

Abstract
Modulation of the activity of tumor suppressor p53 is a key event in the replication of many viruses. We have studied the function of p53 in African swine fever virus (ASFV) infection by determining the expression and activity of this transcription factor in infected cells. p53 levels are increased at early times of infection and are maintained throughout the infectious cycle. The protein is transcriptionally active, stabilized by phosphorylation, and localized in the nucleus. p53 induces the expression of p21 and Mdm2. Strikingly, these two proteins are located at the cytoplasmic virus factories. The retention of Mdm2 at the factory may represent a viral mechanism to prevent p53 inactivation by the protein. The expression of apoptotic proteins, such as Bax or active caspase-3, is also increased following ASFV infection, although the increase in caspase-3 does not appear to be, at least exclusively, p53 dependent. Bax probably plays a role in the induction of apoptosis in the infected cells, as suggested by the release of cytochrome c from the mitochondria. The significance of p21 induction and localization is discussed in relation to the shutoff of cellular DNA synthesis that is observed in ASFV-infected cells.
The tumor suppressor p53 plays a pivotal role in the cellular response to DNA damage, as it controls DNA repair, cell cycle arrest, and apoptosis (24). A major target for p53 is the p21 gene, which encodes an inhibitor of cyclin-dependent kinases (cdks). p21, which also binds to proliferating cell nuclear antigen (PCNA), thus inhibiting PCNA-dependent DNA replication (18), plays important roles in regulating cell cycle arrest or progression, DNA methylation, cell senescence, apoptosis, and differentiation. After DNA damage, this p21-dependent response allows the opportunity for DNA repair before entry of the cell into S phase. p53 also activates apoptosis through transcriptional control of Bax and probably other apoptotic inducers (32).
Under normal conditions, p53 is maintained at low levels by Mdm2 interaction and subsequent ubiquitin-dependent degradation (29), but in response to stress, such as oncogenic activation, hypoxia, DNA damage, or viral infection, p53 is activated (8). Activation of p53 can be modulated at three levels: increase of p53 expression, transformation of the protein from a latent to an active conformation through different mechanisms, and translocation of p53 to the nucleus, where it acts as a transcriptional factor (39, 40). It is well documented that DNA damage leads to phosphorylation on Ser/Thr-Pro motifs and activation of p53 (8, 23). Although the mechanisms of p53 activation are still not fully understood, several kinases have been identified that detect genotoxic stress and initiate signaling pathways through p53 phosphorylation (61). p53 works fundamentally as a transcription factor, and its nuclear import or retention is essential for its function in induction of apoptosis or growth inhibition (2).
For many viruses, replication depends on the induction of S phase, which often leads to increased levels of p53. Since activation of p53 can induce apoptosis, such viruses have evolved strategies for counteracting either p53 activation or programmed cell death. DNA tumor viruses interfere with p53 function through multiple mechanisms. The papillomavirus E6 protein interacts directly with p53, promoting its degradation (46), and Epstein-Barr virus also regulates p53 function through the BZLF1 early protein (28). Many members of the herpesvirus family have also been shown to manipulate p53 for their own purposes, using specific viral proteins: for instance, the Kaposi's sarcoma-associated herpesvirus open reading frame K8 protein (37) and the cytomegalovirus IE2 protein (21) stabilize p53, thus increasing the overall levels of p53 but inhibiting its transactivation ability (13). A similar function has also been described for the human adenovirus E4ORF 6 protein (13). On the other hand, the hepatitis B virus X protein counteracts p53 by preventing its nuclear localization (51) and DNA binding (57). Thus, the modulation of p53 seems to be an important event for the replication of many viruses, since multiple viral strategies have been developed to regulate p53 function.
In this report, we investigate the relation between African swine fever virus (ASFV) and p53 during the viral infection to gain a better understanding of the interaction of the virus with the infected cell and the mechanisms involved in apoptosis, a process associated with both in vivo and in vitro infections (7, 34). ASFV is a complex enveloped deoxyvirus of the family Asfarviridae that infects domestic and wild pigs, causing an acute and frequently fatal disease (53). The analysis of the DNA sequence of the virus has revealed the presence of several genes able to modulate host-virus interactions (59). Among these, A179L, which encodes a 19-kDa protein structurally similar to the members of the Bcl-2/Bax family, and A224L, which encodes a 27-kDa protein homologous to IAP family members, have been shown to inhibit apoptosis (1, 34, 43, 44). It has also been reported that ASFV induces apoptosis in the cell in a postbinding step, during or after virus uncoating, through the activation of caspases (7).
Here, we show that p53 expression is enhanced in Vero cells from early times after ASFV infection. p53, which is stabilized by phosphorylation and is located in the nucleus, is functionally active during infection, inducing the expression of p21 and Mdm2. Bax expression is also increased during infection, probably playing a role in the induction of apoptosis in the infected cell, as suggested by the release of cytochrome c from the mitochondria. Unexpectedly, Mdm2 and p21 are found at the cytoplasmic virus factories, which may suggest the existence of virus-mediated mechanisms to retain these proteins in the cytoplasm. The implications of this finding in relation to p53 function in the infected cell are discussed.
MATERIALS AND METHODS
Cells and virus.
Vero cells were obtained from the American Type Culture Collection and grown in Dulbecco's modified Eagle Medium supplemented with 5% newborn calf serum (Gibco) and containing 2 mM l-glutamine, 100 U of gentamicin per liter, and nonessential amino acids at 37°C in 7% CO2 in air saturated with a water vapor incubator. The Vero-adapted ASFV strain BA71V was propagated and titrated by plaque assay on Vero cells as described previously (15).
Metabolic labeling.
Cultures of Vero cells were mock infected or infected with ASFV at a multiplicity of infection of 5 PFU per cell and labeled at different times after infection with 200 μCi of [35S]methionine-cysteine/ml (1,200 Ci/mmol; Amersham) in cysteine-methionine-free medium for 2 h. The cells were washed twice with cold phosphate-buffered saline (PBS), dissociated in TNT buffer (20 mM Tris-HCl, pH 7.6, 200 mM NaCl, 1% Triton X-100) supplemented with protease inhibitor cocktail tablets (Roche), and analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) in 7 to 20% acrylamide gels as described previously (22). Proteins were detected by fluorography.
Cellular DNA synthesis.
To study the kinetics of cellular DNA synthesis, Vero cells were mock infected or infected with 5 PFU per cell and then pulse-labeled for 2 h with [methyl-3H]thymidine (Amersham) at 10 μCi/ml in Dulbecco's modified Eagle Medium. At different times after infection, the cells were washed and fractionated with NP-40 into nuclear and cytoplasmic fractions as previously described (35). The acid-insoluble radioactivity in the nuclear fractions was determined.
