up to 92%. 3 However there are large interindividual differences in ARVC, according to the clinical phase of the disease. Patients with concealed disease may have a normal right ventricular volume in 50% of cases. A decrease of right ejection fraction has been reported in 60–70% of ARVC patients, but it can be normal in the localized forms. 5
Severe dilatation and reduction of right ventricular ejection fraction have been associated with a worse arrhythmic prognosis. In high‐risk patients with aborted or nonaborted sudden death, recurrent VT, or syncope, Peters et al. 34 demonstrated an increased frequency of right ventricular dilatation as compared to low risk patients. Turrini et al. 35 described a relationship between sustained ventricular arrhythmias and a reduced right ventricular ejection fraction (≤50%). Echocardiography has been used as the first diagnostic tool to evaluate functional or structural right ventricular abnormalities. It allows serial examinations to document progression of the disease, without identifiable risk or discomfort to the patient. The sensitivity and specificity of this technique in the diagnosis of ARVC is high, especially when the disease is diffuse. Risk stratification based on radionuclide angiography showed that the detection of even focal right ventricular abnormalities was the most important independent predictor of sudden death. 14 However, all these patients had severe ventricular arrhythmias during their clinical history and the rate of death was comparable in patients with diffuse or localized right ventricular disease during a mean follow‐up at 45 ± 34 months.
LEFT VENTRICULAR INVOLVEMENT
Although ARVC was initially considered a disease strictly localized to the right ventricle, many studies demonstrated that left ventricular abnormalities were common and frequently associated with a progression of the disease. In the meta‐analysis of Pinamonti and Camerini, 36 of more than 500 patients about one‐third presented a left ventricular involvement. In the 10 major clinical series of ARVC on a total of 347 patients, left ventricular abnormalities were present in 18% of patients, ranging from 0 to 55%. In the pathologic experience left ventricular involvement exceeds that observed in the clinical ground. In the clinicopathologic series of Corrado et al., 4 the left ventricle was anatomically involved in the majority of the patients: only histologically in 36% and histologically and macroscopically in another 40% of the patients. Left ventricular involvement occurred in older patients with a longer clinical history and was progressing during serial echocardiographic examinations. It was associated with clinical arrhythmic events, severe cardiomegaly, inflammatory infiltrates, and heart failure. A left ventricular involvement appears to portend a poor arrhythmic prognosis. Peters et al. 34 reported an association between the risk of sudden death and left ventricular involvement, regardless of its extension. In this study, left ventricular involvement was demonstrated in 83% of the high‐risk patients and in only 12% of the low risk group. Left ventricular involvement was mainly localized and associated with a normal left ventricular function. Only 6% of these patients presented an overall dilatation and global contraction reduction. In the prospective noninvasive risk stratification of Le Guludec et al., 14 left ventricular abnormalities at radionuclide angiography were the second independent predictor of sudden death, after right ventricular abnormalities. Patients with global left systolic dysfunction had a similar prognosis as those with localized left wall motion abnormalities.
RIGHT PRECORDIAL NEGATIVE T WAVES
Although right precordial inverted T wave in people aged more than 12 years and, in the absence of right bundle branch block, is a minor criterion of diagnosis, there are few data about the frequency of repolarization abnormalities in the normal population. Some normal individuals show persistence of right precordial negative T wave in early adulthood, the so‐called “juvenile” t‐wave pattern. However, in a series of 2647 athletes (age range 15–21 years) with normal echocardiogram, 37 only 16 (0.6%) showed a negative T wave in V1–V2 or V1–V3, thus suggesting that the diagnosis of juvenile t‐wave pattern is one of exclusion. In ARVC patients precordial t‐wave inversion has been reported in up to 90% cases. 3 , 11 , 25
The extension of t‐wave inversion on right precordial leads has been directly correlated with right ventricular enlargement. 38 The concomitant presence of negative T wave in the lateral leads V5–V6 may be a marker for left ventricular involvement. 4 Recently, t‐wave inversion has been reported to be a marker for the risk of life‐threatening arrhythmias. 18 , 34 The specificity of this sign depends on the extent of precordial lead involvement. Turrini et al. 18 found that negative T waves beyond V1 had a sensitivity of 85% and a specificity of 42% in predicting the risk of sudden death. Peters et al. 34 demonstrated that when negative T waves were considered beyond V3, the specificity and sensitivity increased to 82% and 62%, respectively.
