

Abstract
The D,DX35E motif characteristic of retroviral integrase enzymes (INs) is expected to bind the required metal cofactors (Mg2+ or Mn2+), but direct evidence for a catalytic role has been lacking. Here we used a metal rescue strategy to investigate metal binding. We established conditions for analysis of an activity of IN, disintegration, in both Mg2+ and Mn2+, and tested IN mutants with cysteine substitutions in each acidic residue of the D,DX35E motif. Mn2+ but not Mg2+ can bind tightly to Cys, so if metal binding at the acidic residues is mechanistically important, it is expected that the Cys-substituted enzymes would be active in the presence of Mn2+ only. Of the three acidic residues, a strong metal rescue effect was obtained for D116C, a weaker rescue was seen for D64C, and no rescue was seen with E152C. Modest rescue could also be detected for D116C in normal integration in vitro. Comparison to Ser and Ala substitutions at D116 established that the rescue was selective for Cys. Further studies of the response to pH suggest that the metal cofactor may stabilize the deprotonated nucleophile active in catalysis, and studies of the response to NaCl titrations disclose an additional role for the metal cofactor in stabilizing the IN-DNA complex.
The D,DX35E phosphotransfer enzymes comprise a large family including the retroviral integrases (INs), long terminal repeat (LTR)-retrotransposon INs, and bacterial transposases (23, 36, 41, 49). The family is defined by a characteristic three-dimensional fold containing mixed alpha helix and beta sheets that brings the three acidic residues of the D,DX35E motif close to one another in space. In some of the available structures of D,DX35E catalytic domains, one or two metal atoms can be visualized bound to the D,DX35E residues (4, 10-12, 16, 18, 47, 50). Mutations of these acidic residues strongly decrease enzyme function (20, 36, 44, 45), and cross-linking studies show that parts of the nucleic acid substrates near the reactive phosphates bind near the D,DX35E motif (22, 24, 32). The central domain of IN contains the conserved D,DX35E motif, and this domain alone can catalyze a permissive relative of the normal integration reaction in vitro (disintegration), indicating that this domain contains the catalytic center (8, 46). In addition, binding of the diketo-acid inhibitors to the IN active site has been proposed to involve binding two metal atoms as well as protein functional groups (30). These data suggest a role for metal atoms bound by the D,DX35E in catalysis, but a direct functional role has not previously been documented.
A linear form of the viral DNA serves as the substrate for IN (Fig. 1A). Prior to integration, IN removes two nucleotides from the 3′ end of each LTR DNA, exposing recessed 3′ hydroxyl groups. IN then catalyzes a nucleophilic attack by the recessed 3′ hydroxyl groups on phosphodiester bonds on each target DNA strand. This results in joining of each viral DNA 3′ end to protruding 5′ ends in the target and concomitant breaking of the target phosphodiester backbone. Gap repair and connection of the remaining DNA strands are probably carried out by host DNA repair systems. The terminal cleavage and first strand transfer reactions can be modeled in vitro with purified IN (Fig. 1B) (5, 7, 9, 14, 35, 43) and the gap repair step modeled with purified cellular DNA repair enzymes (51).
FIG. 1.
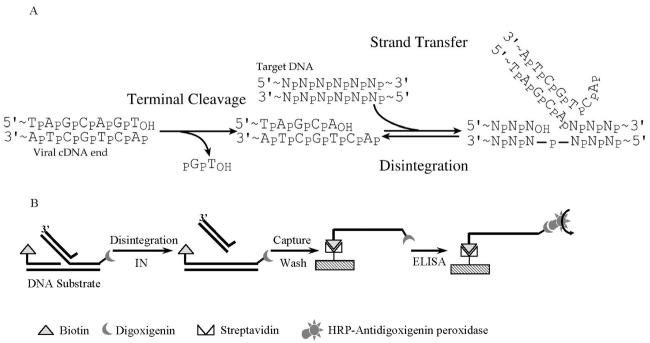
(A) DNA breaking and joining reactions involved in integration. Two nucleotides are removed from the viral cDNA end in the terminal cleavage step (left), then the recessed 3′ hydroxyl generated by cleavage is used to attack a phosphodiester in the target DNA, joining the viral DNA end and cleaving the target (right). Disintegration is the reversal of the strand transfer step. (B) Diagram of the microtiter plate assay for disintegration. The disintegration reaction covalently links a biotin-modified DNA strand to digoxigenin-modified strand. The resulting strand can be captured on a streptavidin-coated microtiter plate and quantified with an antidigoxigenin ELISA. HRP, horseradish peroxidase.
Here we have used a strategy based on metal rescue to investigate the roles of the D,DX35E acidic residues in catalysis. Mn2+ can bind tightly to sulfur atoms while Mg2+ binds much more weakly. In previous studies of ribozyme catalysis, for example, replacement of phosphate oxygens involved in metal binding with phosphorothioates has allowed rescue of catalysis by the addition of Mn2+ instead of the usual Mg2+ (17, 38). Human immunodeficiency virus type 1 (HIV-1) IN is active in the presence of either metal in vitro, so substitution of a metal-binding amino acid by Cys is expected to be tolerated if Mn2+ is the metal cofactor but not if Mg2+ is used.