Western blot analysis.
Mock-infected or ASFV-infected cells were washed twice with PBS and lysed in TNT buffer supplemented with protease inhibitor cocktail tablets. The protein concentration was determined by the bicinchoninic acid spectrophotometric method (Pierce). Cell lysates (30 μg of protein) were fractionated by SDS-10% PAGE and electrophoretically transferred to an Immobilon extra membrane (Amersham), and the separated proteins were reacted with specific primary antibodies raised against p53 (sc-6243; Santa Cruz Biotechnologies), p-p53 (Ser392 and sc-7997; Santa Cruz Biotechnologies), p-p53 (Ser15 and sc-11764-R; Santa Cruz Biotechnologies), Mdm2 (sc-965; Santa Cruz Biotechnologies), active caspase-3 (BD PharMingen), Bax (sc-493; Santa Cruz Biotechnologies), and PCNA (BD PharMingen). The membranes were exposed to horseradish peroxidase-conjugated secondary antibodies (Dako), followed by chemiluminescence (ECL kit; Amersham) detection by autoradiography.
Plasmids.
The p53RE-Luc reporter plasmid contains 14 tandem repeats of the p53 consensus binding motif (Stratagene). The p21-Luc plasmid was constructed by inserting a 2.3-kb genomic Waf 1 DNA fragment (HindIII-HindIII) from plasmid WWP Luc (14) into the HindIII site of pGL3-Basic (Promega) and was a generous gift from M. Serrano (Centro Nacional de Investigaciones Onlogicas, Madrid, Spain). The Mdm2-Luc reporter plasmid contains the mouse Mdm2 P2 promoter inserted into pGL3-Basic and was a generous gift from M. Serrano.
Transfections.
Vero cells were transfected with 100 ng of each specific plasmid per 106 cells by using the Lipofectamine Plus reagent (Invitrogen) according to the manufacturer's instructions. Twenty-four hours after transfection, the cells were lysed with cell culture lysis reagent (Promega) and microcentrifuged at full speed for 5 min at 4°C, and 20 μl of each supernatant was used to determine firefly luciferase activity in a Monolight 2010 luminometer (Analytical Luminescence Laboratory). The results were expressed as the number of luminescence units after normalization of the protein concentration determined by the bicinchoninic acid method.
Immunofluorescence and confocal microscopy.
ASFV-infected cells were grown on coverslips to 2 × 105/cm2 and incubated with 1 μM Mitotracker Red CM-H2Ros (Molecular Probes) to stain mitochondria. The cultures were rinsed three times with PBS and fixed with cold 99.8% methanol (Merck) for 15 min at −20°C before they were rehydrated twice with PBS and blocked with 1% bovine serum albumin in PBS for 10 min at room temperature. The cells were incubated overnight with the specific antibodies, rinsed extensively with PBS, and then incubated with the secondary antibody (Alexa; Molecular Probes) for 1 h at room temperature in the dark. Nuclear and viral DNAs were visualized by staining the cells with DAPI. Finally, the cells were rinsed successively with PBS, distilled water, and ethanol and mounted with a drop of Mowiol on a microslide. Visualization of the stained cultures was performed under a fluorescence Axioskop2 plus (Zeiss) microscope coupled to a color charge-coupled device camera or to Confocal Microradiance (Bio-Rad) equipment. The images were digitized, processed, and organized with Metamorph, Lasershap2000 version 4, Adobe Photoshop version 7.0, Adobe Illustrator version 10, and Microsoft PowerPoint SP-2 software.
RESULTS
Induction of p53 and expression of p53 target genes following ASFV infection.
ASFV induces a blockage of cellular DNA and protein synthesis in the infected cells, as demonstrated by the data shown in Fig. 1A and B. In an attempt to investigate the mechanism involved in the shutoff induced by the virus, we determined whether p53 or p21 accumulate upon infection, since p53 is a transcriptional activator that exerts part of its cytostatic effect through the induction of p21. Extracts of ASFV-infected or mock-infected Vero cells were prepared at various times after infection, and 30 μg from each sample was separated by SDS-PAGE and blotted with specific antibodies to p53 and the p53 target p21 gene. As shown in Fig. 1C, small amounts of p53 and p21 were observed in uninfected cells but increased after infection. While the increase in p53 could be detected 3 h postinfection (p.i.), a delay of several hours in the expression of p21 was observed, as would be expected for a p53-dependent gene. The expression of the proapoptotic Bax gene, another p53-dependent gene, was also examined in infected cells. As can be seen in Fig. 1C, the kinetics of Bax expression closely parallels that of p21, with maximal levels at 13 h p.i., a time at which activation of caspases is observed (7). On the other hand, since PCNA is involved in DNA replication, acting as a processivity factor for DNA polymerases, we also examined the expression of this protein in infected cells. In contrast to p21, the levels of PCNA do not change at any time after infection (Fig. 1C) with respect to those detected in mock-infected cells.
FIG. 1.