RIGHT BUNDLE BRANCH BLOCK
The great majority of patients with right or biventricular failure exhibit a right bundle branch block, whereas patients with a localized form show normal or slightly enlarged QRS complex. Localized prolongation of QRS complex in V1–V3 to more than 110 ms has a sensitivity of 55% and a specificity of 100% for the diagnosis of the disease. 39 This QRS prolongation or the pattern of incomplete or complete right bundle branch block is usually due to intraventricular myocardial defect (parietal block) rather than a specialized conduction system (septal block). 40 However, marked dilatation of the right ventricle in the setting of global right ventricular dysfunction or biventricular failure can dislocate the right bundle branch, leading to incomplete or complete right bundle branch block. In this case, right bundle branch block is a strong noninvasive predictor of heart failure and it can occur years before the development of right ventricular failure. 41 The degree of QRS prolongation is somewhat correlated with the arrhythmic status of ARVC patients. In the series of Turrini et al., 18 patients who died suddenly showed a maximum QRS of 125 ms as compared to patients without (106 ms) and with ventricular tachycardia (113 ms) (Table 2). A prolonged right precordial QRS complex may be associated with the presence of epsilon waves in 30% of the cases. Epsilon waves are potentials of small amplitude just beyond the QRS complex at the beginning of ST segment and reflect delayed ventricular activation of some myocardial fibers of the right ventricle. Sometimes the shape of epsilon waves may look like a smooth potential, forming an atypical prolonged R wave in lead V1–V3. 16
Table 2.
Comparison of QRS Duration and QT‐QRS Dispersion in Three ARVC Groups (from Turrini et al., 18 modified)
| Group I (20 pts) | Pa | Group II (20 pts) | Pb | Group III (20 pts) | |
|---|---|---|---|---|---|
| QRS max (ms) | 125.2 ± 18.3 | 0.08 | 113 ± 21 | N.S. | 106.5 ± 9.8 |
| QTD (ms) | 77.5 ± 10.6 | 0.001 | 64.5 ± 13.9 | <0.0001 | 48 ± 8.9 |
| QRSD (ms) | 45.7 ± 8.1 | <0.0001 | 33.5 ± 8.7 | 0.07 | 28 ± 5.2 |
Group I = patients who died suddenly; Group II = patients with sustained ventricular tachycardia; Group III = patients with no sustained ventricular tachycardia; QRSD = QRS dispersion; QTD = QT dispersion; aGroup I versus Group II; bGroup II versus Group III.
QT AND QRS DISPERSION
In myocardial disease there is an increased electrical heterogeneity that is likely to be responsible for ventricular instability. QT interval dispersion, defined as the difference in duration between the longest and the shortest QT intervals in any lead of surface ECG, has been suggested to reflect regional variations in ventricular repolarization and then provide an indirect measure of the extent of nonuniformity of myocardial repolarization. In the study of Benn et al. 42 QT dispersion was increased in patients with ARVC, but did not change in response to treatment with sotalol; neither was it a predictor of life‐threatening arrhythmias. Also, Peeters et al., 32 analyzing ARVC patients with sustained VT did not find an increased QT dispersion, though they noted a relationship between repolarization abnormalities and ventricular arrhythmias. In contrast, Turrini et al. 18 showed that a QT dispersion > 65 ms had a sensitivity of 85% and a specificity of 77% for predicting the occurrence of sudden death (Fig. 1). Whether QT dispersion reflects the true regional dispersion of repolarization times or it is an index of repolarization abnormalities as well as right negative T waves in precordial leads is still unclear. 43 As QT dispersion has been suggested to reflect regional inhomogeneity of repolarization times, QRS dispersion is likely to represent regional inhomogeneity of depolarization times. In the series of Peters et al. 34 concerning patients at high and low arrhythmic risk, a QRS dispersion ≥ 50 ms was a marker of increased arrhythmogenicity. In 20 patients who died suddenly with ARVC proven at autopsy, Turrini et al. 18 showed that QRS dispersion was the strongest independent predictor of sudden death, with a sensitivity of 90% and a specificity of 77% when ≥ 40 ms (Fig. 2). These data suggest that depolarization abnormalities are more commonly associated with cardiac arrest than repolarization abnormalities in ARVC. 18
Figure 1.