Key to this work was developing conditions in which the relatively permissive disintegration assay (13) was active in Mg2+ as well as Mn2+, since this allowed direct assay of metal rescue. Previous workers demonstrated that certain IN derivatives containing Cys substitutions retained some activity in Mn2+ in vitro (26). However, the lack of conditions permitting efficient disintegration in Mg2+ prevented direct demonstration of metal rescue and consequently prevented a formal demonstration of a functional role for bound metal at the D,DX35E motif. Obtaining disintegration activity in Mg2+ required recognizing that the F185K substitution in IN, often included to improve IN solubility, in fact strongly inhibited reactions in Mg2+ (39). Here we have used these methods to evaluate the roles of metal in IN-mediated reactions.
MATERIALS AND METHODS
Oligodeoxynucleotides.
The following oligodeoxynucleotides were purchased from Integrated DNA Technologies or QIAGEN: HU520B (5′-ACTGCTAGAGATTTTCCACA), HU520T (5′-TGTGGAAAATCTCTAGCAGT), HU518T (5′-TGTGGAAAATCTCTAGCA), DIS1 (5′-ACTGCTAGAGATTTTCCACA), DIS2 (5′-TGTGGAAAATCTCTAGCAGGGGCTATGGCGTCC), DIS3 (5′-GAAAGCGACCGCGCC), and DIS4 (5′-GGACGCCATAGCCCCGGCGCGGTCGCTTTC). The following oligodeoxynucleotides were used in the microtiter assay: ST33bd (5′-digoxigenin-GACCCTTTTAGTCAGTGTGGAAAATCTCTAGCA), ST35tb (5′-ACTGCTAGAGATTTTCCACACTGACTAAAAGGGTC-3′-biotin), Dis2-bio (5′-TGTGGAAAATCTCTAGCAGGGGCTATGGCGTCC-3′-biotin), dig-Dis3 (5′-digoxigenin-GAAAGCGACCGCGCC).
To prepare 5′ 32P-labeled strand transfer substrate HU520, HU520T was 5′ end-labeled with T4 polynucleotide kinase and [γ-32P]ATP (37) and then was annealed with HU520B. Postprocessing strand transfer substrates (HU518), in which two nucleotides had been removed, were formed by annealing the mixture of HU518T and HU520B. Disintegration substrates (DIS) were formed by annealing a mixture of Dis1, Dis2, 5′ 32P-labeled Dis3, and Dis4.
To prepare strand transfer substrate ST33 for microtiter assay, ST33bd and ST35tb were mixed and annealed to form the duplex substrate. Disintegration substrate DISbd for the microtiter assay was formed by annealing the mixture of Dis1, Dis2-bio, dig-Dis3, and Dis4.
IN mutants.
Mutants D64C, D116C, E152C, D116A, D116S, C65S, and D116C+D64C were prepared by introducing each mutation into a synthetic full-length HIV-1 IN sequence that also encoded an amino-terminal His tag and thrombin cleavage site (25). The wild-type IN and all mutants were overexpressed and purified as described previously (24). All of the mutants tested displayed similar behavior during purification, suggesting that the amino acid substitutions did not drastically disrupt normal protein folding.
We also investigated the effect of the His tag on the activities of wild-type IN and several mutants. In some cases, the form with the His tag removed was slightly more active (1.3-fold, data not shown). The metal atom usage profile was identical where checked between IN proteins with and without the His tag (data not shown). The data reported are for proteins with the His tag.
Microtiter assays of IN activity.
Standard reaction mixtures contained 250 nM IN, 6 ng of substrate oligodeoxynucleotide duplex, 20 mM HEPES (pH 7.5), 5% polyethylene glycol, 5 mM MgCl2, 10 mM dithiothreitol, and 0.1 mg of bovine serum albumin/ml. The reactions were carried out at 37°C for 40 min and then stopped by the addition of single-stranded DNA buffer to make a final mixture containing 40 mM Tris-HCl (pH 8.0), 0.4 M NaCl, 20 mM EDTA, and 0.1 mg of sonicated salmon sperm DNA/ml. The mixtures were then transferred to streptavidin-coated microtiter plates and gently shaken on a platform shaker for 1 h at room temperature. Unbound DNA was removed, and wells were washed with alkali wash (30 mM NaOH, 200 mM NaCl, 1 mM EDTA) three times for 5 min, followed by one wash with Tris-EDTA buffer. Fifteen units of antidigoxigenin peroxidase Fab fragments was incubated for 1 h at 37°C. Excess antibody was removed by washing five times with buffer Tris-buffered saline-0.1% Tween 20. 3,3′-5,5′-tetramethylbenzidine was added to each well and incubated at room temperature for 15 min to allow sufficient blue product to accumulate. Reactions were stopped by adding 1 M sulfuric acid. Colorimetric analysis was carried out at 690 nm.
All reactions with 32P-labeled substrates were carried out as described previously (24).
RESULTS
Microtiter assays of disintegration and strand transfer.
To facilitate quantification of disintegration by HIV-1 IN, we established an enzyme-linked immunosorbent assay (ELISA)-based microtiter assay (Fig. 1B). The disintegration substrate used contained four oligodeoxynucleotide strands. One strand contained a biotin on the 5′ end, and another strand contained digoxigenin on the 3′ end. The disintegration reaction covalently connected the 5′ biotin-modified strand to the 3′ digoxigenin-modified strand. After denaturation, this strand could be captured on streptavidin-coated microtiter plates by binding to the 5′ biotin group. The 3′ digoxigenin group could then be detected by ELISA with a horseradish peroxidase-conjugated antidigoxigenin antibody and colorimetric horseradish peroxidase substrate. The previously published microtiter assay for strand transfer similarly used an ELISA method exploiting the reaction to join DNA strands containing 5′ biotin and 3′ digoxigenin (15, 33).