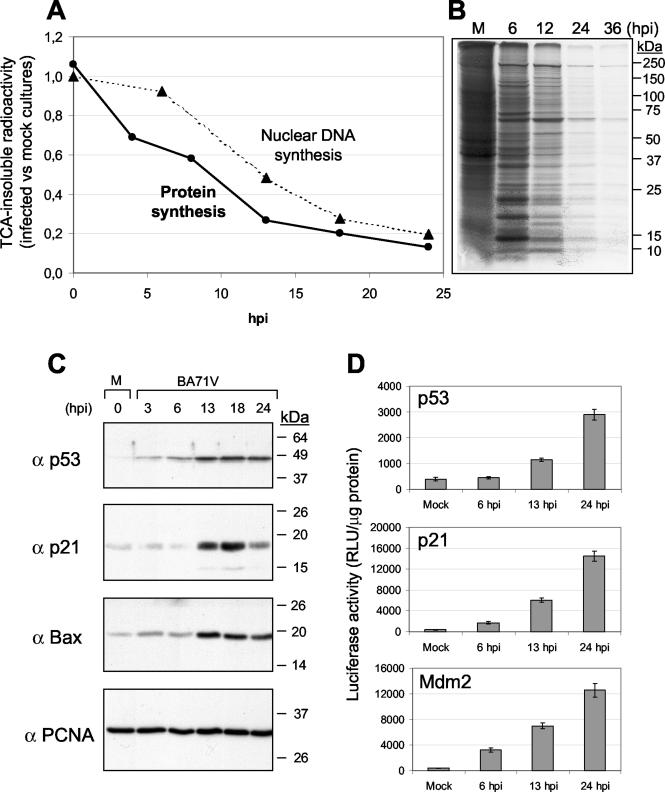
ASFV inhibition of cellular DNA and protein synthesis is concomitant with the expression of cellular proteins that control the cell cycle. (A) Cellular DNA synthesis was determined as described in Materials and Methods. The radioactivity incorporated at the indicated times of infection into nuclear fractions from 2 × 106 cells (the number of cells in the culture at the time of infection) was measured. Protein synthesis was determined by measuring the incorporation of [35S]methionine into acid-insoluble material from 2 × 106 cells. For both DNA and protein synthesis, the ratio between ASFV-infected and mock-infected cultures is represented in the graph. TCA, trichloroacetic acid. (B) Cultures of Vero cells (5 × 105) were mock infected (M) or infected with ASFV and labeled at different times after infection with 200 μCi of [35S]methionine-cysteine/ml in cysteine-methionine-free medium for 2 h. The cells were resuspended in 250 μl of TNT buffer, and cell extracts (10 μl) were analyzed by SDS-PAGE. Proteins were detected by fluorography. (C) Kinetics of p53, p21, Bax, and PCNA expression analyzed by Western blotting in mock-infected (M) or ASFV-infected Vero cells. Times after infection are indicated above the lanes. (D) The p53 transcriptional activity was analyzed in mock-infected or ASFV-infected Vero cells by transient transfection of specific plasmids containing the luciferase gene under the control of p53-binding motif or of p21 or Mdm2 promoters, as indicated in Materials and Methods. The transfected cells were incubated overnight and then infected with BA71V at a multiplicity of infection of 5 PFU/cell. At the indicated times after infection, cell extracts were prepared, and the luciferase activity was measured in a luminometer. RLU, relative light units.
To better understand the role of p53, we studied the p53 transcriptional function during ASFV infection. To achieve this, we transfected into Vero cells a plasmid that contains the luciferase reporter gene cloned under the control of 14 p53-binding DNA sequences. Twelve hours after transfection, the cells were mock infected or infected with ASFV, and at different times postinfection, cell extracts were prepared and luciferase activity was measured in a luminometer. As shown in Fig. 1D, luciferase activity increased during infection, indicating that p53 was expressed in an active form.
As mentioned in the introduction, p53 functions primarily as a transcription factor that triggers cell cycle arrest or apoptosis by inducing a growing number of proteins (55), among them p21 and Mdm2 (40). To examine whether Mdm2 and p21 gene promoters are activated during ASFV infection, we used Vero cells transiently transfected with plasmids containing the luciferase reporter gene cloned under the control of the p21 or Mdm2 gene promoter. Twelve hours after transfection, the cells were mock infected or infected with ASFV, and at different times postinfection, cell extracts were prepared and analyzed for luciferase activity. As shown in Fig. 1D, the activities of Mdm2 and p21 gene promoters increased from 13 h p.i., consistent with the increased activity of p53.
Phosphorylation of p53 and localization at the nucleus.
Since the nuclear localization of p53 is related to its activation (25), we next examined the subcellular localization and the phosphorylation state of p53 upon ASFV infection. The p53 protein is known to be phosphorylated at multiple sites located in both the N- and C-terminal regions. The amino terminus of p53, which contains the transcriptional regulatory domain, has eight phosphorylation sites (4). The carboxyl terminus contains the nuclear import and export signals, the tetramerization domain, and four phosphorylation sites (serines 315, 376, 378, and 392). Although it is not clear whether this elevated p53 phosphorylation is directly involved in the p53 nucleocytoplasmic translocation, it has been speculated that phosphorylation may affect p53 localization and activation. Thus, phosphorylation of p53 appears to enhance p53 transcriptional activity and to render it more resistant to inhibition by Mdm2 (48).
To investigate the localization of p53 following ASFV infection, we incubated mock-infected or ASFV-infected Vero cells with the specific anti-p53 antibody. Using confocal microscopy, we could demonstrate that p53 shows a nuclear localization from 4 h p.i. (Fig. 2A); the nuclear signal is enhanced at later times of infection.
FIG. 2.
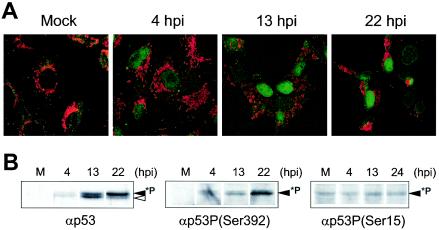
Subcellular localization and phosphorylation of p53 during ASFV infection. (A) Mock-infected or ASFV-infected Vero cells were labeled with Mitotracker (red) and anti-p53 antibody (green) and then examined by confocal microscopy. Shown are images corresponding to one of three independent experiments performed. (B) Nuclear extracts (30 μg) from mock-infected or ASFV-infected Vero cells were subjected to Western blot analysis using an anti-p53 (αp53) antibody (left) or anti-p-p53 antibodies that recognize phosphorylated p53 in Ser392 (center) or in Ser15 (right).
To determine whether the nuclear p53 is phosphorylated, nuclear and cytosolic fractions were obtained from ASFV-infected cells, at different times after infection. The fractions were standardized for protein concentration (30 μg) and analyzed by Western blotting using the specific anti-p53 antibody. As shown in Fig. 2B, two closely migrating bands were recognized by the anti-p53 antibody in the nuclear fraction; the upper band probably represented the phosphorylated form of p53. The intensity of the upper band could also be observed to increase at late times after infection. To confirm that this band corresponded to a phosphorylated form of p53, we incubated the nuclear fractions with a specific antibody against p-p53, Ser392, which recognizes the specific site 392 when phosphorylated. As shown, a band with a molecular weight similar to that of the upper band previously detected with the anti-p53 serum was observed when the anti-p-p53 antibody was used. From this result, we conclude that this band corresponds to phosphorylated p53, which appears in the nuclear fraction from the infected cells and not in mock-infected cells or in cytoplasmic fractions from ASFV-infected cells (not shown). It should be noted that phosphorylation of p53 at Ser392 is an early response to a wide range of stress-inducing conditions (3, 9). Phosphorylation at Ser15 is also a response to DNA damage and viral infections and has been shown to alleviate inhibition of p53 by Mdm2 (48, 52). In the case of ASFV, as shown in Fig. 2B, it seems that phosphorylation at this residue does not increase after ASFV infection, suggesting that this mechanism is not used by the virus to inhibit p53-Mdm2 interaction.