Distribution of QT dispersions in three ARVC groups: group I is composed of 20 patients who died suddenly, group II of 20 patients with sustained ventricular tachycardia, and group III of 20 patients with no sustained ventricular tachycardia. Mean values are indicated by a horizontal line (from Turrini et al., 18 modified).
Figure 2.
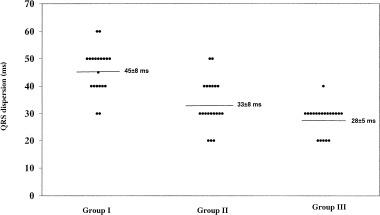
Distribution of QRS dispersions in three ARVC groups: group I is composed of 20 patients who died suddenly, group II 20 of patients with sustained ventricular tachycardia, and group III of 20 patients with no sustained ventricular tachycardia. Mean values are indicated by horizontal line (from Turrini et al, 18 modified).
RIGHT PRECORDIAL ST SEGMENT ELEVATION
The presence of an upward displacement of ST segment elevation has been frequently reported in ARVC. Nava et al. 15 found an incidence of 12% of ST segment elevation in familial cases of ARVC. Fontaine et al. 44 estimated a frequency of 30% in the clinically documented cases of ARVC. They described three categories of ST segment elevation in ARVC. Two are commonly observed during arrhythmias: triangular with a raised J point and dome shaped with a normal J point. The third form with a saddle‐shaped appearance and an ST segment elevation > 1 mm, is typically observed during intercritical periods. In the series of Peters et al. 34 concerning 121 patients with ARVC, a nonspecific upward displacement of ST segment was encountered in 18% of the patients and it was not predictive of the occurrence of sudden death. Also in the study of Turrini et al., 18 an ST segment elevation ≥ 0.5 mm had a comparable frequency in three different groups of patients, that is, those who died suddenly, those with sustained VT, and those with no sustained VT (Table 1). An ST segment elevation >1 mm with a coved or saddle‐back aspect has been associated with a high risk of sudden death. Martini et al. 45 studied six patients with ventricular fibrillation and an apparent normal heart; three of these patients showed at ECG a distinct right precordial ST segment elevation associated with right bundle branch block (complete in two and incomplete in one) and negative right T waves. In these three patients the authors documented a concealed ARVC, confirmed by autopsy in one case. Corrado et al. 46 showed that familial ARVC could produce a syndrome characterized by right bundle branch block, ST segment elevation, and sudden death. They described 16 members of a family affected with ARVC, whose proband died suddenly and was found to have a sclerotic interruption of right bundle branch.
A right precordial ST segment elevation and a right bundle branch block are markers of a high risk for malignant arrhythmias even for patients without cardiac disease. Brugada and Brugada 47 described a syndrome characterized by ST segment elevation in V1–V3 and right bundle branch block associated with a high risk of sudden death in the absence of obvious structural disease. Since then many other patients were recognized to be affected by the Brugada syndrome, which is now defined as an arrhythmogenic disorder associated with a high risk of sudden death in individuals with a structurally normal heart. The disease is characterized by transient right bundle branch block and ST segment elevation (usually ≥ 1 mm) in the right precordial leads. The electrocardiographic features of the Brugada syndrome are dynamic and often concealed, but can be unmasked by sodium channel blockers. Genetic mutations of the cardiac channel gene SCN5A can lead to loss of action potential dome at some epicardial sites. 48 The consequent development of a marked transmural dispersion of repolarization and refractoriness may predispose to VF. The occurrence of cardiac arrest at three years of follow‐up has been estimated to be 30% in symptomatic and asymptomatic patients.
An overlap between ARVC with ST segment elevation and Brugada syndrome has been recently demonstrated by Corrado et al. 49 This subgroup of ARVC patients showed predominant right ventricular anterior wall involvement, ECG changes over time, polymorphic VT, propensity to die from non‐exercise‐related cardiac arrest, all features encountered in Brugada syndrome.
Being patients with a pattern of right bundle branch block, ST segment elevation, and no cardiac disease at risk for malignant ventricular arrhythmias, it is then incumbent on the clinician to carefully exclude organic cardiac disease before accepting the Brugada syndrome as a distinct entity. The modulation of ST segment elevation by the effects of autonomic and antiarrhythmic drugs, in addition, makes it difficult to identify the disease when concealed. The presence of organic cardiac disease, particularly ARVC, familial conduction system disease, and cardiomyopathy or infiltrative cardiomyopathy must be excluded through noninvasive (ECG, Holter monitoring, echocardiogram, MRI) and invasive tests (angiography and endomyocardial biopsy).