Metal rescue of active site cysteine mutants.
To identify potential metal ion binding residues in the D,DX35E motif, we purified IN mutants containing Cys in place of the acidic residues (e.g., D64C, D116C, and E152C) and analyzed their activities. Figure 2 shows the catalytic activities of D64C, D116C, and E152C in the presence of Mg2+, Mn2+, or Zn2+, assessed with the microtiter assays (Fig. 2A and B). No activity was seen in the presence of Mg2+ for the D64C, D116C, and E152C mutants in the strand transfer assay. In the presence of Mn2+, in contrast, a partial rescue could be seen. D116C showed about 15% of the activity of wild-type IN, and D64C and E152C showed about 6% of the activity of wild-type IN. Zn2+ did not support the activity of wild-type or mutant IN derivatives in any assay.
FIG. 2.
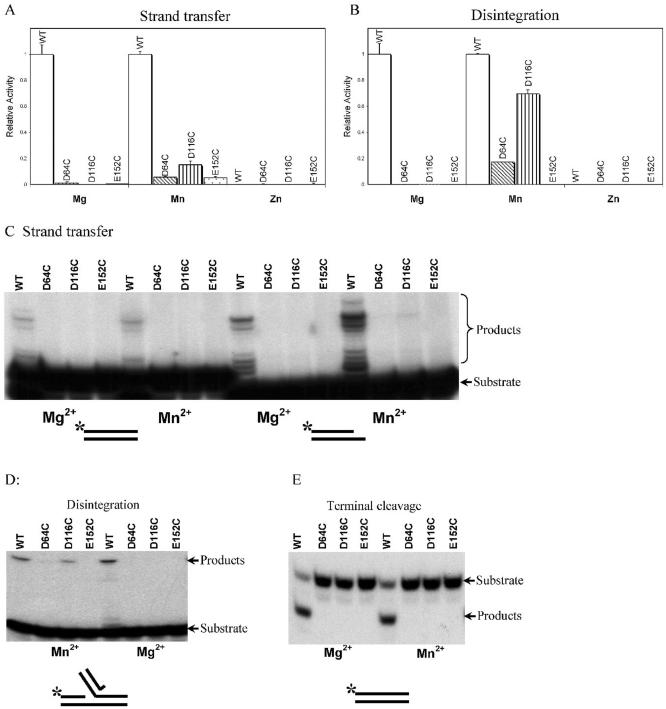
Activities of IN mutants containing substitutions of the D,DX35E motif acidic residues with Cys. In each panel, the metal used in the test reactions is indicated below the graph. (A) Activities of wild-type IN and mutants in the strand transfer assay quantified by the microtiter ELISA assay. The wild-type activity is set at 1; the mutants studied are indicated below the graph. (B) Disintegration activities of mutant and wild-type IN. Markings are as described for panel A. The values (arbitrary PhosphorImager units) were as follows: wild-type, 1874; D64C, 159; D116C, 928; E152C, 11. (C) Strand transfer activity of wild-type and Cys substitution IN mutants measured with end-labeled substrates. Left, activities with a blunt-ended LTR substrate. Right, activities with the postcleavage LTR substrate. (D) Disintegration activities measured with end-labeled substrates. (E) Terminal cleavage activities measured with end-labeled substrates.
In the disintegration assay, in the presence of Mg2+ (Fig. 2B), the three Cys substitution mutants did not display detectable activity. However, in the presence of Mn2+, the D116C mutant displayed fully 70% of the wild-type activity. Characterization of the reaction kinetics showed that the rate of product formation with the D116C mutant was within a factor of two of that with wild-type IN (data not shown). About 17% rescue can be seen in the disintegration reaction containing D64C in the presence of Mn2+. E152C was still inactive in the presence of Mn2+.
To extend the results of the microtiter assays, we used 32P-labeled oligodeoxynucleotides as substrates to investigate the terminal cleavage, strand transfer, and disintegration reactions. After the reaction, DNA products were separated by denaturing polyacrylamide gel electrophoresis and visualized by autoradiography. These reactions are sufficiently time-consuming that they are usually carried out with minimal repetition, leading to greater uncertainty in quantitation, but they do have the advantage of revealing more about the product DNA structure.
Figure 2C displays the products of strand transfer reactions containing wild-type IN, D64C, D116C, and E152C in presence of Mg2+ or Mn2+. Two different strand transfer substrates were compared: one was the blunt-ended DNA found prior to cleavage of the viral DNA termini by IN (left panel) and the other was a postprocessing substrate with the dinucleotide removed (right panel). Most Cys substitution mutants were not detectably active, as in the microtiter assay. However, slight activity could be seen with D116C in the presence of Mn2+ in assays with the postprocessing substrate, indicative of weak metal rescue. On long exposures of the autoradiogram (data not shown), the pattern of strand transfer products was similar for the wild-type IN and D116C, indicating that supplying Mn2+ by binding to Cys rather than Asp did not have a strong effect on selection of target sequences for integration.