Localization of p21, Mdm2, and PCNA.
We have demonstrated above that p21, which inhibits cell cycle progression by binding to G1 cyclin-cdk and PCNA (14, 19), is accumulated after ASFV infection. This raises the possibility that the shutoff of cellular DNA synthesis, which occurs in the infected cells by an unknown mechanism, could be due to the increased levels of p21. Since the cell cycle-inhibitory activity of p21 is intimately correlated with its nuclear localization (5), we investigated the subcellular localization of p21 after ASFV infection by confocal microscopy of mock-infected or ASFV-infected Vero cells, using a specific anti-p21 antibody. Surprisingly, the protein does not localize in the nucleus but accumulates in the cytoplasm of infected cells after 13 h p.i. (Fig. 3A), mainly associated with discrete structures that correspond to the virus factories, as indicated by staining of the viral DNA with DAPI (Fig. 3B). In relation to this finding, it should be mentioned that a short form of p21 is present in the cytoplasm of UV-treated cells (38) and that phosphorylation of p21 by Akt also leads to its cytoplasmic localization (62). However, when the phosphorylation state of p21 was examined by Western blotting using an antibody specific for phosphorylated Ser146, we could not detect an increase in the phosphorylated form of p21 after infection (data not shown).
FIG. 3.
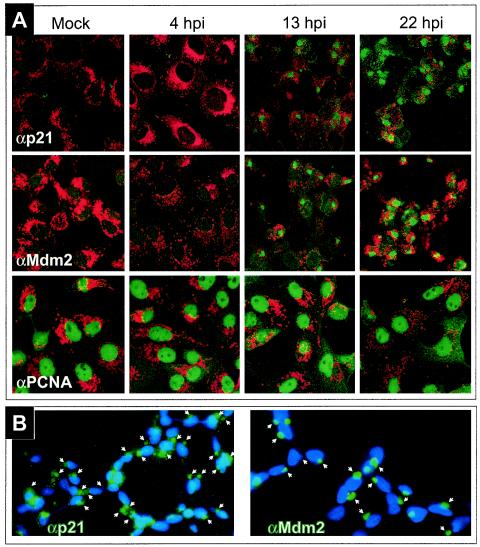
Subcellular localization of p21, Mdm2, and PCNA. (A) Mock-infected or ASFV-infected Vero cells were labeled with Mitotracker (red) and anti-p21, anti-Mdm2, or anti-PCNA antibodies (green) and then examined by confocal microscopy. Shown are images corresponding to one of three independent experiments performed. (B) ASFV-infected Vero cells were stained at 13 h p.i. with DAPI (blue) and anti-p21 or anti-Mdm2 antibodies (green) and examined by fluorescence microscopy. The arrows point to virus factories identified by DAPI staining.
It is known that in normal, nonstressed cells, p53 has a very short half-life due to a feedback mechanism in which the Mdm2 protein plays a key role. Wild-type p53 acts as a transcriptional activator of the Mdm2 gene; in turn, Mdm2 has the ability to interact with p53 and to function as a ubiquitin E3 ligase that promotes the conjugation of p53 to polyubiquitin and its posterior degradation by the proteasome (49). It has also been established that the nuclear localization of Mdm2 is required to inhibit the transcriptional function of p53 (33). Therefore, we investigated the localization of Mdm2 during the viral cycle. For this, we analyzed Vero cells at different times after ASFV infection by confocal microscopy using a specific anti-Mdm2 antibody. Representative fields are shown in Fig. 3A. As can be seen, in mock-infected cells and at early times after infection (4 h p.i.), the localization of Mdm2 is mainly nuclear. However, as the infection progresses, Mdm2 accumulates in the cytoplasm, focalized in areas corresponding to virus factories. As in the case of p21, the colocalization of Mdm2 with these virus structures was confirmed by DAPI staining (Fig. 3B) and supported by the fact that the label corresponding to p21 or Mdm2 in cells infected at 13 and 22 h p.i. appeared to be surrounded by mitochondria (Fig. 3A), as expected for ASFV factories (45).
In contrast with the cytoplasmic localization of p21, PCNA, a protein whose expression does not change at any time after infection, was localized in the nucleus in both mock-infected and ASFV-infected cells (Fig. 3A), a distribution consistent with the function of the protein in DNA replication.
These results might suggest that Mdm2 and p21 are sequestered in the cytoplasm by an unknown viral mechanism. This would impair p53-Mdm2 interaction, allowing the stabilization of p53 and its function as a transcriptional factor. On the other hand, the cytoplasmic localization of p21 indicates that the function of the protein, if any, during ASFV infection would not be mediated through its interaction with PCNA.
Role of p53 in the induction of apoptosis during ASFV infection.
ASFV induction of apoptosis in an early step of infection has been described (7). ASFV-infected Vero cells showed several apoptotic signs after 13 h p.i., including the release of cytochrome c from the mitochondria to the cytosolic fraction as detected by immunoblot analysis. We have now examined this release of cytochrome c by confocal microscopy using specific anti-cytochrome c antibody. Figure 4A shows that in mock-infected cells, cytochrome c colocalizes with the mitochondria. The release of cytochrome c from the organelle starts 4 h after infection, and from 13 to 22 h p.i., most of the protein is localized throughout the cytoplasm, including the virus factory. This free cytochrome induces the activation of effector caspases and therefore the accumulation of the catalytic fragment of caspase-3, which is observed in the infected cells (7, 34).
FIG. 4.
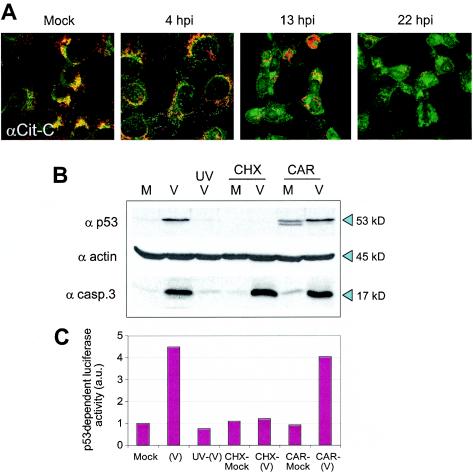
ASFV-induced cytochrome c release and expression of p53 and caspase-3 activation under different infection conditions. (A) Mock-infected or ASFV-infected Vero cells were labeled with Mitotracker (red) and anti-cytochrome c (αCit-C) antibody (green) and then examined by confocal microscopy. Colocalization of mitochondria and cytochrome c yields a yellow signal. Shown are images corresponding to one of two independent experiments performed. (B) Extracts from mock-infected cells (M), ASFV-infected cells (V), cells incubated with UV-inactivated virus (UVV), cells mock infected (CHX/M) or infected (CHX/V) in the presence of CHX (10 μg/ml), and cells mock infected (CAR/M) or infected (CAR/V) in the presence of CAR (40 μg/ml) were analyzed by Western blotting using specific antibodies against p53 (α p53) or caspase-3 (α casp.3). A representative experiment is shown. (C) Luciferase activity in Vero cells transfected with the p53RE-Luc reporter plasmid. Twelve hours after transfection, the cells were mock infected or infected under the conditions described for panel B, and cell extracts were prepared and analyzed for p53 transcriptional activity. The results of a representative experiment are shown. a.u., arbitrary units.