LATE POTENTIALS
The signal‐averaged ECG (SAECG) has been used to detect low‐amplitude, high‐frequency and altered‐frequency components in the terminal QRS complex. These signals are called late potentials (LP) and are considered a noninvasive marker for areas of slow conduction that are a prerequisite for reentrant arrhythmias. In the original series of Marcus et al., 3 among the 16 patients who underwent SAECG 13 (81%) had LP. Blomström‐Lundqvist et al. 50 reported similar results in their study, LP being present in 72% of the 18 patients with ARVC. In the series of Wichter et al., 22 LP were present in 24 of 48 patients (50%), with a higher incidence in those with a classic form of ARVC and more pronounced right ventricular contraction abnormalities. Mehta et al. 51 demonstrated a relationship between the extent of abnormality of SAECG variables and the degree of right ventricular enlargement. Nava et al. 52 confirmed the good correlation between SAECG and the extent of the disease. These findings explain why SAECG can identify patients with ARVC with a high specificity and an intermediate sensitivity. Combining the time‐ and frequency‐domain analysis can increase sensitivity without diminution of specificity. 53
The studies on the role of this technique for arrhythmic stratification did not show good correlation between LP and the occurrence of malignant ventricular arrhythmias. Blomström‐Lundqvist et al. 50 found that LP were not predictive of ventricular arrhythmia in ARVC patients. Also, the follow‐up by repeated SAECG did not appear to be useful in predicting the susceptibility to VT. 54 In the report of Leclercq and Coumel, 55 the prognostic value of LP was uncertain and the absence of LP did not exclude the risk of sudden death. In the three patients with a history of VF, LP were absent. Additionally, the prevalence of LP was similar in patients with or without sustained VT. Turrini et al. 35 demonstrated that LP were a univariate predictor of sustained arrhythmias. However, their multivariate analysis showed that a decreased right ejection fraction was an independent predictor of sustained arrhythmias, whereas LP were not. In the familial form of ARVC, the abnormality on the SAECG was correlated with the severity of the disease, but not with electrical instability. 56 The predictive value of SAECG in this particular subgroup was low: only 44% of subjects with LP had arrhythmias, whereas 76% with arrhythmias had abnormal SAECG. Also for Nava et al., 52 SAECG did not correlate well with ventricular arrhythmias.
All these results suggest that LP are a marker for the pathological substrate, namely fibro‐fatty substitution, rather than an index of vulnerability to arrhythmias.
This study was supported by Progetto Telethon no. 1288, Rome and ARVC/D Project, QLG1‐CT‐2000‐01091 5th Framework Programme European Commission, Bruxelles.
REFERENCES
- 1. Thiene G, Nava A, Corrado D, et al Right ventricular cardiomyopathy and sudden death in young people. N Engl J Med 1988;318: 129–133. [DOI] [PubMed] [Google Scholar]
- 2. Basso C, Thiene G, Corrado D, et al Arrhythmogenic right ventricular cardiomyopathy: dysplasia, dystrophy or myocarditis? Circulation 1996;94: 983–991. [DOI] [PubMed] [Google Scholar]
- 3. Marcus FI, Fontaine GH, Guiraudon G, et al Right ventricular dysplasia: A report of 24 adult cases. Circulation 1982;65: 384–398. [DOI] [PubMed] [Google Scholar]
- 4. Corrado D, Basso C, Thiene G, et al Spectrum of clinicopathologic manifestations of arrhythmogenic right ventricular cardiomyopathy/dysplasia: A multicenter study. J Am Coll Cardiol 1997;30: 1512–1520. [DOI] [PubMed] [Google Scholar]
- 5. Nava A, Rossi L, Thiene G, eds. Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia. Amsterdam , Elsevier, 1997. [Google Scholar]