The disintegration reaction showed the strongest rescue in the assays with end-labeled substrates, as in the microtiter assay (Fig. 2D). Considerable activity was seen with the D116C in the presence of Mn2+, but no detectable activity was seen in the presence of Mg2+. Slight activity was seen with D64C in the presence of Mn2+ but not Mg2+. E152C did not show any activity in either metal. Assays of terminal cleavage did not display any metal rescue with the Cys substitution mutants (Fig. 2E).
For the case of the D116C mutant, the robust activity seen in the presence of Mn2+ could be due either to Mn2+ binding to the Cys residue or to a lack of requirement for the D116 residue under these conditions. Several previous studies have reported that amino acid substitutions in D64, D116, or E152 resulted in a drastic reduction in IN activities (20, 36, 44, 45), consistent with the D116C activity in Mn2+ indeed resulting from metal rescue. However, to test this point under the specific conditions of our assays, we prepared D116A and D116S derivatives of IN and tested them for the above activities. We found that these mutants were inactive (data not shown), supporting the idea that the activity of D116C resulted from metal rescue.
One possible complication in interpreting the metal rescue experiments is that the residue just carboxyl terminal of D64 is a Cys. Thus, we were concerned that competing binding of Mn2+ to this residue might affect the activity of mutants containing Cys substitutions in the D,DX35E motif. For this reason, we prepared a C65S mutant and assayed its activity alone and in the presence of the other D,DX35E Cys substitutions. The C65S substitution did not alter the activity of the wild-type enzyme, nor did it affect the behavior of the D,DX35E mutants (data not shown). Evidently, C65 did not disrupt binding of metal atoms in the Cys substitution mutants.
Given that both D116C and D64C displayed some activity, it was of interest to test an IN derivative with both substitutions. However, the double mutant did not display any detectable disintegration activity (data not shown).
In summary, these data indicate that Cys substitutions in D116 and D64 can show considerable activity in the presence of Mn2+, which can bind to Cys, but not Mg2+, which does not bind Cys. Robust activity of D116C could be detected in the disintegration reaction, and weak activity could be detected in strand transfer with a postprocessing substrate. Slight activity was seen with D64C in disintegration. These data demonstrate that metal binding at D116 and probably D64 are required for catalysis by HIV-1 IN.
pH profile of disintegration by wild-type IN and the D116C mutant.
The terminal cleavage, strand transfer, and disintegration reactions are all reported to proceed by single-step transesterification reactions with either water (terminal cleavage) or the 3′ OH of DNA (strand transfer and disintegration) serving as the nucleophile (21, 26). The active nucleophiles in these reactions are expected to be the deprotonated forms, the hydroxide ion for terminal cleavage or the deprotonated 3′ oxygen atom for strand transfer and disintegration. This would be expected to confer a strong dependence of catalysis on the pH of the reaction buffer. To probe this issue, we characterized the response to pH of disintegration by wild-type IN or D116C.
Wild type IN displayed a different pH optimum in the presence of Mg2+ and Mn2+(Fig. 3). In the presence of Mg2+, IN shows a relatively sharp peak of activity at about pH 8. In the presence of Mn2+, the optimal activity is lowered to about pH 7. This 1-pH unit difference parallels the 1-unit difference in the pKa of water bound to Mg2+ versus Mn2+. The Mg2+-bound water molecule (Mg2+-H2O) has a pKa of 11.4 while Mn2+-H2O has a pKa of 10.6. Thus, the roughly 1-unit difference in optimum pH seen in the disintegration reaction (Fig. 3) suggests that the metal atom is actively involved in the deprotonation step.
FIG. 3.

Response of the disintegration reaction to different pHs in the presence of Mn2+ or Mg2+. The pH values are indicated along the bottom of the graph; the peak activity of wild-type IN in Mn2+ is set at 1.
Like wild-type IN, D116C also displayed an optimum pH in the presence of Mn2+ of about pH 7. This suggests that identical active species were used in the active site for the wild type and D116C, specifically the deprotonated 3′ oxygen. However, the disintegration activity of D116C dropped much more quickly at lower pH than the wild type in the presence of Mn2+, which we attribute to the higher pKa of cysteine (pKa = 8.5) than of aspartic acid (pKa = 4.4). These data together strengthen the idea that the active substrate for disintegration is the deprotonated 3′ oxygen atom and that one role of the bound metal atom at the active site is stabilizing this species.
Role for metal atoms in stabilizing the IN-DNA complex.
Although the above data indicate that the binding of the metal cofactor to the D,DX35E motif is required for catalysis, it is possible that metal atoms also play additional roles in IN multimerization or substrate binding, as suggested previously (2, 19, 27, 48). Here, the ability of metal atoms to stabilize IN-DNA complexes was probed by titration of NaCl in reactions. Figure 4 shows that wild-type IN responds differently in NaCl titrations in the presence of Mg2+ and Mn2+. In the terminal cleavage reaction, in the presence of Mn2+, IN is active in NaCl concentrations up to 115 mM. In the presence of Mg2+, NaCl is tolerated up to only about 75 mM. In addition, at higher NaCl concentrations in the presence of Mg2+, an additional abnormal product becomes visible, cleavage of a single nucleotide from the 3′ end instead of two nucleotides (data not shown). The higher salt concentrations likely disrupted the conformation of the IN-DNA complex, causing the substrate to become mispositioned in the active site and resulting in the inappropriate cleavage. In the disintegration reaction also, the activity persisted at higher salt concentrations in the presence of Mn2+ than Mg2+, and the D116C protein behaved similarly to the wild-type in Mn2+ (data not shown). Thus, the metal cofactor has a strong effect on the response to NaCl titration, providing evidence for a role of metal in stabilizing the IN-DNA complex.