To study the involvement of p53 in the induction of apoptosis and the activation of caspase-3, Vero cells, previously incubated or not with 10 μg of cycloheximide (CHX)/ml, were infected with 5 PFU of BA71V/cell, and at 18 h p.i., cell extracts were prepared and examined by Western blotting using specific antibodies against p53 and the active fragment of caspase-3. Figure 4B shows that p53 is not present in ASFV-infected cells in the presence of CHX; however, the active fragment (17 kDa) of caspase-3 is detected under these conditions, indicating that protein synthesis is not required for the activation of caspase-3, and more relevant, that the activation of caspase-3 is, at least in part, a p53-independent event.
In order to determine the roles of viral proteins in the induction of both p53 expression and caspase-3 activity, we treated the cells with cytosine arabinoside (CAR), which inhibits virus DNA replication and late protein synthesis but allows the synthesis of early proteins. In the presence of CAR (40 μg/ml), two bands, corresponding to nonphosphorylated (lower) and phosphorylated (upper) p53, could be detected in mock-infected cells (Fig. 4B). Upon infection with ASFV, only the phosphorylated form was observed. These results demonstrate that the synthesis of ASFV late proteins is not required for the induction of the phosphorylated form of p53. The 17-kDa band corresponding to active caspase-3 is also detected under these experimental conditions. On the other hand, and as also shown in Fig. 4B, no p53 is detected when the cells are infected with UV-irradiated virus, suggesting that early viral-gene expression is needed for p53 induction. In order to see whether the p53 expressed in the presence of CAR was transcriptionally active, we transiently transfected Vero cells with a plasmid containing the reporter luciferase gene under the control of specific DNA-binding sequences for p53. After 12 h, the cells were mock infected or infected under the different conditions described in the legend to Fig. 4B. The results presented in Fig. 4C show that the activity of p53 induced in cells infected in the presence of CAR was similar to that found in the absence of the inhibitor. No significant p53 activity was detected in cells infected with UV-irradiated virus or in the presence of CHX.
DISCUSSION
The present study provides a number of novel observations pertaining to the regulation and functional role of p53 during ASFV infection. It has been shown that ASFV induces apoptosis after 13 h in the cell, a time at which viral morphogenesis is well under way (7). It was also shown that several ASFV genes are involved in the inhibition of apoptosis using different mechanisms (34, 43, 44), thus demonstrating that programmed cell death during ASFV infection is a tightly regulated process in which the action of inducers is balanced by the expression of antiapoptotic genes. However, less information is available on the process of ASFV-induced apoptosis in terms of the cellular pathways and specific proteins involved. In a recent report (7), it was shown that the apoptotic signal in ASFV-infected Vero cells is activated in the absence of virus replication and before early ASFV protein synthesis by an intracellular pathway probably triggered during the process of virus uncoating.
Here, we report the expression and the transcriptional activity of p53 observed in ASFV-infected Vero cells from 4 h p.i., with a maximum between 18 and 24 h p.i. Using confocal microscopy, we detected p53 in the nuclei of the cells at the same times after infection. Studies of the phosphorylation state of p53 in cytoplasmic and nuclear extracts from ASFV-infected cells demonstrated that phosphorylated p53 accumulates in the nucleus during ASFV infection, thus confirming the rapid import and retention in the nucleus of p53 detected by confocal fluorescence. This supports a role for phosphorylation in p53 nucleocytoplasmic translocation, as has been previously suggested (25). Working principally as a transcriptional factor, p53 nuclear import or retention is essential for its normal function in cell cycle inhibition or apoptosis induction.
Interestingly, Mdm2, an inhibitor of p53 activity, is found in the cytoplasm of ASFV-infected cells, localized within cellular structures corresponding to virus factories. This finding raises the possibility that Mdm2 could be retained in the cytoplasm by a specific virus-mediated mechanism, thus hindering the entry of Mdm2 into the nucleus and its interaction with p53. This would imply that the virus needs to maintain active p53 for its replication. Further studies are required to confirm this attractive hypothesis.
The elucidation of the mechanisms by which p53 is activated when cells are subjected to stress has been an area of intense research (2). It has been well documented that several types of DNA damage, including double-strand breaks in DNA and the presence of DNA repair intermediates (11, 24, 26, 39), induce a rapid increase in the level of p53 and its activation as a transcription factor, which are proportional to the extent of DNA damage. In relation to this, it is interesting that most viruses that induce p53, such as simian virus 40 (16, 30), polyomavirus (12), papillomavirus (36, 46), adenovirus (27, 41), and herpesvirus (37), replicate in the nucleus of the infected cell. ASFV also has an early nuclear stage of replication (17). It is possible that repair events that could play a role in triggering the activation of p53 might occur during this nuclear phase.
It is also possible that p53 activation might be related to the induction of free radicals, probably released from the mitochondria during ASFV infection. Reactive oxygen species-mediated mitochondrion-dependent pathways are suggested as major pathomechanisms contributing to nuclear DNA damage, which eventually may result in increased levels of p53 (56). In this connection, previous data from our laboratory showed the presence of large clusters of mitochondria located in proximity to the virus factories (45). Interestingly, ASFV infection also promotes the induction of the mitochondrial stress-responsive proteins p74 and cpn 60 concomitant with a change in the organelle toward the morphology characteristic of actively respiring mitochondria (45). These activated mitochondria might release reactive oxygen species that could account for the high p53 levels that are maintained at late times postinfection.
On the other hand, p53 can be activated in the absence of DNA damage. For instance, it has been proposed that certain viral oncoproteins can also increase the level of p53 (10). This activation, at least in some situations, is mediated by an increase in the levels of expression of ARF (58), an antagonist of Mdm2 function that forms complexes with p53 and Mdm2 and both stabilizes p53 and enhances its transactivation activity (47). However, this is not the more plausible explanation for the induction of p53 during ASFV infection, because we did not detect ARF expression in ASFV-infected cells by Western blotting (data not shown).