- 6. Corrado D, Fontaine G, Marcus FI, et al Arrhythmogenic right ventricular dysplasia/cardiomyopathy: Need for an international registry. Circulation 2000;101: e101–e106. [DOI] [PubMed] [Google Scholar]
- 7. McKenna WJ, Thiene G, Nava A, et al Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Br Heart J 1994;71: 215–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Corrado D, Basso C, Thiene G. Arrhythmogenic right ventricular cardiomyopathy: Diagnosis prognosis and treatment. Heart 2000;83: 588–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Blomström‐Lundqvist C, Sabel KG, Olsson SB. A long term follow‐up of 15 patients with arrhythmogenic right ventricular dysplasia. Br Heart J 1987;58: 477–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Marcus FI, Fontaine GH, Frank R, et al Long‐term follow‐up in patients with arrhythmogenic right ventricular disease. Eur Heart J 1989;10(Suppl. D):68–73. [DOI] [PubMed] [Google Scholar]
- 11. Reiter MJ, Smith WM, Gallagher JJ. Clinical spectrum of ventricular tachycardia with left bundle branch morfology. Am J Cardiol 1983;51: 113–121. [DOI] [PubMed] [Google Scholar]
- 12. Leclercq JF, Coumel P. Characteristics, prognosis, and treatment of the ventricular arrhythmias of right ventricular dysplasia. Eur Heart J 1989;10(Suppl D):61–67. [DOI] [PubMed] [Google Scholar]
- 13. Daliento L, Turrini P, Nava A, et al Arrhythmogenic right ventricular cardiomyopathy in young versus adult patients: similarities and differences. J Am Coll Cardiol 1995;25: 655–664. [DOI] [PubMed] [Google Scholar]
- 14. Le Guludec D, Gauthier H, Porcher R, et al Prognostic value of radionuclide angiography in patients with right ventricular arrhythmias. Circulation 2001;103: 1972–1976. [DOI] [PubMed] [Google Scholar]
- 15. Nava A, Bauce B, Basso C, et al Clinical profile and long‐term follow‐up of 37 families with arrhythmogenic right ventricular cardiomyopathy. J Am Coll Cardiol 2000;36: 2226–2233. [DOI] [PubMed] [Google Scholar]
- 16. Fontaine G, Fontaliran F, Lascault G, et al Arrhythmogenic Right Ventricular Dysplasia In Zipes DP, Jalife J. (eds.): Cardiac Electrophysiology: From Cell to Bedside. Philadelphia , W B Saunders, 1994, pp. 754–768. [Google Scholar]
- 17. Canu G, Atallah G, Claudel JP, et al Pronostic et évolution à long term de la dysplasie arythmogène du ventricule droit. Arch Mal Coeur 1993;86: 41–48. [PubMed] [Google Scholar]
- 18. Turrini P, Corrado D, Basso C, et al Dispersion of ventricular depolarization‐repolarization: A non invasive marker for risk stratification in arrhythmogenic right ventricular cardiomyopathy. Circulation. 2001;103: 3075–3080. [DOI] [PubMed] [Google Scholar]
- 19. Corrado D, Thiene G, Nava A, et al Sudden death in young competitive athletes: Clinocopathologic correlations in 22 cases. Am J Med 1990;89: 588–596. [DOI] [PubMed] [Google Scholar]
- 20. Leclercq JF, Potenza S, Maison‐Blanche P, et al Determinants of spontaneous occurrence of sustained monomorphic ventricular tachycardia in right ventricular dysplasia. J Am Coll Cardiol 1996;28: 720–724. [DOI] [PubMed] [Google Scholar]
- 21. Haissaguerre M, Le Metayer P, D'Ivernois C, et al Distinctive response of arrhythmogenic right ventricular disease to high dose isoproterenol. PACE 1990;13: 2119–25. [DOI] [PubMed] [Google Scholar]
- 22. Wichter T, Hindricks G, Lerch H, et al Regional myocardial sympathetic dysinnervation in arrhythmogenic right ventricular cardiomyopathy: an analysis using 123I‐Meta‐Iodobenzylguanidine Scintigraphy. Circulation 1994;89: 667–683. [DOI] [PubMed] [Google Scholar]
- 23. Bauce B, Nava A, Rampazzo A, et al Familial effort polymorphic ventricular arrhythmias in arrhythmogenic right ventricular cardiomyopathy map to chromosome 1q42‐43. Am J Cardiol 85: 573–579. [DOI] [PubMed] [Google Scholar]