FIG. 4.

Terminal cleavage by wild-type (WT) IN monitored with end-labeled substrates in the presence of different concentrations of NaCl and Mg2+ or Mn2+. The concentrations of NaCl in each reaction mixture are shown at the top; the metal cofactor present is shown at the bottom.
DISCUSSION
Here we used a metal rescue strategy to investigate the D,DX35E motif of HIV-1 IN. The acidic side chains expected to bind metal were replaced with Cys residues, and activity was compared in Mn2+ and Mg2+. Since Mn2+ but not Mg2+ can bind to sulfur atoms, catalysis of Cys substitution mutants in the presence of Mn2+ alone provides evidence for metal binding to that side chain during the reaction. In the most telling combination, the D116C mutant retained near-wild-type levels of disintegration activity in Mn2+ but no activity in Mg2+. D64C showed modest but clear activity in the presence of Mn2+ but not Mg2+, but E152C showed no convincing activity under any conditions. These data establish that at least D116 and probably D64 contributes to catalysis by binding metal atoms. The role of E152 is unclear (discussed below). The study of the pH dependence of the reaction in the presence of different metals argued for a direct role of the metal cofactor in catalysis (Fig. 3). A further role for metal could also be detected in stabilizing the IN-DNA complex (Fig. 4), as suggested previously (2, 19, 27, 48).
Organization of the IN active site.
Extensive mutagenic and biochemical experiments allow a detailed model for the IN active site to be considered, despite the lack of an X-ray structure of IN bound to DNA, and these help interpret the results of our metal rescue study. Cross-linking and mutagenesis studies have identified a likely path for the viral DNA end on the catalytic domain (Fig. 5A). Amino acid residue K159 cross-links to the A of the 5′-CA-3′ sequence of the viral DNA end that becomes joined to target DNA (22). E152 has been implicated by altered specificity experiments in also contacting the A of 5′-CA-3′ (27). The 2-base 5′ overhang produced by the terminal cleavage reaction can be specifically cross-linked to residues 143, 148, and 62 (22). Importantly, amino acid residue 119 has been implicated in target DNA binding in studies where mutant IN derivatives were tested for altered target sequence preferences (Fig. 5A) (1, 31). Residue 119 is located on the opposite side of the clustered acidic residues from those implicated in binding the viral DNA end. Analysis of the distribution of charged residues on the IN surface reveals tracks of relatively basic residues extending along the IN surface to the active site (Fig. 5B), marking potential docking sites for the phosphate backbone. These data allow specific positions of the viral DNA end and target DNA to be proposed (Fig. 5C). To allow the known contacts, the viral DNA end is partially unpaired for three bases. It is known that DNA distortion at the viral DNA end can promote the reaction (6, 34, 40, 42), tentatively supporting this feature of the model. The target DNA is positioned by the residue 119 contact and adjusted to maximize contact with basic amino acids on the IN surface.
FIG. 5.
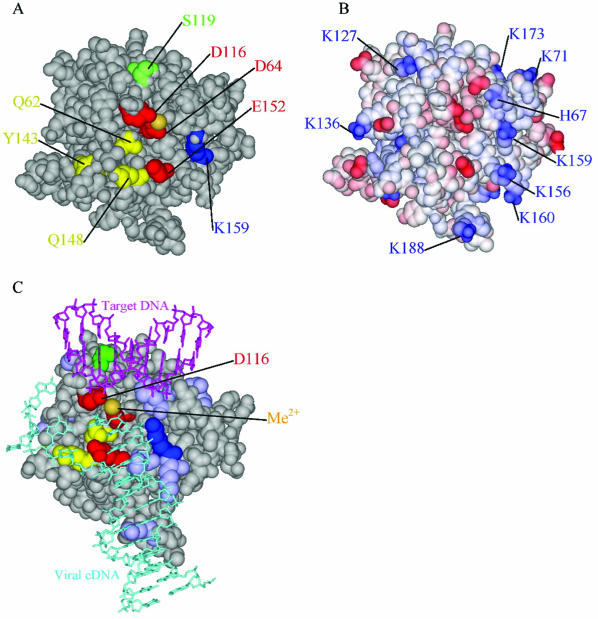
Mapping functional interactions on the IN catalytic domain. (A) Space-filling model of the IN catalytic domain (amino acid residues 50 to 212). The acidic residues of the D,DX35E motif are shown in red. Other colored residues are as follows: 119 (target DNA binding), green; 159 (LTR DNA binding), blue; 148, 143, and 62 (LTR overhang binding), yellow. (B) The IN catalytic domain with basic residues shown blue and acidic residues shown in red. (C) Candidate model of the IN catalytic domain with viral DNA and target DNA docked in locations suggested by mutagenesis and biochemical studies. The viral DNA is shown in cyan, and the target DNA is shown in magenta.
Two structures of the avian sarcoma-leukosis virus (ASLV) IN catalytic domain have been resolved with either two Zn2+ atoms or two Cd2+ atoms bound by the three acidic residues (4). For the HIV IN structures, only one Mg2+ atom has been seen bound, contacting D64 and D116 (4, 28), so only one metal atom is shown in the model. However, it seems likely that two metals are involved mechanistically, as seen in the Zn2+ and Cd2+ structures, since this would parallel many enzymes that are thought to use two metal-type mechanisms (3, 49). In the ASLV structure, the residue equivalent to D64 of HIV IN binds to two metal atoms, with one oxygen of the carboxylate side chain contacting each metal. Each metal atom is also bound by one of the other two acidic residues (the equivalents of D116 and E152) of the D,DX35E motif.