On the other hand, it is worthy of note that some ASFV proteins have been reported to interact with NFκB (42) and NFAT (31) signaling pathways. In relation to this, it has been established (60) that sanglifehrin A, a member of the immunophilin-binding ligands, activates p53 transcription primarily through the activation of NFκB by activating IκB kinase. Our previous finding that A224L, an ASFV protein homologous to the members of the IAP family, induces the activation of NFκB (44) provides still another possible explanation for p53 induction in ASFV-infected cells.
Several lines of evidence support the hypothesis that activation of p53 induces either cell cycle arrest or apoptotic cell death under different stress conditions (50, 55). The cytostatic effect of p53 is mediated by transcriptional activation of p21, which inhibits the cdks (19) and binds to PCNA, thus inhibiting PCNA-dependent DNA replication (18). During ASFV infection, we detected both p21 expression and activation of the p21 gene promoter, although the significance of these events in relation to the shutoff of cellular DNA synthesis is not clear at present. Since we have not been able to demonstrate an interaction between p21 and PCNA, in accordance with the finding that the two proteins are localized in different compartments of the infected cells, a possible effect of p21 on the observed cell cycle arrest cannot be mediated by this mechanism. An alternative possibility would be that p21 could bind and retain in the cytoplasm certain cdk-cyclin complexes that may be involved in the initiation of DNA replication (20). Work is now in progress to explore this possibility.
The mechanisms by which p53 induces apoptosis in cells are still unresolved (54). The apoptotic effect of p53 is mediated by transcriptional activation of the Bax gene and other BH-3 domain-containing genes, which encode downstream activators of apoptosis (6). Regarding the role of p53 in the induction of the apoptosis observed during ASFV infection, the increase detected in the expression of Bax is noteworthy. Furthermore, using confocal microscopy, we could also detect the release of cytochrome c from the mitochondria from 13 h p.i., an event directly related to the overexpression of Bax. The released cytochrome c is mainly found in the virus factory, which may be simply a consequence of the accumulation of the mitochondria around this area at these times postinfection. However, a weaker but significant signal is also detected in the cytoplasm outside the assembly sites. This cytosolic cytochrome c could be responsible for the caspase activation that is observed in infected cells at these times postinfection (34). These findings support the existence of a p53-initiated apoptotic pathway in ASFV-infected cells, although other apoptotic mechanisms may operate as well. This is suggested by the finding that, when the cells are infected in the presence of CHX, caspase-3 activation is observed, but no p53 expression is detected. This could indicate that the apoptosis induced by ASFV is, at least in part, a p53-independent process. Similar results in terms of activation of caspase-3 were obtained during ASFV infection in the presence of CAR, although p53 was active under these conditions. These results, together with the observation that p53 is not detected when the cells are infected with UV-irradiated virus, suggest that p53 induction is probably related to the expression of early proteins of ASFV and that, as with many other viruses, ASFV can also kill cells through the activation of p53-independent mechanisms.
Acknowledgments
We thank M. J. Bustos and R. Ramos for excellent technical assistance.
This work was supported by grants from the Ministerio de Ciencia y Tecnología (BMC2000-1485 and AGL2002-10220-E) and the European Commission (QLRT-2000-02216) and by an institutional grant from the Fundación Ramón Areces. C. Hurtado was a fellow of Fundación Ramón Areces.
REFERENCES
- 1.Afonso, C. L., J. G. Neilan, G. F. Kutish, and D. L. Rock. 1996. An African swine fever virus Bc1-2 homolog, 5-HL, suppresses apoptotic cell death. J. Virol. 70:4858-4863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Agarwal, M. L., W. R. Taylor, M. V. Chernov, O. B. Chernova, and G. R. Stark. 1998. The p53 network. J. Biol. Chem. 273:1-4. [DOI] [PubMed] [Google Scholar]
- 3.Appella, E., and C. W. Anderson. 2001. Post-translational modifications and activation of p53 by genotoxic stresses. Eur. J. Biochem. 268:2764-2772. [DOI] [PubMed] [Google Scholar]
- 4.Appella, E., and C. W. Anderson. 2000. Signaling to p53: breaking the posttranslational modification code. Pathol. Biol. (Paris) 48:227-245. [PubMed] [Google Scholar]
- 5.Asada, M., T. Yamada, H. Ichijo, D. Delia, K. Miyazono, K. Fukumuro, and S. Mizutani. 1999. Apoptosis inhibitory activity of cytoplasmic p21(Cip1/WAF1) in monocytic differentiation. EMBO J. 18:1223-1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Attardi, L. D., S. W. Lowe, J. Brugarolas, and T. Jacks. 1996. Transcriptional activation by p53, but not induction of the p21 gene, is essential for oncogene-mediated apoptosis. EMBO J. 15:3693-3701. [PMC free article] [PubMed] [Google Scholar]
- 7.Carrascosa, A. L., M. J. Bustos, M. L. Nogal, G. Gonzalez de Buitrago, and Y. Revilla. 2002. Apoptosis induced in an early step of African swine fever virus entry into Vero cells does not require virus replication. Virology 294:372-382. [DOI] [PubMed] [Google Scholar]
- 8.Chehab, N. H., A. Malikzay, E. S. Stavridi, and T. D. Halazonetis. 1999. Phosphorylation of Ser-20 mediates stabilization of human p53 in response to DNA damage. Proc. Natl. Acad. Sci. USA 96:13777-13782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cuddihy, A. R., A. H. Wong, N. W. Tam, S. Li, and A. E. Koromilas. 1999. The double-stranded RNA activated protein kinase PKR physically associates with the tumor suppressor p53 protein and phosphorylates human p53 on serine 392 in vitro. Oncogene 18:2690-2702. [DOI] [PubMed] [Google Scholar]