- 24. Buace B, Rampazzo A, Basso C, et al Screening for ryanodine receptor type 2 mutations in families with effort‐induced polymorphic ventricular arrhythmias and sudden death. J Am Coll Cardiol 2002. 17;40: 341–349. [DOI] [PubMed] [Google Scholar]
- 25. Nimkhedkar K, Hilton CJ, Furniss SS, et al Surgery for ventricular tachycardia associated with right ventricular dysplasia: Disarticulation of right ventricle in 9 of 10 cases. J Am Coll Cardiol 1992;19:1079–1084. [DOI] [PubMed] [Google Scholar]
- 26. Wichter T, Block M, Bocker D, et al Cardioverter‐defibrillator therapy in a high‐risk subgroup of patients with arrhythmogenic right ventricular disease. J Am Coll Cardiol 1993;21: 127A. [Google Scholar]
- 27. Aouate P, Fontaliran F, Fontaine G, et al Holter et mort subite: Interet dans un cas de dysplasie ventriculaire droite arythmogene. Arch Mal Coeur 1993;86: 363–367. [PubMed] [Google Scholar]
- 28. Lemery R, Brugada P, Janssen J, et al Non ischemic sustained ventricular tachycardia: clinical outcome in 12 patients with arrhythmogenic right ventricular dysplasia. J Am Coll Cardiol 1989: 14: 96–105. [DOI] [PubMed] [Google Scholar]
- 29. Carlson MD, White RD, Trohman RG, et al Right ventricular outflow tract ventricular tachycardia: detection of previously unrecognized anatomic abnormalities using cine magnetic resonance imaging. J Am Coll Cardiol 1994;24: 720–727. [DOI] [PubMed] [Google Scholar]
- 30. Markowitz SM, Litvak BL, Ramirez de Arellano EA, et al Adenosine‐sensitive ventricular tachycardia: Right ventricular abnormalities delineated by magnetic resonance imaging. Circulation 1997;96: 1192–1200. [DOI] [PubMed] [Google Scholar]
- 31. De Ambroggi L, Aimè E, Ceriotti C, et al Mapping of ventricular repolarization potentials in patients with arrhythmogenic right ventricular dysplasia: Principal component analysis of the ST‐T waves. Circulation 1997;96: 4314–8. [DOI] [PubMed] [Google Scholar]
- 32. Peeters HA, Sippens Groenewegen A, Schoonderwoerd BA, et al Body‐surface QRST integral mapping: arrhythmogenic right ventricular dysplasia versus idiopathic right ventricular tachycardia. Circulation 1997;95: 2668–2676. [DOI] [PubMed] [Google Scholar]
- 33. Pietras R, Lam W, Bauerfeind R, et al Chronic recurrent right ventricular tachycardia in patients without ischemic heart disease: Clinical, hemodynamic and angiographic findings. Am Heart J 1983;105: 357–371. [DOI] [PubMed] [Google Scholar]
- 34. Peters S, Peters H, Thierfelder L. Risk stratification of sudden cardiac death and malignant ventricular arrhythmias in right ventricular dysplasia‐cardiomyopathy. Int J Cardiol 1999;71: 243–250. [DOI] [PubMed] [Google Scholar]
- 35. Turrini P, Angelini A, Thiene G, et al Late potentials and ventricular arrhythmias in arrhythmogenic right ventricular cardiomyopathy. Am J Cardiol 1999;83: 1214–1219. [DOI] [PubMed] [Google Scholar]
- 36. Pinamonti B, Camerini F. Left ventricular involvement, progression of the disease and prognosis In Nava A, Rossi L, Thiene G. (eds.): Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia. Amsterdam , Elsevier, 1997, pp. 46–60. [Google Scholar]
- 37. Mantovani E, Piovesana P, Lafisca N, et al Valutazione clinica, ecocardiografica, ergometrica, ECG dinamica, vettorcardiografica in giovani sportivi con onda T negativa in sede precordiale des In Aulo Gaggi. (ed.):Atti III Congresso SIC‐Sport, Bologna . [Google Scholar]
- 38. Nava A, Canciani B, Buja G, et al Electrovectorcardiographic study of negative T waves on precordial leads in arrhythmogenic right ventricular dysplasia: Relationship with right ventricular volumes. J Electrocardiol 1988;21: 239–245. [DOI] [PubMed] [Google Scholar]