If this metal binding conformation indeed reflects the active arrangement in the HIV IN active site with two bound Mg2+ atoms, then this may explain why D64 displayed only very weak metal rescue in our study. The Cys side chain in D64C is able to bind only one metal atom at a time, so it could not make the two metal contacts simultaneously.
The D116 residue is near both the viral DNA end and the target DNA, making one contact to an Mg2+ atom. This residue displayed the greatest tolerance for the Cys substitution, but the activity was far greater in the disintegration reaction than terminal cleavage or strand transfer. In the disintegration substrate, the connectivity of the substrate holds the reactive 3′ hydroxyl near its DNA target. One possibility is that the D116 side chain normally contacts and aligns the DNA substrates as well as the metal cofactor, and upon substitution with Cys, the substrate contacts are disrupted. Use of the disintegration substrate may make up for such a deficiency. Alternatively, it is possible that the Cys side chain does not place the metal atom in the fully correct position but binding the disintegration substrate helps hold the metal in the proper location.
The role of E152 is unclear. In the X-ray structures of the HIV IN catalytic domain with Mg2+, E152 is not sufficiently close to D64 for both side chains to jointly chelate a metal atom. E152 is located on a flexible protein loop (4, 28, 29, 50), so one possibility is that in the presence of substrate E152 moves to bind metal together with D64. In the native complex, perhaps E152 contacts substrate functional groups as well as metal, as suggested from the altered specificity study (27). Another possibility is that the lack of rescue with E152C is due to the fact that the Glu side chain is longer than that of Cys, so that metal bound by E152C is not correctly positioned. It is also consistent with our data, however, that E152 does not bind metal at all but contributes to catalysis by some other mechanism.
There are many unresolved issues in the above model. Catalysis could involve a single active site or multiple active sites. The cross-linking data and mutagenesis data are all interpreted in terms of a single monomer, though it is possible that the measured interactions are contributed by multiple monomers. It is possible that the arrangement of Zn2+ and Cd2+ atoms in the ASLV active site does not accurately reflect the biological arrangement of Mn2+ and Mg2+ atoms with bound substrates. However, the data summarized in Fig. 5 do allow potential functions for the Cys substituted acidic residues to be considered.
Acknowledgments
We thank Leslie Orgel and members of the Bushman laboratory for help and comments on the manuscript.
This work was supported by grants AI34786 and AI53820 from the National Institutes of Health, the James B. Pendleton Charitable Trust, the Berger Foundation, the Margaret Morris Foundation, and Frederic and Robin Withington. K.G. acknowledges support from the UCSD Center for AIDS Research (NIAID 2 P30 AI 36214-09A1).
REFERENCES
- 1.Appa, R. S., C. G. Shin, P. Lee, and S. A. Chow. 2001. Role of the nonspecific DNA-binding region and alpha helices within the core domain of retroviral integrases in selecting target DNA sites for integration. J. Biol. Chem. 276:45848-45855. [DOI] [PubMed] [Google Scholar]
- 2.Asante-Appiah, E., and A. M. Skalka. 1997. A metal-induced conformational change and activation of HIV-1 integrase. J. Biol. Chem. 272:16196-16205. [DOI] [PubMed] [Google Scholar]
- 3.Beese, L. S., and T. A. Steitz. 1991. Structural basis for the 3′-5′ exonuclease activity of Escherichia coli DNA polymerase I: a two metal ion mechanism. EMBO J. 10:25-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bujacz, G., J. Alexandratos, A. Wlodawer, G. Merkel, M. Andrake, R. A. Katz, and A. M. Skalka. 1997. Binding of different divalent cations to the active site of avian sarcoma virus integrase and their effects on enzymatic activity. J. Biol. Chem. 272:18161-18168. [DOI] [PubMed] [Google Scholar]
- 5.Bushman, F. D., and R. Craigie. 1991. Activities of human immunodeficiency virus (HIV) integration protein in vitro: specific cleavage and integration of HIV DNA. Proc. Natl. Acad. Sci. USA 88:1339-1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bushman, F. D., and R. Craigie. 1992. Integration of human immunodeficiency virus DNA: adduct interference analysis of required DNA sites. Proc. Natl. Acad. Sci. USA 89:3458-3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bushman, F. D., and R. Craigie. 1990. Sequence requirements for integration of Moloney murine leukemia virus DNA in vitro. J. Virol. 64:5645-5648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bushman, F. D., A. Engelman, I. Palmer, P. Wingfield, and R. Craigie. 1993. Domains of the integrase protein of human immunodeficiency virus type 1 responsible for polynucleotidyl transfer and zinc binding. Proc. Natl. Acad. Sci. USA 90:3428-3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bushman, F. D., T. Fujiwara, and R. Craigie. 1990. Retroviral DNA integration directed by HIV integration protein in vitro. Science 249:1555-1558. [DOI] [PubMed] [Google Scholar]