- 10.Das, S., W. S. El-Deiry, and K. Somasundaram. 2003. Regulation of the p53 homolog p73 by adenoviral oncogene E1A. J. Biol. Chem. 278:18313-18320. [DOI] [PubMed] [Google Scholar]
- 11.Dasika, G. K., S. C. Lin, S. Zhao, P. Sung, A. Tomkinson, and E. Y. Lee. 1999. DNA damage-induced cell cycle checkpoints and DNA strand break repair in development and tumorigenesis. Oncogene 18:7883-7899. [DOI] [PubMed] [Google Scholar]
- 12.Dey, D., J. Dahl, S. Cho, and T. L. Benjamin. 2002. Induction and bypass of p53 during productive infection by polyomavirus. J. Virol. 76:9526-9532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dobner, T., N. Horikoshi, S. Rubenwolf, and T. Shenk. 1996. Blockage by adenovirus E4orf6 of transcriptional activation by the p53 tumor suppressor. Science 272:1470-1473. [DOI] [PubMed] [Google Scholar]
- 14.el-Deiry, W. S., T. Tokino, V. E. Velculescu, D. B. Levy, R. Parsons, J. M. Trent, D. Lin, W. E. Mercer, K. W. Kinzler, and B. Vogelstein. 1993. WAF1, a potential mediator of p53 tumor suppression. Cell 75:817-825. [DOI] [PubMed] [Google Scholar]
- 15.Enjuanes, L., A. L. Carrascosa, M. A. Moreno, and E. Vinuela. 1976. Titration of African swine fever (ASF) virus. J. Gen. Virol. 32:471-477. [DOI] [PubMed] [Google Scholar]
- 16.Fromm, L., W. Shawlot, K. Gunning, J. S. Butel, and P. A. Overbeek. 1994. The retinoblastoma protein-binding region of simian virus 40 large T antigen alters cell cycle regulation in lenses of transgenic mice. Mol. Cell. Biol. 14:6743-6754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garcia-Beato, R., M. L. Salas, E. Vinuela, and J. Salas. 1992. Role of the host cell nucleus in the replication of African swine fever virus DNA. Virology 188:637-649. [DOI] [PubMed] [Google Scholar]
- 18.Gibbs, E., Z. Kelman, J. M. Gulbis, M. O'Donnell, J. Kuriyan, P. M. Burgers, and J. Hurwitz. 1997. The influence of the proliferating cell nuclear antigen-interacting domain of p21(CIP1) on DNA synthesis catalyzed by the human and Saccharomyces cerevisiae polymerase delta holoenzymes. J. Biol. Chem. 272:2373-2381. [DOI] [PubMed] [Google Scholar]
- 19.Harper, J. W., G. R. Adami, N. Wei, K. Keyomarsi, and S. J. Elledge. 1993. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell 75:805-816. [DOI] [PubMed] [Google Scholar]
- 20.Henneke, G., S. Koundrioukoff, and U. Hubscher. 2003. Multiple roles for kinases in DNA replication. EMBO Rep. 4:252-256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jault, F. M., J. M. Jault, F. Ruchti, E. A. Fortunato, C. Clark, J. Corbeil, D. D. Richman, and D. H. Spector. 1995. Cytomegalovirus infection induces high levels of cyclins, phosphorylated Rb, and p53, leading to cell cycle arrest. J. Virol. 69:6697-6704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680-685. [DOI] [PubMed] [Google Scholar]
- 23.Lambert, P. F., F. Kashanchi, M. F. Radonovich, R. Shiekhattar, and J. N. Brady. 1998. Phosphorylation of p53 serine 15 increases interaction with CBP. J. Biol. Chem. 273:33048-33053. [DOI] [PubMed] [Google Scholar]
- 24.Levine, A. J. 1997. p53, the cellular gatekeeper for growth and division. Cell 88:323-331. [DOI] [PubMed] [Google Scholar]
- 25.Liang, S. H., and M. F. Clarke. 2001. Regulation of p53 localization. Eur. J. Biochem. 268:2779-2783. [DOI] [PubMed] [Google Scholar]
- 26.Lu, H., Y. Taya, M. Ikeda, and A. J. Levine. 1998. Ultraviolet radiation, but not gamma radiation or etoposide-induced DNA damage, results in the phosphorylation of the murine p53 protein at serine-389. Proc. Natl. Acad. Sci. USA 95:6399-6402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martin, M. E., and A. J. Berk. 1998. Adenovirus E1B 55K represses p53 activation in vitro. J. Virol. 72:3146-3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mauser, A., S. Saito, E. Appella, C. W. Anderson, W. T. Seaman, and S. Kenney. 2002. The Epstein-Barr virus immediate-early protein BZLF1 regulates p53 function through multiple mechanisms. J. Virol. 76:12503-12512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mayo, L. D., and D. B. Donner. 2002. The PTEN, Mdm2, p53 tumor suppressor-oncoprotein network. Trends Biochem. Sci. 27:462-467. [DOI] [PubMed] [Google Scholar]
- 30.McCarthy, S. A., H. S. Symonds, and T. Van Dyke. 1994. Regulation of apoptosis in transgenic mice by simian virus 40 T antigen-mediated inactivation of p53. Proc. Natl. Acad. Sci. USA 91:3979-3983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miskin, J. E., C. C. Abrams, L. C. Goatley, and L. K. Dixon. 1998. A viral mechanism for inhibition of the cellular phosphatase calcineurin. Science 281:562-565. [DOI] [PubMed] [Google Scholar]
- 32.Miyashita, T., S. Krajewski, M. Krajewska, H. G. Wang, H. K. Lin, D. A. Liebermann, B. Hoffman, and J. C. Reed. 1994. Tumor suppressor p53 is a regulator of bcl-2 and bax gene expression in vitro and in vivo. Oncogene 9:1799-1805. [PubMed] [Google Scholar]
- 33.Momand, J., G. P. Zambetti, D. C. Olson, D. George, and A. J. Levine. 1992. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 69:1237-1245. [DOI] [PubMed] [Google Scholar]
- 34.Nogal, M. L., G. Gonzalez de Buitrago, C. Rodriguez, B. Cubelos, A. L. Carrascosa, M. L. Salas, and Y. Revilla. 2001. African swine fever virus IAP homologue inhibits caspase activation and promotes cell survival in mammalian cells. J. Virol. 75:2535-2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ortin, J., and E. Vinuela. 1977. Requirement of cell nucleus for African swine fever virus replication in Vero cells. J. Virol. 21:902-905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pan, H., and A. E. Griep. 1994. Altered cell cycle regulation in the lens of HPV-16 E6 or E7 transgenic mice: implications for tumor suppressor gene function in development. Genes Dev. 8:1285-1299. [DOI] [PubMed] [Google Scholar]