- 39. Fontaine G, Umemura J, Di Donna P, et al La duree des complexes QRS dans la dysplasie ventriculaire droite arythmogene: Un noveau marqueur diagnostique non invasif. Ann Cardiol Angeiol 1993;42: 399–405. [PubMed] [Google Scholar]
- 40. Fontaine G, Frank R, Guiraudon C, et al Signification des troubles de conduction intraventriculaire observes dans la dysplasie ventriculaire droite arythmogene. Arch Mal Coeur 1984;77: 872–879. [PubMed] [Google Scholar]
- 41. Peters S, Peters H, Thierfelder L. Heart failure in arrhythmogenic right ventricular dysplasia‐cardiomyopathy. Int J Cardiol 1999;71: 251–256. [DOI] [PubMed] [Google Scholar]
- 42. Benn M, Hansen PS, Pedersen AK. QT dispersion in patients with arrhythmogenic right ventricular dysplasia. Eur Heart J 1999;20: 764–770. [DOI] [PubMed] [Google Scholar]
- 43. Malik M, Batchvarov VN. Measurements, interpretation and clinical potential of QT dispersion. J Am Coll Cardiol 2000;36: 1749–1766. [DOI] [PubMed] [Google Scholar]
- 44. Fontaine G, Piot O, Sohal P, et al Dérivations en précordiales droites et mort subit: relations avec la dysplasie ventriculaire droite arythmogène. Arch Mal Coer 1996;86: 1323–1329. [PubMed] [Google Scholar]
- 45. Martini B, Nava A, Thiene G, et al Ventricular fibrillation without apparent heart disease: Description of six cases. Am Heart J 1989;118: 1203–1209. [DOI] [PubMed] [Google Scholar]
- 46. Corrado D, Nava A, Buja G, et al Familial cardiomyopathy underlies syndrome of right bundle branch block, ST segment elevation and sudden death. J Am Coll Cardiol 1996;27: 443–448. [DOI] [PubMed] [Google Scholar]
- 47. Brugada P, Brugada J. Right bundle brunch block, persistent ST segment elevation and sudden cardiac death: A distinct clinical and electrocardiographic syndrome. J Am Coll Cardiol 1992;20: 1391–1396. [DOI] [PubMed] [Google Scholar]
- 48. Chen Q, Kirsh GE, Zhang D, et al Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature 1998;392: 293–296. [DOI] [PubMed] [Google Scholar]
- 49. Corrado D, Basso C, Buja G, et al Right bundle branch block, right precordial ST‐segment elevation, and sudden death in young people. Circulation 2001;103: 710–717. [DOI] [PubMed] [Google Scholar]
- 50. Blomström‐Lundqvist C, Hirsch I, Olsson SB, et al Quantitative analysis of the signal averaged QRS in patients with arrhythmogenic right ventricular dysplasia. Eur Heart J 1988;9: 301–312. [DOI] [PubMed] [Google Scholar]
- 51. Mehta D, Goldman M, David O, et al Value of quantitative measurements of signal averaged electrocardiographic variables in arrhythmogenic right ventricular dysplasia: Correlation with echocardiographic right ventricular cavity dimensions. J Am Coll Cardiol 1996;28: 713–719. [DOI] [PubMed] [Google Scholar]
- 52. Nava A, Folino AF, Bauce B, et al Signal‐averaged electrocardiogram in patients with arrhythmogenic right ventricular cardiomyopathy and ventricular arrhytmias. Eur Heart J 2000;21: 58–65. [DOI] [PubMed] [Google Scholar]
- 53. Kinoshita O, Fontaine G, Rosas F, et al Time and frequency domain analyses of signal‐averaged ECG in patients with arrhhythmogenic right ventricular dysplasia. Circulation 1995;91: 715–721. [DOI] [PubMed] [Google Scholar]
- 54. Blomström‐Lundqvist C, Olsson SB, Edvardsson N. Follow‐up by repeated signal‐averaged surface QRS in patients with the syndrome of arrhythmogenic right ventricular dysplasia. Eur Heart J 1989;10(Suppl D):54–60. [DOI] [PubMed] [Google Scholar]
- 55. Leclercq JF, Coumel P. Late potentials in arrhythmogenic right ventricular dysplasia. Prevalence, diagnostic and prognostic values. Eur Heart J 1993;14: 80–83. [DOI] [PubMed] [Google Scholar]
- 56. Oselladore L, Nava A, Buja G, et al Signal‐averaged electrocardiography in familial form of arrhythmogenic right ventricular cardiomyopathy. Am J Cardiol 1995;75(Suppl E):1038–1041. [DOI] [PubMed] [Google Scholar]