- 10.Cai, M., R. Zheng, M. Caffrey, R. Craigie, G. M. Clore, and A. M. Gronenborn. 1997. Solution structure of the N-terminal zinc binding domain of HIV-1 integrase. Nat. Struct. Biol. 4:567-577. [DOI] [PubMed] [Google Scholar]
- 11.Chen, J. C.-H., J. Krucinski, L. J. W. Miercke, J. S. Finer-Moore, A. H. Tang, A. D. Leavitt, and R. M. Stroud. 2000. Crystal structure of the HIV-1 integrase catalytic core and C-terminal domains: a model for viral DNA binding. Proc. Natl. Acad. Sci. USA 97:8233-8238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen, Z., Y. Yan, S. Munshi, Y. Li, J. Zugay-Murphy, B. Xu, M. Witmer, P. Felock, A. Wolfe, V. Sardana, E. A. Emini, D. Hazuda, and L. C. Kuo. 2000. X-ray structure of simian immunodeficiency virus integrase containing the core and C-terminal domain (residues 50-293)-an initial glance of the viral DNA binding platform. J. Mol. Biol. 296:521-533. [DOI] [PubMed] [Google Scholar]
- 13.Chow, S. A., K. A. Vincent, V. Ellison, and P. O. Brown. 1992. Reversal of integration and DNA splicing mediated by integrase of human immunodeficiency virus. Science 255:723-726. [DOI] [PubMed] [Google Scholar]
- 14.Craigie, R., T. Fujiwara, and F. Bushman. 1990. The IN protein of Moloney murine leukemia virus processes the viral DNA ends and accomplishes their integration in vitro. Cell 62:829-837. [DOI] [PubMed] [Google Scholar]
- 15.Craigie, R., K. Mizuuchi, F. D. Bushman, and A. Engelman. 1991. A rapid in vitro assay for HIV DNA integration. Nucleic Acids Res. 19:2729-2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dyda, F., A. B. Hickman, T. M. Jenkins, A. Engelman, R. Craigie, and D. R. Davies. 1994. Crystal structure of the catalytic domain of HIV-1 integrase: similarity to other polynucleotidyl transferases. Science 266:1981-1986. [DOI] [PubMed] [Google Scholar]
- 17.Eckstein, F. 1985. Nucleoside phosphorothioates. Annu. Rev. Biochem. 54:367-402. [DOI] [PubMed] [Google Scholar]
- 18.Eijkelenboom, A. P. A. M., F. M. I. van den Ent, A. Vos, J. F. Doreleijers, K. Hard, T. Tullius, R. H. A. Plasterk, R. Kaptein, and R. Boelens. 1997. The solution structure of the amino-terminal HHCC domain of HIV-2 integrase; a three-helix bundle stabilized by zinc. Curr. Biol. 1:739-746. [DOI] [PubMed] [Google Scholar]
- 19.Ellison, V., and P. O. Brown. 1994. A stable complex between integrase and viral DNA ends mediates human immunodeficiency virus integration in vitro. Proc. Natl. Acad. Sci. USA 91:7316-7320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Engelman, A., and R. Craigie. 1992. Identification of conserved amino acid residues critical for human immunodeficiency virus type 1 integrase function in vitro. J. Virol. 66:6361-6369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Engelman, A., K. Mizuuchi, and R. Craigie. 1991. HIV-1 DNA integration: mechanism of viral DNA cleavage and DNA strand transfer. Cell 67:1211-1221. [DOI] [PubMed] [Google Scholar]
- 22.Esposito, D., and R. Craigie. 1998. Sequence specificity of viral end DNA binding by HIV-1 integrase reveals critical regions for protein-DNA interaction. EMBO J. 17:5832-5843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fayet, O., P. Ramond, P. Polard, M. F. Prere, and M. Chandler. 1990. Functional similarities between the IS3 family of bacterial insertion elements? Mol. Microbiol. 4:1771-1777. [DOI] [PubMed] [Google Scholar]
- 24.Gao, K., S. L. Butler, and F. D. Bushman. 2001. Human immunodeficiency virus type 1 integrase: arrangement of protein domains in active cDNA complexes. EMBO J. 20. [DOI] [PMC free article] [PubMed]
- 25.Gerton, J. L., and P. O. Brown. 1997. The core domain of HIV-1 integrase recognizes key features of its DNA substrates. J. Biol. Chem. 272:25809-25815. [DOI] [PubMed] [Google Scholar]
- 26.Gerton, J. L., D. Herschlag, and P. O. Brown. 1999. Stereospecificity of reactions catalyzed by HIV-1 integrase. Biochemistry 274:33480-33487. [DOI] [PubMed] [Google Scholar]
- 27.Gerton, J. L., S. Ohgi, M. Olsen, J. Derisi, and P. O. Brown. 1998. Effects of mutations in residues near the active site of human immunodeficiency virus type 1 integrase on specific enzyme-substrate interactions. J. Virol. 72:5046-5055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goldgur, Y., F. Dyda, A. B. Hickman, T. M. Jenkins, R. Craigie, and D. R. Davies. 1998. Three new structures of the core domain of HIV-1 integrase: an active site that binds magnesium. Proc. Natl. Acad. Sci. USA 95:9150-9154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Greenwald, J., V. Le, S. L. Butler, F. D. Bushman, and S. Choe. 1999. Mobility of an HIV-1 integrase active site loop is correlated with catalytic activity. Biochemistry 38:8892-8898. [DOI] [PubMed] [Google Scholar]