- 37.Park, J., T. Seo, S. Hwang, D. Lee, Y. Gwack, and J. Choe. 2000. The K-bZIP protein from Kaposi's sarcoma-associated herpesvirus interacts with p53 and represses its transcriptional activity. J. Virol. 74:11977-11982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Poon, R. Y., and T. Hunter. 1998. Expression of a novel form of p21Cip1/Waf1 in UV-irradiated and transformed cells. Oncogene 16:1333-1343. [DOI] [PubMed] [Google Scholar]
- 39.Prives, C., and P. A. Hall. 1999. The p53 pathway. J. Pathol. 187:112-126. [DOI] [PubMed] [Google Scholar]
- 40.Qian, H., T. Wang, L. Naumovski, C. D. Lopez, and R. K. Brachmann. 2002. Groups of p53 target genes involved in specific p53 downstream effects cluster into different classes of DNA binding sites. Oncogene 21:7901-7911. [DOI] [PubMed] [Google Scholar]
- 41.Querido, E., J. G. Teodoro, and P. E. Branton. 1997. Accumulation of p53 induced by the adenovirus E1A protein requires regions involved in the stimulation of DNA synthesis. J. Virol. 71:3526-3533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Revilla, Y., M. Callejo, J. M. Rodriguez, E. Culebras, M. L. Nogal, M. L. Salas, E. Vinuela, and M. Fresno. 1998. Inhibition of nuclear factor κB activation by a virus-encoded IκB-like protein. J. Biol. Chem. 273:5405-5411. [DOI] [PubMed] [Google Scholar]
- 43.Revilla, Y., A. Cebrian, E. Baixeras, C. Martinez, E. Vinuela, and M. L. Salas. 1997. Inhibition of apoptosis by the African swine fever virus Bcl-2 homologue: role of the BH1 domain. Virology 228:400-404. [DOI] [PubMed] [Google Scholar]
- 44.Rodriguez, C. I., M. L. Nogal, A. L. Carrascosa, M. L. Salas, M. Fresno, and Y. Revilla. 2002. African swine fever virus IAP-like protein induces the activation of nuclear factor kappa B. J. Virol. 76:3936-3942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rojo, G., M. Chamorro, M. L. Salas, E. Vinuela, J. M. Cuezva, and J. Salas. 1998. Migration of mitochondria to viral assembly sites in African swine fever virus-infected cells. J. Virol. 72:7583-7588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Scheffner, M., B. A. Werness, J. M. Huibregtse, A. J. Levine, and P. M. Howley. 1990. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 63:1129-1136. [DOI] [PubMed] [Google Scholar]
- 47.Sherr, C. J. 2001. The INK4a/ARF network in tumour suppression. Nat. Rev. Mol. Cell Biol. 2:731-737. [DOI] [PubMed] [Google Scholar]
- 48.Shieh, S. Y., M. Ikeda, Y. Taya, and C. Prives. 1997. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell 91:325-334. [DOI] [PubMed] [Google Scholar]
- 49.Sionov, R. V., E. Moallem, M. Berger, A. Kazaz, O. Gerlitz, Y. Ben-Neriah, M. Oren, and Y. Haupt. 1999. c-Abl neutralizes the inhibitory effect of Mdm2 on p53. J. Biol. Chem. 274:8371-8374. [DOI] [PubMed] [Google Scholar]
- 50.Soussi, T., Y. Legros, R. Lubin, K. Ory, and B. Schlichtholz. 1994. Multifactorial analysis of p53 alteration in human cancer: a review. Int. J. Cancer 57:1-9. [DOI] [PubMed] [Google Scholar]
- 51.Takada, S., N. Kaneniwa, N. Tsuchida, and K. Koike. 1997. Cytoplasmic retention of the p53 tumor suppressor gene product is observed in the hepatitis B virus X gene-transfected cells. Oncogene 15:1895-1901. [DOI] [PubMed] [Google Scholar]
- 52.Takaoka, A., S. Hayakawa, H. Yanai, D. Stoiber, H. Negishi, H. Kikuchi, S. Sasaki, K. Imai, T. Shibue, K. Honda, and T. Taniguchi. 2003. Integration of interferon-alpha/beta signalling to p53 responses in tumour suppression and antiviral defence. Nature 424:516-523. [DOI] [PubMed] [Google Scholar]
- 53.Vinuela, E. 1985. African swine fever virus. Curr. Top. Microbiol. Immunol. 116:151-170. [DOI] [PubMed] [Google Scholar]
- 54.Vogelstein, B., D. Lane, and A. J. Levine. 2000. Surfing the p53 network. Nature 408:307-310. [DOI] [PubMed] [Google Scholar]
- 55.Vousden, K. H., and X. Lu. 2002. Live or let die: the cell's response to p53. Nat. Rev. Cancer 2:594-604. [DOI] [PubMed] [Google Scholar]
- 56.Wang, X., D. Michael, G. de Murcia, and M. Oren. 2002. p53 Activation by nitric oxide involves down-regulation of Mdm2. J. Biol. Chem. 277:15697-15702. [DOI] [PubMed] [Google Scholar]
- 57.Wang, X. W., K. Forrester, H. Yeh, M. A. Feitelson, J. R. Gu, and C. C. Harris. 1994. Hepatitis B virus X protein inhibits p53 sequence-specific DNA binding, transcriptional activity, and association with transcription factor ERCC3. Proc. Natl. Acad. Sci. USA 91:2230-2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xirodimas, D., M. K. Saville, C. Edling, D. P. Lane, and S. Lain. 2001. Different effects of p14ARF on the levels of ubiquitinated p53 and Mdm2 in vivo. Oncogene 20:4972-4983. [DOI] [PubMed] [Google Scholar]
- 59.Yanez, R. J., J. M. Rodriguez, M. L. Nogal, L. Yuste, C. Enriquez, J. F. Rodriguez, and E. Vinuela. 1995. Analysis of the complete nucleotide sequence of African swine fever virus. Virology 208:249-278. [DOI] [PubMed] [Google Scholar]
- 60.Zhang, L. H., H. D. Youn, and J. O. Liu. 2001. Inhibition of cell cycle progression by the novel cyclophilin ligand sanglifehrin A is mediated through the NFκB-dependent activation of p53. J. Biol. Chem. 276:43534-43540. [DOI] [PubMed] [Google Scholar]
- 61.Zheng, H., H. You, X. Z. Zhou, S. A. Murray, T. Uchida, G. Wulf, L. Gu, X. Tang, K. P. Lu, and Z. X. Xiao. 2002. The prolyl isomerase Pin1 is a regulator of p53 in genotoxic response. Nature 419:849-853. [DOI] [PubMed] [Google Scholar]
- 62.Zhou, B. P., Y. Liao, W. Xia, B. Spohn, M. H. Lee, and M. C. Hung. 2001. Cytoplasmic localization of p21Cip1/WAF1 by Akt-induced phosphorylation in HER-2/neu-overexpressing cells. Nat. Cell Biol. 3:245-252. [DOI] [PubMed] [Google Scholar]