- 30.Grobler, J. A., K. Stillmock, B. Hu, M. Witmer, P. Felock, A. S. Espeseth, A. Wolfe, M. Egbertson, M. Bourgeois, J. Melamed, J. S. Wai, S. Young, J. Vacca, and D. J. Hazuda. 2002. Diketo acid inhibitor mechanism and HIV-1 integrase: implications for metal binding in the active site of phosphotransferase enzymes. Proc. Natl. Acad. Sci. USA 99:6661-6666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Harper, A. L., L. M. Skinner, M. Sudol, and M. Katzman. 2001. Use of patient-derived human immunodeficiency virus type 1 integrases to identify a protein residue that affects target site selection. J. Virol. 75:7756-7762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Heuer, T. S., and P. O. Brown. 1998. Photo-cross-linking studies suggest a model for the architecture of an active human immunodeficiency virus type-1 integrase-DNA complex. Biochemistry 37:6667-6678. [DOI] [PubMed] [Google Scholar]
- 33.Hwang, Y., D. Rhodes, and F. D. Bushman. 2000. Rapid microtiter assays for poxvirus topoisomerase, mammalian type IB topoisomerase, and HIV integrase: application to inhibitor isolation. Nucleic Acids Res. 28:4884-4892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Katz, R. A., P. DiCandeloro, G. Kukolj, and A. M. Skalka. 2001. Role of DNA end distortion in catalysis by avian sarcoma virus integrase. J. Biol. Chem. 276:34213-34220. [DOI] [PubMed] [Google Scholar]
- 35.Katzman, M., R. A. Katz, A. M. Skalka, and J. Leis. 1989. The avian retroviral integration protein cleaves the terminal sequences of linear viral DNA at the in vivo sites of integration. J. Virol. 63:5319-5327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kulkosky, J., K. S. Jones, R. A. Katz, J. P. G. Mack, and A. M. Skalka. 1992. Residues critical for retroviral integrative recombination in a region that is highly conserved among retroviral/retrotransposon integrases and bacterial insertion sequence transposases. Mol. Cell. Biol. 12:2331-2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maniatis, T., E. F. Fritsch, and J. Sambrook. 1982. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
- 38.Piccirilli, J. A., J. S. Vyle, M. H. Caruthers, and T. R. Cech. 1993. Metal ion catalysis in the tetrahymena ribozyme reaction. Nature 361:85-88. [DOI] [PubMed] [Google Scholar]
- 39.Podtelezhnikov, A. A., K. Gao, F. D. Bushman, and A. McCammon. 2003. Modelling HIV-1 integrase complexes based on their hydrodynamic structure. Biopolymers 68:110-120. [DOI] [PubMed] [Google Scholar]
- 40.Pruss, D., F. D. Bushman, and A. P. Wolffe. 1994. Human immunodeficiency virus integrase directs integration to sites of severe DNA distortion within the nucleosome core. Proc. Natl. Acad. Sci. USA 91:5913-5917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rowland, S.-J., and K. G. H. Dyke. 1990. Tn552, a novel transposable element from Staphylococcus aureus. Mol. Microbiol. 4:961-975. [DOI] [PubMed] [Google Scholar]
- 42.Scottoline, B. P., S. Chow, V. Ellison, and P. O. Brown. 1997. Disruption of the terminal base pairs of retroviral DNA during integration. Genes Dev. 11:371-382. [DOI] [PubMed] [Google Scholar]
- 43.Sherman, P. A., and J. A. Fyfe. 1990. Human immunodeficiency virus integration protein expressed in Escherichia coli possesses selective DNA cleaving activity. Proc. Natl. Acad. Sci. USA 87:5119-5123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van den Ent, F. M., A. Vos, and R. H. Plasterk. 1998. Mutational scan of the human immunodeficiency virus type 2 integrase protein. J. Virol. 72:3916-3924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van Gent, D. C., A. A. M. Oude Groneneger, and R. H. A. Plasterk. 1993. Identification of amino acids in HIV-2 integrase involved in site-specific hydrolysis and ancoholysis of viral DNA termini. Nucleic Acids Res. 21:3373-3377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vink, C., A. M. Oude Groeneger, and R. H. A. Plasterk. 1993. Identification of the catalytic and DNA-binding region of the human immunodeficiency virus type 1 integrase protein. Nucleic Acids Res. 21:1419-1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang, J. Y., H. Ling, W. Yang, and R. Craigie. 2001. Structure of a two-domain fragment of HIV-1 integrase: implications for domain organization in the intact protein. EMBO J. 20:7333-7343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wolfe, A. L., P. J. Felock, H. C. Hastings, C. U. Blau, and D. J. Hazuda. 1996. The role of manganese in promoting multimerization and assembly of human immunodeficiency virus type 1 integrase as a catalytically active complex on immobilized long terminal repeat substrates. J. Virol. 70:1424-1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang, W., and T. A. Steitz. 1995. Recombining the structures of HIV integrase, RuvC, and RNAse H. Curr. Biol. 3:131-134. [DOI] [PubMed] [Google Scholar]
- 50.Yang, Z. N., T. C. Mueser, F. D. Bushman, and C. C. Hyde. 2000. Crystal structure of an active two-domain derivative of Rous sarcoma virus integrase. J. Mol. Biol. 296:535-548. [DOI] [PubMed] [Google Scholar]
- 51.Yoder, K., and F. D. Bushman. 2000. Repair of gaps in retroviral DNA integration intermediates. J. Virol. 74:11191-11200. [DOI] [PMC free article] [PubMed] [Google Scholar]